

Abstract
Epithelial cell polarity regulator Crumbs3 (Crb3), a mammalian homolog within the Drosophila Crb gene family, was initially identified as an essential embryonic development factor. It is recently implicated in tumor suppression, though its specific functions are controversial. We here demonstrate that Crb3 strongly promotes tumor invasion and metastasis of human colon adenocarcinoma cells. Crb3 centrality to tumor migration was supported by strong expression at invasive front and metastatic foci of colonic adenocarcinoma of the patient tissues. Accordingly, two different Crb3‐knockout (KO) lines, Crb3‐KO (Crb3 −/−) DLD‐1 and Crb3‐KO WiDr from human colonic adenocarcinomas, were generated by the CRISPR‐Cas9 system. Crb3‐KO DLD‐1 cells exhibited loss of cellular mobility in vitro and dramatic suppression of liver metastases in vivo in contrast to the wild type of DLD‐1. Unlike DLD‐1, Crb3‐KO WiDr mobility and metastasis were unaffected, which were similar to wild‐type WiDr. Proteome analysis of Crb3‐coimmunopreciptated proteins identified different respective fibroblast growth factor receptor (FGFR) isotypes specifically bound to Crb3 isoform a through their intracellular domain. In DLD‐1, Crb3 showed membranous localization of FGFR1 leading to its functional activation, whereas Crb3 bound to cytoplasmic FGFR4 in WiDr without FGFR1 expression, leading to cellular growth. Correlative expression between Crb3 and FGFR1 was consistently detected in primary and metastatic colorectal cancer patient tissues. Taking these together, Crb3 critically accelerates cell migration, namely invasion and metastasis of human colon cancers, through specific interaction to FGFR1 on colon cancer cells.
Keywords: Crumbs3, cell polarity, colon cancer, adenocarcinoma, invasion, metastasis
Short abstract
What's new?
Epithelial cell polarity regulator Crumbs3 (Crb3) was initially identified as an essential embryonic development factor. More recently, it has been implicated in tumor suppression, though its specific functions remain controversial. Here, the authors demonstrate that Crb3 strongly promotes tumor invasion and metastasis of human colon adenocarcinoma cells. They identify among the binding partners of Crb3 the FGF receptors family, which is pivotal to tumor cell dynamics including proliferation, migration, and differentiation. Crb3 colocalizes with FGFR1 to activate downstream signaling and critically accelerate tumor migration and metastasis of human colon cancers.
Abbreviations
- Crb3
Crumbs3
- Crb3a
Crb3 isoform a
- Crb3b
Crb3 isoform b
- ERK
extracellular signal‐regulated kinase
- FGFR
fibroblast growth factor receptor
- HIS
histidine‐tagged
- KO
knockout
- Par‐3
partitioning defective‐3
- por
poorly differentiated
- STAT
signal transducer and activator of transcription
- tub
tubular
Introduction
Malignancy in tumor progression is typified by an initial invasion event, ensuing metastasis and attendant organ failure. The liver, as well as the lung, is one of the most frequently targeted organs in colorectal cancer metastases. Liver metastasis is observed in 15% of operable colorectal adenocarcinoma patients, and in over 50% of inoperable patients.1 As to the cancer ontogeny, the current understanding is that the metastatic condition develops as an aggressive cellular transit from the primary lesion, eventual contact with and entry into blood or lymphatic vessels, and migration to distal sites via small tumor nests in vivo. But this basic understanding of invasion/metastasis must be deepened by careful study of the underlying mechanistic complexities to spur novel therapeutics.
Cell polarity in the growing embryo is necessary for normal tissue formation in multicellular organisms. Epithelial cell polarity is reported to be regulated by three different protein complexes—partitioning defective‐3 (Par‐3)‐complex for the tight junction, Scribble‐complex for the lateral membrane and Crumbs3 (Crb3)‐complex for the apical membrane.2 Par‐3 and Scribble reportedly suppress tumor proliferation in genetically engineered artificial models.3, 4 Par‐3 suppressed proliferation of mutant H‐Ras‐introduced murine mammary cell via activating atypical protein kinase C and janus kinase/signal transducer and activator of transcription (STAT) signaling in the mouse model,5 whereas Scribble inhibited tumorigenesis in the mutant Ras‐transgene Drosophila embryo.6 By contrast, in a pathology study using human surgical material, Scribble was expressed in many types of tumors, leaving uncertain the biological role of these genes in human tumors.7
A single transmembrane protein Crb3 was described as expressed at the apical plasma membrane of epithelial cells of diverse origins.8, 9 The Crb3 locus generates two alternative‐spliced isoforms, Crb3 isoform a (Crb3a) and Crb3 isoform b (Crb3b), reported to be involved in different biological processes. Crb3a participated in epithelial polarity formation and suppression, while Crb3b functions in primary ciliogenesis.4, 10 Rather than an embryogenesis role, Crb3a was previously stated to correlate with tumor progression.11, 12 For example, Crb3a overexpression suppresses cellular growth and migration of Rb1 and p53‐dificient murine kidney cells implanted in Balb/c nu/nu mice.13 Since those functional investigations were confined to cellular and non‐human in vivo studies using Drosophila or mice, the biological function of Crb3 in human malignancies remains poorly defined.
The fibroblast growth factor receptor (FGFR) family is pivotal to tumor cell dynamics including proliferation, migration, and differentiation through regulating downstream signaling such as Ras‐mitogen activated kinase‐mediated pathways. The family consists of four genes and its tyrosine kinase activity is regulated in a context‐dependent manner.14, 15 Tumor patient tissue etiology also revealed that FGFR signaling component activation was the most commonly observed.16, 17 Hence, FGFR signaling profoundly concerns cancer progression, so as to prioritize examining the FGFR activation mechanism for therapeutic potential.
Here, we report novel features of Crb3 expression in human tumor tissue experiments using anti‐human Crb3a‐specific monoclonal antibody and Crb3‐knockout (KO) human colon adenocarcinoma cells, describe pertinent‐specific Crb3/FGFR isoform interactions, and offer molecular insights from the range of our in vitro and in vivo studies of cellular invasion and metastasis in colon cancer.
Materials and Methods
Cell culture
Cell lines were obtained from ATCC. For immunoblots, all tumor cell lines were maintained in RPMI1640 medium (#189‐02025, Wako Pure Chemical Industries, Japan) supplemented with 10% fetal bovine serum (FBS, #SH30071, Thermo Fisher Scientific, USA) and Pen/Strep (#15140‐148, Thermo Fisher Scientific). DLD‐1 and WiDr cells were authenticated by short tandem repeat analysis using GenePrint 10 System (Promega, USA).
Plasmid and cloning
For Crb3 gene KO by the CRISPR‐Cas9 system, gRNA cloning vector (plasmid #41824) and hCas9 (plasmid #41815) were obtained from Addgene. Crb3a (Accn#AF503290) and Crb3b (Accn# AY358684) were amplified by PCR from a HEK293T cDNA library. FGFR1 (Accn#NM_023110), FGFR2 (Accn#XM_006717710) and FGFR4 (Accn#XM_011534464) genes were amplified by PCR from DLD‐1 cDNA. PrimeSTAR Max DNA Polymerase (#R045A, TaKaRa Bio, Japan) was used for all PCR in plasmid constructions. Lentiviral expression, packaging and envelope plasmids (pWPI, pMD2.G and psPAX2) were kindly provided by Didier Trono (Addgene #12254, #12259 and #12260). Insert genes were amplified from pcDNA3 constructions and cloned into the PmeI site of pWPI using In‐Fusion HD Cloning Kit (#639648, Clontech, USA). All PCR primers used in our study were shown in supporting information (Table 1).
Transfection
Plasmid transfection was carried out using Lipofectamine LTX (#15338100, Thermo Fisher Scientific) by following manufacturer's protocol. Silencer Select Predesigned siRNAs (Thermo Fisher Scientific) targeting human mRNAs encoding Crb3 (#s40936 and #s195567), FGFR1 (#s5165), FGFR4 (#s5176 and #s5177) or FGFR1 (#L‐003131‐00‐0005, Dharmacon, USA) and control siRNA were transfected at 10 nM into cells using Lipofectamine RNAi MAX (Cat# 13778075, Thermo Fisher Scientific) by reverse transfection protocol. Target mRNA sequence of siRNAs were listed in Table S1. To establish stably expressing cells lentiviral transduction was performed. Lentiviruses were produced by following the Trono lab protocol (https://tronolab.epfl.ch/page-148635-en.html) with some modification.
Generation of Crb3 KO colon cancer cells
Crb3 KO cell line was established using CRISPR‐Cas9‐based genome engineering technology. To target the Crb3 allele, gRNA vector including target sequence (CCGTTCCTGCTGGCCCGCTGggg) was prepared by following the depositor's instruction. Lower case indicates Proto‐spacer Adjacent Motif (PAM). hCas9 and Crb3 gRNA vector were co‐transfected into cells using Lipofectamine LTX by manufacturer's reverse transfection protocol. Growth medium was replaced with selection medium including 500 μg/mL geneticin (#10131‐35, Thermo Fisher Scientific) at 24 hr after transfection, and cultured for 72 hr. Single cell cloning was achieved by serial dilution, and genotype of each clone was analyzed by genomic PCR followed by DNA sequencing as shown in Figure S2b .
MTT assay
MTT assay was performed by seeding 2 × 104 cells into each well of 24 well plates. The values indicate mean ± SD of triplicated experiments. Asterisks (p < 0.05) and daggers (p < 0.01) represent statistical significance. Statistical significance was tested using unpaired two‐tailed student's t‐test.
Transwell chamber assay
For transwell assay, 5 × 104 DLD‐1 cells suspended in 200 μL serum free opti‐MEMI (#31985‐070, Thermo Fisher Scientific) were loaded into the upper compartment of the transwell chamber (#3422, Corning, USA) in triplicate. A 500 μL of opti‐MEMI supplemented with 10% FBS was poured into the lower chamber. Twenty‐four hours after loading cells, cells in the upper chamber were removed with a cotton swab, and the remaining cells which migrate through the transwell membrane were stained with Hoechst 33342 (#H‐3570, Thermo Fisher Scientific). Fluorescent image was acquired with fluorescent inverted microscope (IX71, Olympus, Japan). The number of nuclei was counted in three different field using ImageJ software. The data indicate mean ± SD of triplicated experiments, and asterisks (p < 0.05) and daggers (p < 0.01) represent statistical significance. Statistical significance was tested using unpaired two‐tailed student's t‐test.
Wound healing assay
Wound healing assay was performed as follows: 5 × 104 cells in 80 μL RPMI1640 supplemented with 10% FBS were loaded into each well of culture‐insert (#80209, Ibidi, Germany) attached in 3.5 cm tissue culture dishes. Culture inserts were removed at 24 hr after loading and 2 mL fresh culture medium was added. Cells were cultured for 24 hr in a 5% CO2 incubator to observe cell migration.
Immunoblot
Anti‐human Crb3a monoclonal antibody was raised against a peptide mimicking the C‐terminus of Crb3a (Accn#NP_631900, VGARVPPTPNLKLPPEERLI). The following antibodies were obtained commercially, anti‐beta‐tubulin (#PM054‐7, Medical & Biological Laboratories, Japan), anti‐beta‐actin (#013‐24553, Wako Pure Chemical Industries, Japan), anti‐FGFR1 (#9740, Cell Signaling Technology, USA), anti‐FGFR4 (Cat# 8562, Cell Signaling Technology), anti‐phosphor‐extracellular signal‐regulated kinase (ERK) (#4370, Cell Signaling Technology) and anti‐total‐ERK antibody (#10359, Immuno‐Biological Laboratories, Japan). To investigate Crb3a expression in cancer cells, freshly cultured cells were lysed with radioimmunoprecipitation assay buffer including protease inhibitor cocktail. Protein concentration was determined with bicinchoninic acid protein assay reagent (#23227, Thermo Fisher Scientific) and normalized by adding lysis buffer. Finally, normalized protein samples were mixed with 4× Laemmli's sample buffer. Samples of 20 μg protein were analyzed using 1% SDS‐PAGE/tris‐glycine buffer. Protein transferred PVDF membranes were blocked with PBST (PBS/0.1% Tween 20) including 0.2% bovine serum albumin in for an hour at room temperature, and immunoreaction of anti‐Crb3a (2 μg/mL) was carried out in PBST. Other antibodies used in PBST with the following dilution factor of primary antibodies were as follows: anti‐beta‐tubulin (1:2000), anti‐FGFR1 (1:1000), anti‐FGFR4 (1:1000), anti‐phosphor‐ERK (1:1000), anti‐total‐ERK (1:2000) and 1% nonfat milk was employed for blocking. To perform the absorption test, 2.0 μg of anti‐Crb3a antibody and 0.75 μg of Crb3a peptide (1:25 M ratio), which was employed to generate Crb3a antibody, were mixed in 1.5 mL of PBST, and incubated for 30 min at 4°C before starting the primary antibody reaction.
FGFR‐histidine‐tagged (HIS) pull‐down assay
A 1 μg of pcDNA3‐FGFR‐HIS or pcDNA3‐FGFRΔcyto‐HIS and pcDNA3–FLAG‐Crb3a or the empty vector was co‐transfected into 5 × 105 of HEK293T cells by reverse transfection protocol using polyethylenimine,18 and cultured in the six‐well tissue culture plate. Cells were harvested by adding 1 mL of lysis buffer including 20 mM Tris–HCl (pH 8.0), 0.1% NP‐40, 150 mM NaCl, 1 mM MgCl2, 0.1 mM CaCl2 and protease inhibitor cocktail. The suspension was centrifuged at 15,000g for 10 min at 4°C, and supernatant was transferred to new tubes. A 20 μL each of TALON Metal Affinity Resin (#635501, Clontech) was washed with lysis buffer and added to the suspension. The pull‐down assay was performed for 1 hr at 4°C on rotation shaker. Affinity resins were washed five times in 1 mL lysis buffer, and removed buffer as much as possible. A 30 μL of lysis buffer including 500 mM Imidazole was added to each tube and incubated for 15 min at room temperature. Supernatants were transferred to new tubes, and 10 μL of 4× Laemmli's sample buffer was added. After incubation at 95°C for 3 min, 10 μL samples were analyzed by immunoblot.
Phosphor‐ERK detection
2 × 105 cells were plated in 12‐well plates 24 hr before stimulation. The cells are washed with PBS twice, and pretreated with 20 μM U0126 in opti‐MEMI without serum for 30 min in 5% CO2 incubator. Stimulation was begun by switching opti‐MEMI including 100 ng/mL recombinant human FGF1 protein (#064‐04781, Wako Pure Chemical Industries) for 10 min. The stimulated cells are lysed in 100 μL lysis buffer (100 mM Tris–HCl (pH 6.8), 1% Triton X‐100 and 8 M urea) and slightly sonicated in 1.5 mL tubes. Finally, protein samples were mixed with 4× Laemmli's sample buffer, then subjected to 12% SDS‐PAGE followed by immunoblot analysis.
Immunohistochemistry
Immunohistological experiments using surgically obtained tumor tissues from the patients were approved by the research ethics committee of the Niigata University (authorization number; #2015‐2334, 2016‐0089), and informed consent was obtained from all patients to perform the analyses. Immunohistochemistry was implemented using paraffin‐embedded section. Deparaffinization of paraffin‐embedded section was accompanied by antigen retrieval by using Pascal pressure chamber (Dako, USA) in sodium citrate buffer. Antigen‐retrieved sections were incubated with anti‐human Crb3a monoclonal (1 μg/mL, generated in our study) and/or anti‐FGFR1 (1:100 dilution, #sc‐121, Santa Cruz Biotechnology, USA) in PBS including 1.0% bovine serum albumin (#017‐25771, Wako Pure Chemical Industries), and stained using EnVision+ reagent (#K400011, Dako). DAB‐stained sections were counterstained with Mayer's hematoxylin (#30004, Muto Pure Chemicals, Japan). Fluorescent dye‐labeled secondary antibodies were employed for double staining.
Immunofluorescence
Freshly cultured cells were fixed with 4% paraformaldehyde in PBS for 15 min at room temperature. Fixed cells were washed and permeabilized in PBS supplemented with 0.1% Triton X‐100 or 0.1% saponin (#30502‐42, Nacalai Tesque, Japan). Blocking was done in PBST including 1% bovine serum albumin for 30 min at room temperature. The following antibodies were employed for immunofluorescence: anti‐Crb3a (2 μg/mL), anti‐FGFR1 (#9740, Cell Signaling Technology) and anti‐FGFR4 (1:100 dilution). Antibodies were diluted in PBS and immunoreactions were carried out overnight at 4°C in a humid chamber. The fluorescent dye‐conjugated secondary antibody reaction was performed for 1 hr at room temperature. Counterstain with Rhodamine‐Phalloidin (1:1,000 dilution, #PHDR1, Cytoskeleton, USA) and/or Hoechst 33342 was done simultaneously with the secondary antibody reaction. The fluorescent image was acquired with a fluorescent inverted microscope (IX71, Olympus) or confocal microscope (LSM 510 Meta, Zeiss, Germany).
Mouse model
This investigation has been conducted in accordance with the ethical standards, with the Declaration of Helsinki and with the national and international guidelines, and has been approved by the Experimental Animal Committees of Niigata University (authorization number; #SA27713). For the liver metastasis model, 1 × 106 cells of each human colonic cancer line (DLD‐1, WiDr) was injected intrasplenically into 7 week‐old non‐obese diabetic/severe combined immunodeficient (NOD/SCID) mice. Mice bearing DLD‐1 tumors were sacrificed and immediately assessed for tumor development in vivo at 60 days postimplantation. The model of intraperitoneal dissemination was obtained by injecting 2 × 106 cells in 400 μL medium intraperitoneally into NOD/SCID mice, and tumor development was assessed at 40 days postimplantation.
Semi‐quantification of immunohistochemistry
The detailed procedures are described in Supporting Information Materials and Methods.
Colony forming assay
The detailed procedures are described in Supporting Information Materials and Methods.
Sample preparation and immunoprecipitation for mass spec analysis
The detailed procedures are described in Supporting Information Materials and Methods.
Detection of FGFR1 phosphorylation
The detailed procedures are described in Supporting Information Materials and Methods.
Results
Crb3a is expressed in adenocarcinoma cells
Focusing on Crb3a function, a specific monoclonal antibody was generated against Crb3a using an encoded peptide arising only from Crb3a. Antibody specificity was confirmed by absorption test with the Crb3a peptide, which abrogated its immunoreactivity (Fig. S1a ). In addition, the antibody was demonstrated to be Crb3a‐specific by immunoblots of FLAG‐Crb3a‐transfected cell lysate in comparison with that of the Crb3b‐transfected cell (Fig. S1b ). The molecular weight of unmodified Crb3a is approximately 10 kDa without signal peptides, but the Crb3a extracellular domain is considered N‐glycosylated at multiple sites including asparagine 36.19 Actually, hyperglycosylated forms of Crb3a were detected as major bands around 25–35 kDa in adenocarcinoma line (Fig. S1a ). First, we screened endogenous expression of Crb3a in various malignancies with diverse origins using tumor cell lysates and tissue arrays. Consequently, adenocarcinomas was especially revealed to show prominent expression of Crb3a (Iioka et al., unpublished data).
Based on the result, we focused to investigate the specific biological function of Crb3a on colorectal adenocarcinomas. Crb3a showed unique expression at the apical site of luminal epithelium in normal colonic crypts (Fig. 1 a, left column), and this expression pattern was maintained in tubular (tub) adenocarcinomas with well‐ and moderately differentiated types (Fig. 1 a, middle two columns). In the poorly differentiated (por) adenocarcinoma, Crb3a expression was retained, but localized both to the plasma membrane and cytoplasm (Fig. 1 a, right column). Colorectal tub adenoma also showed luminal Crb3a expression as well as cancer foci and normal crypt in the same tissues, suggesting that Crb3a is consistently required during colon adenocarcinoma development (Fig. 1 b). In connection with advanced colon tumor progression, we examined Crb3a expression in metastatic liver foci, lymph nodal metastasis and vascular invasions, which showed prominent Crb3a expression in cancer cells (Fig. 1 c). The semi‐quantification of intensity of Crb3 expression on immunostained tissue specimens revealed that both tumor cells at primary and metastatic site (liver metastasis) showed increased intensity of Crb3a expression in comparison with that of normal crypt cells both in tub1 and por cases (tub1: p < 0.01; por: p < 0.01). Furthermore, Crb3 expression was higher in metastatic site than in primary tumor site in all three cases (p < 0.01; Figs. S1 c–S1e). Mean intensity of Crb3 expression was 1.00 in normal crypt, 1.46 in primary site and 2.41 in liver metastatic site of tub1 adenocarcinoma case. In the case of por adenocarcinoma, 1.00 in normal crypt and 3.32 in primary site. Thus, Crb3a expression might be constitutively required even in tumor cells after transformation.
Figure 1.
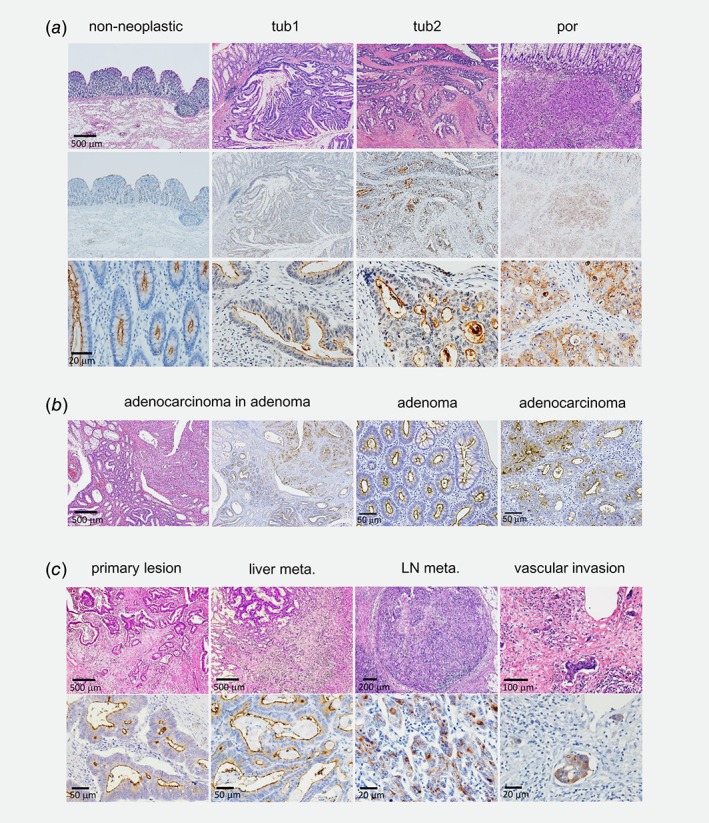
Crb3a is expressed in colon adenocarcinomas. (a) Immunohistochemcal analyses of Crb3a expression in normal colon crypts or colon cancer tissues (tub, tubular adenocarcinoma; por, poorly differentiated adenocarcinoma). Top panels are showing hematoxylin and eosin (HE) staining. Middle and bottom panels show immunohistochemistry using anti‐Crb3a antibody followed by hematoxylin staining. (b) Crb3a is expressed in both colon adenoma and adjacent adenocarcinoma. (c) Crb3a is expressed in both primary colon adenocarcinoma and metastatic foci in liver, lymph node and vascular invasion. Top panels indicate HE staining.
Crb3 deficiency affected cellular dynamics in human colon adenocarcinoma cells
We next analyzed Crb3a function in colon cancer cells. First, we examined subcellular Crb3a localization by immunofluorescence of colon adenocarcinoma cell lines, DLD‐1 and WiDr. Confocal microscopy revealed that Crb3a mainly localized to the cell membrane and submembrane portion of DLD‐1 cells, while WiDr cells predominantly expressed cytoplasmic Crb3a (Fig. S2a ). Notably, distinct membranous accumulation of Crb3a was observed at cell‐to‐cell interfaces, particularly in DLD‐1 cells. They proliferate in vitro with prominent adhesive‐cell morphology (Fig. S2a ).
Using the CRISPR‐Cas9 system, Crb3‐KO clones derived from DLD‐1 and WiDr were established. The gRNA design, matching immediately downstream to the initiation codon, is indicated in Figure 2 a. Genomic sequences around the gRNA target site of Crb3a‐negative cells were validated by DNA sequencing (Fig. S2b ), and expression loss of Crb3a in each clone was confirmed by immunoblot (Figs. 2 b and 2c). Three KO clones (Crb3−/−, namely Crb3a and Crb3b −/−) of DLD‐1 showed no significant change in cellular proliferation and colony‐formation ability in comparison with the wild‐type (wt) DLD‐1 (Fig. 2 d and Fig. S2c ). In WiDr, cell growth retardation up to 15–33% was observed in the Crb3‐KO clones (Fig. 2 e), but nonsignificant difference was showed in colony formation between wt and Crb3‐KO clones (Fig. S2d ). Alternatively, wt DLD‐1 cells were highly motile and showed diffusion in culture, while Crb3‐KO DLD‐1 cells lost their cellular motility and showed agglutinate proliferation (Fig. 2 f). Both the wound healing assay and the cell migration assay using transwell chambers corroborated loss of cellular motility in Crb3‐KO cells (over 90% reduction in numbers of migratory cells; † p < 0.01) (Figs. 2 g and 2h). Similar results were obtained from invasion assay using matrigel‐coated transwell chamber (Fig. S2e ). This phenomenon was also supported by Crb3 siRNA knockdown in DLD‐1 cells (Fig. 2 i). In contrast, WiDr motility in culture was originally very low, and no significant difference in cellular migration was observed by transwell chamber assay (data not shown).
Figure 2.
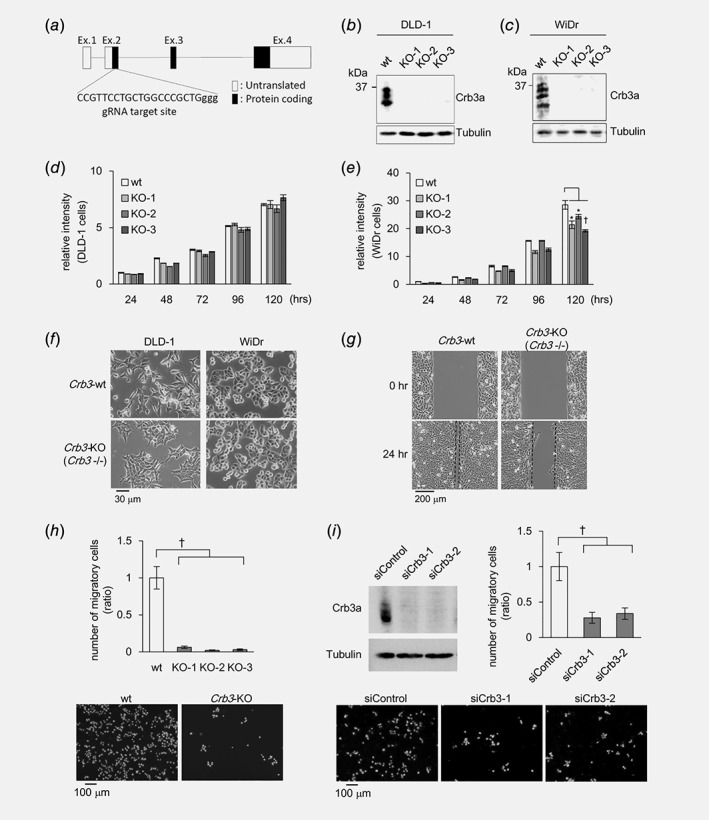
Crb3 affects colon adenocarcinoma cell behavior. (a) Genomic structure of human Crb3 and gRNA targeting site. (b, c) Validation of Crb3‐KO colon cancer cell lines by immunoblot. (d, e) Cell proliferation of wild type (wt) and Crb3‐KO DLD‐1 or WiDr cells was analyzed by MTT assay. (f) Morphology of wt and Crb3‐KO cells. (g) Wound healing assay using wt and Crb3‐KO DLD‐1 cell. (h) Transwell chamber assay of wt and Crb3‐KO DLD‐1 cells. The migratory cells were stained with Hoechst 33342 (bottom). (i) Crb3 protein expression and cell motility of DLD‐1 cell treated with Crb3 siRNAs were analyzed by Western blot and transwell chamber assay.
Loss of tumor metastasis in Crb3‐KO DLD‐1 cells in vivo
Based on the result of cellular assay, we further examined in vivo DLD‐1 dynamics with or without Crb3 using tumor cell‐xenografted mouse models. Either Crb3‐KO or wt DLD‐1 cells were implanted by splenic injection into NOD/SCID mice, and 60 days after implant, liver metastasis was examined in both mouse models by fresh autopsy. Mice injected with wt DLD‐1 cell developed multiple liver metastasis, whereas they were almost abrogated in Crb3‐KO cell‐implanted mice (Fig. 3 a). Pronounced membranous Crb3a expression was observed in liver metastatic tumors (Fig. 3 b), supporting the result of cellular assays and pathological examination of patients' colon cancer tissues.
Figure 3.
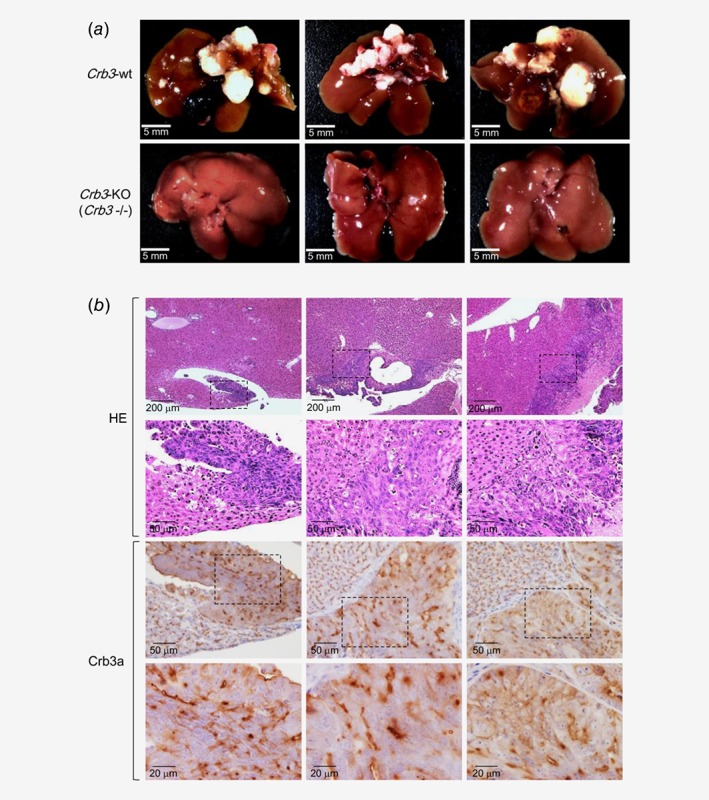
Deficiency of Crb3 in DLD‐1 inhibits liver metastasis in vivo. (a) Crb3 deficiency was examined in DLD‐1 xenograft in NOD/SCID mice. Suspension of wt or Crb3‐KO DLD‐1 cells were intrasplenically injected. (b) Immunohistochemistry of metastatic foci in the liver from wt DLD‐1 cell injected mice. Thin lined hatched boxes indicate liver metastatic lesions formed in wt DLD‐1 injected mice.
According to the liver metastatic models, similar phenomena were observed between mice with intraperitoneal injection of wt DLD‐1 and those injected with the Crb3‐KO clone. Namely, mice treated with wt DLD‐1 cell developed aggressive intra‐abdominal dissemination, but Crb3‐KO cell‐implant mice showed drastic decreases in disseminated foci (Fig. S3a ). Total tumor weight in Crb3‐KO cell‐injected mice yielded a 94% decrease in comparison with wt DLD‐1 injected mice (Fig. S3b ). Immunohistochemistry revealed pronounced membranous Crb3a expression in metastatic tumors (Fig. S3c ), supporting the in vitro assays and colon cancer tissue pathology.
By contrast, Crb3‐KO WiDr cells, which had been affected in cellular growth by lack of Crb3, did not show significant alteration in metastatic potential (Figs. S4a and S4 b ), even as tumors retained Crb3a expression in wt WiDr‐implant mice (Fig. S4c ).
Crb3a specifically interact with FGFR subtype in cellular dynamic synergy
Although several Crb3a interacting proteins have been reported in studies using overexpressed MDCK and MCF10A cells, all of these were tight junction‐related proteins.9, 20, 21 To identify specific tumor regulatory molecules interacting with Crb3a, proteomic analyses combined with immunoprecipitation and subsequent mass spectrometry were performed. A revertant DLD‐1 clone stably expressing FLAG‐tagged Crb3a using Crb3‐KO DLD‐1 cells as a backbone was established. This clone showed over 60% recovery in cellular motility compared to the parental Crb3‐KO clone (Fig. 4 a). Coimmunoprecipitation was carried out by employing anti‐FLAG affinity magnetic beads from the cellular membrane fraction. Several specific bands were thus observed as binding partner candidates in the silver‐stained gel (Fig. 4 b). We isolated the bands ranging from 75 to 110 kDa in molecular weight for mass spectrometry (LC/MS/MS) analysis. FGFR4 was retrieved as a novel candidate for the binding partner in addition to MPP5/Pals1, an already known Crb3a‐binding protein.19, 20 Based on the LC/MS/MS analysis, the precipitated fragment was part of the cytoplasmic domain of FGFR4 that shares a highly homologous region with all isotypes of the FGFR family molecules, and consequently we further focused on FGFR expression (FGFR1, 2, 3, 4). According to RT‐PCR assay and immunoblot analysis, FGFR1 (M.W. of glycosylated FGFR1; 120–145 kDa) was strongly expressed in DLD‐1, whereas they expressed at almost undetectable level of FGFR4 (M.W. of glycosylated FGFR4; 95–110 kDa). Inversely, WiDr cell highly expressed FGFR4 (Figs. 4 c and 4d). FGFR2 and FGFR3 were not significantly expressed, or not efficiently coprecipitated in the four lines (Fig. S5a ). FGFR1 and FGFR4 deletion mutants lacked the cytoplasmic domain (Δcyto) lost Crb3a interaction, indicating that Crb3a bound a particular site(s) in the FGFR1 or FGFR4 intracellular domain (Fig. 4 e and Fig. S5b ). When endogenously expressed FGFR1 and Crb3a were examined by double immunofluorescence, unique colocalizations of these proteins were detected at the plasma membrane of the cell‐to‐cell contact surface of the original DLD‐1 cells (Fig. 4 f, upper panel). However, colocalization of Crb3a and FGFR4 was mainly detected in the cytoplasm of WiDr cells (Fig. 4 f, lower panel).
Figure 4.
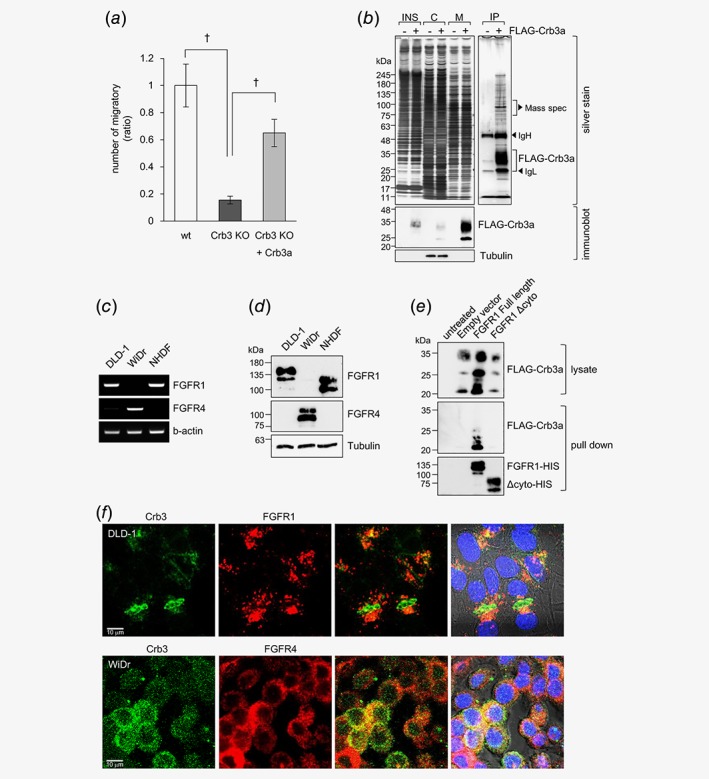
FGFRs interact with Crb3a in colon cancer cell lines. (a) Transwell chamber assay using wt, Crb3‐KO and Crb3‐KO stably expressing FLAG‐Crb3a DLD‐1 cells. (b) Crb3‐KO with or without expressing FLAG‐Crb3a were biochemically fractionated into insoluble (INS), cytoplasmic (C) and membrane (M) fractions. Immunoprecipitation was performed using membrane fraction. The details of this method were described in Supporting Information Materials and Methods. Top panels indicate silver staining, and bottom panels indicate immunoblot to qualify the fractionation. (c) Expression of FGFR1, FGFR4 and b‐actin in colon adenocarcinoma cell lines were analyzed by conventional RT‐PCR. (d) Protein expression of FGFR1 and FGFR4 in colon adenocarcinoma cell lines was analyzed by immunoblot. (e) The interaction between Crb3a and FGFR1 was examined by HIS pull‐down assay. FGFR1 interacts with Crb3a through the cytoplasmic domain in vitro. (f) Immunofluorescent analysis of endogenously expressed Crb3a and FGFRs in DLD‐1 or WiDr cells.
Knockdown assays with two different siRNA sequences against FGFR1 or FGFR4 suggested endogenous functions of FGFR1 or FGFR4 in DLD‐1 or WiDr, respectively. Namely, siFGFR1‐treated DLD‐1 cells were not significantly impacted in cellular proliferation (Figs. 5 a and 5b), but they were substantially attenuated in cellular migration according to the invasion assay using the transwell chamber (Fig. 5 c). siFGFR4‐treated WiDr cells yielded ~20% growth retardation (Figs. 5 d and 5e). FGFR1 or FGFR4 knockdowns altered cell motility or proliferation and were phenocopied by respective Crb3 KOs in DLD‐1 or WiDr cells, suggesting that Crb3a regulates tumor cell dynamics in conjunction with FGFRs. Both wt and Crb3‐KO DLD‐1 cells exogenously overexpressing FGFR4 showed increase in proliferation and cell migration (Figs. S5c–S5e). On the other hand, FGFR1‐overexpressing WiDr cells exhibited no acceleration of cell proliferation and motility in WiDr cells (Figs. S5f and S5g). Immunofluorescence using anti‐FGFR1 or anti‐FGFR4 Ab revealed that the introduced FGFR1 and FGFR4 were retained at the cytoplasm in Crb3‐KO clones of DLD‐1 and WiDr, while they were localized at the plasma membrane in wt cells (Fig. S5h), suggesting that ectopically expressed FGFR isotypes function in a context‐dependent manner. To investigate correlation between Crb3a and FGFR1 precisely, Crb3a and/or FGFR1 stably expressing clones were established. Expression of FLAG‐Crb3a, FGFR1‐HIS and FGFR1 Δcyto‐HIS was confirmed by immunoblot, which demonstrated that the transgenes were equally expressed in those cell lines (Fig. 5 f). In cell migration assays for invasive potential, the stably Crb3a‐FGFR1‐coexpressing DLD‐1 clone (generated by using Crb3‐KO DLD‐1 cell as a backbone) most efficiently augmented cellular migration, while FGFR1 lacking the cytoplasmic domain eliminated cell migration enhancement even under Crb3a overexpression (Fig. 5 g). siFGFR1‐treated Crb3‐KO DLD‐1 cells exhibited further decrease in cellular migration in comparison with that of Crb3‐KO cells treated with control siRNA, suggesting that full activation of FGFR1 might be regulated not only by Crb3 (Fig. S5i ), although increase of the motility was maximum in the Crb3a and FGFR1‐stably coexpressing clone as shown in Figure 5 g. Thus, cooperative Crb3a/FGFR1 interaction additively enhances colon cancer cell invasiveness.
Figure 5.
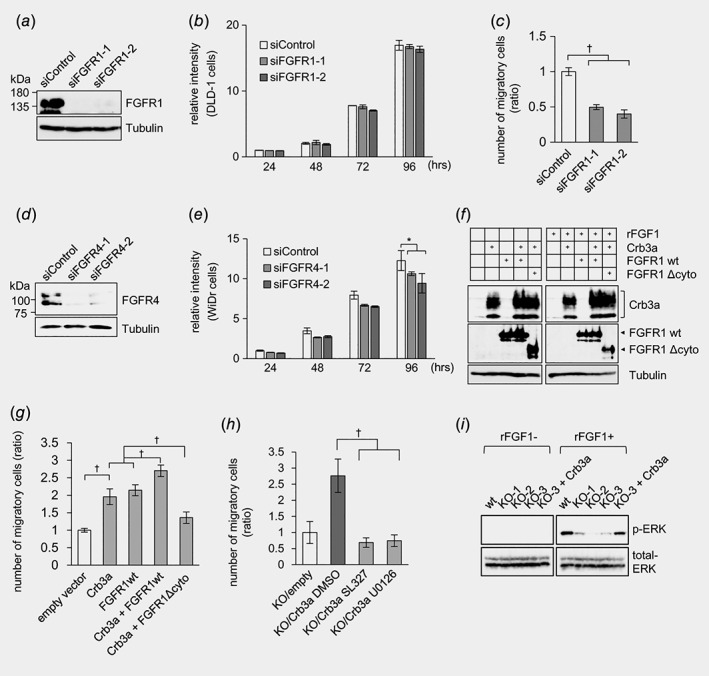
FGFR1 correlates with Crb3a in colon cancer cells. (a, d) Knockdown of FGFR1 or FGFR4 using siRNAs in (a) DLD‐1 or (d) WiDr cells were validated by immunoblot. (b, e) MTT assay using siFGFR treated (b) DLD‐1 or (e) WiDr cells. (c) Cell migration of siFGFR1 treated DLD‐1 cells was examined by transwell chamber assay. (f) Expression of FLAG‐Crb3a, FGFR1‐HIS, FGFR1Δcyto‐HIS and Tubulin in stably expressing cells was validated by immunoblot. (g) Cell migration of Crb3‐KO DLD‐1 cell stably expressing Crb3 and/or FGFR1 constructions was evaluated by transwell chamber assay. (h) Cell migration of Crb3a‐revertant DLD‐1 cells was examined by transwell chamber assay in the presence of 20 μM MEK inhibitors (SL327 or U0126). (i) ERK phosphorylation by recombinant FGF1 treatment decreased in Crb3‐KO DLD‐1 cells, and forced expression of Crb3a in Crb3‐KO cell recovered ERK phosphorylation.
Crb3 deficiency attenuates FGFR1 activation in DLD‐1 cells
ERK 1/2 is a downstream target of various receptor tyrosine kinases including FGFRs, and is phosphorylated in active status.22 Phosphor‐ERK is described as a key indicator for invasion including epithelial–mesenchymal transition (EMT).23, 24 To examine if the ERK activation is essential to the cellular migration, transwell chamber assay was performed using Crb3a‐revertant DLD‐1 clone with or without the mitogen‐activated protein kinase (MEK) inhibitor SL327 or U0126. Consequently, both inhibitors significantly decreased Crb3a‐induced cellular migration (Fig. 5 h). When DLD‐1 cells stimulated with recombinant FGFR‐ligand FGF1, a marked decrease of phosphorylated ERK (phosphor‐ERK T202 and Y204) expression was observed only in siFGFR1‐treated samples in contrast to siControl‐treated cells (Fig. S5j ). Similar results were confirmed in Crb3‐KO DLD‐1 clones, and the Crb3a‐revertant restored phosphor‐ERK expression (Fig. 5 i), whereas siFGFR4‐treated WiDr cells retained phosphor‐ERK expression, suggesting that FGFR4 is at least not key to invasiveness (Fig. S5k ). The phosphorylation of FGFR1 at Tyr‐653/654, which is essential for the catalytic activity,25 was drastically augmented in the Crb3a‐revertant clone in comparison with the parental Crb3‐KO DLD‐1 clones (Fig. S6a ). RT‐PCR assay revealed that Crb3b was expressed as well as Crb3a in DLD‐1 and WiDr cells (Fig. S6b ). Then, we generated the Crb3 mutant lacking unique sequence encoded by 3a or 3b at C‐terminus (Crb3ΔCter). In pull‐down assay of 293T cells co‐transfected with FLAG‐Crb3b and FGFR1‐HIS, or with FLAG‐Crb3ΔCter and FGFR1‐HIS, both Crb3b and Crb3ΔCter were detected suggesting that Crb3b is capable of binding to FGFR1 as well as Crb3a (Figs. S6c and S6 d ). Their coupling facilitated phosphorylation of FGFR1 in CRC cells (DLD‐1) as well as Crb3a (Fig. S6e ). The motility of Crb3‐KO DLD‐1 cell was increased by stable expression of Crb3b or Crb3ΔCter. (Fig. S6f ). Taking these together, both Crb3a and Crb3b isoforms activate FGFR signaling via common protein domains. Expression of EMT‐related protein, E‐cadherin and Snail1/2, were not altered in Crb3‐KO DLD‐1 clones, suggesting that Crb3a might regulate the motility of colon adenocarcinoma cells as being independent of EMT (Fig. S6g ). Crb3 switched its binding partner to FGFR4 in WiDr which is not expressing FGFR1, weakly affecting cell proliferation but its diverse role still remains unknown.
Correlative of Crb3a/FGFR1 expression in human colon adenocarcinoma tissues
Collectively, our results merited examining interrelated expression of Crb3a and FGFR1 in colon adenocarcinoma tissue from the patients. In conventional immunohistochemistry, both Crb3a and FGFR1 expressions were observed in the patients' tissues including primary sites, liver metastatic lesions and vascular invasion sites of colon adenocarcinomas (Fig. S7). Double immunofluorescence using laser‐scanning confocal microscopy revealed that Crb3a and apical part of FGFR1 on tumor glands coordinately expressed in primary tumors and their metastases from the patients. Namely, in three cases of well‐differentiated tub adenocarcinoma, Crb3a and FGFR1 were colocalized at the lumen site of tumor glands (Fig. 6 a; patient 1–3). In the case of por adenocarcinoma, they colocalized at the cell junction in the invasive tumor nest as well as in the nest of lymph nodal metastasis (Fig. 6 a; patient 4). Similar patterns were also observed in liver metastatic model of mice (Fig. 6 b). These findings indicated the functional coordination between Crb3a and FGFR1 in vivo.
Figure 6.
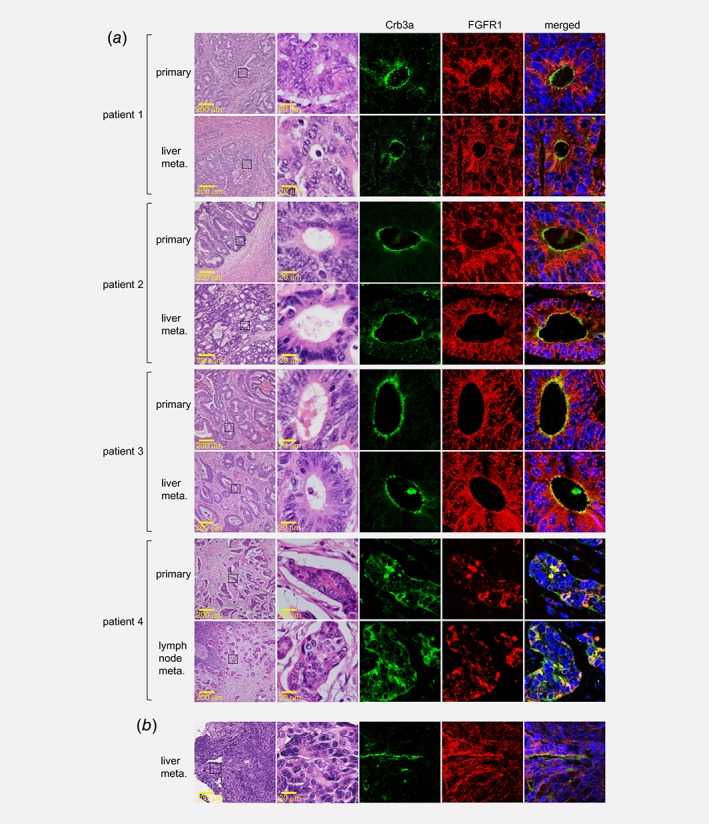
Correlative expression of Crb3a and FGFR1 in tumor tissues. Expression of Crb3a (green) and FGFR1 (red) in primary of metastatic lesions of colon adenocarcinoma derived from four independent patients (a), or in the metastatic lesion formed in a mice liver metastasis model (b) were analyzed by dual‐stained fluorescent immunohistochemistry. Left panels indicate HE staining on the same visual field in serial sections.
Discussion
Crb3 is recognized as essential for regulating epithelial cell polarity, and for tub formation during embryogenesis as evinced in developmental biology studies,8, 21 where all such prior research, we note, has been in non‐human models. Here, we uncovered a novel Crb3 biologic role in human cancer, namely a major accelerant of invasion/metastasis cooperating with FGFR1 in cologenic cancers.
We demonstrated that Crb3 facilitated cancer invasion and metastasis, while previous studies reported that Crb3a expression suppressed cell migration and metastasis of immortalized baby mouse kidney epithelial cells or canine cells in vitro.11, 13 This contradiction may be due to differences in cellular materials, the species, especially due to the point that preliminary characterization of experimental cells chosen was not enough (not well defined unlike human adenocarcinoma lines) for in vivo or in vitro assay.
Development of anti‐human Crb3a monoclonal antibody available to the formalin‐fixed paraffin‐embedded (FFPE) tissue specimen enabled unprecedented information retrieval for in vivo human tumors including colorectal cancers. Crb3a expression was observed in cancers of diverse origin, as in colorectal adenocarcinomas, and its expression was well conserved at tub structures in tumor tissues, though such retention was found even in por cancers with disparate expression patterns (Fig. 1 a). FFPE analysis of metastatic tissues also retained prominent Crb3a expression, correlating Crb3 expression with colon cancer metastasis (Fig. 1 c and Fig. S6). Semiquantitative analysis of the relative intensity of Crb3a expression revealed that Crb3a expression was found to be higher in primary tumor sites than that in normal crypt. Moreover, tumor cells of the metastatic site (liver metastasis) even increased when comparing with the primary site. This might be due to a positive selection of stronger Crb3‐expressing cells occurred among the total CRC cells at the primary lesion for their metastasis (Fig. S1d ). Our pilot experiment using tissue microarray showed Crb3a expression in the lung, stomach, pancreas, breast and prostate adenocarcinomas (data not shown). The Crb3 expression at normal glands and crypts may reflect Crb3 function in tub morphogenesis, but it was also detected in colorectal adenomas (non‐malignant), and in adenocarcinomas with well to poorly differentiated type, suggesting diverse Crb3 function in vivo (Fig. 1 b). Crb3 functional details for each neoplasm will warrant further investigation.
Given our immunostaining findings, we pursued Crb3 functional analysis in colon adenocarcinomas. Crb3 affected DLD‐1 cell migration (augmented invasive potential) and WiDr cell proliferation to some extent under in vitro assay (Fig. 2). Crb3‐KO cell‐xenografted mice revealed a Crb3 requirement in colon adenocarcinoma metastasis (Fig. 3). In seeking a counterpart molecule, LC/MS/MS analysis of immunoprecipitated proteins identified candidate FGFRs through the fragment of FGFR4 cytoplasmic domain. We thereupon established FGFR1 and FGFR4 as Crb3 binding partners in further molecular assays (Fig. 4). Examining the cause of LC/MS/MS analysis having missed FGFR1 fragment, we excised protein gel bands between 75 and 110 kDa, but the glycosylated FGFR1 is at 120–145 kDa (and glycosylated FGFR4 is at 95–110 kDa), so that FGFR1 was likely omitted from MS analysis. Despite this omission, Crb3 was demonstrated to bind FGFR1 and functional assays indicated that Crb3 efficiently induced activation of FGFR1, ultimately promoting cancer cell invasiveness (Fig. 5).
We found that FGFR subtypes are differentially expressed among colon cancer lines, and show reciprocal expression between FGFR1 and FGFR4 in DLD‐1 and WiDr. Remarkably, WiDr predominantly expresses FGFR4, and facilitative coordination of Crb3 with FGFR4 augments proliferation of FGFR4‐positive cancer cells, whereas Crb3/FGFR1 coupling strongly enhances cancer cell motility (Figs. 4 and 5; Figs. S3 and S4). Immunohistochemical examination with anti‐FGFR family antibody showed that over 90% of colorectal adenocarcinomas were FGFR1 positive, and only 14% of cases were FGFR4 positive (a few cases expressed both FGFR1 and FGFR4) (data not shown), making FGFR1 the predominant FGFR subtype in colorectal adenocarcinomas. Prior investigations described enhanced expression of FGFR1 correlated with liver metastasis,16, 26 and FGFR4 was reported to promote tumor cell growth and metastasis in colon adenocarcinomas.27, 28 However, the details of the activation mechanism and other functions, and of operative differences between FGFR subtypes in tumor progression are only partially understood.
We further demonstrated that Crb3 bound to the cytoplasmic domain of FGFR1 or FGFR4, and that its coupling recruited FGFR to the plasma membrane (Figs. 4 e and 6; Figs. S5a and S8) and consequently activated FGFR1, leading to downstream phosphorylation of ERK. The Crb3 mutant lacking C‐terminal domain (Crb3ΔCter) is still capable of coupling to FGFR1 and facilitated its phosphorylation, suggesting that known binding partners of Crb3, such as Pals1 and Par‐6, may not be essential to activate FGFR1 by forming the complex. As for the interaction of Crb3 with FGFR4, there still remains unknown biological role; further investigation should be required as the next step.
While the biological role of Crb3 might be diverse in human cancers of different origins, as it plays essential role in embryogenesis, the present study revealed that Crb3 is critically involved in triggering metastasis/invasion of cologenic cancers. In this light Crb3 is accordingly an excellent candidate for a critical molecular target in the pursuit of future advancements in colon cancer therapeutics.
Authors’ contributions
Conception and design: H.I. and E.K.; Development of methodology: H.I., K.S., T.T., and E.K.; Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): H.I., M.S., K.S., K.H., and E..; Writing, review and/or revision of the manuscript: H.I. and E.K.
Supporting information
Appendix S1: Supporting Information
Supplemental Figures
Acknowledgements
The authors thank Eiichi Morii (Department of Pathology, Osaka University Graduate School of Medicine) for providing tissue specimens. The authors thank Kyl Myrick for critical reading of the manuscript. This work was also supported by the Nitto Foundation, the Promotion and Mutual Aid Corporation for Private Schools of Japan, the Aichi Medical University Aikeikai Foundation, the Kato Memorial Bioscience Foundation and the Takeda Science Foundation to H.I. This work was also supported by the Japan Society for Promotion of Sciences (JSPS) Grant‐in‐Aid for Young Scientists (B) #26870692 and Scientific Research (C) #17K08737 to H.I.
Conflicts of interest: The authors declare that they have no potential conflicts of interest.
[Correction added on May 9 after first online publication: Figure 3 was updated.]
Contributor Information
Hidekazu Iioka, Email: hiioka@med.niigata-u.ac.jp.
Eisaku Kondo, Email: ekondo@med.niigata-u.ac.jp.
References
- 1. Manfredi S, Lepage C, Hatem C, et al. Epidemiology and management of liver metastases from colorectal cancer. Ann Surg 2006;244:254–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Shin K, Fogg VC, Margolis B. Tight junctions and cell polarity. Annu Rev Cell Dev Biol 2006;22:207–35. [DOI] [PubMed] [Google Scholar]
- 3. Royer C, Lu X. Epithelial cell polarity: a major gatekeeper against cancer? Cell Death Differ 2011;18:1470–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Martin‐Belmonte F, Perez‐Moreno M. Epithelial cell polarity, stem cells and cancer. Nat Rev Cancer 2012;12:23–38. [DOI] [PubMed] [Google Scholar]
- 5. McCaffrey LM, Montalbano J, Mihai C, et al. Loss of the Par3 polarity protein promotes breast tumorigenesis and metastasis. Cancer Cell 2012;22:601–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wu M, Pastor‐Pareja JC, Xu T. Interaction between Ras(V12) and scribbled clones induces tumour growth and invasion. Nature 2010;463:545–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Vaira V, Faversani A, Dohi T, et al. Aberrant overexpression of the cell polarity module scribble in human cancer. Am J Pathol 2011;178:2478–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Whiteman EL, Fan S, Harder JL, et al. Crumbs3 is essential for proper epithelial development and viability. Mol Cell Biol 2014;34:43–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lemmers C, Michel D, Lane‐Guermonprez L, et al. CRB3 binds directly to Par6 and regulates the morphogenesis of the tight junctions in mammalian epithelial cells. Mol Biol Cell 2004;15:1324–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fan S, Fogg V, Wang Q, et al. A novel Crumbs3 isoform regulates cell division and ciliogenesis via importin beta interactions. J Cell Biol 2007;178:387–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Whiteman EL, Liu C‐J, Fearon ER, et al. The transcription factor snail represses Crumbs3 expression and disrupts apico‐basal polarity complexes. Oncogene 2008;27:3875–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Spaderna S, Schmalhofer O, Wahlbuhl M, et al. The transcriptional repressor ZEB1 promotes metastasis and loss of cell polarity in cancer. Cancer Res 2008;68:537–45. [DOI] [PubMed] [Google Scholar]
- 13. Karp CM, Tan TT, Mathew R, et al. Role of the polarity determinant crumbs in suppressing mammalian epithelial tumor progression. Cancer Res 2008;68:4105–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Powers CJ, MclEskey SW, Wellstein A. Fibroblast growth factors, their receptors and signaling. Endocr Relat Cancer 2000;7:165–97. [DOI] [PubMed] [Google Scholar]
- 15. Turner N, Grose R. Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer 2010;10:116–29. [DOI] [PubMed] [Google Scholar]
- 16. Helsten T, Elkin S, Arthur E, et al. The FGFR landscape in cancer: analysis of 4,853 tumors by next‐generation sequencing. Clin Cancer Res 2016;22:259–67. [DOI] [PubMed] [Google Scholar]
- 17. Greenman C, Stephens P, Smith R, et al. Patterns of somatic mutation in human cancer genomes. Nature 2007;446:153–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Longo PA, Kavran JM, Kim MS, et al. Transient mammalian cell transfection with polyethylenimine. Methods Enzymol 2014;529:227–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Makarova O, Roh MH, Liu CJ, et al. Mammalian Crumbs3 is a small transmembrane protein linked to protein associated with Lin‐7 (Pals1). Gene 2003;302:21–9. [DOI] [PubMed] [Google Scholar]
- 20. Roh MH, Fan S, Liu C‐J, et al. The Crumbs3‐Pals1 complex participates in the establishment of polarity in mammalian epithelial cells. J Cell Sci 2003;116:2895–906. [DOI] [PubMed] [Google Scholar]
- 21. Fogg VC, Liu C‐J, Margolis B. Multiple regions of Crumbs3 are required for tight junction formation in MCF10A cells. J Cell Sci 2005;118:2859–69. [DOI] [PubMed] [Google Scholar]
- 22. Cobb MH, Boulton TG, Robbins DJ. Extracellular signal‐regulated kinases: ERKs in progress. Cell Regul 1991;2:965–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Javle MM, Gibbs JF, Iwata KK, et al. Epithelial‐mesenchymal transition (EMT) and activated extracellular signal‐regulated kinase (p‐Erk) in surgically resected pancreatic cancer. Ann Surg Oncol 2007;14:3527–33. [DOI] [PubMed] [Google Scholar]
- 24. Doehn U, Hauge C, Frank SR, et al. RSK is a principal effector of the RAS‐ERK pathway for eliciting a coordinate promotile/invasive gene program and phenotype in epithelial cells. Mol Cell 2009;35:511–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mohammadi M, Dikic I, Sorokin A, et al. Identification of six novel autophosphorylation sites on fibroblast growth factor receptor 1 and elucidation of their importance in receptor activation and signal transduction. Mol Cell Biol 1996;16:977–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sato T, Oshima T, Yoshihara K, et al. Overexpression of the fibroblast growth factor receptor‐1 gene correlates with liver metastasis in colorectal cancer. Oncol Rep 2009;21:211–6. [PubMed] [Google Scholar]
- 27. Desnoyers LR, Pai R, Ferrando RE, et al. Targeting FGF19 inhibits tumor growth in colon cancer xenograft and FGF19 transgenic hepatocellular carcinoma models. Oncogene 2008;27:85–97. [DOI] [PubMed] [Google Scholar]
- 28. Liu R, Li J, Xie K, et al. FGFR4 promotes stroma‐induced epithelial‐to‐mesenchymal transition in colorectal cancer. Cancer Res 2013;73:5926–35. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supporting Information
Supplemental Figures