

Abstract
Darolutamide is a novel androgen receptor (AR) antagonist with a distinct chemical structure compared to other AR antagonists and currently in clinical Phase 3 trials for prostate cancer. Using cell‐based transactivation assays, we demonstrate that darolutamide, its diastereomers and its main metabolite keto‐darolutamide are strong, competitive antagonists for AR wild type, and also for several mutants identified in prostate cancer patients for which other AR antagonists show reduced antagonism or even agonism. Darolutamide, its two diastereomers and main metabolite are also strong antagonists in assays measuring AR N/C interaction and homodimerization. Molecular modeling suggests that the flexibility of darolutamide allows accommodation in the W742C/L mutated AR ligand‐binding pocket while for enzalutamide the loss of the important hydrophobic interaction with W742 leads to reduced AR interaction. This correlates with an antagonistic pattern profile of coregulator recruitment for darolutamide. In vitro efficacy studies performed with androgen‐dependent prostate cancer cell lines show that darolutamide strongly reduces cell viability and potently inhibits spheroid formation. Also, a marked down‐regulation of androgen target genes paralleled by decreased AR binding to gene regulatory regions is seen. In vivo studies reveal that oral dosing of darolutamide markedly reduces growth of the LAPC‐4 cell line‐derived xenograft and of the KuCaP‐1 patient‐derived xenograft. Altogether, these results substantiate a unique antagonistic profile of darolutamide and support further development as a prostate cancer drug.
Keywords: prostate cancer, androgen receptor, antagonist
Short abstract
What's new?
Comparison of genomic landscapes from primary prostate cancer and metastatic tumor shows that resistance mechanisms are centered on androgen signaling and increased synthesis. Here, the novel androgen receptor (AR) antagonist darolutamide shows strong in vitro and in vivo efficacy in different prostate cancer models. Darolutamide retains its antagonistic properties at elevated androgen levels and for several AR mutants identified in therapy‐resistant patients. A unique binding profile inside the AR ligand‐binding domain linked to the flexibility of darolutamide is proposed. Altogether, these results substantiate a unique antagonistic profile of darolutamide and support further development as a prostate cancer drug.
Abbreviations
- AR
androgen receptor
- cFBS
charcoal‐stripped fetal bovine serum
- DHT
dihydrotestosterone
- LBD
ligand‐binding domain
Introduction
Prostate adenocarcinoma is one of the most frequent male malignancies and an important cause of cancer‐related deaths worldwide.1 Treatment options for early disease stages include surgery, local irradiation and androgen deprivation therapy. Most men under androgen deprivation therapy will develop castration‐resistant prostate cancer and these tumors usually still respond to androgen receptor (AR) antagonists and androgen synthesis inhibitors.2, 3 Unfortunately, therapy resistance, due to a variety of genomic alterations and adaptive responses of the tumor, is often observed.2, 3 Comparison of genomic landscapes from primary and metastatic tumor samples shows that resistance mechanisms are centered on androgen signaling and include overexpression, rearrangement of the gene locus, enhancer hijacking, ligand‐binding domain (LBD) mutations and aberrant splice variants of the AR.4, 5, 6, 7, 8 In addition, increased androgen synthesis linked to overexpression of steroidogenesis enzymes in tumor tissue is observed.9 These findings vindicate ongoing efforts toward the identification of more efficacious AR signaling blockers.
Darolutamide is a novel oral AR antagonist which has recently completed a pivotal Phase 3 clinical study to determine its efficacy in nonmetastatic castration‐resistant prostate cancer.10, 11 It has a unique chemical structure, exists as two pharmacologically active diastereomers, (S,R)‐ and (S,S)‐darolutamide,12 and forms keto‐darolutamide as main metabolite in patients.13, 14 Darolutamide strongly impairs androgen binding to the AR and androgen‐induced nuclear translocation.12 It shows strong in vivo efficacy in the vertebral cancer of the prostate (VCaP) xenograft model which expresses high levels of AR wild type and of the V7 splice variant,12 and in the enzalutamide‐resistant MR49F model which contains the AR mutations F877L and T878A.15
Here, we determined the activity of darolutamide in several cellular assays and prostate cancer models. Strong inhibition of transactivation, N‐ and C‐terminal domain interaction and homodimerization were seen for AR wild type and W742C/L mutants. Molecular modeling revealed a unique positioning of darolutamide inside the AR LBD which may explain why it retains its antagonistic properties for the W742C/L forms. Also, an antagonistic pattern profile of coregulator interaction was identified for darolutamide bound to AR W742C. A marked reduction of prostate cancer cell viability and spheroid formation was measured after darolutamide treatment. Concordantly, potent in vivo efficacy was observed for the LAPC‐4 and KuCaP‐1 models, which harbor AR wild type and the W742C form,16 respectively.
Materials and Methods
Reagents, plasmids and cell lines
Darolutamide, its diastereomers and main metabolite keto‐darolutamide were synthesized by Orion Corporation (Espoo, Finland). Enzalutamide, apalutamide and bicalutamide were obtained from Selleckchem (Munich, Germany) or synthesized in‐house. The chemical structures are shown in Supporting Information Figure 1. R1881 was synthesized in‐house.
Human AR expression plasmids and reporter vectors have been described.17 The mammalian two‐hybrid kit (Agilent Technologies, Santa Clara, CA) was used for the AR N/C interaction assay. The AR region 2–554 was introduced in‐frame into the pCMV‐AD plasmid, the 665–918 region into the pCMV‐BD plasmid, and the pFR‐Luc vector used as reporter. The NanoBiT protein:protein interaction system (Promega, Madison, WI) was utilized for dimerization assays, using the vectors TK/LgBiT‐AR and TK/AR‐SmBiT which contain human AR. Site‐directed mutagenesis was performed with the QuickChange II XL kit (Agilent Technologies) and appropriate primer pairs.
The cell lines used were: VCaP (CRL‐2876, American Type Culture Collection (ATCC), Manassas, VA), LAPC‐4 (CRL‐13009, ATCC), LNCaP (ACC‐256, Deutsche Sammlung von Mikroorganismen und Zellkulturen (DSMZ), Braunschweig, Germany), PC‐3 (ACC‐465, DSMZ) and HCT‐116 (CCL‐247, ATCC). They were routinely grown in the recommended medium in the presence of 10% (v/v) fetal bovine serum (Life Technologies, Carlsbad, CA) at 37°C and 5% CO2. Authentication was performed by the DSMZ using short tandem repeat DNA typing analysis. Cell lines were confirmed to be free of mycoplasma using the MycoAlert Mycoplasma Detection Assay (Lonza, Basel, Switzerland).
Cellular transfection assays
For the transactivation assays, PC‐3 cells grown in medium supplemented with charcoal‐stripped fetal bovine serum (cFBS) were transfected with expression vectors for AR wild type or mutants, together with the pGL4‐MMTV reporter plasmid (Promega). Luciferase activity was measured 24 hr later, as described.17, 18 The average value of six wells treated in parallel was taken. Experiments were repeated at least three times independently.
For the AR N/C interaction assay, HCT‐116 cells were seeded into 6‐well plates and transfected 1 day later with 2 μg of pFR‐Luc reporter vector, and 250 ng of pCMV‐AD‐AR(2‐554) and pCMV‐BD‐AR(665‐918) plasmids. The cells were reseeded 1 day later in 384‐well plates and R1881 was added at 10 nM final concentration, together with a dose‐range of AR antagonists. The luciferase assay was performed 24 hr later using the Steadylite plus Reporter Gene Assay System and the Victor X3 device (Perkin Elmer, Waltham, MA).
For the AR dimerization assay, the NanoBiT system was used according to the manufacturer's protocol (Promega). HCT‐116 cells were seeded into 96‐well plates and transfected 24 hr later with 50 ng of LgBiT‐AR and 50 ng of AR‐SmBiT plasmids. Nano‐Glo Living Cell Substrate (Promega) was added 24 hr later and basal luminescence measured. Then R1881 was added at 10 nM final concentration together with AR antagonists, and luminescence was determined in a Victor X3 device (Perkin Elmer) 30 min later.
Molecular modeling
The model of the antagonistic AR conformer was generated using SwissModel19 based on the antagonistic conformation of the progesterone receptor (PDB entry 2ovh,20). A three‐dimensional low‐energy conformation of darolutamide was generated using the Discovery Studio Suite 2017. Receptor‐ligand modeling was done using Coot version 0.8.8,21 figures were prepared using PyMOL (The PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC, Cambridge, MA).
Coregulator interaction assay
HCT‐116 cells exhibited the highest transfection rate in a panel of AR‐negative cell lines (not shown) and were selected for the assay. Five million cells were seeded into 75‐cm2 flasks in medium supplemented with cFBS, and then transfected with 25 μg pSG5‐based expression plasmids for AR wild type or W742C. After 24 hr the cells were treated with 10 nM dihydrotestosterone (DHT) or 10 μM AR antagonist for 30 min. Cell pellets were then prepared after trypsinization and stored at −80°C. After quantification and quality control, normalized samples were hybridized in triplicate to the Nuclear Receptor PamChip® microarray with 154 peptide motifs originating from 66 nuclear receptor coregulators, using the PamStation® 96 (MARCoNI assay, PamGene International BV, Den Bosch, The Netherlands). The binding of AR wild type or mutant to peptide motifs was determined on three biological replicates after image acquisition of individual wells. AR image analysis was performed using the BioNavigator6 software package (PamGene). Results for the 5 amino acid‐long FQNLF motif found in the AR were discarded as the antibody used to detect the AR covers a region including this motif. Further calculations and statistics were performed using the R software environment (www.r-project.org/). Ligand‐modulated binding of transfected cell extracts to each coregulator peptide under various treatments was compared to controls after log‐transformation and fitting to a linear model.
Cell viability and spheroid formation assays
The experiments were performed in 384‐well plates with VCaP, LAPC‐4 and LNCaP prostate cancer cells grown in cFBS. The cells were stimulated with 0.1, 10 or 1 nM R1881, respectively, and treated with a dose‐range of AR antagonists. Viability was measured 6 days later from triplicate samples using the CellTiter‐Glo® assay (Promega) in a Victor X3 device. C0 was defined as the signal measured for cells treated only with R1881 and CI as the signal measured for cells grown without R1881, at Day 6. IC50 values were calculated using the E‐workbook software (IDBS, Munich, Germany).
For spheroid formation assays, single‐cell suspensions were plated in cFBS‐supplemented medium into ultralow attachment 96‐well spheroid microplates (Corning Life Sciences, Amsterdam, The Netherlands). A total of 10,000 VCaP cells or 5,000 LAPC‐4 cells were plated in 100 μl medium. After 24 hr, R1881 at 1 nM final concentration and different compound concentrations were added. Culture medium was changed every 3–4 days. Spheroid formation was determined in triplicate at different time points by microscopy using the ImageJ software and the formula (V [106 μm3] = π × d max × d min 2/6,000,000).
Gene expression analysis
Cells were grown in 12‐well plates in medium supplemented with cFBS for 2 (VCaP) or 3 (LAPC‐4) days and then treated with 1 nM R1881 and AR antagonists for 20 hr. RNA was then extracted from 64,000 (VCaP) or 32,000 (LAPC‐4) cells using the RNeasy Plus Mini kit (Qiagen, Hilden, Germany). For tumor xenografts, 30 mg snap‐frozen tissue was cut into small slices, disrupted and homogenized before RNA extraction. Synthesis of cDNA was performed with the SuperScript® III First Strand Synthesis SuperMix for qRT‐PCR (Thermo Fisher Scientific, Waltham, MA). Real‐time PCR was performed with the TaqMan® Fast Advanced Master Mix (Applied Biosystems, Foster City, CA) and appropriate primers. The results were normalized to human cyclophilin A (PPIA Control Mix Applied Biosystems). A 7900 HT Fast Real‐Time PCR system (Applied Biosystems) was used for measurement.
AR ELISA
VCaP cells were seeded in complete medium and treated 1 day later with AR antagonists for 3 days. LAPC‐4 cells were seeded in medium supplemented with cFBS and treated 4 days later with 10 nM R1881 and AR antagonists for 1 day. Proteins were extracted using the M‐PER Mammalian Protein Extraction Reagent (Thermo Fisher Scientific). For total AR determination, 5 μg of whole cell lysate was analyzed with a sandwich AR ELISA kit (Active Motif, Carlsbad, CA) and a TECAN microplate reader (Zurich, Switzerland).
Chromatin immunoprecipitation
VCaP and LAPC‐4 cells were grown for 48 hr in phenol‐red free medium supplemented with 10% cFBS and then stimulated with 1 nM R1881 and treated with 2 μM darolutamide. The cells were harvested 20 hr later and processed as described previously.22 Briefly, cell nuclei were extracted with buffer A (150 mM NaCl, 20 mM EDTA (pH 8), 50 mM Tris (pH 7.5), 0.5% NP‐40, 1% Triton‐X‐100, 20 mM NaF) and the DNA was sheared in buffer B (150 mM NaCl, 1% NP‐40, 0.5% sodium deoxycholate, 50 mM Tris–HCl (pH 8), 20 mM EDTA, 20 mM NaF, 0.13% SDS) to approximately 300 base‐pair‐long fragments using the Bioruptor Pico with sonication beads (Diagenode, Liège, Belgium). Immunoprecipitation was performed with 2.5 μg AR‐specific antibody (06‐680, Millipore Corporation, Billerica, MA) in buffer B and protein‐antibody complexes were isolated with 30 μl of magnetic Protein A bead suspension (Invitrogen, Carlsbad, CA). Overnight incubation and washing steps were done with the IPStar device (Diagenode) using wash buffers 1–4 and elution buffer. Immunoprecipitation samples were treated with 1 μl RNase A (10 mg/ml) and 1 μl proteinase K (20 mg/ml), and incubated for 30 min at 37°C. The samples were then shaken at 65°C for 16 hr. DNA was extracted with the PCR purification kit (Qiagen) and AR occupancy levels determined by qRT‐PCR using appropriate primers (Supporting Information Table 1).
Animal studies
Approval for all in vivo experiments was obtained from the internal animal welfare committee of the company and included an ethical evaluation of the proposed procedures. Official permission was granted by the Governmental Animal Care and Use Office (Landesamt für Gesundheit und Soziales, Germany).
For the LAPC‐4 model,23 testosterone‐supplemented CB17‐SCID male mice were inoculated subcutaneously with 2 × 106 cells. Treatment started on Day 26 post tumor inoculation at a mean tumor size range of 137–150 mm3. Efficacy was evaluated on Day 56. For the KuCaP‐1 model,16 CB17‐SCID male mice were inoculated subcutaneously with 5 × 5 mm tumor fragments. Treatment started on Day 35 post tumor inoculation at a mean tumor size of about 150 mm3. Treatment schedules for the different AR antagonists were chosen based on their respective pharmacokinetic properties.12, 24 Efficacy was evaluated on Day 68. Tumor size and body weight loss were monitored at least twice weekly. Tumor size was measured with calipers and converted into volume using the formula: (length × width2)/2. Efficacy was evaluated by calculating the % ΔT/ΔC which takes into account tumor size at treatment start25 and is defined as: [(mean tumor volume in the treated group on final day − mean tumor volume in the treated group at start of therapy)/(mean tumor volume in the vehicle control group on final day − mean tumor volume in the vehicle control group at start of therapy)] × 100. Efficacy was defined as % ΔT/ΔC ≤ 40%, according to NCI criteria.26 The statistics are as follows: ****p = 0.0001, *p = 0.04 versus vehicle using one‐way ANOVA, Dunnett's method on Log‐transformed tumor volumes at the end of the study.
Results
Darolutamide is a strong antagonist for AR wild type and several LBD mutants
First we determined the antagonistic properties of the different compounds in a cell‐based transactivation assay using an MMTV‐driven luciferase reporter. For darolutamide, the (S,R)‐ and (S,S)‐diastereomers, and the main in vivo metabolite keto‐darolutamide we found a strong antagonistic activity against AR wild type, when stimulating with 1 nM R1881. This was reduced but still significantly better than observed for other AR antagonists when increasing the androgen level used for stimulation to 10 nM (Table 1). Strong antagonism was also measured for darolutamide, its diastereomers and for keto‐darolutamide when testing the W742C and W742L forms (Table 1). We also analyzed additional AR mutants identified in PCa patients and found in most cases the IC50 values for darolutamide to be below or around 200 nM after stimulation with 0.1 nM R1881 (Supporting Information Table 2). Importantly, darolutamide was a potent antagonist for AR F877L which is resistant to enzalutamide and apalutamide.27 Marked antagonism was additionally measured for the M896T and M896V forms for which enzalutamide had only reduced activity (Supporting Information Table 2). As reported earlier,12 darolutamide lost some antagonistic activity in presence of the T878A mutation (Supporting Information Table 2).
Table 1.
Effects of AR antagonists on AR wild type and W742C/L mutants in cell‐based transactivation assays
| Compound | R1881 (nM) | AR wild type | AR W742C | AR W742L |
|---|---|---|---|---|
| Darolutamide | 0.1 | 80 ± 103 | 170 ± 701,3 | 200 ± 301,2,3 |
| 1 | 460 ± 201,2,3 | 700 ± 2901,2,3 | 860 ± 1801,2,3 | |
| 10 | 6,560 ± 1,430 | 7,530 ± 2,3603 | 9,400 ± 7803 | |
| (S,R)‐darolutamide | 0.1 | 60 ± 101,2,3 | 120 ± 201,3 | 190 ± 801,2,3 |
| 1 | 400 ± 801,2,3 | 490 ± 1101,2,3 | 1,110 ± 2201,2,3 | |
| 10 | 5,880 ± 2,920 | 6,800 ± 1,8603 | 9,400 ± 7803 | |
| (S,S)‐darolutamide | 0.1 | 100 ± 30 | 210 ± 601,3 | 290 ± 1401,3 |
| 1 | 650 ± 120 | 750 ± 1401,2,3 | 1,420 ± 2201,3 | |
| 10 | 7,560 ± 2,430 | 8,570 ± 1,7601,2,3 | >10,000 | |
| Keto‐darolutamide | 0.1 | 80 ± 203 | 160 ± 101,3 | 300 ± 301,2,3 |
| 1 | 510 ± 901,2,3 | 600 ± 6801,2,3 | 1,250 ± 2501,3 | |
| 10 | 5,670 ± 1,8203 | 5,140 ± 1,1501,2,3 | 8,760 ± 2,1403 | |
| Enzalutamide | 0.1 | 100 ± 10 | 530 ± 150 | 1,350 ± 390 |
| 1 | 740 ± 40 | 3,530 ± 1,630 | 7,460 ± 2,200 | |
| 10 | 8,450 ± 1,570 | >10,000 | >10,000 | |
| Apalutamide | 0.1 | 90 ± 10 | 200 ± 160 | 590 ± 130 |
| 1 | 710 ± 80 | 1,560 ± 220 | 3,380 ± 1,780 | |
| 10 | 8,290 ± 1,600 | >10,000 | >10,000 | |
| Bicalutamide | 0.1 | 240 ± 90 | Agonism | Agonism |
| 1 | 2,330 ± 750 | 90 ± 10% | 100 ± 10% | |
| 10 | >10,000 |
Notes: Mean IC50 ± SD values for at least three biological replicates are given in nM. Cells were treated with the indicated R1881 concentrations (in nM) and the mentioned AR antagonist. In some cases agonism was found and the % activity measured in the presence of 1 μM compound, in comparison to 1 nM R1881 which was set to 100%, is given in bold. Statistical analysis was performed with t‐test on average pIC50 values. Superscripts indicate antagonism significantly stronger compared to enzalutamide1, apalutamide2 or bicalutamide3.
Enzalutamide is approved for metastatic and nonmetastatic castration‐resistant prostate cancer.3, 28, 29 It showed weaker activity in comparison to darolutamide in several instances, for example, for the mutations W742C/L and M896V/T (Table 1 and Supporting Information Table 2). When increasing the androgen concentrations, enzalutamide antagonism was reduced for AR wild type and several mutants. Agonistic activity was determined by treating the cells only with compound and in the case of AR F877L we found that 1 μM enzalutamide led to a 20% stimulation, when compared to the activity seen with 1 nM R1881 (Supporting Information Table 2).
Apalutamide was recently approved for nonmetastatic castration‐resistant prostate cancer.3, 30 Its activity was comparable to enzalutamide with however a stronger antagonistic activity for the W742C/L and M896V/T mutations (Table 1 and Supporting Information Table 2). Here also, there was a marked loss of antagonism at elevated androgen levels. Agonism was observed for AR F877L as 1 μM apalutamide treatment led to 40% stimulation, compared to the effect of 1 nM R1881 (Supporting Information Table 2).
Bicalutamide was generally a weaker antagonist than the newer compounds. For AR modified at positions W742 or M896, agonistic activities of 90–100% or 50–60%, respectively, were observed upon treatment with 1 μM bicalutamide (Table 1 and Supporting Information Table 2).
Darolutamide inhibits AR wild type as well as the W742C and W742L forms in the cellular N/C domain interaction and homodimerization assays
In view of the strong antagonistic properties of darolutamide observed in transactivation assays, we established orthogonal assays to better understand the underlying mechanisms. The interaction between the AR N‐terminal region and C‐terminal LBD is essential to impart transcriptional activity upon androgen stimulation and this can be measured using a mammalian two‐hybrid assay.31, 32 The N/C interaction transfection assay was established in HCT‐116 cells. We first determined the optimal R1881 concentration for stimulation to be 10 nM, and then evaluated the respective effects of AR antagonists (Supporting Information Table 3). For wild type AR, we found darolutamide, and also its two diastereomers and keto‐darolutamide, to have comparably strong antagonistic effects. This was maintained for AR W742C and W742L. In contrast, enzalutamide and apalutamide had similar activity for AR wild type but strongly lost activity for AR W742C and W742L. As observed in the transactivation assay, 1 μM bicalutamide treatment was agonistic for these two mutants.
Homodimerization is an essential feature necessary for AR function and a novel dimerization interface has been identified in the LBD.33 A cellular assay based on the reversible interaction of a two‐subunit luminescent enzyme has recently been established to measure AR homodimerization.34 We first determined the optimal conditions for a strong AR dimerization and found treatment with 10 nM R1881 for 30 min to give the best measurement window (not shown). We then tested the impact of AR antagonists and found darolutamide, its two diastereomers and keto‐darolutamide to all strongly impair AR wild type homodimerization (Table 2). In comparison, enzalutamide and especially bicalutamide had less inhibitory activity than darolutamide. We performed similar homodimerization assays with AR W742C and W742L. We saw a weaker activity of darolutamide and its derivatives for the mutants, especially the W742C form (Table 2). The loss of activity was, however, significantly higher for enzalutamide and apalutamide. Bicalutamide turned into a strong agonist.
Table 2.
Inhibition of AR dimerization by AR antagonists
| Compound | AR wild type | AR W742C | AR W742L |
|---|---|---|---|
| Darolutamide | 100 ± 10 | 420 ± 140 | 180 ± 30 |
| (S,R)‐darolutamide | 70 ± 30 | 380 ± 90 | 160 ± 10 |
| (S,S)‐darolutamide | 120 ± 20 | 520 ± 130 | 190 ± 90 |
| Keto‐darolutamide | 120 ± 30 | 580 ± 140 | 190 ± 70 |
| Enzalutamide | 180 ± 80 | 2,090 ± 1301 | 2,180 ± 2201 |
| Apalutamide | 130 ± 40 | 1,450 ± 501 | 1,040 ± 1801 |
| Bicalutamide | 340 ± 701 | Ago: 130 ± 10% | Ago: 120 ± 30% |
Notes: Mean IC50 ± SD values from three biological replicates are given in nM. Cells were treated with 10 nM R1881. In case agonistic activity was observed, the value is shown in bold and gives the % activity measured with 10 μM compound only, in comparison to treatment with 10 nM R1881 which was set to 100%. Statistical analysis was performed with t‐test on average pIC50 values. Superscript indicates inhibitory activity that is significantly lower than that of darolutamide. Ago, agonism.
Structural interpretation of the darolutamide antagonistic profile
In order to interpret the experimentally observed inhibition profile of darolutamide in comparison to other AR antagonists in structural terms, we visualized the putative interaction mode to the AR, based on known binding features of structurally related ligands studied previously by X‐ray crystallography. In the absence of an AR crystal structure in antagonistic conformation, we constructed a homology model for the AR LBD with distorted helix 12 (Fig. 1 a) based on the antagonistic conformer of the progesterone receptor LBD, as performed in a previous study.35 Darolutamide was placed in the AR ligand‐binding pocket (Fig. 1 b) while giving special care to the preserved interaction of the cyano group of the CF3‐/Cl‐substituted benzonitrile moiety to R753, which was identified as key interaction in other structural and modeling studies.36 Inspection of the proposed binding mode of darolutamide to the antagonistic AR wild type conformer suggested a face‐to‐face stacking interaction of the eastern pyrazole moiety to the indole group of W742 (Fig. 1 c). This interaction can also be expected to occur for the aromatic counterparts found in the eastern part of the other studied AR antagonists. Crystal structure analysis of bicalutamide bound to the W742L form showed AR to adopt an agonistic conformation.37 In this complex, bicalutamide takes up the space occupied by the indole group of W742 in the AR wild type agonistic conformer and thereby stabilizes the agonistic conformer of AR W742L.37 In contrast to bicalutamide, antagonism was maintained for the other studied compounds, but darolutamide showed the most potent activity, especially when compared to enzalutamide. This can be interpreted in structural terms by a better ability of darolutamide to fit into the pocket created by this modification. This is possibly due to the higher flexibility introduced by the isopropylamine linker which allowed maintaining van der Waals contacts to the leucine side chain (Fig. 1 d). Enzalutamide and apalutamide possess a more rigid imidazolidine ring. This structural restraint likely prevented a similar adaptation to compensate for the loss of van der Waals contacts resulting from the amino acid exchange. A similar antagonistic profile was observed for darolutamide in presence of AR W742C. According to our model, the preserved antagonistic activity could be explained by the formation of a hydrogen bond between the cysteine SH group and the alcohol functionality of darolutamide (Fig. 1 e). This may compensate for the loss of van der Waals contacts to the indole of W742 in AR wild type. Importantly, the results from the transactivation, N/C interaction and homodimerization assays for the darolutamide diastereomers and for keto‐darolutamide are in line with the proposed hydrogen bond formation and suggest the ligand oxygen to act as hydrogen bond acceptor, given the observation that the hydroxyl and keto derivatives are nearly equipotent. In summary, our proposed binding model of darolutamide along with the comparison of the molecular constitution of the other studied ligands provides a structural hypothesis for the strong antagonistic activity of darolutamide against AR W742L/C. As to date none of the clinically used antagonists, except bicalutamide,38 has been cocrystallized with the AR LBD or its mutants; further experimental studies to validate the proposed binding features will be of high importance.
Figure 1.
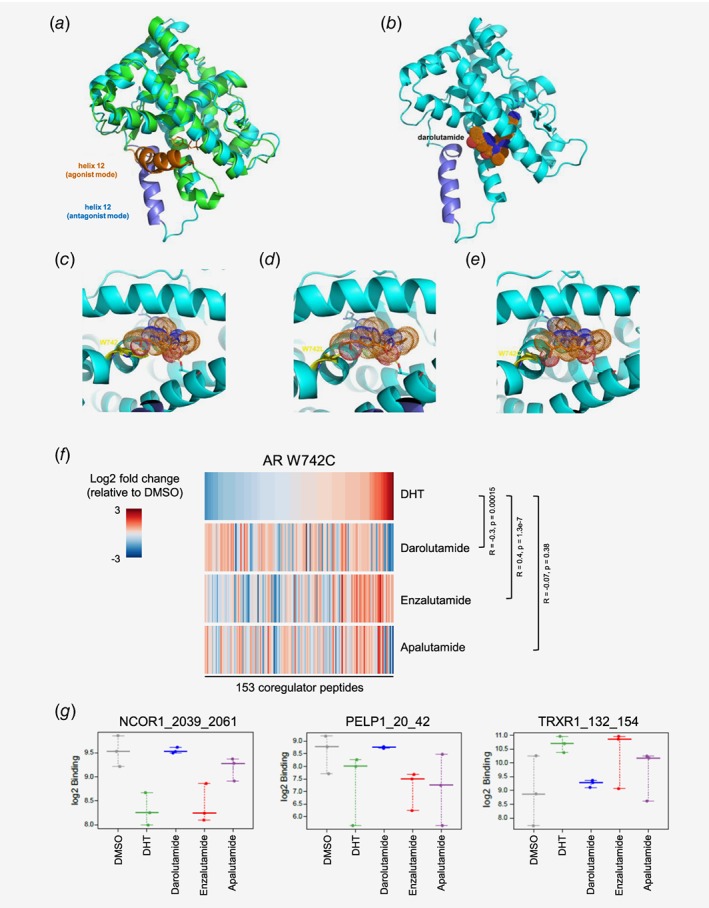
Visualization of antagonist binding to the AR LBD wild type or mutated at position W742, and impact on coregulator recruitment. (a) Superimposition of AR LBD crystal structure (pdb entry 4ojb, green) complex to bicalutamide (orange sticks) with helix 12 in agonistic conformation (orange) and structural model (cyan) with helix 12 in antagonistic mode (dark blue). (b) Darolutamide (dotted surface) modeled into the AR LBD in antagonist conformation. (c) Binding model of darolutamide (orange, sticks and dotted surface) suggests a stacking interaction of its pyrazole moiety to the indole of W742 (yellow sticks). (d) Binding model of darolutamide to AR W742L. (e) Binding model of darolutamide to AR W742C suggests formation of an H‐bond to the hydroxyethyl moiety. (f) Heatmap showing the overall pattern of coregulator peptide binding to the AR W742C after treatment with DHT or with different AR antagonists and sorted according to the effects observed after DHT treatment. Increased (red) and reduced (blue) recruitment are observed. (g) Examples of coregulator peptides for which a differential binding pattern for AR W742C was observed after treatment with different AR antagonists. [Color figure can be viewed at wileyonlinelibrary.com]
Darolutamide has a unique coregulator interaction profile after AR W742C interaction
Full functionality of the AR necessitates recruitment of several coregulators.39 In order to find out whether the unique antagonistic profile of darolutamide for AR W742C related to a differential recruitment of cofactors, we performed the Microarray Assay for Real‐time Coregulator Nuclear receptor Interaction.40 We compared the binding profiles of coregulator peptides to AR W742C after treatment with DHT or with AR antagonists (Fig. 1 f). The profile of peptide binding upon darolutamide treatment was negatively correlated to the binding profile upon DHT treatment (R = −0.3, p = 0.00015), which is in line with the antagonistic effects of this compound for AR W742C. Conversely, upon treatment with enzalutamide, the W742C form exhibited a cofactor binding profile very similar to that seen with DHT treatment (fold changes are positively correlated: R = 0.4, p = 1.3e‐7), which fits with the reduced antagonism for this AR mutant. Apalutamide treatment does not lead to a peptide binding profile similar to DHT, nor does it reverse it (R = −0.07, p = 0.38). Examples of coregulator peptides for which the effect of darolutamide was opposite to that of DHT, while enzalutamide was similar to DHT, are shown in Figure 1 g. They include NCoR1, a corepressor that negatively regulates AR activity and is recruited upon antagonist binding;38, 41 PELP1, a member of chromatin remodeling complexes;42 and TRXR1, which is upregulated in proliferating prostate cancer cells.43
Altogether, these results are in line with the potent antagonistic activity of darolutamide and weak activity of enzalutamide for AR W742C, and strongly suggest that darolutamide binding leads to a unique coregulator recruitment profile that favors antagonistic AR function and likely translates into antitumor efficacy, as observed in vitro and in vivo (see below).
Darolutamide shows strong in vitro efficacy in androgen‐dependent prostate cancer cell lines
We next determined the efficacy of darolutamide, its two diastereomers and keto‐darolutamide in different prostate cancer cell lines. The compounds all showed a comparable efficacy in androgen‐stimulated prostate cancer cells, demonstrating a consistent, high pressure on AR (Table 3). In LAPC‐4 cells, darolutamide displayed a stronger activity than enzalutamide or apalutamide did. In LNCaP cells, darolutamide had a less pronounced effect, in line with its weaker activity for AR T878A which is found in these cells.12
Table 3.
Inhibition of prostate cancer cell viability by AR antagonists
| Compound | VCaP | LAPC‐4 | LNCaP |
|---|---|---|---|
| Darolutamide | 410 ± 150 | 500 ± 220 | 5,260 ± 2,510 |
| (S,R)‐darolutamide | 250 ± 60 | 440 ± 110 | 4,710 ± 1,010 |
| (S,S)‐darolutamide | 380 ± 90 | 840 ± 350 | 5,230 ± 1,940 |
| Keto‐darolutamide | 500 ± 140 | 660 ± 170 | 3,210 ± 1,010 |
| Enzalutamide | 440 ± 130 | 680 ± 460 | 770 ± 3602 |
| Apalutamide | 390 ± 130 | 1,100 ± 6001 | 5,900 ± 2,560 |
| Bicalutamide | 4,950 ± 1,8401 | 1,300 ± 7701 | 3,470 ± 2,4502 |
Notes: Mean IC50 ± SD values from at least five biological replicates are given in nM. Cells were stimulated with 0.1 nM (VCaP), 10 nM (LAPC‐4) or 1 nM (LNCaP) R1881. Statistical analysis was performed with t‐test on average pIC50 values. Superscripts indicate inhibitory activity that is significantly lower1 or higher2 compared to the corresponding darolutamide value.
Compared to monolayer culture, 3D growth models better simulate the in vivo tumor microenvironment. We established spheroid formation assays for the VCaP and LAPC‐4 models, and observed a strong stimulation after androgen treatment (Figs. 2 a and 2b). In the case of VCaP cells, darolutamide application prevented androgen‐induced spheroid formation mainly at the two highest doses tested (Fig. 2 a). This was seen already 5 days after treatment start and maintained for at least 15 days. Enzalutamide and especially apalutamide had a weaker activity. In LAPC‐4 cells, a similar pattern was observed with darolutamide and enzalutamide having the strongest impact and apalutamide showing the least activity (Fig. 2 b).
Figure 2.
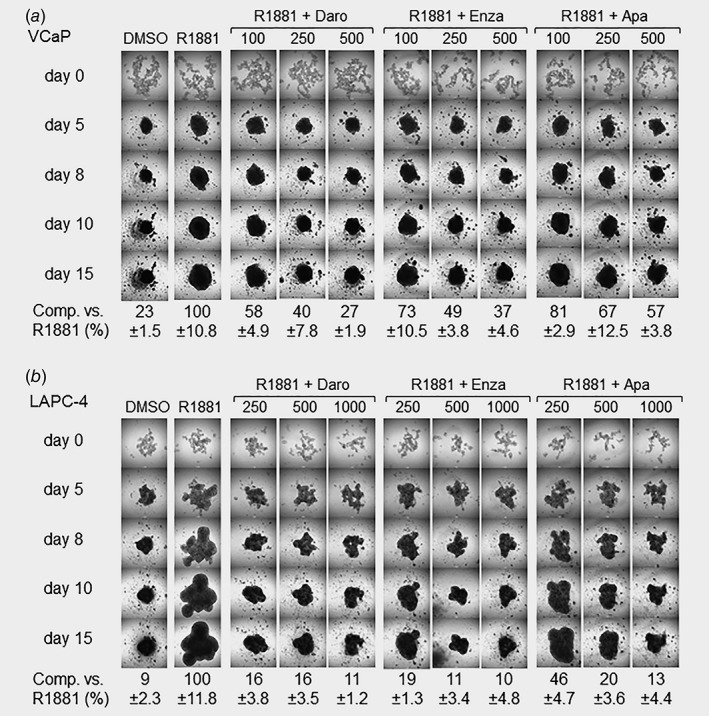
Impact of AR antagonists on spheroid formation. (a) Inhibition of VCaP spheroid formation. (b) Inhibition of LAPC‐4 spheroid formation. Single‐cell suspensions were plated into ultralow attachment microplates, treated with R1881 and different AR antagonist concentrations (as indicated in nM) for up to 15 days. Spheroid formation was determined by microscopy using the ImageJ software. Values measured at Day 15 for three biological replicates ± SD are given in comparison to the signal measured for R1881‐stimulated cells, which was set to 100%. Daro, darolutamide; Enza, enzalutamide; Apa, apalutamide.
Darolutamide reduces androgen target gene expression and local AR binding
Treatment of VCaP cells with darolutamide led to reduction of androgen‐stimulated FKBP5, KLK3, KLK2 and TMPRSS2 expression, down to nonstimulated levels at the highest concentrations tested (Fig. 3 a). The effects were more pronounced in LAPC‐4 cells where the expression levels of these genes were nearly down to the nonandrogen treated ones already in presence of 500 nM darolutamide. Enzalutamide showed weaker effects in VCaP cells and in LAPC‐4 cells, here only in the case of KLK3 expression (Fig. 3 a). For apalutamide the overall impact was altogether comparable to that of darolutamide (Fig. 3 a). We next determined AR binding to the FKBP5 and KLK3 gene regulatory regions and found it to be markedly reduced by darolutamide, down to levels observed in absence of androgen stimulation (Fig. 3 b). The situation was similar after enzalutamide or apalutamide treatment (not shown).
Figure 3.
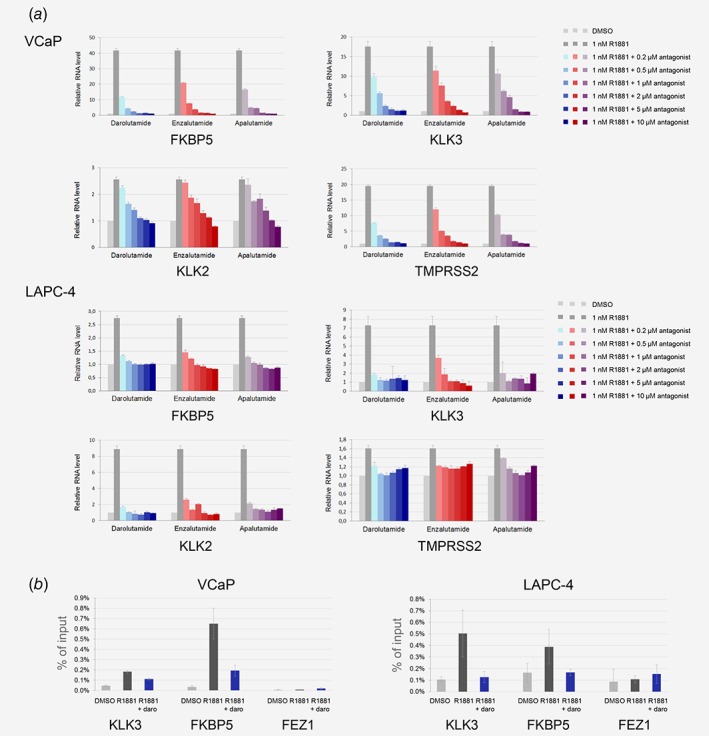
Impact of AR antagonists on downstream target genes. (a) Inhibition of androgen target gene expression. Cells were treated with R1881 and AR antagonists, and RNA was extracted 20 hr later. Real‐time qPCR was performed with appropriate primers and the results were normalized to human cyclophilin A expression. (b) Reduction of AR binding at gene regulatory regions. Nuclei were purified from cells treated with R1881 and darolutamide, and immunoprecipitation performed using an AR‐specific antibody. AR occupancy was determined by qRT‐PCR using appropriate primers. [Color figure can be viewed at wileyonlinelibrary.com]
We furthermore looked at the impact of the antagonists on AR protein levels by ELISA. In VCaP cells, treatment with any of the three compounds led to slightly elevated total AR protein levels after 3 days of treatment, compared to cFBS treatment only (Supporting Information Fig. 2). The situation was similar for the AR‐V7 splice variant. Conversely, in LAPC‐4 cells, there was a marked reduction of total AR protein levels, down to nonandrogen‐treated levels at the highest darolutamide and apalutamide concentrations used (Supporting Information Fig. 2). The effects were less pronounced for enzalutamide as higher concentrations were needed to see a comparable decrease of AR protein levels (Supporting Information Fig. 2).
Darolutamide shows strong in vivo efficacy in the LAPC‐4 prostate cancer xenograft which harbors AR wild type
In order to determine whether the superior in vitro efficacy of darolutamide in LAPC‐4 cells also translated in vivo, a corresponding xenograft experiment was performed. When dosing darolutamide orally, daily or bi‐daily, a significant reduction of LAPC‐4 tumor growth was observed, compared to vehicle‐treated mice (Fig. 4 a). At Day 56, the ΔT/ΔC was 25% for the 100 mg/kg bi‐daily dose and 23% for the 200 mg/kg once daily dose (Fig. 4 b). The impact on mean tumor volumes is depicted in Supporting Information Figure 3. All the treatment schedules were well tolerated, as indicated by the limited body weight loss observed (2–3% at nadir). Gene expression analysis revealed a 50% reduction of FKBP5 expression for the highest treatment doses in the LAPC‐4 tumors (Fig. 4 c).
Figure 4.
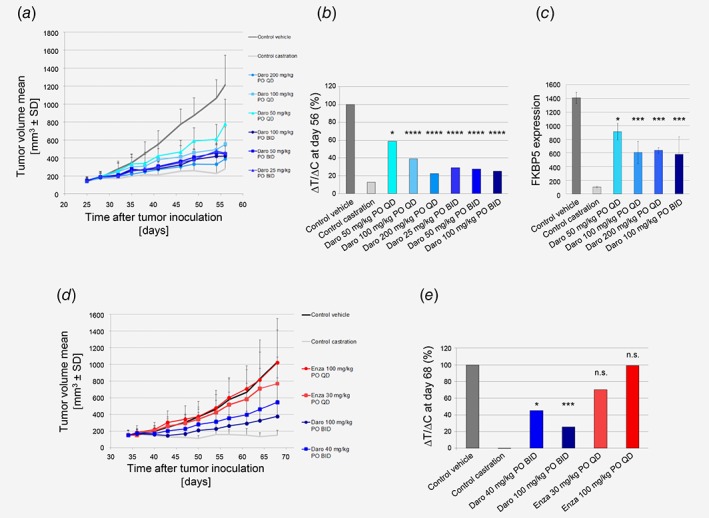
In vivo efficacy studies in prostate cancer models. (a) Darolutamide strongly inhibits LAPC‐4 tumor growth inhibition as seen by measuring mean tumor volume during treatment. (b) % ΔT/ΔC volume at Day 56. Significance versus control vehicle group was determined at Day 56 using one‐way ANOVA with Dunnett's post hoc test on Log‐transformed tumor volumes: *p = 0.04; ****p = 0.0001. (c) FKBP5 gene expression levels in tumors harvested 4 hr after final dosing (n = 3). Significance versus control vehicle group was determined using unpaired t‐test. *p = 0.02; ***p < 0.005. (d) Darolutamide strongly inhibits KuCaP‐1 tumor growth as seen by measuring mean tumor volume during treatment. (e) % ΔT/ΔC volume at Day 68. Significance versus control vehicle group was determined at Day 68 using one‐way ANOVA with Dunnett's post hoc test on Log‐transformed tumor volumes: *p = 0.0169; ***p = 0.0001, n.s., not significant. Daro, darolutamide; Enza, enzalutamide. [Color figure can be viewed at wileyonlinelibrary.com]
Darolutamide shows strong in vivo efficacy in the KuCaP‐1 prostate cancer xenograft which harbors the AR W742C mutation
As darolutamide retained strong antagonism for the W742 mutations in different cellular assays, we next determined whether this was also the case in vivo and tested efficacy in the human KuCaP‐1 model, which harbors AR W742C. A reduction of KuCaP‐1 tumor growth was observed when dosing 40 or 100 mg/kg darolutamide orally, bi‐daily, compared to vehicle‐treated mice (Fig. 4 d). At Day 68, the ΔT/ΔC tumor volume was 26% for the 100 mg/kg bi‐daily dose and 45% for the 40 mg/kg bi‐daily dose. In contrast, the ΔT/ΔC tumor volume values were 99 and 70% for the 100 and 30 mg/kg enzalutamide daily treatment arms, respectively (Fig. 4 e). The impact on mean tumor volumes is depicted in Supporting Information Figure 3. Darolutamide treatments were well tolerated, as indicated by the limited body weight loss (maximal values were 6 and 8% for the 100 and 40 mg/kg dose, respectively), compared to the 10% tumor‐induced body weight loss in the vehicle group.
Discussion
Extensive in vitro profiling of darolutamide, its diastereomers and its main metabolite showed strong antagonistic activity for AR wild type. This was well maintained upon stimulation with elevated androgen levels, in line with the strong binding of darolutamide to the AR.12 This should be of advantage in tumors where steroid biosynthesis is upregulated, an important resistance mechanism observed in patients.9
Darolutamide also exhibited strong antagonism for several AR mutants for which other antagonists had only reduced activity. A prime example is the AR W742C/L forms which are stimulated by bicalutamide, thus activating an androgenic gene expression program resembling that of an AR agonist.44 Here we show that darolutamide remained a strong antagonist whereas enzalutamide and apalutamide had only reduced antagonist properties for these AR mutants. This superior activity of darolutamide was furthermore evidenced in the AR N/C interaction and homodimerization assays. AR mutations are found in about 15–20% of castration‐resistant prostate cancer patients4, 18, 45, 46 and represent a clinically relevant mechanism for therapy resistance after treatment with an AR antagonist or a CYP17A1 inhibitor.4, 18, 45, 46 AR LBD mutations have also been described in earlier, nonmetastatic castration‐resistant prostate cancer patients who have only undergone androgen deprivation therapy,47 which further portends the essentiality of androgen signaling in several stages of prostate cancer. These modifications have different impacts, depending on the individual amino acid changes48 and in several cases they are aberrantly activated by other steroids or adopt an agonistic conformation upon antagonist binding.46 As no AR LBD structure in the antagonistic conformation is available, the antagonistic progesterone receptor LBD structure was used for modeling, due to the high similarity between both receptors. Our results suggest that the reason why darolutamide can accommodate the W742L modification is the higher flexibility introduced by the isopropylamine linker and the maintained van der Waals contacts to the LBD leucine side chain. In contrast, enzalutamide and apalutamide possess a more rigid imidazolidine ring which probably prevents these contacts. A definitive confirmation of this model and of the precise interactions of darolutamide within the AR LBD will likely necessitate the identification of mutants that can be crystallized for X‐ray structure determination, as previously accomplished for bicalutamide bound to the AR LBD with the W742L mutation.37
Importantly, the differing interactions of antagonists inside the AR LBD led to a differential coregulator recruitment pattern for the W742C form. In agreement with the binding of darolutamide to AR W742C in an antagonistic conformational state and an orientation of helix 12 which did not lead to coregulator binding, our results revealed that the coregulator peptide recruitment pattern of the darolutamide‐treated group resembled that of the DMSO control group. A striking example was that the reduced NCoR1 peptide recruitment which follows DHT binding was entirely abrogated by darolutamide, but not by enzalutamide, and this may be the molecular basis for the maintained strong antagonistic action of darolutamide. Additional studies to further dissect the differential recruitment of NCoR1 and its impact on androgen signaling will help to better understand the significance of this mutation for prostate cancer progression. An altered recruitment of coregulators has previously been reported for AR H875Y treated with DHT49 and the AR F877L/T878A double mutant bound by enzalutamide.36 Interestingly, altered transcriptomes and cofactor recruitment profiles have also been documented for clinically relevant estrogen receptor mutants.50
Altogether, our results demonstrate the strong antagonistic activity of darolutamide, its diastereomers and the keto‐derivative in different prostate cancer models. Importantly, darolutamide possessed marked inhibitory effects on androgen‐stimulated prostate cancer cell viability and spheroid formation, which was also reflected by the strong impact on hallmark androgen target gene expression and impairment of AR binding to regulatory regions. This translated into significant in vivo efficacy for two different prostate cancer models harboring either AR wild type or the W742C alteration.
In conclusion, these new findings enhance our understanding and extend previous research on the activity of darolutamide in preclinical prostate cancer models. The broad efficacy of darolutamide, together with the previously reported absence of stimulation of the androgen feedback loop and the reduced brain penetration in comparison to approved AR antagonists12 further support the clinical evaluation of this novel compound in different stages of prostate cancer.
Supporting information
Supplementary Figure 1 Chemical structure of darolutamide and derivatives, and of the other AR antagonists analyzed. The molecular weight of each compound is indicated in parentheses.
Supplementary Figure 2. Impact of AR antagonists on AR protein levels. Lysates of cells treated with different AR antagonist concentrations were analyzed using a sandwich AR ELISA kit.
Supplementary Figure 3. (a) In vivo efficacy in LAPC‐4 model. Mean tumor volume ± SD at day 56 is shown. Significance vs. control vehicle group was determined at day 56 using one‐way ANOVA with Dunnett's post hoc test on Log‐transformed tumor volumes: *, p = 0.04; ****, p = 0.0001. (b) In vivo efficacy in KuCaP‐1 model. Mean tumor volume ± SD at day 68 is shown. Significance vs. control vehicle group was determined at day 68 using one‐way ANOVA with Dunnett's post hoc test on Log‐transformed tumor volumes: *, p = 0.0169; ***, p = 0.0001, n.s.: not significant. Daro: darolutamide, Enza: enzalutamide.
Supplementary Table 1. Primers used for ChIP‐qPCR analysis.
Supplementary Table 2. Transactivation assays for AR mutants. Mean IC50 ± SD values from 3 to 6 biological replicates are given in nM. Cells were treated with the indicated R1881 concentrations (in nM) and the mentioned AR antagonist. In some cases agonism was found and the % activity measured in the presence of 1 μM compound, in comparison to 1 nM R1881 treatment which was set to 100%, is given in bold. Statistical analysis was performed with t‐test on average pIC50 values. Superscripts indicate antagonism significantly lower1 or higher2 compared to darolutamide. Abbreviation: n.d., not determined.
Supplementary Table 3. Effects of AR antagonists on AR wild‐type and mutant N/C interaction. Mean IC50 ± SD values from at least 2 biological replicates are given in nM. Cells were treated with 10 nM R1881. In case agonistic activity was observed, the value is shown in bold and gives the % activity measured with 10 μM compound only, in comparison to treatment with 10 nM R1881 which was set to 100%. Statistical analysis was performed with t‐test on average pIC50 values. A superscript indicates inhibitory activity that is significantly lower than that of darolutamide. Ago: agonism.
Acknowledgements
Darolutamide is jointly developed by Bayer AG (Germany) and Orion Corporation (Finland). We thank the project team for continuous support. The excellent technical help of B. Hartmann, H. Muckwar and F. Knoth is gratefully acknowledged. Many thanks to D. Pijnenburg, R. van Beuningen and R. Houtman (PamGene International BV) for performance of the MARCoNI assay and support in data interpretation, to the VTT Technical Research Centre for LAPC‐4 cells and to O. Ogawa and T. Inoue (University of Kyoto, Japan) for the KuCaP‐1 model.
Conflict of interest: All authors are employees and/or shareholders of Bayer AG.
References
- 1. Torre LA, Bray F, Siegel RL, et al. Global cancer statistics, 2012. CA Cancer J Clin 2015;65:87–108. [DOI] [PubMed] [Google Scholar]
- 2. Yap TA, Smith AD, Ferraldeschi R, et al. Drug discovery in advanced prostate cancer: translating biology into therapy. Nat Rev Drug Discov 2016;15:699–718. [DOI] [PubMed] [Google Scholar]
- 3. Nevedomskaya E, Baumgart SJ, Haendler B. Recent advances in prostate cancer treatment and drug discovery. Int J Mol Sci 2018;19:E1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Robinson D, Van Allen EM, Wu YM, et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015;162:454. [DOI] [PubMed] [Google Scholar]
- 5. Armenia J, Wankowicz SAM, Liu D, et al. The long tail of oncogenic drivers in prostate cancer. Nat Genet 2018;50:645–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Takeda DY, Spisak S, Seo JH, et al. A somatically acquired enhancer of the androgen receptor is a noncoding driver in advanced prostate cancer. Cell 2018;174:422.e13–32.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Viswanathan SR, Ha G, Hoff AM, et al. Structural alterations driving castration‐resistant prostate cancer revealed by linked‐read genome sequencing. Cell 2018;174:433.e19–47.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Quigley DA, Dang HX, Zhao SG, et al. Genomic hallmarks and structural variation in metastatic prostate cancer. Cell 2018;174:758.e9–69.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Stuchbery R, McCoy PJ, Hovens CM, et al. Androgen synthesis in prostate cancer: do all roads lead to Rome? Nat Rev Urol 2017;14:49–58. [DOI] [PubMed] [Google Scholar]
- 10. Fizazi K, Smith MR, Tombal B. Clinical development of darolutamide: a novel androgen receptor antagonist for the treatment of prostate cancer. Clin Genitourin Cancer 2018;16:332–40. [DOI] [PubMed] [Google Scholar]
- 11. Shore ND. Darolutamide (ODM‐201) for the treatment of prostate cancer. Expert Opin Pharmacother 2017;18:945–52. [DOI] [PubMed] [Google Scholar]
- 12. Moilanen AM, Riikonen R, Oksala R, et al. Discovery of ODM‐201, a new‐generation androgen receptor inhibitor targeting resistance mechanisms to androgen signaling‐directed prostate cancer therapies. Sci Rep 2015;5:12007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Matsubara N, Mukai H, Hosono A, et al. Phase 1 study of darolutamide (ODM‐201): a new‐generation androgen receptor antagonist, in Japanese patients with metastatic castration‐resistant prostate cancer. Cancer Chemother Pharmacol 2017;80:1063–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fizazi K, Massard C, Bono P, et al. Activity and safety of ODM‐201 in patients with progressive metastatic castration‐resistant prostate cancer (ARADES): an open‐label phase 1 dose‐escalation and randomised phase 2 dose expansion trial. Lancet Oncol 2014;15:975–85. [DOI] [PubMed] [Google Scholar]
- 15. Borgmann H, Lallous N, Ozistanbullu D, et al. Moving towards precision urologic oncology: targeting enzalutamide‐resistant prostate cancer and mutated forms of the androgen receptor using the novel inhibitor darolutamide (ODM‐201). Eur Urol 2018;73:4–8. [DOI] [PubMed] [Google Scholar]
- 16. Terada N, Shimizu Y, Yoshida T, et al. Antiandrogen withdrawal syndrome and alternative antiandrogen therapy associated with the W741C mutant androgen receptor in a novel prostate cancer xenograft. Prostate 2010;70:252–61. [DOI] [PubMed] [Google Scholar]
- 17. Faus H, Meyer HA, Huber M, et al. The ubiquitin‐specific protease USP10 modulates androgen receptor function. Mol Cell Endocrinol 2005;245:138–46. [DOI] [PubMed] [Google Scholar]
- 18. Sugawara T, Lejeune P, Kohr S, et al. BAY 1024767 blocks androgen receptor mutants found in castration‐resistant prostate cancer patients. Oncotarget 2016;7:6015–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Biasini M, Bienert S, Waterhouse A, et al. SWISS‐MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res 2014;42:W252–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Madauss KP, Grygielko ET, Deng SJ, et al. A structural and in vitro characterization of asoprisnil: a selective progesterone receptor modulator. Mol Endocrinol 2007;21:1066–81. [DOI] [PubMed] [Google Scholar]
- 21. Emsley P, Lohkamp B, Scott WG, et al. Features and development of coot. Acta Crystallogr D Biol Crystallogr 2010;66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Baumgart SJ, Najafova Z, Hossan T, et al. CHD1 regulates cell fate determination by activation of differentiation‐induced genes. Nucleic Acids Res 2017;45:7722–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Craft N, Shostak Y, Carey M, et al. A mechanism for hormone‐independent prostate cancer through modulation of androgen receptor signaling by the HER‐2/neu tyrosine kinase. Nat Med 1999;5:280–5. [DOI] [PubMed] [Google Scholar]
- 24. Clegg NJ, Wongvipat J, Joseph JD, et al. ARN‐509: a novel antiandrogen for prostate cancer treatment. Cancer Res 2012;72:1494–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Alley MC, Hollingshead MG, Dykes DJ, et al. Human tumor xenograft models in NCI drug development In: Teicher BA, Andrews PA, eds Anticancer drug development guide: preclinical screening, clinical trials, and approval. New York: Humana Press Inc, 2004. 125–52. [Google Scholar]
- 26. Liu M, Hicklin D. Human tumor xenograft efficacy models In: Teicher BA, ed. Tumor models in cancer research, 2nd edn. New York: Humana Press Inc, 2011. 99–124. [Google Scholar]
- 27. Joseph JD, Lu N, Qian J, et al. A clinically relevant androgen receptor mutation confers resistance to second‐generation antiandrogens enzalutamide and ARN‐509. Cancer Discov 2013;3:1020–9. [DOI] [PubMed] [Google Scholar]
- 28. Hussain M, Fizazi K, Saad F, et al. Enzalutamide in men with nonmetastatic, castration‐resistant prostate cancer. N Engl J Med 2018;378:2465–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ritch C, Cookson M. Recent trends in the management of advanced prostate cancer. F1000Res 2018;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Smith MR, Saad F, Chowdhury S, et al. Apalutamide treatment and metastasis‐free survival in prostate cancer. N Engl J Med 2018;378:1408–18. [DOI] [PubMed] [Google Scholar]
- 31. Haendler B. Androgen‐selective gene regulation in the prostate. Biomed Pharmacother 2002;56:78–83. [DOI] [PubMed] [Google Scholar]
- 32. Ikonen T, Palvimo JJ, Janne OA. Interaction between the amino‐ and carboxyl‐terminal regions of the rat androgen receptor modulates transcriptional activity and is influenced by nuclear receptor coactivators. J Biol Chem 1997;272:29821–8. [DOI] [PubMed] [Google Scholar]
- 33. Nadal M, Prekovic S, Gallastegui N, et al. Structure of the homodimeric androgen receptor ligand‐binding domain. Nat Commun 2017;8:14388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dixon AS, Schwinn MK, Hall MP, et al. NanoLuc complementation reporter optimized for accurate measurement of protein interactions in cells. ACS Chem Biol 2016;11:400–8. [DOI] [PubMed] [Google Scholar]
- 35. Voet A, Helsen C, Zhang KY, et al. The discovery of novel human androgen receptor antagonist chemotypes using a combined pharmacophore screening procedure. ChemMedChem 2013;8:644–51. [DOI] [PubMed] [Google Scholar]
- 36. Prekovic S, van Royen ME, Voet AR, et al. The effect of F877L and T878A mutations on androgen receptor response to enzalutamide. Mol Cancer Ther 2016;15:1702–12. [DOI] [PubMed] [Google Scholar]
- 37. Bohl CE, Gao W, Miller DD, et al. Structural basis for antagonism and resistance of bicalutamide in prostate cancer. Proc Natl Acad Sci U S A 2005;102:6201–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hodgson MC, Shen HC, Hollenberg AN, et al. Structural basis for nuclear receptor corepressor recruitment by antagonist‐liganded androgen receptor. Mol Cancer Ther 2008;7:3187–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Baumgart SJ, Haendler B. Exploiting epigenetic alterations in prostate cancer. Int J Mol Sci 2017;18:E1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Koppen A, Houtman R, Pijnenburg D, et al. Nuclear receptor‐coregulator interaction profiling identifies TRIP3 as a novel peroxisome proliferator‐activated receptor gamma cofactor. Mol Cell Proteomics 2009;8:2212–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Berrevoets CA, Umar A, Trapman J, et al. Differential modulation of androgen receptor transcriptional activity by the nuclear receptor co‐repressor (N‐CoR). Biochem J 2004;379:731–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Girard BJ, Daniel AR, Lange CA, et al. PELP1: a review of PELP1 interactions, signaling, and biology. Mol Cell Endocrinol 2014;382:642–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Singh SS, Li Y, Ford OH, et al. Thioredoxin reductase 1 expression and castration‐recurrent growth of prostate cancer. Transl Oncol 2008;1:153–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. O'Neill D, Jones D, Wade M, et al. Development and exploitation of a novel mutant androgen receptor modelling strategy to identify new targets for advanced prostate cancer therapy. Oncotarget 2015;6:26029–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Marcelli M, Ittmann M, Mariani S, et al. Androgen receptor mutations in prostate cancer. Cancer Res 2000;60:944–9. [PubMed] [Google Scholar]
- 46. McCrea E, Sissung TM, Price DK, et al. Androgen receptor variation affects prostate cancer progression and drug resistance. Pharmacol Res 2016;114:152–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rathkopf DE, Smith MR, Ryan CJ, et al. Androgen receptor mutations in patients with castration‐resistant prostate cancer treated with apalutamide. Ann Oncol 2017;28:2264–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hay CW, McEwan IJ. The impact of point mutations in the human androgen receptor: classification of mutations on the basis of transcriptional activity. PLoS One 2012;7:e32514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Duff J, McEwan IJ. Mutation of histidine 874 in the androgen receptor ligand‐binding domain leads to promiscuous ligand activation and altered p160 coactivator interactions. Mol Endocrinol 2005;19:2943–54. [DOI] [PubMed] [Google Scholar]
- 50. Jeselsohn R, Bergholz JS, Pun M, et al. Allele‐specific chromatin recruitment and therapeutic vulnerabilities of ESR1 activating mutations. Cancer Cell 2018;33:173.e5–86.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1 Chemical structure of darolutamide and derivatives, and of the other AR antagonists analyzed. The molecular weight of each compound is indicated in parentheses.
Supplementary Figure 2. Impact of AR antagonists on AR protein levels. Lysates of cells treated with different AR antagonist concentrations were analyzed using a sandwich AR ELISA kit.
Supplementary Figure 3. (a) In vivo efficacy in LAPC‐4 model. Mean tumor volume ± SD at day 56 is shown. Significance vs. control vehicle group was determined at day 56 using one‐way ANOVA with Dunnett's post hoc test on Log‐transformed tumor volumes: *, p = 0.04; ****, p = 0.0001. (b) In vivo efficacy in KuCaP‐1 model. Mean tumor volume ± SD at day 68 is shown. Significance vs. control vehicle group was determined at day 68 using one‐way ANOVA with Dunnett's post hoc test on Log‐transformed tumor volumes: *, p = 0.0169; ***, p = 0.0001, n.s.: not significant. Daro: darolutamide, Enza: enzalutamide.
Supplementary Table 1. Primers used for ChIP‐qPCR analysis.
Supplementary Table 2. Transactivation assays for AR mutants. Mean IC50 ± SD values from 3 to 6 biological replicates are given in nM. Cells were treated with the indicated R1881 concentrations (in nM) and the mentioned AR antagonist. In some cases agonism was found and the % activity measured in the presence of 1 μM compound, in comparison to 1 nM R1881 treatment which was set to 100%, is given in bold. Statistical analysis was performed with t‐test on average pIC50 values. Superscripts indicate antagonism significantly lower1 or higher2 compared to darolutamide. Abbreviation: n.d., not determined.
Supplementary Table 3. Effects of AR antagonists on AR wild‐type and mutant N/C interaction. Mean IC50 ± SD values from at least 2 biological replicates are given in nM. Cells were treated with 10 nM R1881. In case agonistic activity was observed, the value is shown in bold and gives the % activity measured with 10 μM compound only, in comparison to treatment with 10 nM R1881 which was set to 100%. Statistical analysis was performed with t‐test on average pIC50 values. A superscript indicates inhibitory activity that is significantly lower than that of darolutamide. Ago: agonism.