

Abstract
Upadacitinib is a selective Janus kinase 1 inhibitor being developed for the treatment of several inflammatory autoimmune diseases, including rheumatoid arthritis. Upadacitinib is a nonsensitive substrate for metabolism by cytochrome P450 3A enzymes. This open‐label, single‐dose, multicenter study assessed the pharmacokinetics of upadacitinib following oral administration of a single 15‐mg dose of the upadacitinib extended‐release formulation in subjects with mild (n = 6) and moderate (n = 6) hepatic impairment relative to demographically matched healthy subjects (n = 6). Subjects were assigned to 1 of the 3 groups according to the Child‐Pugh classification. Relative to subjects with normal hepatic function, the ratios (90% confidence intervals) of upadacitinib area under the plasma concentration‐versus‐time profile from time 0 to infinity (AUCinf) for subjects with mild and moderate hepatic impairment were 1.28 (0.91‐1.79) and 1.24 (0.87‐1.76), respectively. The central ratios of upadacitinib maximum observed concentration (Cmax) were 1.04 (0.77‐1.39) and 1.43 (1.05‐1.95) in subjects with mild and moderate hepatic impairment, respectively, compared with subjects with normal hepatic function. No clinically significant changes in vital signs or hematology measurements were observed, and no new safety events were identified in this study. These results indicate that mild and moderate hepatic impairment has no clinically relevant effect on upadacitinib pharmacokinetics.
Keywords: ABT‐494, clinical pharmacology, hepatic impairment, Janus kinase inhibitor, pharmacokinetics, upadacitinib
The clinical implications of the JAK/STAT pathway in the regulation of innate immunity, orchestration of adaptive immune mechanisms, and constraining immune and inflammatory responses have led to the development of upadacitinib (ABT‐494).1 Upadacitinib is an oral selective Janus kinase 1 (JAK 1) inhibitor being developed for the treatment of several inflammatory diseases, including rheumatoid arthritis (RA), psoriatic arthritis, ankylosing spondylitis, giant‐cell arteritis, Crohn's disease, ulcerative colitis, and atopic dermatitis. Upadacitinib potently inhibits JAK 1 but is less potent against the other isoforms, JAK 2, JAK 3, and Tyk2.1 The enhanced selectivity of upadacitinib for JAK 1 may provide an improved benefit‐risk profile.2 Upadacitinib demonstrated favorable efficacy and acceptable safety profiles in 2 phase 2 studies and in 5 phase 3 studies in subjects with moderate to severe RA.3, 4, 5, 6, 7, 8, 9 In addition, upadacitinib recently demonstrated favorable efficacy in subjects with Crohn's disease, atopic dermatitis, and ulcerative colitis in phase 2 trials.10, 11, 12
Upadacitinib pharmacokinetics have been characterized in healthy subjects following the administration of 1‐ to 48‐mg doses of the immediate‐release (IR) formulation and 15‐ to 30‐mg doses of the extended‐release (ER) formulation.1, 13 In addition, upadacitinib pharmacokinetics were characterized in subjects with RA following the administration of multiple doses of the IR and the ER formulations. The ER formulation of upadacitinib was developed to enable once‐daily dosing in phase 3 studies. Upadacitinib plasma exposure was approximately dose proportional and time independent over the range of IR and ER doses evaluated in clinical studies.1, 13, 14, 15 The bioavailability of the ER formulation used in this study as well as in phase 3 studies is 76% relative to the IR formulation.13, 15 No significant accumulation was observed with multiple once‐daily dosing relative to a single dose of the upadacitinib ER formulation.13 Approximately 20% of the upadacitinib IR dose is eliminated unchanged in urine,1 and approximately 30% is eliminated in urine and feces as metabolites (data on file at AbbVie). Upadacitinib is a nonsensitive substrate for cytochrome P450 (CYP) 3A, which was evaluated in a phase 1 drug‐interaction study with ketoconazole. Results indicated that coadministration of ketoconazole with the upadacitinib IR formulation resulted in increased upadacitinib plasma Cmax and AUC by 70% and 75%, respectively, relative to administration of upadacitinib alone.16 Food had no clinically relevant effect on upadacitinib plasma exposure (approximately 20% increase in upadacitinib exposure with a high‐fat meal).16 Upadacitinib was administered in phase 3 trials without regard to food. In phase 3 studies in subjects with moderate to severe RA, upadacitinib was evaluated at doses of 15 and 30 mg once daily using the ER formulation. Based on phase 3 results, upadacitinib 15 mg once daily is the optimal dose in RA patients, as it provided maximum efficacy across different studies.5, 6, 7, 13, 17 The most common adverse events associated with upadacitinib treatment in subjects with moderate to severe RA in phase 3 studies were upper respiratory tract infections and nasopharyngitis.
Abnormal liver function has been reported in patients with autoimmune diseases, with therapy‐induced hepatotoxicity and chronic viral infections contributing to this comorbidity.18, 19, 20 Impaired hepatic function is documented in up to 6% of patients with RA, emerging with abnormal elevations of alkaline phosphatase, bilirubin, alanine aminotransferase (ALT), aspartate aminotransferase (AST), and serum gamma‐glutamyltransferase and low albumin levels.18, 21, 22 Abnormal liver function has also been attributed to use of disease‐modifying antirheumatic drugs, which are commonly used for treatment of immune‐mediated inflammatory diseases.23
Because of the contribution of CYP metabolism to upadacitinib clearance, it is important to characterize the impact of hepatic impairment on upadacitinib pharmacokinetics to inform if dose adjustments are required. The objective of this study was to evaluate the pharmacokinetics and safety of upadacitinib following administration of a single dose in subjects with mild and moderate hepatic impairment (according to the Child‐Pugh classification) compared with subjects with normal hepatic function.
Methods
Ethics
The study protocols and consent forms were approved by institutional review boards (IntegReview IRB, Austin, Texas, and Western IRB, Puyallup, Washington) prior to subject enrollment, and all subjects provided written informed consent before participating in the study. The study was conducted at 2 sites (Orlando Regional Medical Center Clinical Lab, Orlando, Florida, and Reliable Research Laboratory, Miami Gardens, Florida) in accordance with Good Clinical Practice guidelines and ethical principles that have their origin in the Declaration of Helsinki.
Study Design and Study Population
This was an open‐label, single‐dose study conducted at 2 sites in the United States. Subjects eligible for enrollment in the study were men and women aged 18 to 75 years, inclusive, with a body mass index of 18.0 to 38.0 kg/m2. Subjects were assigned to 1 of 3 groups (6 subjects per group) according to the Child‐Pugh classification. The 3 groups were a mild hepatic impairment group (Child‐Pugh category A), a moderate hepatic impairment group (Child‐Pugh category B), and a normal hepatic function group.24, 25 Enrollment of subjects with normal hepatic function was staggered with respect to the mild and moderate hepatic impairment groups to ensure similar demographics in the groups with respect to age, weight, sex, and ethnicity. Based on medical history, physical examination, vital sign assessment, laboratory profile, and a 12‐lead electrocardiogram (ECG), subjects with normal hepatic function were required to be in general good health, and subjects with hepatic impairment were required to be in stable condition. Use of known inhibitors or inducers of drug‐metabolizing enzymes or hormonal contraceptives within 30 days of study drug administration and through the end of study was prohibited.
Eligible subjects were admitted for confinement 1 day prior to dosing. Following a 10‐hour fast, subjects were administered a 15‐mg dose of upadacitinib ER formulation with 240 mL of water. Subjects continued to fast for 4 hours after dosing. Subjects were confined to the study site for 7 days (to ensure adequate pharmacokinetic sample collection during the terminal elimination phase in case hepatic impairment resulted in significant prolongation of upadacitinib half‐life), beginning on day ‐1 and completing after the 120‐hour pharmacokinetic sample on day 6.
Pharmacokinetic Sampling and Bioanalytical Methods
Blood samples for the assay of upadacitinib in plasma were collected by venipuncture in 3‐mL evacuated K2‐ethylenediaminetetraacetic acid‐containing collection tubes prior to dosing (0 hour) and 0.5, 1, 1.5, 2, 3, 4, 6, 8, 10, 12, 16, 24, 36, 48, 72, 96, and 120 hours after dosing. Upadacitinib concentrations in plasma were assessed by a validated liquid chromatography‐tandem mass spectrometry method as previously described.1 The lower limit of quantitation for upadacitinib was established at 0.05 ng/mL. The coefficient of variation (% CV) was ≤4.1%, and the mean absolute bias was ≤4.7% for upadacitinib assay in plasma.
Pharmacokinetics and Statistical Analyses
Pharmacokinetic parameter values for upadacitinib were estimated by noncompartmental methods using SAS version 9.3 (SAS Institute, Inc., Cary, North Carolina). Pharmacokinetic parameters included the maximum observed plasma concentration (Cmax), time to Cmax (peak time, Tmax), area under the plasma concentration‐time curve (AUC) from time 0 to the time of last measurable concentration (AUCt), AUC from time 0 to infinite time (AUCinf), terminal‐phase elimination rate constant (β), terminal‐phase elimination half‐life (t1/2), and apparent oral plasma clearance (CL/F, where F is bioavailability). The Cmax and Tmax were determined directly from the plasma concentration‐time data.
The statistical analysis was performed using SAS software, version 9.3 (SAS Institute Inc., Cary, North Carolina). An analysis of covariance (ANCOVA) was performed for Tmax, the terminal‐phase elimination rate constant (β), and the natural logarithms of Cmax and AUC. Body weight, age, sex, and smoking status were considered possible covariates (P < .10). Within the framework of the ANCOVA, the effect of each hepatic impairment group was estimated and compared with the normal hepatic function group at a significance level of 0.05. For Cmax and AUC, estimates and 90% confidence intervals, were provided for the ratio of each impaired hepatic function group relative to that of the normal hepatic function group.
Safety Assessments
Safety and tolerability were assessed for all subjects during the study and included adverse event monitoring, hematology, blood chemistry, vital signs, ECGs, and physical examinations. The number and percentage of subjects reporting treatment‐emergent adverse events were tabulated for each hepatic function group using the Medical Dictionary for Regulatory Activities (MedDRA) system organ class and preferred term. The tabulations were also provided with a breakdown by severity rating and relationship to the study drug. Subjects reporting more than 1 adverse event for a given MedDRA preferred term were counted only once for that term using the most severe incident.
Results
Participants
A total of 18 participants (15 men, 3 women) were enrolled in the study. The demographics of subjects with mild and moderate hepatic impairment were similar to the subjects with normal hepatic function (Table 1). All participants completed the study.
Table 1.
Subject Demographics
| Degree of Hepatic Impairment | |||
|---|---|---|---|
| Characteristic | Normal (n = 6) | Mild (n = 6) | Moderate (n = 6) |
| Age (years)a | 54.7 ± 10.4 (36‐66) | 50.7 ± 10.4 (31‐62) | 56.0 ± 6.8 (49‐66) |
| Weight (kg)a | 78.5 ± 10.5 (64‐95) | 78.7 ± 21.9 (54‐115) | 81.7 ± 6.9 (75‐95) |
| Height (cm)a | 173 ± 7.6 (165‐187) | 172 ± 7.2 (164‐182) | 170 ± 6.7 (158‐178) |
| Sex | |||
| Maleb | 5 (83%) | 5 (83%) | 5 (83%) |
| Femaleb | 1 (17%) | 1 (17%) | 1 (17%) |
| Race | |||
| Whiteb | 5 (83%) | 4 (67%) | 5 (83%) |
| Blackb | 1 (17%) | 2 (33%) | 1 (17%) |
| Ethnicity | |||
| Hispanic or Latinob | 3 (50%) | 3 (50%) | 4 (67%) |
| Not Hispanic or Latinob | 3 (50%) | 3 (50%) | 2 (33%) |
| Smoking status | |||
| Userb | 2 (33%) | 5 (83%) | 0 (0%) |
Mean ± standard deviation (minimum‐maximum).
Number (percentage).
Pharmacokinetics
The mean plasma concentration‐versus‐time profiles following administration of a single dose of 15 mg upadacitinib are presented on linear and log‐linear scales in Figure 1. The mean pharmacokinetic parameters following a single 15‐mg dose of upadacitinib are shown for each group in Table 2.
Figure 1.
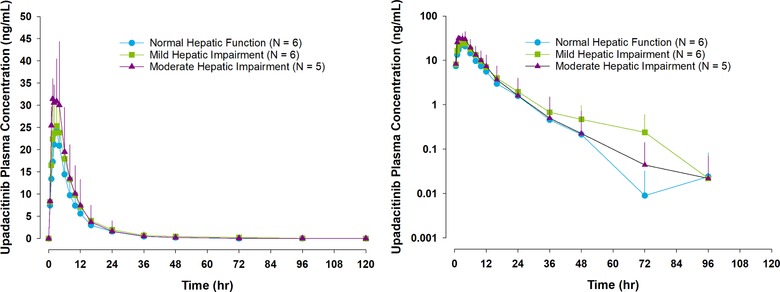
Mean ± SD upadacitinib plasma concentration‐versus‐time profiles (log and log‐linear scales) in subjects with normal hepatic function and those with mild or moderate hepatic impairment. Sensitivity analysis excluding the subject with moderate hepatic impairment who had distinctively low upadacitinib exposure. The upadacitinib plasma concentration value for the 120‐hour time was below the assay limit of quantification across all subjects and all groups.
Table 2.
Pharmacokinetic Parameters (Mean ± SD) of Upadacitinib in Subjects With Normal and Impaired Hepatic Function
| Study Group | |||
|---|---|---|---|
| Parameter | Normal Hepatic Function (n = 6) | Mild Hepatic Impairment (n = 6) | Moderate Hepatic Impairment (n = 5)a |
| Cmax (ng/mL) | 26.6 (8.39) | 27.3 (6.98) | 37.2 (8.94) |
| Tmax (h)b | 2.5 (1.5 to 3.0) | 2.5 (1.5 to 3.0) | 1.5 (1.5 to 4.0) |
| AUCt (ng·h/mL) | 212 (56.5) | 270 (75.0) | 289 (141.0) |
| AUCinf (ng·h/mL) | 215 (56.1) | 274 (74.5) | 290 (141.0) |
| t1/2 (h)c | 8.9 (4.9) | 8.0 (4.6) | 4.1 (1.5) |
| CL/F (L/h) | 74.5 (21.6) | 58.1 (15.4) | 64.1 (32.9) |
Cmax, maximum observed plasma concentration; Tmax, time to Cmax; AUCt, area under the plasma concentration‐time curve from time zero to last measurable concentration; AUCinf, area under the plasma concentration‐time curve from time zero to infinite time; t1/2, terminal elimination half‐life; CL/F, oral plasma clearance.
Conservative analysis excluding 1 subject who had distinctively low upadacitinib exposure in the moderate hepatic impairment group.
Median (minimum to maximum).
Harmonic mean ± pseudo‐standard deviation.
The estimates of the ratios and 90% confidence intervals (CIs) for Cmax, AUCt, and AUCinf by hepatic function category (mild or moderate) relative to subjects with normal hepatic function are presented in Figure 2. In this analysis, 1 subject in the moderate hepatic impairment group had approximately 70% lower upadacitinib AUC compared with the mean AUC in subjects with normal hepatic function. The subject's Cmax and AUC were also notably lower than all other subjects within the moderate hepatic impairment group. This subject was excluded from the analyses to ensure a conservative estimate for the effect of hepatic impairment on upadacitinib exposure. Relative to subjects with normal hepatic function, the estimates of ratios (90% confidence intervals) of upadacitinib area under the plasma concentration‐versus‐time profile from time 0 to infinity (AUCinf) for subjects with mild and moderate hepatic impairment were 1.28 (0.91‐1.79) and 1.24 (0.87‐1.76), respectively. The estimates of ratios (90% confidence intervals) of upadacitinib maximum observed concentration (Cmax) were 1.04 (0.77‐1.39) and 1.43 (1.05‐1.95) in subjects with mild and moderate hepatic impairment, respectively, compared with subjects with normal hepatic function. The terminal‐phase elimination half‐life (t1/2; harmonic mean ± pseudo‐standard deviation) was 7.99 ± 4.87 hours in the mild hepatic impairment group, 4.14 ± 1.46 hours in the moderate hepatic impairment group, and 8.93 ± 4.87 hours in the normal hepatic function group.
Figure 2.
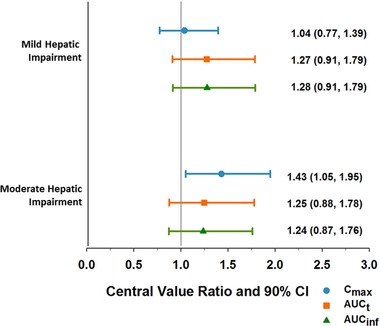
Central value and 90%CIs for upadacitinib Cmax, AUCt, and AUCinf in subjects with hepatic impairment compared with those with normal hepatic function.
The central values for upadacitinib Tmax, Cmax, AUCt, and AUCinf in subjects with mild and moderate hepatic impairment did not statistically significantly differ from the corresponding values in subjects with normal hepatic function. There was no statistically significant difference in β (as a measure for terminal t1/2) between subjects with mild hepatic impairment and subjects with normal hepatic function. The central value for β was significantly higher (indicating shorter t1/2) in subjects with moderate hepatic impairment compared with subjects with normal hepatic function.
Safety
Upadacitinib 15 mg was well tolerated by subjects in this study, and no subject had a severe or serious adverse event, discontinued participating in the study because of an adverse event, or had a clinically relevant change in vital signs, hematology and chemistry laboratory values, or ECGs. In the mild hepatic impairment group, 1 subject reported diarrhea and headache, considered by the investigator to be moderate in severity, and another subject reported nausea and dizziness, considered by the investigator as mild in severity. These events were the only events reported in this study and were assessed by the investigator as having a reasonable possibility of being related to the study drug.
Discussion
This study was conducted to characterize the effect of hepatic impairment on the pharmacokinetics, safety, and tolerability of a single 15‐mg dose of upadacitinib. The 15‐mg dose of upadacitinib ER formulation administered in this study is 1 of the 2 doses (15 and 30 mg) that were evaluated in phase 3 clinical trials in rheumatoid arthritis and is the dose that optimized efficacy across studies in rheumatoid arthritis patients.5, 6, 7, 17, 26, 27 In this study, there was no statistically significant difference in upadacitinib Cmax and AUC in subjects with mild and moderate hepatic impairment compared with subjects with normal hepatic function. The upadacitinib AUC central value was 28% and 24% higher in subjects with mild and moderate hepatic impairment, respectively, compared with subjects with normal hepatic function. The central value of upadacitinib Cmax was similar in subjects with mild hepatic impairment and 43% higher in subjects with moderate hepatic impairment compared with subjects with normal hepatic function. These results were based on an analysis that excluded an outlier with low upadacitinib exposure in the moderate hepatic impairment group. A sensitivity analysis was conducted including the outlier subject (data not shown); exclusion of the outlier subject provided a more conservative estimate from the effect of moderate impairment on upadacitinib exposures. Single‐dose administration of upadacitinib was well tolerated in all subjects in the study.
This study was conducted in accordance with the Food and Drug Administration guidance on conducting pharmacokinetic studies in subjects with impaired hepatic function.28 Because upadacitinib exhibits linear and time‐independent pharmacokinetics at the doses evaluated in phase 1, phase 2, and phase 3 clinical studies,1, 13, 14, 15 a single‐dose assessment was considered sufficient to provide adequate information about the impact of the compound on hepatic impairment. Considering that upadacitinib is not extensively bound to plasma proteins (data on file at AbbVie), changes in the free fraction associated with decreased protein binding in hepatic impairment were not anticipated to have a relevant impact on upadacitinib clearance or total exposure. Treatment with upadacitinib was anticipated to be not recommended in subjects with severe hepatic impairment; therefore, the impact of severe hepatic impairment on upadacitinib exposure was not evaluated in this study. Upadacitinib was administered under fasting conditions in this study to minimize any potential confounding sources of variability in the pharmacokinetics between subjects (as a standard practice). Given the lack of a clinically relevant effect of food on upadacitinib exposure, results from this study are applicable to the administration of upadacitinib under fed or fasting conditions.
The study was conducted in subjects with mild and moderate hepatic impairment, based on Child‐Pugh classification, and a matching control group with normal liver function. The effect of hepatic impairment on the upadacitinib pharmacokinetics characterized in this study is considered applicable to subjects with immunologic disorders (eg, rheumatoid arthritis). In the population pharmacokinetic analyses across phase 1 to 3 studies encompassing healthy subjects and subjects with RA,15 baseline AST, ALT, and bilirubin had no impact on upadacitinib exposure, consistent with lack of a clinically meaningful impact of mild and moderate hepatic impairment on the upadacitinib exposure observed in this dedicated phase 1 study. In addition, the effect of renal impairment on upadacitinib exposure was similar in RA patients and non‐RA patients.14, 15, 29
Loss of CYP activity has been documented to correlate with reduced hepatic function in a Child‐Pugh score‐dependent manner.30 Because there is a metabolic component in clearance of upadacitinib that is mediated through CYP3A4 with a minor possible contribution of CYP2D6, it was possible to assume that upadacitinib exposure may increase with reduced liver function. Previous studies established that the CYP2D6 metabolic phenotype was not correlated with upadacitinib clearance.14 The relatively small increase in upadacitinib exposure (<30% for AUC and <45% for Cmax) in subjects with mild and moderate hepatic impairment can be attributed in part to changes in CYP3A activity in subjects with hepatic impairment. The effects of mild and moderate hepatic impairment on upadacitinib exposure are not considered clinically relevant and therefore do not warrant dose adjustment in patients with either mild or moderate hepatic impairment and an autoimmune disease, such as RA; this is further supported by a upadacitinib safety profile characterized in phase 3 studies in RA5, 6, 7, 13, 17 and results of exposure‐response analyses of upadacitinib safety and effects on laboratory parameters; detailed reports of these analyses are forthcoming.
Additional assessments were conducted to characterize the effect of other intrinsic factors on upadacitinib pharmacokinetics. Assessment of the effect of renal impairment on upadacitinib exposure demonstrated that upadacitinib AUC was 18%, 33%, and 44% higher in subjects with mild, moderate, and severe renal impairment, respectively, and Cmax was similar compared with subjects with normal renal function.29 In addition, population pharmacokinetic analyses demonstrated that age, weight, sex, race, and ethnicity have no clinically relevant effect on upadacitinib exposure.14 Taken together, results from individual studies as well as population and exposure‐response analyses across multiple studies indicate that no dose adjustment is warranted for upadacitinib based on the evaluated intrinsic factors of the patients.
Conclusions
Mild and moderate hepatic impairment have no clinically relevant effect on upadacitinib exposure; thus, dose adjustment is not warranted in RA patients with mild or moderate hepatic impairment.
Declaration of Conflicting Interests
Drs. Trueman, Mohamed, Feng, Lacerda, and Othman are employees of AbbVie and may hold AbbVie stock or stock options. Dr. Marbury is an employee and equity owner of Orlando Clinical Research Center and reports no other conflicts of interest.
Acknowledgments
The authors thank the study participants and the investigators and site personnel who conducted the study. Medical writing support was provided by Wesley Wayman, an employee of AbbVie Inc.
Funding
This work was supported by AbbVie. AbbVie contributed to the study design, research, and interpretation of the data and the writing, review, and approval of the manuscript.
Data‐Sharing Statement
AbbVie is committed to responsible data sharing regarding the clinical trials we sponsor. This includes access to anonymized, individual, and trial‐level data (analysis data sets), as well as other information (eg, protocols and clinical study reports), as long as the trials are not part of an ongoing or planned regulatory submission. This includes requests for clinical trial data for unlicensed products and indications.
These clinical trial data can be requested by any qualified researchers who engage in rigorous independent scientific research and will be provided following review and approval of a research proposal and statistical analysis plan (SAP) and execution of a data‐sharing agreement (DSA). Data requests can be submitted at any time, and the data will be accessible for 12 months, with possible extensions considered. For more information on the process, or to submit a request, visit the following link: https://www.abbvie.com/our-science/clinical-trials/clinical-trials-data-and-information-sharing/data-and-information-sharing-with-qualified-researchers.html.
Dr. Ahmed A Othman is a fellow of the American College of Clinical Pharmacology.
References
- 1. Mohamed MF, Camp HS, Jiang P, Padley RJ, Asatryan A, Othman AA. Pharmacokinetics, safety and tolerability of ABT‐494, a novel selective JAK 1 inhibitor, in healthy volunteers and subjects with rheumatoid arthritis. Clin Pharmacokinet. 2016;55(12):1547‐1558. [DOI] [PubMed] [Google Scholar]
- 2. Norman P. Selective JAK inhibitors in development for rheumatoid arthritis. Expert Opin Investig Drugs. 2014;23(8):1067‐1077. [DOI] [PubMed] [Google Scholar]
- 3. Kremer JM, Emery P, Camp HS, et al. A phase IIb study of ABT‐494, a selective JAK‐1 inhibitor, in patients with rheumatoid arthritis and an inadequate response to anti‐tumor necrosis factor therapy. Arthritis Rheumatol. 2016;68(12):2867‐2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Genovese MC, Smolen JS, Weinblatt ME, et al. Efficacy and safety of ABT‐494, a selective JAK‐1 inhibitor, in a phase IIb study in patients with rheumatoid arthritis and an inadequate response to methotrexate. Arthritis Rheumatol. 2016;68(12):2857‐2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Burmester GR, Kremer JM, Van den Bosch F, et al. Safety and efficacy of upadacitinib in patients with rheumatoid arthritis and inadequate response to conventional synthetic disease‐modifying anti‐rheumatic drugs (SELECT‐NEXT): a randomised, double‐blind, placebo‐controlled phase 3 trial. Lancet. 2018;391(10139):2503‐2512. [DOI] [PubMed] [Google Scholar]
- 6. Genovese MC, Fleischmann R, Combe B, et al. Safety and efficacy of upadacitinib in patients with active rheumatoid arthritis refractory to biologic disease‐modifying anti‐rheumatic drugs (SELECT‐BEYOND): a double‐blind, randomised controlled phase 3 trial. Lancet. 2018;391(10139):2513‐2524. [DOI] [PubMed] [Google Scholar]
- 7. Smolen JCS, Emery P, Rigby W, et al. Upadacitinib as monotherapy: a phase 3 randomized controlled double‐blind study in patients with active rheumatoid arthritis and inadequate response to methotrexate. Ann Rheum Dis. 2018;77(suppl 2):67‐68. [Google Scholar]
- 8. Higgins JPT, Green S. (editors). Cochrane Handbook for Systematic Reviews of Interventions, version 5.1.0 [updated March 2011]. The Cochrane Collaboration; 2011. www.cochrane-handbook.org. [Google Scholar]
- 9. Atsukawa M, Tsubota A, Koushima Y, et al. Efficacy and safety of ombitasvir/paritaprevir/ritonavir in dialysis patients with genotype 1b chronic hepatitis C. Hepatol Res. 2017;47(13):1429‐1437. [DOI] [PubMed] [Google Scholar]
- 10. Sandborn WJ, Feagan BG, Panes J, et al. Safety and efficacy of ABT‐494 (upadacitinib), an oral JAK1 inhibitor, as induction therapy in patients with Crohn's disease: results from Celest. Gastroenterology. 2017;152(5):S1308‐S1309. [Google Scholar]
- 11. Guttman‐Yassky E, Silverberg JI, Thaci D, et al. Primary results from a phase 2b, randomized, placebo‐controlled trial of upadacitinib for patients with atopic dermatitis. American Academy of Dermatology Annual Meeting; February 16‐20, 2018; San Diego, CA. Abstract 6533. [Google Scholar]
- 12. Sandborn W, Ghosh S, Panes J, et al. Efficacy and safety of upadacitinib as an induction therapy for patients with moderately‐to‐severely active ulcerative colitis: data from the phase 2b study u‐achieve [OP 195]. United European Gastroenterology Week; October 20‐24, 2018; Vienna, Austria.
- 13. Mohamed M‐F, Zeng T, Marroum PJ, Song I, Othman AA. Pharmacokinetics of upadacitinib with the clinical regimens of the extended‐release formulation utilized in rheumatoid arthritis phase 3 trials. Clin Pharmacol Drug Develop. 2019;8(2):208‐216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Klunder B, Mohamed MF, Othman AA. Population pharmacokinetics of upadacitinib in healthy subjects and subjects with rheumatoid arthritis: analyses of phase i and ii clinical trials. Clin Pharmacokinet. 2018;57(8):977‐988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Klünder B, Mittapalli RK, Mohamed M‐EF, et al. Population pharmacokinetics of upadacitinib using the immediate‐release and extended‐release formulations in healthy subjects and subjects with rheumatoid arthritis: analyses of phase I–III clinical trials [published online ahead of print 2019]. Clin Pharmacokinet. 10.1007/s40262-019-00739-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mohamed MF, Jungerwirth S, Asatryan A, Jiang P, Othman AA. Assessment of effect of CYP3A inhibition, CYP induction, OATP1B inhibition, and high‐fat meal on pharmacokinetics of the JAK1 inhibitor upadacitinib. Br J Clin Pharmacol. 2017;83(10):2242‐2248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Van Vollenhoven R, Pangan AL, Friedman A, et al. A phase 3, randomized, controlled trial comparing upadacitinib monotherapy to MTX monotherapy in MTX‐naïve patients with active rheumatoid arthritis [abstract]. Arthritis Rheumatol. 2018;70(suppl 10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cojocaru M, Cojocaru IM, Silosi I, Vrabie CD. Liver involvement in patients with systemic autoimmune diseases. Maedica (Buchar). 2013;8(4):394‐397. [PMC free article] [PubMed] [Google Scholar]
- 19. Selmi C, De Santis M, Gershwin ME. Liver involvement in subjects with rheumatic disease. Arthritis Res Ther. 2011;13(3):226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Joseph AM. Treatment of rheumatoid arthritis in patients with concomitant chronic hepatitis C infection. Ther Adv Musculoskelet Dis. 2012;4(1):35‐40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Amital H, Arnson Y, Chodick G, Shalev V. Hepatotoxicity rates do not differ in patients with rheumatoid arthritis and psoriasis treated with methotrexate. Rheumatology (Oxford). 2009;48(9):1107‐1110. [DOI] [PubMed] [Google Scholar]
- 22. Olago‐Rakuomi A, Oyoo GO, Kamaue E, Okalebo F, Ogutu E. Prevalence of abnormal liver function tests in rheumatoid arthritis. Afr J Rheumatol. 2017;5(1):70‐75. [Google Scholar]
- 23. Ogdie A, Grewal SK, Noe MH, et al. Risk of incident liver disease in patients with psoriasis, psoriatic arthritis, and rheumatoid arthritis: a population‐based study. J Invest Dermatol. 2018;138(4):760‐767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pugh RN, Murray‐Lyon IM, Dawson JL, Pietroni MC, Williams R. Transection of the oesophagus for bleeding oesophageal varices. Br J Surg. 1973;60(8):646‐649. [DOI] [PubMed] [Google Scholar]
- 25. Child CG, Turcotte JG. Surgery and portal hypertension. Major Probl Clin Surg. 1964;1:1‐85. [PubMed] [Google Scholar]
- 26. Genovese MCFR, Combe B, Hall S, et al. Upadacitinib (ABT‐494) in patients with active rheumatoid arthritis and inadequate response or intolerance to biological Dmards: a phase 3 randomized, placebo‐controlled, double‐blind study of a selective JAK‐1 inhibitor [abstract]. Arthritis Rheumatol. 2017;69(suppl 10). [Google Scholar]
- 27. Fleischmann R, Pangan AL, Mysler E, et al. A phase 3, randomized, double‐blind study comparing upadacitinib to placebo and to adalimumab, in patients with active rheumatoid arthritis with inadequate response to methotrexate [abstract]. Arthritis Rheumatol. 2018;70(suppl 10). [DOI] [PubMed] [Google Scholar]
- 28. United States Food and Drug Administration . Guidance for Industry‐ Pharmacokinetics in Patients with Impaired Hepatic Function: Study Design, Data Analysis, and Impact on Dosing and Labeling. 2003. https://www.fda.gov/ucm/groups/fdagov-public/@fdagov-drugs-gen/documents/document/ucm072123.pdf. Accessed March 13, 2019.
- 29. Mohamed M‐EF, Trueman S, Feng T, Anderson J, Marbury TC, Othman AA. Characterization of the effect of renal impairment on upadacitinib pharmacokinetics. J Clin Pharmacol. 2019;59(6):856‐862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Albarmawi A, Czock D, Gauss A, et al. CYP3A activity in severe liver cirrhosis correlates with Child‐Pugh and model for end‐stage liver disease (MELD) scores. Br J Clin Pharmacol. 2014;77(1):160‐169. [DOI] [PMC free article] [PubMed] [Google Scholar]