

Abstract
Aberrant activation in fibroblast growth factor signaling has been implicated in the development of various cancers, including squamous cell lung cancer, squamous cell head and neck carcinoma, colorectal and bladder cancer. Thus, fibroblast growth factor receptors (FGFRs) present promising targets for novel cancer therapeutics. Here, we evaluated the activity of a novel pan‐FGFR inhibitor, rogaratinib, in biochemical, cellular and in vivo efficacy studies in a variety of preclinical cancer models. In vitro kinase activity assays demonstrate that rogaratinib potently and selectively inhibits the activity of FGFRs 1, 2, 3 and 4. In line with this, rogaratinib reduced proliferation in FGFR‐addicted cancer cell lines of various cancer types including lung, breast, colon and bladder cancer. FGFR and ERK phosphorylation interruption by rogaratinib treatment in several FGFR‐amplified cell lines suggests that the anti‐proliferative effects are mediated by FGFR/ERK pathway inhibition. Furthermore, rogaratinib exhibited strong in vivo efficacy in several cell line‐ and patient‐derived xenograft models characterized by FGFR overexpression. The observed efficacy of rogaratinib strongly correlated with FGFR mRNA expression levels. These promising results warrant further development of rogaratinib and clinical trials are currently ongoing (ClinicalTrials.gov Identifiers: NCT01976741, NCT03410693, NCT03473756).
Keywords: fibroblast growth factor receptor, rogaratinib, cancer, colorectal cancer, preclinical models
Short abstract
What's new?
Deregulated fibroblast growth factor receptor (FGFR) signaling is involved in tumorigenesis and cancer progression. Here, the authors report on a novel pan‐FGFR inhibitor, rogaratinib, that potently and highly selectively prevents the activity of FGFRs 1, 2, 3, and 4. Rogaratinib inhibits cell proliferation in various FGFR‐addicted cancers in vitro, including colon, lung, and bladder cancer. Rogaratinib also exhibits strong in vivo efficacy in several cell line‐ and patient‐derived xenograft models characterized by FGFR mRNA overexpression with good tolerability. Altogether, these data warrant the further development of rogaratinib for treatment of cancers with FGFR alterations, and clinical trials are currently ongoing.
Abbreviations
- AKT
protein kinase B
- ANOVA
analysis of variance
- CISH
chromogenic in situ hybridization
- CSF1R
colony‐stimulating factor 1 receptor
- DAG
diacylglycerol
- EGFR
epidermal growth factor receptor
- ERK
extracellular signal‐regulated kinase
- ESCC
esophageal squamous cell carcinoma
- FGF
fibroblast growth factor
- FGFR
fibroblast growth factor receptor
- FRS2
fibroblast growth factor receptor substrate 2
- HNSCC
head and neck squamous cell carcinoma
- HUVEC
human umbilical vein endothelial cell
- IP3
inositol triphosphate
- Kd
dissociation constant
- MAPK
mitogen‐activated protein kinase
- NGS
next‐generation sequencing
- PDGFR
platelet‐derived growth factor receptor
- PI3K
phosphatidylinositol 3‐kinase
- PKB
protein kinase B
- PKC
protein kinase C
- PLC
phospholipase C
- RTK
receptor tyrosine kinase
- SCC
squamous cell carcinoma
- SOC
standard of care
- STAT
signal transducer and activator of transcription
- Tie2
tyrosine kinase with Ig and EGF homology domains‐2
- TKI
tyrosine kinase inhibitor
- TR‐FRET
time‐resolved fluorescence resonance energy transfer
- VEGF‐A
vascular endothelial growth factor A
- VEGFR
vascular endothelial growth factor receptor, IC50 half maximal inhibitory concentration
Introduction
Fibroblast growth factor receptor (FGFR) signaling plays an essential role in angiogenesis and normal tissue homeostasis during embryonic development and in adulthood.1 FGFR family members 1–4 (FGFR1‐4) represent transmembrane receptor tyrosine kinases (RTKs) which bind extracellular ligands of the fibroblast growth factor (FGF) family to induce a complex intracellular signaling cascade in a cell‐dependent manner. This includes the MAPK/ERK pathway, which regulates several distinct mitogen‐activated protein kinases (MAPK) including the extracellular signal–regulated kinases ERK1/2, the PI3K/AKT pathway including the serine/threonine‐specific protein kinase AKT1 (also known as protein kinase B, PKB), and the activation of phospholipase Cγ (PLCγ) which triggers the inositol‐1,4,5‐trisphosphate (IP3)–Ca2+ and diacylglycerol (DAG)–protein kinase C (PKC) signaling cascades.2 As a consequence of its key role in cell proliferation, survival, differentiation and migration, deregulated FGFR signaling is involved in tumorigenesis and cancer progression in various human cancers. A recent study of 4,853 solid tumors found that genetic alterations in FGFR encoding genes were present in 7.1% of human malignancies including urothelial (32%), breast (18%), endometrial (13%), lung (squamous) (13%) and ovarian (9%) cancer.3 Various genetic FGFR alterations have been linked to different types of cancers in several studies. FGFR1 gene amplification, for example, has been associated with squamous cell lung cancer4, 5, 6, 7 as well as with head and neck squamous cell carcinoma (HNSCC),8, 9 while bladder cancers have been shown to frequently harbor FGFR3‐activating mutations.10, 11, 12, 13 As the majority of FGFR aberrations identified to date are related to gain‐of‐function, targeting these cancers with FGFR inhibitors has become an attractive strategy.14 , 15
As oncogenic alterations need to be translated into functional activation in order to be able to drive tumor development or growth, analysis of FGFR expression levels could complement mutational analysis in identifying FGFR‐dependent tumors more specifically. Indeed, recent data support the role of expression levels of FGFR subtypes in cancer development and sensitivity to drug‐induced inhibition of FGFR signaling.16, 17
Several tyrosine kinase inhibitors (TKI) have been identified or optimized to inhibit FGFRs and some of them have entered clinical development. However, most of these molecules are not selective FGFR inhibitors and show significant activity against other RTKs such as vascular endothelial growth factor receptors (VEGFRs), platelet‐derived growth factor receptor (PDGFR), and c‐Kit, which may limit their potential utility due to side effects associated with the inhibition of those kinases.18
Here, we report on the preclinical pharmacology of a novel, highly selective small molecule inhibitor of pan‐FGFR kinase activity, rogaratinib (BAY 1163877), which specifically inhibits FGFR1, 2, 3 and 4 in vitro and in vivo. In fact, selective inhibition of FGFR signaling by rogaratinib results in robust monotherapy efficacy in cancers with alterations leading to FGFR overexpression. Furthermore, rogaratinib demonstrates additive activity with standard‐of‐care (SOC) therapy in lung and colorectal cancer models. Importantly, rogaratinib is well‐tolerated in multiple rodent cancer models. Altogether, these data support the further development of rogaratinib as a new anticancer therapy in currently ongoing clinical studies which include patient stratification based on the overexpression of FGFR1‐3 mRNA in relevant tumor biopsies (ClinicalTrials.gov Identifiers: NCT01976741, NCT03410693, NCT03473756).
Materials and Methods
Compounds
Rogaratinib, BAY 1163877 (4‐{[4‐amino‐6‐(methoxymethyl)‐5‐(7‐methoxy‐5‐methyl‐1‐benzothiophen‐2‐yl)pyrrolo[2,1‐f][1,2,4]triazin‐7‐yl]methyl}piperazin‐2‐one) was identified and synthesized at Bayer AG (Germany; Fig. 1).19 In in vitro studies, 100% DMSO was used as solvent. For certain in vivo studies the HCl salt of rogaratinib (BAY 1213802) was used. No difference in efficacy was detected between the two forms, and all data shown are calculated for the free base of the compound. The in vivo vehicle used for both BAY 1163877 and BAY 1213802 was 10% ethanol, 40% Solutol® HS 15, 50% water at pH 4 (HCl). Carboplatin (Teva, USA), docetaxel (Jiangsu Hengrui Medicine Co., Ltd, China) or paclitaxel (Iapharm GmbH, Germany), formulated in NaCl solution (9 g/l), were used in the in vivo combination studies.
Figure 1.
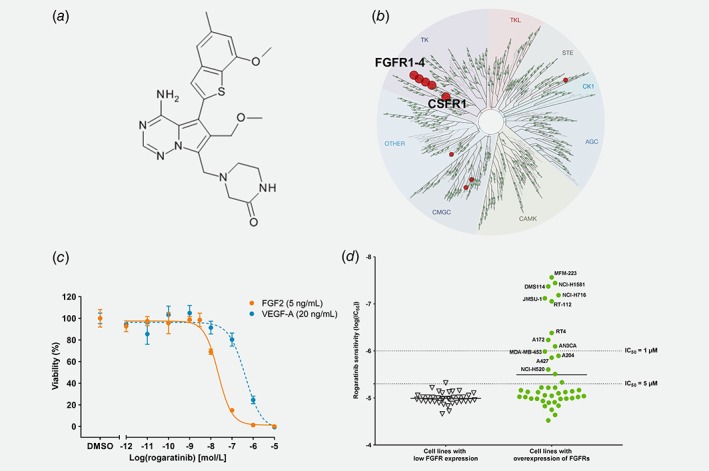
Structure and in vitro activity of rogaratinib. (a) Chemical structure of rogaratinib (BAY 1163877). (b) TREEspot™ interaction map of KINOMEscan™ assay results with 100 nM rogaratinib. (c) Viability of HUVEC cells stimulated with growth factors FGF2 (5 ng/mL) or VEGF‐A (20 ng/mL) in minimal medium after a 72‐h treatment with increasing concentrations of rogaratinib. (d) Correlation of FGFR mRNA levels (sum of Z‐scores for subtypes with Z‐score > 1, derived from CCLE database) and sensitivity (IC50 of inhibition of cell proliferation expressed as LogIC50) to rogaratinib in a panel of cancer cell lines covering various cancer types. Dotted lines indicate IC50 levels.
Tumor cell lines and primary cells
MDA‐MB‐453, NCI‐H1581, NCI‐H520, UM‐UC‐3, NCI‐H716 and DMS‐114 cells were obtained from American Type Culture Collection (ATCC, USA); RT‐112 and JMSU1 cells were obtained from Leibniz Institute Collection of Microorganisms and Cell Cultures (DSMZ, Germany) and MFM‐223 cells were obtained from the European Collection of Authenticated Cell Cultures (ECACC, UK). C51 cells were provided by Dario Neri (Philogen, Italy). Human umbilical vein endothelial cells (HUVEC) were acquired from CellSystems Biotechnologie Vertrieb GmbH (Germany).
All cell lines were regularly subjected to identity verification by DNA fingerprinting at DSMZ to ensure correct use of the cell lines. Mycoplasma contamination tests were done in‐house using MycoAlert (Lonza, USA).
Animals
All animal experiments were performed under European Animal Welfare Law and approved by local authorities, or the guidelines approved by the Institutional Animal Care and Use Committee (IACUC) of CrownBio (China) following the guidance of the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC). Studies with patient‐derived tumors were conducted at Oncotest GmbH (Germany) or CrownBio with written informed consent from each patient and the approval of local ethical committees.
Subcutaneous tumor growth was monitored by measuring tumor volume (0.5 × length × width2) using a caliper. Animal body weight was monitored as one indicator of treatment‐related toxicity. Measurement of tumor volume and body weight was performed two to three times per week. Individual animals were sacrificed when showing signs of toxicity (e.g. >20% body weight loss) or when tumors reached a maximum size of ~1,000 mm3. At study termination, the animals were sacrificed by decapitation under narcosis, tumors were excised and tumor wet weights were determined. T/C (treatment/control) ratios were calculated using the mean final tumor volume (T/C volume) and tumor weight values (T/C weight) respectively. In addition, treatment responses were evaluated by means of clinically used RECIST20 criteria; response rates were calculated as the percentage of animals with a complete or partial response.
Biochemical assays for inhibition of FGFR1‐4
The inhibition of FGFR1, FGFR3, FGFR4 and 22 other protein kinases by rogaratinib was assessed in‐house by TR‐FRET‐based in vitro kinase assays which quantify the phosphorylation of the peptide Biotin‐Ahx‐AAEEEYFFLFAKKK‐NH2 by recombinant GST‐fusion proteins of the cytoplasmic kinase domains (purchased from Proqinase, Germany). The FGFR subtype profile and kinase selectivity of rogaratinib were assessed for half maximal inhibitory concentration (IC50) in radiometric kinase assays at Merck Millipore (USA) (IC50Profiler™, KinaseProfiler™) and for binding affinity in active site‐directed competition binding assays at DiscoverX (KINOMEscan™, USA); in addition the safety profile of rogaratinib has been analyzed in a SafetyScreen at Eurofins Panlabs (77 Enzyme and Radioligand Binding assays) all according to manufacturers’ instructions.
Assessment of inhibition of cell proliferation
The capacity of rogaratinib to selectively inhibit the proliferation of cancer cells addicted to the FGFR pathway was tested in a cell‐based assay in an Oncoline™ (NTRC, Netherlands) panel of cell lines21 and in‐house in NCI‐H1581, DMS‐114, MFM‐223, MDA‐MB‐453, C51, NCI‐H716, NCI‐H520, RT‐112, JMSU1 and UM‐UC‐3 cancer cells following a 72‐h drug exposure in normal medium. Cell viability was determined using CellTiter‐Glo® (CTG, cat#G7571/2/3, Promega, USA) according to manufacturer's instructions and as described in the Supporting Information Methods.
The ability of rogaratinib to inhibit FGF‐related signaling pathways and the selectivity of rogaratinib against VEGFRs were assessed in human umbilical vein endothelial cells (HUVEC). The inhibition of cell proliferation by rogaratinib was measured in HUVECs cultured in FGF2‐ or VEGF‐A‐containing minimal medium after a 72‐h exposure to the compound as described in the Supporting Information Methods.
Evaluation of molecular mechanism‐of‐action in rogaratinib‐sensitive cancer cell lines and tumor tissue
The capacity of rogaratinib to inhibit main downstream MAPK effector pathways in cancer cell lines and tumor tissue was evaluated by Western blot analysis of ERK phosphorylation after rogaratinib treatment. The inhibition of FGF2‐induced phosphorylation of ERK or AKT in DMS‐114 small cell lung cancer, MFM‐223 breast cancer and C51 colon cancer cells was measured using MSD‐ELISA technology as described in the Supporting Information Methods.
Antitumor efficacy in cell line and patient‐derived xenograft models
Details of all in vivo studies are provided in Supporting Information Methods. Briefly, female BALB/cJ mice (18–21 g, 5–6 weeks) from Janvier (France) were injected subcutaneously (s.c.) in the left inguinal region with 1 × 106 syngeneic C51 colon cancer cells as suspension in 0.1 mL 50% Matrigel/50% medium. The mice were randomized (n = 9–10/group) on day 6 (average tumor volume 104–117 mm3) and treated orally (p.o.) for 13 days with vehicle or rogaratinib (25, 50 or 75 mg/kg QD, or 25 mg/kg, BID). For PK/PD studies, blood samples were collected at study termination 1, 3, 7 and 24 h after the last dose.
Female immunocompromised Hsd:RH‐Foxn1 rnu rats (80–100 g, 5–6 weeks) from Harlan (Netherlands) were injected s.c. with 1 × 106 C51 mouse colon cancer cells as described above. The rats were randomized (n = 8/group) on day 7 (average tumor volume 212 mm3) and treated for 12 days with vehicle or rogaratinib (10 or 50 mg/kg, QD, p.o.).
Female immunocompromised NMRInu/nu mice from Janvier were injected s.c. with 3 × 106 NCI‐H716 human colon cancer cells, randomized (n = 8/group) on day 22 (average tumor volume 63–99 mm3) and treated for 17 days with vehicle or rogaratinib (35, 50 or 65 mg/kg, BID, p.o.). Female NMRInu/nu nude mice from Janvier were injected s.c. with 2 × 106 DMS‐114 human SCLC cells, randomized (n = 8/group) on day 13, and treated for 10 days with vehicle, rogaratinib, chemotherapy or combinations.
Female nu/nu mice (Vr:NU‐Foxn1nu) from Vital River (China) were implanted s.c. with 2–3 mm diameter LU299 human lung cancer fragments. The mice were randomized (n = 10/group) on day 22 and treated for a period of 33 days with vehicle, rogaratinib, docetaxel or the combination.
Female NMRInu/nu mice from Janvier were implanted s.c. with 2 mm diameter LXFL1121 human NSCLC tumor fragments. The mice were randomized (n = 10/group) on day 26 and treated for a period of 22 days with vehicle, rogaratinib, docetaxel or combinations.
Ex vivo analyses of FGFR1‐4 mRNA levels and gene copy numbers in tumors
For analysis of mRNA expression and gene copy numbers, total RNA and genomic DNA, respectively, were isolated from xenograft tumors and subjected to Nanostring analysis or TaqMan assays (Life Biotechnologies, USA), respectively, as described in the Supporting Information Methods.
Statistical analyses
Descriptive statistics for all groups in in vivo studies were performed on tumor volumes and weights. All analyses were performed using linear models estimated with generalized least squares and with separate variance parameters for each study group. Mean comparisons between the treatment and control groups were performed using the estimated linear model and corrected for family‐wise error rate using Dunnett's method. p Values <0.05 were considered statistically significant. All analyses were performed using the statistical programming language R, version 3.4.3 (https://www.R-project.org/).
Results
Rogaratinib selectively inhibits FGFR1‐4
Rogaratinib (BAY 1163877) is a small molecule RTK inhibitor designed to reversibly occupy the ATP‐binding pocket of the kinase domain of FGFRs 1–4 (Fig. 1 a).19 KINOMEscan™ profiling of 468 kinase targets demonstrated that rogaratinib is highly selective for FGFRs. Of 403 non‐mutant kinases, only 4 or 18 additional kinases besides FGFR1‐4 resulted in competition binding >65% at 100 nM or 1 μM rogaratinib, respectively (Fig. 1 b, Supporting Information Table S1). Rogaratinib potently binds to all four FGFR subtypes with Kd values of 1.6, 5.0, 7.8 and 7.6 nM for FGFRs 1, 2, 3 and 4, respectively. In radiometric kinase activity assays, rogaratinib inhibited FGFRs 1, 2, 3 and 4 with high potency in a similar concentration range with IC50 values of 1.8, <1, 9.2 and 1.2 nM. Binding and inhibition was highly selective vs. related receptor tyrosine kinases. For instance, the IC50 for kinase inhibition of CSF1R, Tie2 or VEGFR3 was 166 nM, 1,300 nM or 130 nM, respectively, although KINOMEscan™ profiling showed strong inhibition of rogaratinib binding to CSF1R at 100 nM (Fig. 1 b, Supporting Information Table S1). The evaluation of rogaratinib in a set of 77 non‐kinase targets (PanLabs Safety screen) showed only 3 significant hits supporting the high selectivity (Supporting Information Table S2).
The Binding affinity to VEGFRs was about 100‐fold lower (Kd (VEGFR1) = 190 nM, Kd (VEGFR3) = 150 nM) compared to FGFR1 (Kd = 1.6 nM).
Selectivity vs. VEGFRs was also observed in cellular assays. In endothelial cells (HUVEC), rogaratinib selectively and potently inhibited FGF2‐stimulated growth with an IC50 of 16 nM (−logIC50 7.8 ± 0.3; n = 10) while the IC50 for inhibition of VEGF‐A‐stimulated growth was 28‐fold higher with 453 nM (−logIC50 6.3 ± 0.3; n = 11) (Fig. 1 c).
Rogaratinib has selective anti‐proliferative activity against FGFR‐overexpressing cancer cell lines
The anti‐proliferative effect of rogaratinib was assessed in the Oncoline™ panel comprised of 66 human cancer cell lines of different histological origins and in additional cell lines reported to be sensitive to FGFR inhibition, including NCI‐H520,22 UMUC3,23 NCI‐H1581,24 NCI‐H716,25 DMS‐114,26 MFM‐223,27 JMSU‐128 and RT‐11229 cells using a standard CellTiter‐Glo® proliferation assay. The IC50 values ranged from 27 nM to >30 μM with nine cell lines showing IC50 values <1 μM. Proliferation inhibition was compared to available cellular FGFR expression and genomic mutation data (Supporting Information Table S3). The sensitivity of the cell lines towards rogaratinib correlated with overall FGFR mRNA expression levels, which were described as the sum of Z‐scores of the individual FGFR subtypes (median‐based Z‐score,30). All cell lines with IC50 < 1 μM showed elevated expression for at least one FGFR subtype (Z‐score > 1) (Fig. 1 d). Cancer cell lines with IC50 > 1 μM either lacked overexpression (Z‐score < 1) of FGFRs or harbored known activating mutations and/or gene amplifications in genes described to confer resistance to FGFR inhibition, e.g. KRAS, NRAS, HRAS, PIK3CA, BRAF, PTEN or EGFR genes, or resisted FGFR inhibition by rogaratinib through a currently unknown mechanism (Supporting Information Table S3).
Rogaratinib inhibits FGFR phosphorylation and downstream signaling in human and murine cancer cell lines
Inhibition of the FGFR pathway by rogaratinib treatment was analyzed by Western blot in rogaratinib‐sensitive as well as ‐insensitive cell lines from various cancer types. In FGFR4‐overexpressing MDA‐MB‐453 breast cancer cells, rogaratinib inhibited auto‐phosphorylation of FGFR4 in a concentration‐dependent manner and caused inhibition of downstream signaling by preventing ERK1/2 phosphorylation in the same concentration range (Fig. 2 a). Similarly, rogaratinib effectively inhibited auto‐phosphorylation of FGFR2 as well as phosphorylation of ERK1/2 at 100 nM in NCI‐H716 colon cancer cells (Fig. 2 b). Furthermore, potent inhibition of ERK1/2 phosphorylation by rogaratinib was observed in the rogaratinib‐sensitive RT‐112 bladder cancer and NCI‐H1581 lung cancer cell lines while UM‐UC‐3 bladder cancer and NCI‐H520 lung cancer cells were less sensitive to rogaratinib‐mediated inhibition of downstream phosphorylation, consistent with the differentiated anti‐proliferative activity of rogaratinib in these cell lines (Fig. 2 c, Supporting Information Table S3). In FGFR1‐overexpressing DMS‐114 lung cancer and FGFR2‐overexpressing MFM‐223 breast cancer cells, rogaratinib potently inhibited downstream signaling through ERK1/2 and AKT as determined by MSD‐ELISA technology. The IC50 values for inhibition of ERK and AKT phosphorylation for DMS‐114 (20 and 26 nM, respectively) and MFM‐223 cells (11 and 19 nM, respectively) corresponded well with the IC50 values for proliferation inhibition (42 nM for DMS‐114; 27 nM for MFM‐223). Rogaratinib also inhibited phosphorylation of ERK1/2 in FGFR1‐overexpressing murine C51 colon cancer cells with an IC50 of 280 nM (Fig. 2 c). In these cell lines the potency of inhibition of phosphorylation of ERK1/2 correlates directly with the sensitivity of these cell lines to treatment with rogaratinib (Supporting Information Table S4). Furthermore, the inhibition of ERK1/2 phosphorylation by rogaratinib treatment shown ex vivo (Supporting Information Fig. S1) corresponds well with the rogaratinib sensitivity of the respective cell lines (Supporting Information Table S3).
Figure 2.
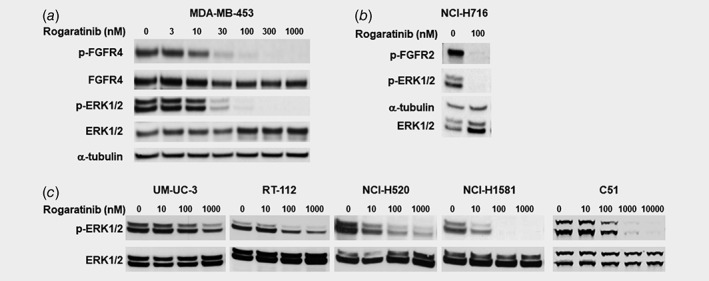
Effects of rogaratinib treatment on phosphorylation of FGFR and ERK in various cell lines as determined by Western blotting. (a) p‐FGFR4, total FGFR, p(T202/Y204,T185/Y187)‐ERK1/2, and total ERK1/2 expression in MDA‐MB‐453 cells after treatment with 0–1,000 nM rogaratinib. (b) p‐FGFR2, α‐tubulin, p(T202/Y204,T185/Y187)‐ERK1/2, and total ERK1/2 expression in NCI‐H716 cells after treatment with 0 or 100 nM rogaratinib. (c) p(T202/Y204,T185/Y187)‐ERK1/2 and total ERK1/2 expression in UM‐UC‐3, RT‐112, NCI‐H520, NCI‐H1581 and C51 cells after treatment with 0–10,000 nM rogaratinib.
Rogaratinib potently inhibits growth of murine colon C51 tumors in mice and rats
The syngeneic C51 mouse colon cancer model with high mRNA expression of FGFR1 and, to a lower extent, FGFR2 was identified as highly sensitive to inhibition of the FGFR pathway. Therefore, C51 was used in vivo for testing the efficacy of rogaratinib in FGFR‐overexpressing cancer models. The maximally tolerated dose for rogaratinib was determined at 75 mg/kg, QD and 50 mg/kg, BID in Balb/cJ mice. In a first experiment, monotherapy of the C51 model in Balb/cJ mice was performed with different doses and schedules of rogaratinib followed by a pharmacokinetic analysis after the last treatment (Fig. 3).
Figure 3.
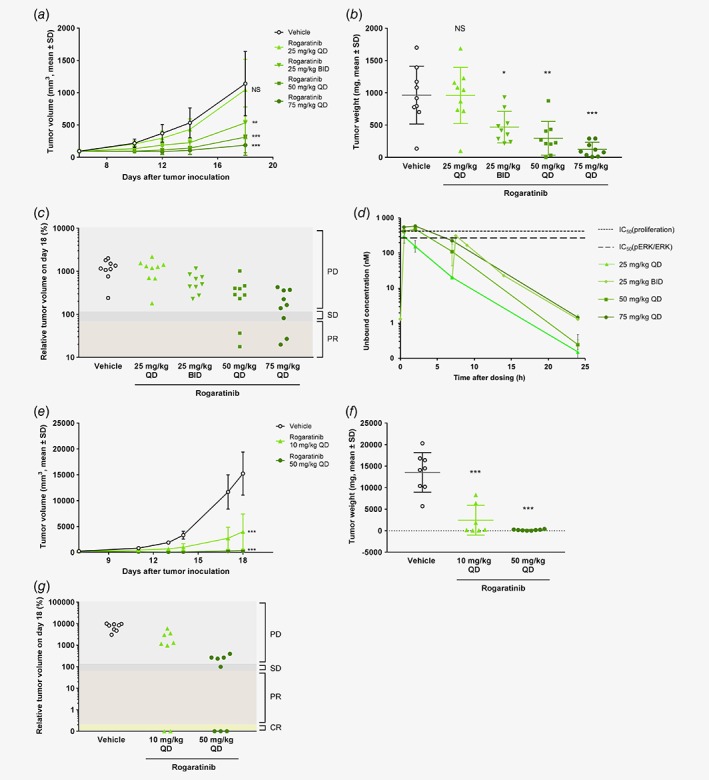
Antitumor activity of rogaratinib in the C51 syngeneic colon cancer model in immunocompetent BALB/c mice and the C51 xenogeneic model in nude rats. (a) Growth curves of C51 colon tumors treated p.o. with vehicle or rogaratinib (25, 50 or 75 mg/kg, QD; or 25 mg/kg BID) in BALB/c mice as measured by tumor volumes (mm3, mean +/− SD) over time. (b) Weights of C51 tumors in mice described in (a) at the end of the study (day 18). (c) Relative volumes (100% equals volume at start of treatment) of C51 tumors described in (a) at the end of the study (day 18) analyzed using RECIST criteria. PD, progressive disease; SD, stable disease; PR, partial response; CR, complete response. (d) Pharmacokinetic profile of rogaratinib in plasma of C51 tumor‐bearing BALB/c mice, shown as unbound concentration over 24 h after the last dose of rogaratinib and simulated exposure levels for the twice daily application of 25 mg/kg. The dotted lines denote the IC50 values of rogaratinib (280 and 430 nM) in C51 cells for inhibition of p‐ERK and proliferation, respectively. (e) Growth curves of C51 tumors in nude rats treated p.o. with vehicle or rogaratinib (10 or 50 mg/kg, QD) as measured by tumor volumes (mm3, mean ± SD) over time. (f) Weight of C51 tumors in rats described in (e) at the end of the study. (g) Relative volumes of C51 tumors described in (e) at the end of the study analyzed using RECIST criteria. PD, progressive disease; SD, stable disease; PR, partial response. Stars in a, b, e and f denote statistical difference compared to vehicle group. *, p < 0.05; **, p < 0.01; ***, p < 0.001; NS, non‐significant. [Color figure can be viewed at wileyonlinelibrary.com]
Rogaratinib demonstrated strong antitumor efficacy in a dose‐dependent manner with significant effects at doses reaching functional (pERK) IC50 of C51 cells. Oral treatment at 50 and 75 mg/kg (QD) reduced tumor growth compared to the vehicle group with T/C volume ratios of 0.27 and 0.16, respectively (Figs. 3 a–3b, Supporting Information Table S5). In both groups, a 22% partial response (PR) rate (2/9 mice) was observed according to RECIST criteria20 (Fig. 3 c). In addition, 1/9 mice (11%) had stable disease (SD) in the 75 mg/kg treatment group. While daily treatment at 25 mg/kg was not efficacious, twice‐daily application of 25 mg/kg resulted in a decrease of tumor growth with a T/C volume ratio of 0.47.
The pharmacokinetic (PK) profile of rogaratinib is presented in Figure 3 d and Supporting Information Table S6 as unbound rogaratinib concentration in plasma over 24 h after the last dose of rogaratinib, and as simulated exposure levels for the twice‐daily application of 25 mg/kg. AUC and Cmax were dose‐proportional in the range of 25–75 mg/kg and tumor growth reduction correlated well with exposure. Unbound rogaratinib levels were in the range of or above the C51 cell IC50 values (proliferation and pERK inhibition) up to 4 to 6 h after administration of the efficacious doses of 50 and 75 mg/kg QD while IC50 levels were barely reached with the non‐efficacious dose of 25 mg/kg QD. This indicated that an exposure to unbound rogaratinib concentrations close to the IC50 levels over several hours is adequate in this model to reach good antitumor efficacy. The significant but moderate anti‐tumor effect of the 25 mg/kg BID dose of rogaratinib suggests that twice daily dosing of 50 mg/kg may also result in better efficacy than once daily application due to longer exposure above IC50.
The efficacy of rogaratinib was further evaluated in the C51 colon cancer model in nude rats to determine efficacy and to support the human dose prediction by extending the analyses to another species. Daily oral treatment with 10 or 50 mg/kg rogaratinib significantly reduced tumor growth with T/C volume ratios of 0.26, and 0.02, respectively (Figs. 3 e–3f, Supporting Information Table S5). In the 10 mg/kg group 2/8 (25%) complete responses (CR) and in the 50 mg/kg group 3/8 (37.5%) CR and 1/8 (12.5%) SD were observed (Fig. 3 g). Treatment with rogaratinib monotherapy was generally well‐tolerated with no body weight losses above 10% or any fatal toxicity in mice or rats (Supporting Information Figs. S2A‐B).
Rogaratinib has antitumor activity in an FGFR2 over‐expressing colorectal carcinoma cell line‐derived model
In order to determine the antitumor activity of rogaratinib in a cell line‐derived model of human cancer, its in vivo efficacy was assessed in an NCI‐H716 human colon cancer model with FGFR2 amplifications and highly elevated levels of FGFR2 mRNA, with a PK/PD study performed at the end of the experiment. Similarly to the syngeneic C51 colon model, twice‐daily rogaratinib treatment at 35, 50 or 65 mg/kg showed notable, dose‐dependent antitumor efficacy with T/C volume ratios of 0.17, 0.14 and 0.09, respectively. PRs were observed in 1/8 (12.5%) and 5/8 (63.5%) mice in the two highest dose groups (Figs. 4 a–4b).
Figure 4.
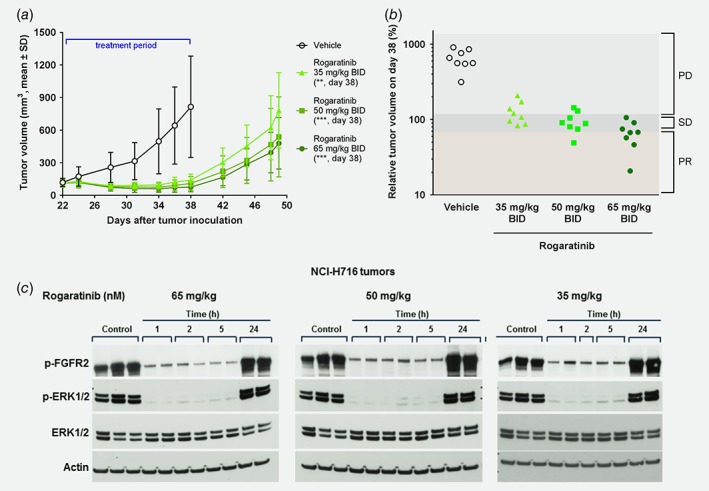
In vivo antitumor efficacy and mechanism of action of rogaratinib in the human NCI‐H716 colorectal xenograft model in immunocompromised NMRInu/nu mice. (a) Growth curves of human NCI‐H716 colorectal tumors treated p.o. with vehicle or rogaratinib (35, 50 or 65 mg/kg, BID) from days 22 to 38 after tumor inoculation as measured by tumor volumes (mm3, mean ± SD) over time. Stars denote statistical difference compared to vehicle group. **, p < 0.01; ***, p < 0.001. (b) Relative volumes of NCI‐H716 tumors described in (a) on the last treatment day (day 38). PD, progressive disease; SD, stable disease; PR, partial response. (c) Expression of p‐FGFR2, p‐ERK, total ERK and actin in NCI‐H716 tumor tissue after rogaratinib treatment. After a drug‐free observation period of 10 days, mice of the three rogaratinib‐dose groups (35, 50 or 65 mg/kg) in (a) received a single respective dose of rogaratinib or vehicle. Plasma and tumors were collected 1, 2, 5 or 24 h after treatment for PK/PD analysis and phosphorylation of FGFR2 and ERK1/2 was determined by Western blotting. [Color figure can be viewed at wileyonlinelibrary.com]
Unbound Cmax concentrations of rogaratinib in plasma, measured after a 10‐day drug‐free observation period followed by a last single dose of 35, 50 or 65 mg/kg rogaratinib, reached 123, 294 and 357 nM, respectively, exceeding the proliferation IC50 of 65.4 nM for NCI‐H716 cells by two–six‐fold.
PK/PD analysis of FGFR2 and ERK1/2 phosphorylation levels clearly showed strong inhibition of FGFR signaling by rogaratinib for at least five hours when rogaratinib levels still reached unbound plasma concentrations (127, 204 and 238 nM) 2–4 times above the proliferation IC50. At the 24‐h time point, phosphorylation of FGFR2 and ERK1/2 had returned to levels similar to the vehicle control, consistent with plasma drug levels <1 nM (Fig. 4 c).
Antitumor activity of rogaratinib in various FGFR2 over‐expressing lung cancer models
In vivo antitumor efficacy of rogaratinib alone and in combination with SOCs was investigated in cell line‐ and patient‐derived models of lung cancer. In the DMS‐114 model, which displays FGFR1 amplification and overexpression, rogaratinib (50 mg/kg, BID) showed marked antitumor efficacy (T/C volume = 0.34) (Figs. 5 a–5b). Treatment with either docetaxel (30 mg/kg, Q7D) or carboplatin/paclitaxel combination (80 mg/kg, Q7D/24 mg/kg, Q7D) showed potent antitumor efficacy with T/C volume ratios of 0.27 and 0.31, respectively. Although the combination therapy with 50 mg/kg (BID) rogaratinib and docetaxel (30 mg/kg, Q7D) showed only minor additional benefit compared to docetaxel monotherapy alone in terms of T/C ratios, the PR rate was increased from 40% to 70% and one CR was observed (Fig. 5 c). Similarly, combining 50 mg/kg (BID) rogaratinib with carboplatin/paclitaxel (80 mg/kg, Q7D/24 mg/kg, Q7D, T/C volume 0.31) resulted in a T/C volume ratio of 0.20 and increased the PR rate from 30% to 70% compared to both of the chemotherapies alone.
Figure 5.
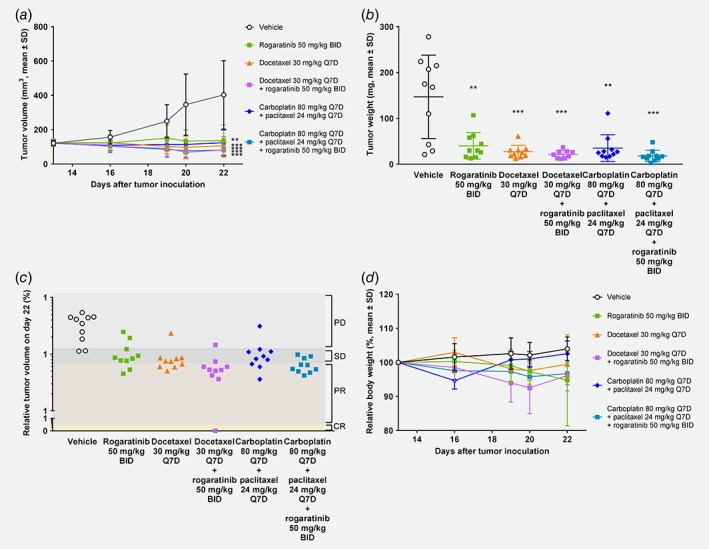
In vivo antitumor efficacy of rogaratinib in the DMS‐114 lung cancer xenograft model. (a) Growth curves of DMS‐114 lung tumors treated with vehicle, rogaratinib (50 mg/kg, p.o., BID), docetaxel (30 mg/kg, i.v., Q7D), combination of rogaratinib (50 mg/kg, BID)/docetaxel (30 mg/kg, Q7D), combination of carboplatin (80 mg/kg, i.p., Q7D)/paclitaxel (24 mg/kg, Q7D) or combination of carboplatin (80 mg/kg, Q7D)/paclitaxel (24 mg/kg, i.p., Q7D)/rogaratinib (50 mg/kg, BID), as measured by tumor volumes (mm3, mean ± SD) over time. (b) Weights of DMS‐114 tumors described in (a) at the end of the study. (c) Relative volumes of DMS‐114 tumors described in (a) at the end of the study. PD, progressive disease; SD, stable disease; PR, partial response; CR, complete response. (d) Body weights of mice described in (a) relative to body weights at treatment start. Stars denote statistical difference compared to vehicle group. **, p < 0.01; ***, p < 0.001.
As observed in the syngeneic C51 model, rogaratinib treatment was generally well‐tolerated also in the xenogeneic NCI‐H716 and DMS‐114 models with maximum body weight losses of 5.2% and 5%, respectively (Fig. 5 d). Maximal body weight loss in the two chemotherapy groups was 2.4% for docetaxel and 5.4% for carboplatin/paclitaxel. In the combination group of rogaratinib with docetaxel body weight loss above 20% was observed in one mouse (out of 10) for unknown reasons, while all others showed no signs of acute toxicity until the end of study. The combination of rogaratinib with carboplatin/paclitaxel did not result in any additional body weight loss and was well‐tolerated.
To investigate the importance of elevated FGFR expression, the efficacy of rogaratinib was assessed in two patient‐derived lung cancer models, LU299 and LXFL1121, which overexpress FGFR1 mRNA but do not harbor high FGFR gene amplifications (Supporting Information Table S7). At a dose of 50 mg/kg (BID), rogaratinib showed significant antitumor efficacy as monotherapy (LU299, T/C volume = 0.33; LXFL1121, T/C volume = 0.26) (Fig. 6, Supporting Information Figs. S3A–B). In the LU299 model, the combination of rogaratinib (50 mg/kg) and docetaxel showed a trend towards superior tumor growth inhibition compared to treatment with docetaxel alone (Figs. 6 a–6b). Evaluation of the treatment response according to RECIST criteria20 resulted in 3/10 (30%) CRs and 7/10 (70%) PRs in the rogaratinib and docetaxel combination treatment group, compared to 6/10 (60%) PRs in the docetaxel monotherapy group (Fig. 6 b). A notable improvement in the duration of response was observed in the rogaratinib/docetaxel combination group resulting in only 1 of 10 tumors with regrowth for up to 50 days post treatment (Fig. 6 d).
Figure 6.
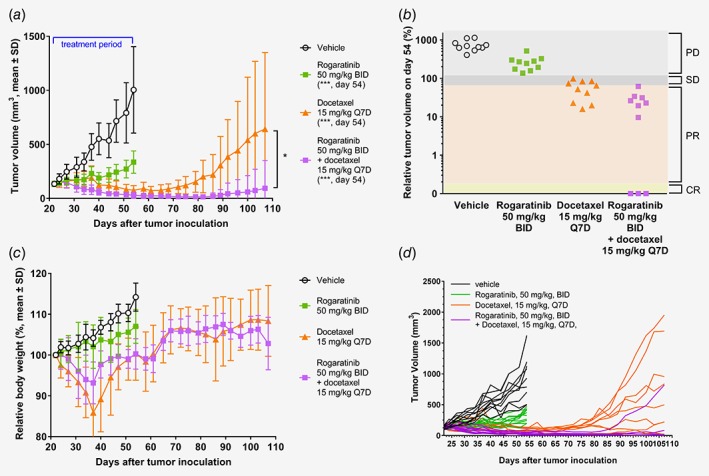
In vivo antitumor efficacy of rogaratinib in the patient‐derived LU299 lung cancer xenograft model. (a) Growth curves of LU299 lung tumors treated with vehicle, rogaratinib (50 mg/kg, BID), docetaxel (15 mg/kg, i.v., Q7D), or combination of rogaratinib and docetaxel for 33 days followed by a drug‐free observation period of 53 days, as measured by tumor volumes (mm3, mean ± SD) over time. Stars denote statistical difference compared to vehicle group. *, p < 0.05; ***, p < 0.01. (b) Relative volumes of tumors described in (a) at the last treatment day (day 54 after tumor inoculation). PD, progressive disease; SD, stable disease; PR, partial response; CR, complete response. (c) Relative body weights of LU299 mice described in (a). Body weight loss in docetaxel groups recovered after dosing break at day 37. (d) Growth curves of single mice of LU299 lung tumors treated as noted above (a).
In the LXFL1121 model, the rogaratinib/docetaxel combination also resulted in prolonged tumor response post‐treatment, whereas tumor regrowth was observed in the docetaxel monotherapy group (Supporting Information Fig. S3).
Rogaratinib treatment was well‐tolerated with no body weight losses over 10% in either of the studied PDX models (Fig. 6 c, Supporting Information Fig. S3C). Docetaxel monotherapy showed maximum body weight losses of 14.1% in the LU299 model, but no additional body weight loss in combination with rogaratinib (Fig. 6 c). In the LXFL1121 model, combination therapy with docetaxel/rogaratinib showed a maximum body weight loss of 18.9% (Supporting Information Fig. S3C).
Discussion
Here, we present for the first time the pharmacological profile of rogaratinib, an orally bioavailable, highly potent and selective novel FGFR1‐4 kinase inhibitor,19 which inhibits tumor growth in various FGFR‐driven cell line and patient‐derived xenograft cancer models both as monotherapy and in combination with SOC treatments.
The potency of FGFR inhibition was confirmed in biochemical kinase domain binding and activity assays. Rogaratinib binds to all four FGFR receptor subtypes with single digit nM affinity and potently inhibits FGFR1‐4 kinase activity with IC50 values in the nanomolar range. KINOMEscan™ profiling of 468 kinase targets showed very favorable selectivity scores with binding and inhibition being highly selective compared to related receptor tyrosine kinases such as VEGFRs, PDGFRs or Tie2. Biochemical selectivity against VEGFR2 was demonstrated in cellular assays, as rogaratinib potently inhibited FGF2 vs. VEGF‐A stimulated HUVEC proliferation. Rogaratinib had selective antiproliferative activity against cancer cell lines with increased FGFR expression, i.e., elevated mRNA levels of at least one FGFR subtype including cell lines such as A172 which do not harbor FGFR amplifications. This is in agreement with reports showing that FGFR expression rather than gene amplification is a determinant of sensitivity to FGFR inhibition.16, 17 While some FGFR1‐amplified lung cancers are sensitive to FGFR inhibition, initial clinical results of the pan‐FGFR‐inhibitor BGJ398 showed a response rate of only 11% in FGFR1‐amplified squamous NSCLC.31 Screening for FGFR overexpression may therefore be a more suitable selection marker for sensitive cancers and potentially for patient stratification.32 However, not all cell lines overexpressing one of the FGFR subtypes were sensitive to FGFR inhibition, suggesting that other oncogenes or oncogenic pathways besides FGFR signaling may be dominant oncogenic drivers in these cells. Furthermore, the dependence of activation of wildtype FGFRs by specific ligands can impact pathway activation in two ways, either overactivation with increased levels of ligand and physiologically expressed receptor, or lack of activation despite overexpression of receptor if the specific ligand is not expressed. The presented studies do not include such analysis and this will be part of future activities.33, 34
Together, our data demonstrate that elevated expression of at least one FGFR subtype is essential for conferring sensitivity to rogaratinib but it is not sufficient as shown for several tumor cell lines. In agreement with previous observations on the sensitivity of cancer cell lines with FGFR amplification or overexpression to the FGFR inhibitor JNJ‐42756493,35 many of the FGFR‐overexpressing but rogaratinib‐insensitive cell lines carry either mutations in RAS family members or in the PI3K pathway. This indicates that mutations downstream of FGFR may be able to bypass dependency on the receptor and activate alternative signaling cascades. Similar observations have been reported for EGFR and KRAS mutations36 with the clinical implication that the use of anti‐EGFR agents like cetuximab is not indicated in colorectal cancer patients with KRAS alterations.37 Alternatively, genetic aberrations, e.g. activating mutations or overexpression of other RTKs, may confer resistance to rogaratinib despite FGFR overexpression.
Activation of FGFRs leads to phosphorylation of adaptor or directly interacting key proteins of four major intracellular signaling pathways, RAS/MAPK/ERK, PI3K/AKT, PLCγ and STAT.38 Inhibition of ERK1/2 phosphorylation was established as a suitable surrogate marker for cellular FGFR inhibition. In line with this, inhibition of ERK phosphorylation by rogaratinib mirrors inhibition of FGFR4 phosphorylation in MDA‐MB‐453 cells or inhibition of FGFR2 phosphorylation in NCI‐H716 tumors and is consistent with sensitivity to proliferation inhibition in the tested cell lines as well as with inhibition of tumor growth in vivo. FGFR‐mediated activation of RAS/MAPK/ERK is initiated by phosphorylation of FRS2α, which also leads to activation of PI3K/AKT signaling.39, 40 PI3K/AKT kinase activity rather than ERK activation may play a key role in certain cancers addicted to FGFR.41, 42 Rogaratinib caused inhibition of downstream phosphorylation of ERK as well as AKT with similar nM potency in cell lines overexpressing either FGFR1 (DMS‐114) or FGFR2 (MFM‐223).
In vivo profiling of rogaratinib was initially performed using C51 colon cancer cells either subcutaneously inoculated into syngeneic immunocompetent mice or into xenogeneic immunocompromised rats. Rogaratinib caused dose‐dependent reduction of tumor growth with partial responses observed at 50 and 75 mg/kg daily dosing in mice. PK/PD analysis showed that exposure correlated with in vivo antitumor efficacy and suggested that increasing the exposure time above IC50 by dosing twice daily may result in better efficacy. In vivo assessment of pharmacodynamic biomarkers for up to 24 h was performed in mice bearing FGFR2‐driven NCI‐H716 tumors after a single dose of rogaratinib. A reduction was observed in the phosphorylation of both FGFR2 and downstream ERK1/2 for 1 to 5 h, which is consistent with unbound rogaratinib levels exceeding IC50 values by a factor of 2 to 4. In summary, the PK/PD data suggest that rogaratinib levels above tumor cell IC50 for proliferation or downstream signaling (pERK) over several hours, resulting in concentrations that completely block FGFR signaling in cancer cells, are sufficient to reach significant antitumor efficacy in FGFR‐driven cancers.
As a pan‐FGFR inhibitor, rogaratinib targets FGFRs on cancer cells as well as on stromal cells. For instance, FGF2‐induced growth of endothelial cells is potently inhibited by rogaratinib with an IC50 of 16 nM. Antitumor efficacy may therefore be mediated by several simultaneous mechanisms such as inhibition of cancer cell proliferation and survival combined with targeting paracrine FGF signaling in the tumor microenvironment.
Rogaratinib demonstrated efficacy in a number of in vivo xenograft models and was tolerated well at doses up to 75 mg/kg QD or 50 mg/kg BID. Durable partial and complete responses were observed in several cell line‐ and patient‐derived models that are driven by FGFR overexpression, such as the FGFR1‐overexpressing C51 colon cancer, DMS‐114 lung cancer and LU299 and LXFL1121 PDX lung cancer models, or the FGFR2‐overexpressing NCI‐H716 colon cancer model.
Additionally, as shown using the DMS‐114 model, combination treatment with SOC compounds docetaxel or paclitaxel/cisplatin resulted in enhanced efficacy compared to rogaratinib monotherapy or the SOC treatments alone. Similarly, combination treatment with the SOC docetaxel in the PDX lung cancer models LU299 and LXFL1121 resulted in better efficacy or prolonged duration of response compared to treatment with rogaratinib or docetaxel alone. Combination treatment with SOCs was tolerated well in the models tested.
Importantly, the rogaratinib‐sensitive cell lines and the studied in vivo models in which rogaratinib caused tumor growth inhibition overexpress one or several FGFR subtypes, irrespective of the underlying mechanism. Thus, the effects on cell proliferation and tumor growth strongly suggest that rogaratinib inhibits FGFR‐activation driven cancer growth in a dose‐dependent manner with excellent efficacy in cancers with high FGFR expression. This observation suggests that it may be beneficial to use tumor FGFR mRNA expression as a stratification biomarker in clinical trials.
In summary, rogaratinib is a highly potent and selective pan‐FGFR inhibitor with a unique selectivity profile that supports good tolerability and safety. The compound demonstrates robust efficacy in vivo in cancers with altered FGFR mRNA expression both as monotherapy and when combined with SOC treatments and it is well‐tolerated, making it a viable candidate for further development. Several clinical studies with rogaratinib using FGFR mRNA expression for prospective patient selection are currently ongoing (ClinicalTrials.gov Identifiers: NCT01976741, NCT02592785, NCT03410693).
Supporting information
Figure S1 Correlation of pERK levels to FGFR subtype expression to response / resistance towards treatment with rogaratinib in selected invivo models
Figure S2 Relative body weights of C51 mice and rats treated with rogaratinib.
Figure S3 In vivo antitumor efficacy of rogaratinib in the patient‐derived LXFL1121 NSCLC xenograft model in mice.
Table S1. Kinase selectivity of rogaratinib. The table shows kinases with % inhibition as determined by KINOMEscan™ at 1 μM rogaratinib.
Table S2. Non‐kinase selectivity of rogaratinib. Procent inhibition of targets at 10 μM rogaratinib according to PanLabs screen
Table S3. Inhibition of cell proliferation by rogaratinib in a variety of cell lines including the Oncoline™ panel. The corresponding FGFR mRNA expression levels and alterations in FGFR1‐4, PIK3CA, KRAS, NRAS, HRAS and EGFR genes are presented (CCLE database), sorted by IC50. (table corrected)
Table S4. IC50 values of rogaratinib for downstream phosphorylation in cell lines with FGFR alterations.
Table S5. In vivo efficacy of rogaratinib.
Table S6. PK/PD correlations from study on efficacy of rogaratinib in syngeneic C51 colon cancer model in Balb/cJ mice.
Table S7. Molecular characteristics of two PDX models.
Acknowledgements
We would like to thank all team members for their support and expert technical assistance. Aurexel Life Sciences Ltd (www.aurexel.com) is acknowledged for editorial support funded by Bayer AG.
Conflict of interest: All authors are employees of Bayer AG. Authors SG, KZ, KL DM, MH, SI, PE, UB and UT have an ownership interest in Bayer AG.
References
- 1. Katoh M. Therapeutics targeting FGF signaling network in human diseases. Trends Pharmacol Sci 2016;37:1081–96. [DOI] [PubMed] [Google Scholar]
- 2. Turner N, Grose R. Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer 2010;10:116–29. [DOI] [PubMed] [Google Scholar]
- 3. Helsten T, Elkin S, Arthur E, et al. The FGFR landscape in cancer: analysis of 4,853 tumors by next‐generation sequencing. Clin Cancer Res 2016;22:259–67. [DOI] [PubMed] [Google Scholar]
- 4. Cihoric N, Savic S, Schneider S, et al. Prognostic role of FGFR1 amplification in early‐stage non‐small cell lung cancer. Br J Cancer 2014;110:2914–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Heist RS, Mino‐Kenudson M, Sequist LV, et al. FGFR1 amplification in squamous cell carcinoma of the lung. J Thorac Oncol 2012;7:1775–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Preusser M, Berghoff AS, Berger W, et al. High rate of FGFR1 amplifications in brain metastases of squamous and non‐squamous lung cancer. Lung Cancer 2014;83:83–9. [DOI] [PubMed] [Google Scholar]
- 7. Weiss J, Sos ML, Seidel D, et al. Frequent and focal FGFR1 amplification associates with therapeutically tractable FGFR1 dependency in squamous cell lung cancer. Sci Transl Med 2010;2:62ra93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Goke F, Bode M, Franzen A, et al. Fibroblast growth factor receptor 1 amplification is a common event in squamous cell carcinoma of the head and neck. Mod Pathol 2013;26:1298–306. [DOI] [PubMed] [Google Scholar]
- 9. Koole K, Brunen D, van Kempen PM, et al. FGFR1 is a potential prognostic biomarker and therapeutic target in head and neck squamous cell carcinoma. Clin Cancer Res 2016;22:3884–93. [DOI] [PubMed] [Google Scholar]
- 10. Cappellen D, De Oliveira C, Ricol D, et al. Frequent activating mutations of FGFR3 in human bladder and cervix carcinomas. Nat Genet 1999;23:18–20. [DOI] [PubMed] [Google Scholar]
- 11. Gust KM, McConkey DJ, Awrey S, et al. Fibroblast growth factor receptor 3 is a rational therapeutic target in bladder cancer. Mol Cancer Ther 2013;12:1245–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Morales‐Barrera R, Suarez C, de Castro AM, et al. Targeting fibroblast growth factor receptors and immune checkpoint inhibitors for the treatment of advanced bladder cancer: new direction and new Hope. Cancer Treat Rev 2016;50:208–16. [DOI] [PubMed] [Google Scholar]
- 13. Tomlinson DC, Baldo O, Harnden P, et al. FGFR3 protein expression and its relationship to mutation status and prognostic variables in bladder cancer. J Pathol 2007;213:91–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dienstmann R, Rodon J, Prat A, et al. Genomic aberrations in the FGFR pathway: opportunities for targeted therapies in solid tumors. Ann Oncol 2014;25:552–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Katoh M. Fibroblast growth factor receptors as treatment targets in clinical oncology. Nat Rev Clin Oncol 2018;16:105–22. [DOI] [PubMed] [Google Scholar]
- 16. Goke F, Franzen A, Hinz TK, et al. FGFR1 expression levels predict BGJ398 sensitivity of FGFR1‐dependent head and neck squamous cell cancers. Clin Cancer Res 2015;21:4356–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wynes MW, Hinz TK, Gao D, et al. FGFR1 mRNA and protein expression, not gene copy number, predict FGFR TKI sensitivity across all lung cancer histologies. Clin Cancer Res 2014;20:3299–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Katoh M. FGFR inhibitors: effects on cancer cells, tumor microenvironment and whole‐body homeostasis (review). Int J Mol Med 2016;38:3–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Collin M‐P, Lobell M, Hübsch W, et al. Discovery of Rogaratinib (BAY 1163877): a pan‐FGFR inhibitor. ChemMedChem 2018;13:1–10. [DOI] [PubMed] [Google Scholar]
- 20. Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 2009;45:228–47. [DOI] [PubMed] [Google Scholar]
- 21. Uitdehaag JCM, de Roos JADM, Prinsen MBW, et al. Cell panel profiling reveals conserved therapeutic clusters and differentiates the mechanism of action of different PI3K/mTOR, Aurora kinase and EZH2 inhibitors. Mol Cancer Ther 2016;15:3097–3109. [DOI] [PubMed] [Google Scholar]
- 22. Brower M, Carney DN, Oie HK, et al. Growth of cell lines and clinical specimens of human non‐small cell lung cancer in a serum‐free defined medium. Cancer Res 1986;46:798–806. [PubMed] [Google Scholar]
- 23. Grossman HB, Wedemeyer G, Ren L, et al. Improved growth of human Urothelial carcinoma cell cultures. J Urol 1986;136:953–9. [DOI] [PubMed] [Google Scholar]
- 24. Goke A, Goke R, Ofner A, et al. The FGFR inhibitor NVP‐BGJ398 induces NSCLC cell death by activating caspase‐dependent pathways as well as Caspase‐independent apoptosis. Anticancer Res 2015;35:5873–9. [PubMed] [Google Scholar]
- 25. Mathur A, Ware C, Davis L, et al. FGFR2 is amplified in the NCI‐H716 colorectal cancer cell line and is required for growth and survival. PLoS One 2014;9:e98515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pettengill OS, Sorenson GD, Wurster‐Hill DH, et al. Isolation and growth characteristics of continuous cell lines from small‐cell carcinoma of the lung. Cancer 1980;45:906–18. [DOI] [PubMed] [Google Scholar]
- 27. Hackenberg R, Lüttchens S, Hofmann J, et al. Androgen sensitivity of the new human breast cancer cell line MFM‐223. Cancer Res 1991;51:5722–7. [PubMed] [Google Scholar]
- 28. Tomlinson DC, Lamont FR, Shnyder SD, et al. Fibroblast growth factor receptor 1 promotes proliferation and survival via activation of the mitogen‐activated protein kinase pathway in bladder cancer. Cancer Res 2009;69:4613–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Guagnano V, Furet P, Spanka C, et al. Discovery of 3‐(2,6‐dichloro‐3,5‐dimethoxy‐phenyl)‐1‐{6‐[4‐(4‐ethyl‐piperazin‐1‐yl)‐phenylamin o]‐pyrimidin‐4‐yl}‐1‐methyl‐urea (NVP‐BGJ398), a potent and selective inhibitor of the fibroblast growth factor receptor family of receptor tyrosine kinase. J Med Chem 2011;54:7066–83. [DOI] [PubMed] [Google Scholar]
- 30. Barretina J, Caponigro G, Stransky N, et al. The cancer cell line encyclopedia enables predictive modeling of anticancer drug sensitivity. Nature 2012;483:603–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nogova L, Sequist LV, Perez Garcia JM, et al. Evaluation of BGJ398, a fibroblast growth factor receptor 1‐3 kinase inhibitor, in patients with advanced solid tumors harboring genetic alterations in fibroblast growth factor receptors: results of a global phase I, dose‐escalation and dose‐expansion study. J Clin Oncol 2017;35:157–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kotani H, Ebi H, Kitai H, et al. Co‐active receptor tyrosine kinases mitigate the effect of FGFR inhibitors in FGFR1‐amplified lung cancers with low FGFR1 protein expression. Oncogene 2016;35:3587–97. [DOI] [PubMed] [Google Scholar]
- 33. Beenken A, Mohammadi M. The FGF family: biology, pathophysiology and therapy. Nat Rev Drug Discov 2009;8:235–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhao X, Xu F, Dominguez NP, et al. FGFR4 provides the conduit to facilitate FGF19 signaling in breast cancer progression. Mol Carcinog 2018;0;57:1616–25. [DOI] [PubMed] [Google Scholar]
- 35. Perera TP, Jovcheva E, Mevellec L, et al. Discovery and pharmacological characterization of JNJ‐42756493 (erdafitinib), a functionally selective small molecule FGFR family inhibitor. Mol Cancer Ther 2017;16:1010–20. [DOI] [PubMed] [Google Scholar]
- 36. Benvenuti S, Sartore‐Bianchi A, Di Nicolantonio F, et al. Oncogenic activation of the RAS/RAF signaling pathway impairs the response of metastatic colorectal cancers to anti‐epidermal growth factor receptor antibody therapies. Cancer Res 2007;67:2643–8. [DOI] [PubMed] [Google Scholar]
- 37. Lievre A, Bachet JB, Boige V, et al. KRAS mutations as an independent prognostic factor in patients with advanced colorectal cancer treated with cetuximab. J Clin Oncol 2008;26:374–9. [DOI] [PubMed] [Google Scholar]
- 38. Haugsten EM, Wiedlocha A, Olsnes S, et al. Roles of fibroblast growth factor receptors in carcinogenesis. Mol Cancer Res 2010;8:1439–52. [DOI] [PubMed] [Google Scholar]
- 39. Ong SH, Hadari YR, Gotoh N, et al. Stimulation of phosphatidylinositol 3‐kinase by fibroblast growth factor receptors is mediated by coordinated recruitment of multiple docking proteins. Proc Natl Acad Sci USA 2001;98:6074–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ornitz DM, Itoh N. The fibroblast growth factor signaling pathway. Wiley Interdiscip Rev Dev Biol 2015;4:215–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hu Y, Lu H, Zhang J, et al. Essential role of AKT in tumor cells addicted to FGFR. Anticancer Drugs 2014;25:183–8. [DOI] [PubMed] [Google Scholar]
- 42. Starska K, Forma E, Lewy‐Trenda I, et al. Fibroblast growth factor receptor 1 and 3 expression is associated with regulatory PI3K/AKT kinase activity, as well as invasion and prognosis, in human laryngeal cancer. Cell Oncol 2018;41:253–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Correlation of pERK levels to FGFR subtype expression to response / resistance towards treatment with rogaratinib in selected invivo models
Figure S2 Relative body weights of C51 mice and rats treated with rogaratinib.
Figure S3 In vivo antitumor efficacy of rogaratinib in the patient‐derived LXFL1121 NSCLC xenograft model in mice.
Table S1. Kinase selectivity of rogaratinib. The table shows kinases with % inhibition as determined by KINOMEscan™ at 1 μM rogaratinib.
Table S2. Non‐kinase selectivity of rogaratinib. Procent inhibition of targets at 10 μM rogaratinib according to PanLabs screen
Table S3. Inhibition of cell proliferation by rogaratinib in a variety of cell lines including the Oncoline™ panel. The corresponding FGFR mRNA expression levels and alterations in FGFR1‐4, PIK3CA, KRAS, NRAS, HRAS and EGFR genes are presented (CCLE database), sorted by IC50. (table corrected)
Table S4. IC50 values of rogaratinib for downstream phosphorylation in cell lines with FGFR alterations.
Table S5. In vivo efficacy of rogaratinib.
Table S6. PK/PD correlations from study on efficacy of rogaratinib in syngeneic C51 colon cancer model in Balb/cJ mice.
Table S7. Molecular characteristics of two PDX models.