

Abstract
Maintenance of a balanced expression of the two isoforms of the transcription factor GATA‐1, the full‐length protein (GATA‐1FL) and a shorter isoform (GATA‐1 S), contributes to control hematopoiesis, whereas their dysregulation can alter the differentiation/proliferation potential of hematopoietic precursors thereby eventually leading to a variety of hematopoietic disorders. Although it is well established that these isoforms play opposite roles in these remarkable processes, most of the molecular pathways involved remain unknown.
Here, we demonstrate that GATA‐1FL and GATA‐1S are able to differently influence intracellular redox states and reactive oxygen species (ROS) compartmentation in the erythroleukemic K562 cell line, thus shedding novel mechanistic insights into the processes of cell proliferation and apoptosis resistance in myeloid precursors. Furthermore, given the role played by ROS signaling as a strategy to escape apoptosis and evade cell‐mediated immunity in myeloid cells, this study highlights a mechanism through which aberrant expression of GATA‐1 isoforms could play a role in the leukemogenic process.
Keywords: GATA‐1, mitochondria remodeling, myeloid leukemia, oxidative stress, succinate dehydrogenase subunit C (SDHC)
Abbreviations
- AML
acute myeloid leukemia
- ROS
reactive oxygen species
- SDH
succinate dehydrogenase
- TD
terminal transactivation domain
1. INTRODUCTION
Complex transcription factor networks play a crucial role in the orchestration of myeloid cell fate and in the differentiation block observed in many myeloid leukemias (Paul et al., 2015; Rosmarin, Yang & Resendes, 2005). In this regard, dysregulated expression of GATA‐1, a master regulatory transcription factor of key hematopoietic genes in several myeloid cell types, is emerging as a key factor in malignant hematopoiesis (Bresnick, Katsumura, Lee, Johnson & Perkins, 2012; Gao, Chen & Peterson, 2015; Lentjes et al., 2016). GATA‐1 exists at least as two naturally occurring isoforms that play opposite roles in the differentiation and proliferation processes of various hematopoietic lineages: the full‐length protein (GATA‐1FL), which comprises an N‐terminal transactivation domain (TD), two conserved zinc finger domains and a C‐terminal domain, and a shorter isoform, named GATA‐1S, initiated from a downstream alternative start site (Met84) and thus lacking the N‐terminal TD (Halsey et al., 2012). A balanced GATA‐1FL/GATA‐1S ratio plays an important role in determining cell fate during hematopoiesis with the short isoform mainly involved in the maintenance of the proliferative potency of hematopoietic precursors whereas, conversely, GATA‐1FL is required to promote the terminal differentiation of megakaryocytic‐erythroid lineages through the orchestration of sophisticated transcriptional networks (Chlon, McNulty, Goldenson, Rosinski, & Crispino, 2015; Doré & Crispino, 2011; Halsey et al., 2012; Kaneko, Kobayashi, Yamamoto & Shimizu, 2012). Therefore, not surprisingly, correlations have been found between unbalanced GATA‐1FL/GATA‐1S expression and several hematopoietic disorders including different subtypes of acute and chronic myeloid leukemia commonly characterized by a prevalent expression of GATA‐1S with respect to its full‐length isoform (Crispino, 2005; Petruzziello et al., 2013). Also, elevated GATA‐1S levels have been recognized as a poor prognostic factor, thus further emphasizing the proleukemic role of this isoform in hematological malignancies (Burda, Laslo & Stopka, 2010; Cantor, 2016; Ciovacco, Raskind & Kacena, 2008; Gao et al., 2015, Khan, Malinge & Crispino, 2011; Shimamoto et al., 1995). However, in spite of all these molecular and clinical evidence, further studies are still required to assess whether GATA‐1S has a unique role in hematopoiesis and how its abnormal expression contributes to pathogenetic processes.
Recent progress in leukemia has revealed that impaired homeostasis of reactive oxygen species (ROS) is strongly involved in the development of hematological malignancies and in mechanisms of drug resistance of leukemic cells thus contributing to fuel a widespread interest in therapies based on ROS modulation as a new promising strategy for treatment of patients with relapsed or refractory disease (Hole, Darley & Tonks, 2011; F. L. Zhou et al., 2010; F. Zhou, Shen & Claret, 2013). This is of high clinical relevance in acute myeloid leukemia (AML), an aggressive malignancy characterized by an abnormal expansion of uncontrollably proliferating myeloid progenitors that have lost the capacity to differentiate into mature cells. Indeed, although many AML patients initially respond to chemotherapy, the majority relapse and develop a drug‐resistant disease (Döhner, Weisdorf & Bloomfield, 2015; Sriskanthadevan et al., 2015). Consequently, there is a great need for novel therapeutic strategies that could exclusively target malignant cells (Kuntz et al., 2017; Wouters & Delwel, 2016).
Nevertheless, in spite of a growing body of knowledge in this field, more exhaustive studies are required to better elucidate the molecular mechanisms underlying ROS production, their contribution to the leukemogenic process and their prognostic significance.
In recent years, according to the evidence that the transition from normal to cancerous phenotype is associated with biochemical, molecular and morphological changes, great efforts have focused toward the implementation of optical techniques capable of determining dimension, shape, or other cell physical properties for early detection of neoplastic changes so as to bring improvements in survival and clinical outcomes in cancer patients (Bellisola & Sorio, 2012). Based on these evidence, we recently exploited the use of optical techniques in an attempt to identify novel oncometabolites and biomarkers linked to dysregulated GATA‐1 expression in the myeloid leukemia K562 cell line. This approach eventually allowed us to highlight the different impact of GATA‐1 isoforms on the production of mitochondrial and cytoplasmatic ROS as well as on the modulation of the expression levels of subunit C of the succinate dehydrogenase complex (SDHC). Given the role played by ROS in myeloid leukemogenesis, these observations also prompted us to evaluate the correlations between oxidative stress conditions triggered by GATA‐1 isoforms and cell proliferation and apoptotic resistance.
Taken as a whole, this study indicates that GATA‐1S contributes to enhance apoptotic resistance and proliferation rate in hematopoietic precursors by differently modulating intracellular redox state and ROS compartmentation with respect to its full‐length counterpart. Furthermore, our findings also shed light on the mechanism through which its abnormal expression could promote leukemogenesis. Even more importantly, unraveling the role of GATA‐1 in ROS production and in the structural and functional modulation of mitochondrial activity also promises to pave the way to novel and more effective ROS‐based therapies in patients with leukemia with relapsed or refractory disease.
2. MATERIALS AND METHODS
2.1. Cell culture
The human erythroleukemia K562 cell line was maintained in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS) plus 4 mM glutamine, 10 U/ml penicillin and 10 mg/ml streptomycin (all reagents from Gibco, Thermo Fisher Scientific Inc, Waltham, MA) at 37°C in a humidified 5% CO2‐containing atmosphere. Cells were kept sub‐confluent for transient transfection experiments.
2.2. Transient transfection
K562 cells were transiently transfected with a mix containing 1 μg of p3XFLAG‐GATA‐1FL, p3XFLAG‐GATA‐1S, or p3XFLAG‐CMV empty vector (mock control) and 5 μl of Lipofectamine 2000 as transfection reagent (Invitrogen, Carlsbad, CA). Two hours before transfection, cells were plated into six‐well plates at a density of 5 × 105 in 2 ml of Optimem medium (Invitrogen) without FBS. Five hours after transfection, the medium was supplemented with 10% FBS in each well. Forty‐eight hours after transfection, cells were harvested for total RNA and protein extraction or were used for evaluation of oxidative stress and for 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide (MTT) and Annexin V/propidium iodide (PI) assays.
2.3. Spectroscopy analysis
Absorbance data were taken by a double‐grating double‐channel Perkin Elmer Lambda 900 UV‐Vis‐NIR spectrophotometer (Perkin Elmer, Monza, Italy). Optical transmission spectra of samples were measured by scanning in the wavelength range of 300 to 800 nm: the sample suspension of cells at a density of 8 × 105 in 400 μl of Optimem medium was contained in quartz cuvette of 1 cm optical path length while a similar cuvette, containing Optimem medium, was inserted in the reference channel.
2.4. Assessment of cell viability and apoptosis
Cell viability was determined using the MTT assay. Briefly, after transient transfection, K562 cells were seeded into a 96‐well plate at a concentration of 1.5 × 104 cells/100 μl. At 24, 48, and 72 hr after transfection, respectively, 10 μl of MTT labeling reagent provided by the Cell Proliferation Kit 1 (Roche, Mannheim, Germany) was added to each well according to the procedures recommended by the manufacturer. Measurement of the soluble formazan product in each well was carried out by photometric reading at 570/690 nm on a Synergy H1 Hybrid Multi‐Mode Microplate Reader (BioTek, Winooski, VT). The experiments were repeated three times for each transfection.
Apoptosis resistance was assessed with an Annexin V‐FITC Apoptosis Detection Kit 1 (BD Biosciences, San Diego, CA) according to the manufacturer's protocol.
Forty‐eight hours after transfection, cells were treated for 16 hr with low and high doses of cisplatin (10 and 20 μM) to induce apoptosis and were analyzed using an Accuri C6 flow cytometer (BD Biosciences, San Jose, CA) and BD ACCURI C‐Flow software.
2.5. Evaluation of ROS levels and compartmentation
Transiently transfected K562 cells were analyzed by flow cytometry and fluorescence microscopy for quantification of cytoplasmatic and mitochondrial ROS levels. Cytoplasmatic ROS were measured with CellROX deep red oxidative stress reagent (Thermo Fisher Scientific), a fluorogenic probe designed to reliably measure cytoplasmatic ROS in living cells. Cells were incubated in 5 μM of CellROX reagent for 30 min at 37°C, according to the manufacturer's protocol, and analyzed by flow cytometry.
Cytoplasmatic ROS were also measured in wild‐type K562 cells treated with 10 and 20 μM cisplatin, to verify if the proapoptotic effect of this drug was mediated by an increase in ROS production. Treatment with menadione (Sigma‐Aldrich, St. Louis, MO), a well‐known ROS inducer (Steinmeier & Dringen, 2019), was used at 10 μM concentration for 16 hr as a positive control for ROS production.
Determination of the production of superoxide radicals in mitochondria was performed using the MitoSOX red mitochondrial superoxide reagent (Thermo Fisher Scientific) that is specifically targeted to mitochondria where it is rapidly oxidized by superoxide anion but not by other reactive oxygen species. Cells were incubated with 5 μM MitoSOX reagent for 10 min at 37°C. Assessment of mitochondria mass was performed by incubating cells for 15 min in 100 nM MitoTracker Green FM reagent (Thermo Fisher Scientific) which stains mitochondria in a trans‐membrane potential‐independent manner and allows quantitative analysis by flow cytometry. After washing and resuspension in phosphate‐buffered saline (PBS), all samples were analyzed using an Accuri C6 flow cytometer (Accuri C6) and BD ACCURI C‐Flow software. Green (530/30 nm), orange (585/42 nm), and red ( > 650 nm) fluorescence emissions were evaluated using logarithmic amplification. Data from 10,000 cells were collected, a sufficient number of events for the statistical analysis of relatively small changes in relative fluorescence intensity (RFI). Mean RFI was determined after exclusion of debris and PI‐positive events from the list mode data set.
All samples were also examined by fluorescence microscope analysis (DM14000; Leica Microsystems, Wetzlar, Germany).
2.6. Glutathione determination
Measurement of total glutathione (GSH+GSSG) or oxidized glutathione (GSSG) was accomplished with the GSH/GSSG‐Glo Assay kit (Promega, Madison, WI). The GSH/GSSG ratio was calculated from net relative luminescence units according to the procedures recommended by the manufacturer. For each sample, analysis was performed in triplicate using 10,000 cells/well.
2.7. Quercetin treatment
Forty‐eight hours after transfection, cells were treated with 50, 100, and 150 μM quercetin (Sigma‐Aldrich) for 3 and 24 hr. After treatments, cells were collected and used for evaluation of glutathione levels and for cell viability and apoptosis assays. To perform cotreatments with quercetin and cisplatin, 48 hr after transfection, cells were initially treated with 50 or 150 μM quercetin for 8 hr and for additional 16 hr with 10 μM cisplatin.
2.8. Protein extraction
For protein extraction, K562 cells were collected 48 hr after transfection and washed twice with 3 to 4 ml of cold 1X PBS by centrifugation at 3,000g for 10 min at 4°C. Pellets were resuspended in 50 µl of lysis buffer (10% glycerol, 50 mM Tris‐HCl pH 8.0, 150 mM NaCl, 0.1% NP‐40, 1 mM EDTA pH 8, 0.5 μl of protein inhibitor cocktail mixture (Sigma‐Aldrich) and incubated for 30 min on ice. Samples were then centrifuged at 10,000g for 30 min at 4°C and the supernatant containing the total protein extract was collected. Evaluation of protein concentration was performed by spectrophotometer analysis, according to the Bradford method with the Bio‐Rad protein assay reagent (Bio‐Rad Laboratories, Hercules, CA). Protein extraction from bone marrow specimens from a patient with AML and from three healthy controls was performed using the Qiazol (Qiagen GmbH, Hilden, Germany) procedure according to the manufacturer's instructions. Informed consent for genetic studies was obtained from the investigated subjects in agreement with the Declaration of Helsinki.
2.9. Real‐time PCR analysis
Total RNA was extracted from K562 cells with Qiazol reagent (Qiagen) according to the manufacturer's protocol. After spectrophotometric quantization, RNA quality was verified by gel electrophoresis on a 1.5% denaturing agarose gel in MOPS 1X buffer (20 mM MOPS pH 7.0, 8 mM sodium acetate, 1 mM EDTA pH 8.0).
To quantitatively determine the mRNA expression levels of SDHC, real‐time PCR was performed using a CFX96 real‐time system (Bio‐Rad Laboratories). cDNA was synthesized from 250 ng of total RNA using the QuantiTect Reverse Transcription Kit (Qiagen) and 2 μl of 7xgDNA wipeout buffer in a final volume of 14 μl to remove any traces of genomic DNA. The reaction was performed according to the kit protocol and subsequently used for quantitative real‐time PCR procedures. The following primers were used to detect the expression of SDHC and GAPDH (endogenous control):
SDHC (sense): 5′‐CCCAAGATGGCTGCGCTGTT‐3′,
SDHC (antisense): 5′‐TCAAAGCAATACCAGTGCCACG‐3′,
GAPDH (sense): 5′‐GAGCCACATCGCTCAGACAC‐3′,
GAPDH (antisense): 5′‐ GGCAACAATATCCACTTTACCA ‐3′.
Each real‐time PCR was performed for triplicate measurements in a 20 μl reaction mix containing 10 μl of 2× SsoAdvanced Universal SYBR Green supermix (Bio‐Rad Laboratories), 0.38 μl of a 20 μM primer mix, 2 μl of cDNA (1/10 volume of RT‐PCR product), and 7.62 μl of nuclease‐free water.
The cycling conditions consisted of an initial denaturation step at 95°C for 3 min, followed by 40 cycles (95°C for 15 s, 60°C for 30 s) and 80 cycles performed according to standard protocols for melting curve analysis. The calibration curve for assessing the efficiency of the PCR reaction was performed on at least three serial dilutions (1:10) of the reverse transcriptase products. CT values were determined by automated threshold analysis and data were analyzed by the CFX Manager 3.0 software (Bio‐Rad Laboratories) according to the manufacturer's specifications.
2.10. Quantification of mitochondrial DNA
Total DNA was purified from cells using a conventional phenol‐chloroform extraction method. Relative quantification of mitochondrial DNA (mtDNA) copy number was performed by a real‐time PCR method using a CFX96 real‐time system (Bio‐Rad Laboratories). Quantitative PCR was performed using primers and conditions as previously described (Refinetti, Warren, Morgenthaler & Ekstrøm, 2017).
2.11. Western blot analysis
Western blot analysis was performed on 30 µg of total protein extracts according to the protocol previously described (Petruzzelli et al., 2010). The following primary antibodies were used: anti‐FLAG antibody (1:10,000 dilution; Sigma‐Aldrich), GATA‐1 (4F5, 1:1,000 dilution; Sigma‐Aldrich), VDAC1 (sc‐390996, 1:500 dilution; Santa Cruz Biotechnology, Dallas, TX), SOD1 (sc‐17767, 1:1,000 dilution; Santa Cruz Biotechnology), SOD2 (MA1–106, 1:10,000 dilution; Thermo Fisher Scientific), DRP1 (1:4,000 dilution; Cell Signaling Technology, Leiden, The Netherlands), MFN2 (1:5,000 dilution; Cell Signaling), SDHA (2E3GC12FB2AE2, 1:10,000 diluition; Abcam, Cambridge, UK), SDHB (21A11AE7, 1:10,000 diluition; Abcam), SDHC (EPR110 35, 1:10,000 diluition; Abcam), SDHD (H1; 1:2,000 dilution; Thermo Fisher Scientific).
Filters were incubated at 4°C for 1.30 hr with the anti‐FLAG antibody or O.N. with the other primary antibodies. Filters were washed three times with 1x TBS‐Tween 20 buffer for 5 min and incubated for 45 min with respective secondary antibodies conjugated to peroxidase (Sigma‐Aldrich). The antigen‐antibody complexes were then detected using the ECL Immobilon Western Chemiluminescent HRP‐substrate system (Millipore, Darmstadt, Germany) and autoradiography, according to the manufacturer's instructions. Signals were subsequently normalized with an antibody anti‐α‐actin (dilution 1:10,000; Santa Cruz Biotechnology, Santa Cruz, CA). Western blots bands were quantified using the ImageJ software.
2.12. Statistical analysis
All data are reported as the mean ± standard deviation of three separate experiments. Statistical differences between mock control and treated cells were calculated using the one‐way analysis of variance procedure followed by Dunnett's multiple comparison test, where appropriate. Differences were considered significant when p < 0.05 (*) (#) and highly significant when p < 0.0001 (**) (##) versus each respective mock control or untreated control group.
3. RESULTS
3.1. Spectral differences in K562 cells overexpressing different GATA‐1 isoforms
We first recorded absorbance spectra obtained from K562 cells overexpressing GATA‐1FL (GATA‐1FL cells) and GATA‐1S (GATA‐1S cells), respectively, in the 300 to 800 nm wavelength range (Figure 1). Spectra of the light absorbance from K562 living cells were normalized to the culture medium. Absorbance values from K562 cells overexpressing GATA‐1FL were found to be slightly higher compared with those obtained from either K562 cells overexpressing GATA‐1S or mock control in the considered spectral range. Furthermore, the absorbance spectra from K562 cells overexpressing GATA‐1FL exhibited a small peak at 412.5 nm and a dip at 560 nm. This observed peak is associated with a slight increase of absorbance compared with the general spectral trend while the absorbance becomes relatively less pronounced at the dip of 560 nm.
Figure 1.
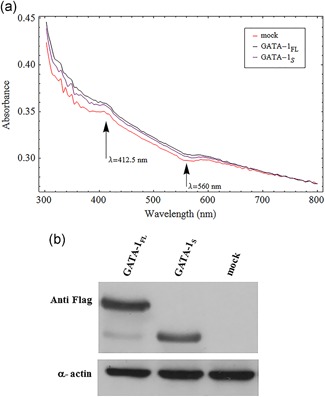
Absorbance spectra of transfected cells with expression vectors for GATA‐1FL or GATA‐1S. (a) Spectra of the light absorbance from K562 living cells which were normalized to the culture medium. Spectrum wavelengths (nm) are represented on the abscissa and the relative intensity is plotted on the ordinate scale. The figure shows representative results of one of at least three independent experiments. (b) Western blot analysis of the expression levels of GATA‐1 isoforms in total protein lysates from K562 cells transiently transfected with either FLAG‐tagged GATA‐1FL (48 kD), GATA‐1S (38 kD) isoforms or empty vector (mock) [Color figure can be viewed at wileyonlinelibrary.com]
As shown in Figure 1a, both the peak at 412.5 nm and the dip at 560 nm become more pronounced in the absorbance spectra from K562 cells overexpressing GATA‐1S and from mock control compared with the spectra from K562 cells overexpressing GATA‐1FL.
Notably, we observed that spectral features at 560 nm and 412.5 nm could resemble the absorption spectra of the reduced and oxidized form of cytochrome b‐560, that, as previously reported, shows absorption maxima at 279, 412.5, and 533 nm in the oxidized form, and at 427, 530, and 560 nm in the reduced form (Doi, Takamiya & Nishimura, 1982).
Therefore, the combined absorbance spectra variations at both 412.5 and 560 nm prompted thus us to hypothesize that the expression of specific GATA‐1 isoforms could be associated with different redox states of this cytochrome, a component of complex II of the mitochondrial respiratory chain that is responsible for transferring electrons from succinate to ubiquinone (Yu, Xu, Haley & Yu, 1987).
3.2. Oxidative stress and ROS levels in K562 cells overexpressing GATA‐1 specific isoforms
To verify the hypothesis that the expression of GATA‐1 isoforms could differently affect the redox states of cytochrome b‐560 and according to recent reports indicating that dysregulation of cytochrome b‐560 redox activity may be linked to electron recycling and abnormal ROS production (Mailloux, 2015; Quinlan et al., 2012), we next asked whether the putative changes in the redox state of cytochrome b‐560 in cells overexpressing different GATA‐1 isoforms might lead to variations in cytoplasmatic and/or mitochondrial ROS production. To this aim, we first evaluated by cytofluorometry the levels of total cytoplasmic ROS in transiently transfected K562 cells overexpressing GATA‐1FL or GATA‐1S isoforms. Results showed a dramatic increase of ROS levels in GATA‐1FL cells with respect to mock or GATA‐1S cells, thus suggesting that GATA‐1FL may promote a more oxidizing intracellular environment in hematopoietic progenitors. This condition may eventually drive redox‐sensitive signaling processes involved in the antiproliferative and prodifferentiation activities of the full‐length isoform of GATA‐1 (Figure 2a). Furthermore, given the role described for complex II in superoxide anion production, cells were incubated with MitoSox reagent to specifically measure mitochondrial superoxide anion levels that unexpectedly were found remarkably higher in GATA‐1S cells with respect to GATA‐1FL cells (Figure 2b). In an effort to better elucidate the mechanisms giving rise to this differential ROS production, we next asked whether the different superoxide production in these cells could be related to variations in their mitochondrial mass. Cells were thus stained with the Mitotracker green dye. A dramatic increase of the mitochondrial mass was found in GATA‐1S cells that is in agreement with recent studies reporting increased mitochondrial mass in myeloid leukemia cells with respect to their normal counterpart (Di Marcantonio et al., 2018; Farge et al., 2017; Geethakumari et al., 2017; Ye, Zhang, Townsend & Tew, 2015). Notably, this finding is even more relevant if compared with GATA‐1FL cell behavior whose mitochondrial mass content is even lower than that of mock cells (Figure 2c). Nonetheless, when we normalized the superoxide levels to the mitochondrial mass, a higher ratio was found in GATA‐1FL cells (Figure 2d). This data is also consistent with the elevated cytoplasmic ROS levels detected in these cells with respect to GATA‐1S cells, given the well established direct correlation between cytoplasmic ROS and mitochondrial superoxide concentrations (Sena & Chandel, 2012; Shadel & Horvath, 2015). Furthermore, to verify that the superoxide signals revealed by cytofluorometric analysis derived from mitochondria, cell samples incubated with MitoSox, Mitotracker, or DAPI were also observed by fluorescence microscopy. As shown in Figure 2e, the merged microscopy images showed colocalization of MitoSox red fluorescence with the green fluorescence of Mitotracker (Figure 2d, panel iv), thus confirming the cytofluorimetric data. The merged images also clearly showed the prevalence of the MitoSox red fluorescence signal in GATA‐1FL cells and conversely of the Mitotracker green signal in GATA‐1S cells, thereby providing further strong support to the data reported in Figure 2d. Furthermore, microscopy images are also suggestive of structural remodeling of mitochondria that apparently could resemble dynamic fission and/or fusion processes since in GATA‐1S cells they are consistently enlarged as compared with GATA‐1FL cells whose mitochondria appear smaller and more widely dispersed. Taken as a whole, these data allowed us to hypothesize that specific GATA‐1 isoforms could take part to mechanisms of mitochondrial remodeling and ROS compartmentation that, in turn, could contribute to modulate the cellular redox environment to sustain either proliferation or differentiation programs in hematopoietic cells.
Figure 2.
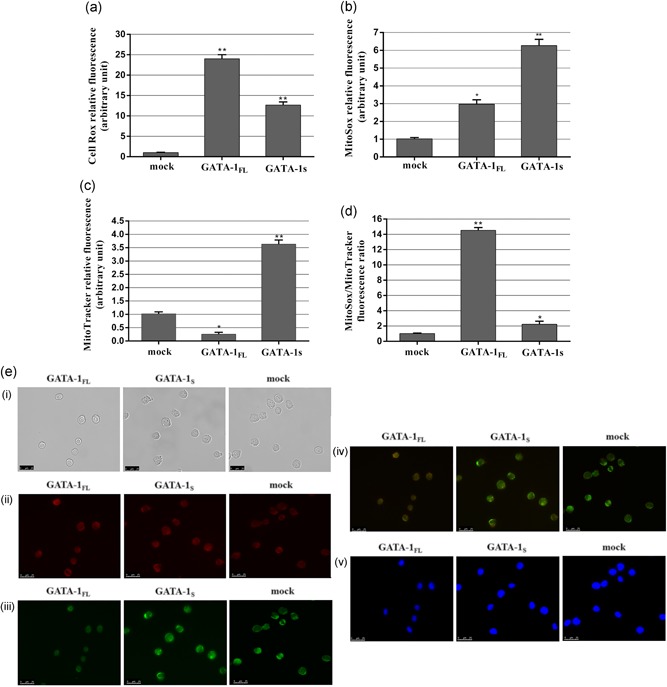
Evaluation of ROS production in K562 cells overexpressing GATA‐1FL and GATA‐1S isoforms. (a) Cytoplasmatic ROS levels detected by flow cytometry analysis in cells overexpressing GATA‐1 isoforms and in mock control after CellRox staining. (b) Mitochondrial superoxide levels detected by flow cytometry analysis in cells overexpressing GATA‐1 isoforms and in mock control stained with MitoSOX red mitochondrial superoxide reagent. (c) Total mitochondrial mass detected by flow cytometry analysis in cells overexpressing GATA‐1 isoforms and in mock cells stained with MitoTracker green FM reagent. (d) Mitochondrial superoxide/mitochondrial mass ratio in cells overexpressing GATA‐1 isoforms and in mock control. All data are presented as the means ± SD (n = 3 in each group). Statistical analysis was performed by one‐way ANOVA, followed by Dunnett's multiple comparison test where appropriate. Differences were considered significant when p < 0.05 and highly significant when p < 0.0001. *p < 0.05; **p < 0.0001, versus mock control. (e) Fluorescent microscopy images of cells overexpressing GATA‐1 isoforms and mock cells. (i) bright field images; (ii) fluorescence images of K562 stained with MitoSox red mitochondrial dye; (iii) fluorescence images of K562 stained with MitoTracker green FM dye; (iv) merged images; (v) nuclei stained with 4′,6′‐diamidino‐2‐phenylindole (DAPI). All data shown are representative of three independent experiments. ANOVA: analysis of variance; ROS: reactive oxygen species; SD: standard deviation [Color figure can be viewed at wileyonlinelibrary.com]
3.3. Changes in DNA contents and in mitochondrial dynamics‐related proteins
On the basis of these observations, we next asked whether the differences in mitochondrial mass found in cells overexpressing GATA‐1 isoforms directly reflected changes in mitochondria size and/or number. Toward this aim, we first proceeded with the quantitation of mtDNA/ChrDNA ratio. Real‐time PCR analysis showed that the mtDNA relative copy number was markedly reduced in GATA‐1FL cells and, conversely, almost unaltered in GATA‐1S cells with respect to the mock control (Figure 3a), thus allowing us to exclude that the increased mitochondrial mass in GATA‐1S cells was associated with a higher number of mitochondria. To further address this issue, we also examined the expression levels of VDAC1, the most abundant protein in the mitochondrial outer membrane (Gonçalves, Buzhynskyy, Prima, Sturgis & Scheuring, 2007). In this case, VDAC1 levels were found reduced in GATA‐1FL cells and markedly elevated in GATA‐1S cells (Figure 3b), thus further confirming that the higher mitochondrial mass detected in GATA‐1S cells is to be related to the increased size of these organelles, whereas, on the other hand, the reduced VDAC1 levels found in GATA‐1FL cells well correlate with the reduced mitochondrial mass detected in these cells.
Figure 3.
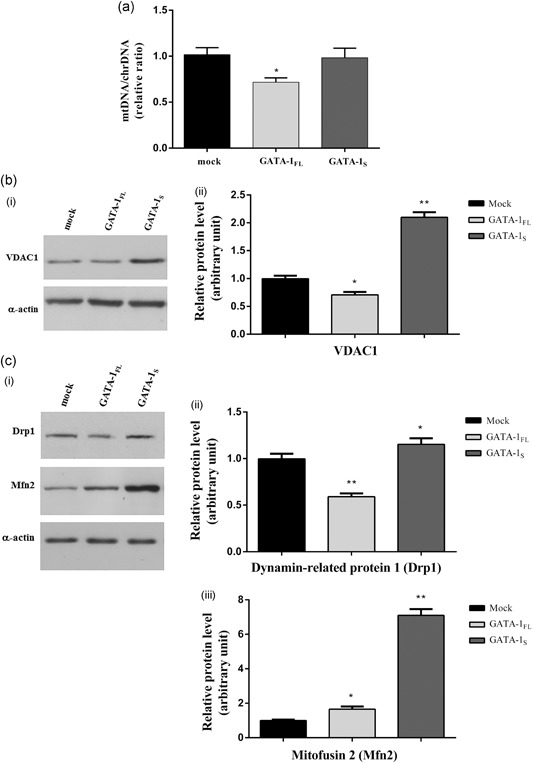
Changes in mitochondrial DNA contents and in the levels of mitochondrial dynamics‐related proteins. (a) Real‐time PCR quantitative analysis of mitochondrial DNA (mtDNA) content relatively to the nuclear DNA (chrDNA) in K562 cells overexpressing GATA‐1 isoforms. Data are presented as fold‐changes relative to the mock control. (b) (i) Western blot analysis of VDAC1 expression levels in total protein lysates from mock control and from cells overexpressing GATA‐1FL and GATA‐1S, respectively. The figure shows representative results of three independent experiments; (ii) densitometric analysis of western blot results. (c) (i) Western blot analysis of dynamin‐related protein 1 (Drp1) and mitofusin 2 (Mfn2) expression levels in total protein lysates from mock control and from cells overexpressing GATA‐1 isoforms. Representative results of three independent experiments are shown; (ii) densitometric analysis of western blot results. For all western blotting data, band intensities from three independent experiments were quantified and normalized to α‐actin used as a loading control. All data were analyzed by one‐way ANOVA, followed by Dunnett's multiple comparison test, where appropriate. Differences were considered significant when p < 0.05 and highly significant when p < 0.0001. *p < 0.05; **p < 0.0001, versus mock control. ANOVA: analysis of variance
To further evaluate the dynamic mechanisms involved in these events, the mitochondrial fission or fusion proteins dynamin‐related protein 1 (Drp1) and mitofusin 2 (Mfn2) were also monitored (Ni, Williams & Ding, 2015). Western blot analysis cells revealed that the most relevant changes affecting GATA‐1FL cells involved the mitochondria fission protein Drp1 whose levels were found significantly reduced in these cells in marked contrast with the mitochondria fusion protein Mfn2 that was found dramatically increased only in GATA‐1S cells (Figure 3c,d).
Collectively, these findings are thus consistent with those reported in Figure 2c,e indicating that the increased mitochondrial mass detected in GATA‐1S cells should be mainly due to mitochondrial fusion mechanisms, thus shedding light on the different roles played by GATA‐1 isoforms in the dynamic remodeling of mitochondria.
3.4. Cell viability and apoptosis in cells expressing GATA‐1 isoforms
In recent years, a substantial and growing body of evidence indicates that variations in ROS levels and in their compartmentation are able to affect cells’ fate and to play a critical role in the transition from a pluripotent to a differentiated state. Indeed, mild oxidative stress is reported to provide beneficial effects to support proliferative and prosurvival pathways whereas excessive ROS production eventually leads to growth arrest and cell death (Sena & Chandel, 2012; Shadel & Horvath, 2015; Sies, 2015; Ye et al., 2015).
According to these observations, we next focused our attention on the correlation between different intracellular redox states triggered by GATA‐1 isoforms and cell viability or apoptosis susceptibility. Cell viability was determined by the MTT assay, whereas early and late apoptosis was measured by flow cytometry with the Annexin V/PI detection kit in cells treated or untreated with the proapoptotic drug cisplatin.
The MTT assay indicated that GATA‐1S cells display increased cell viability and proliferation rate with respect to GATA‐1FL and mock cells, as particularly evident 72 hr posttransfection, whereas in striking contrast, GATA‐1FL expression is associated with progressive reduced cell viability even with respect to the mock counterpart (Figure 4a).
Figure 4.
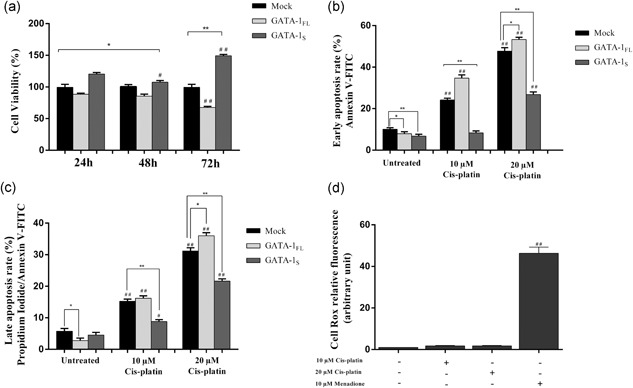
Flow cytometric analysis of cell viability and apoptosis rate in K562 cells overexpressing GATA‐1 isoforms. (a) Cell viability assessed by the MTT assay in mock control and in GATA‐1 overexpressing K562 cells at 24, 48, and 72 hr after transfection. (b) Early apoptosis rate detected with Annexin V staining in mock control and in K562 cells overexpressing GATA‐1 isoforms 48 hr after transfection. (c) Late apoptosis rate detected with Annexin V/propidium iodide staining in mock control and in K562 cells overexpressing GATA‐1 isoforms 48 hr after transfection. Apoptosis was evaluated in untreated cells and in cells treated for 16 hr with 10 and 20 μM cisplatin, respectively. (d) Cytoplasmatic ROS levels detected by flow cytometry analysis in K562 cells stained with CellRox dye after 10 and 20 μM cisplatin exposure. Menadione treatment (10 μM) was used as a positive control for cytoplasmatic ROS production. All data shown represent the mean ± SD of three independent experiments. Statistical analysis was performed by one‐way ANOVA, followed by Dunnett's multiple comparison test where appropriate. Differences were considered significant when p < 0.05 and highly significant when p < 0.0001. # p < 0.05, ## p < 0.0001 versus untreated control group, *p < 0.05, **p < 0.0001 versus mock control. ANOVA: analysis of variance; ROS: reactive oxygen species; SD: standard deviation
On the other hand, Annexin V and PI staining revealed only slight variations in the percentage of basal apoptosis in mock and K562 transfected cells whereas cells treated with cisplatin revealed considerable differences in the apoptosis rate between GATA‐1FL and GATA‐1S cells, with a strong reduction of both early and late apoptotic GATA‐1S cells even at high drug dose as compared to both mock and GATA‐1FL cells (Figure 4b,c). To verify if the proapoptotic effect exerted by cisplatin could be mediated by an increase in ROS production, cytoplasmic ROS levels were evaluated in wild‐type K562 cells after exposure to low and high concentrations of cisplatin, using treatment with menadione as a positive control of ROS production. Interestingly, no variations were found in ROS levels after cisplatin treatment, thus allowing us to exclude that the apoptotic mechanism triggered by cisplatin may involve changes in oxidative stress conditions (Figure 4d).
Taken as a whole, these data are completely in agreement with our hypothesis that a fundamental decrease in cytoplasmatic ROS levels, as observed in GATA‐1S cells, may confer the increased susceptibility to mitogenic signaling and apoptotic resistance.
3.5. Differential antioxidant capacity in cells expressing GATA‐1 isoforms
On the basis of the different ROS levels detected in GATA‐1 cells (as also shown in Figure 2a), we next asked whether variations in oxidative stress conditions in these cells could be related to different changes in their antioxidant systems. First, we evaluated the protein levels of cytoplasmatic (SOD1) and mitochondrial (SOD2) superoxide dismutase (Zelko, Mariani & Folz, 2002; Figure 5a). Interestingly, in GATA‐1FL cells only slight variations were found for both SOD1 and SOD2 with respect to the mock control, whereas, on the contrary, GATA‐1S cells showed a slight increase in SOD1 levels and a marked reduction of SOD2, according to the higher mitochondrial superoxide content detected in these cells (Figure 2b).
Figure 5.
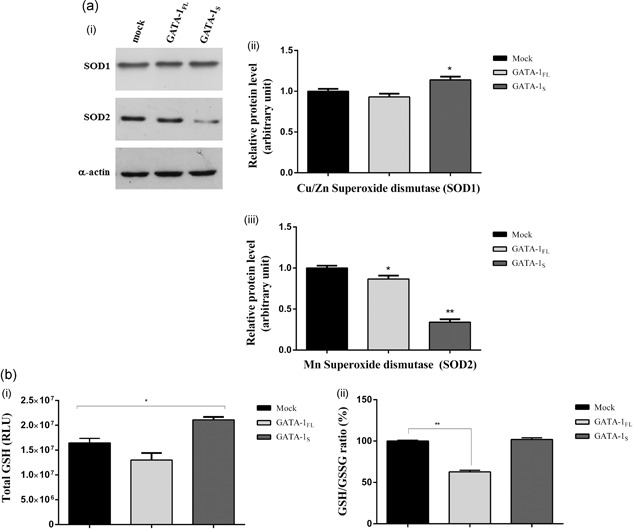
Evaluation of the antioxidant capacity in cells expressing GATA‐1 isoforms. (a) (i) Western blot analysis of the expression levels of Cu/ZnSOD (SOD1) and MnSOD (SOD2) in total protein lysates from mock control and from cells overexpressing GATA‐1FL and GATA‐1S. Each blotting is representative of three independent experiments; (ii,iii) Densitometric analysis of western blot results. Band intensities were quantified and normalized to α‐actin used as loading control. Data are presented as fold‐changes relative to the mock control. Statistical analysis was performed by one‐way ANOVA, followed by Dunnett's multiple comparison test, where appropriate. Differences were considered significant when p < 0.05 and highly significant when p < 0.0001. *p < 0.05; **p < 0.0001, versus mock control. (b) (i) Total glutathione levels (GSH+GSSG) determined in K562 cells 48 hr after transfection with expression vectors for GATA‐1 isoforms or with empty vector (mock). Results represent the net luminescence (as relative luminescence units, RLU) after background subtraction. The mean ± SD of three independent experiments were plotted on the graph; (ii) relative GSH/GSSG ratio for mock control or GATA‐1FL and GATA‐1S cells were obtained using the following formula: (Net transfected cells total glutathione RLU − Net transfected cells GSSG RLU)/(Net transfected cells GSSG RLU/2). Mean ± SD of three independent experiments were plotted on the graph as a relative percentage versus mock control cells. Statistical analysis was performed by one‐way ANOVA, followed by Dunnett's multiple comparison test, where appropriate. Differences were considered significant when p < 0.05 and highly significant when p < 0.0001. *p < 0.05, **p < 0.0001 versus mock control. ANOVA: analysis of variance; GSH: glutathione; GSSG: oxidized glutathione; RLU: relative luminescence units; SD: standard deviation
Measurements of total GSH and of the ratio of its reduced to oxidized form (GSH/GSSG) were also used as indicators of the oxidative stress status in these cells (Figure 5b). Total GSH levels were found reduced in GATA‐1FL cells and increased in GATA‐1S cells, thus indicating an enhanced antioxidant capacity in this latter cell type. Furthermore, in GATA‐1FL cells we also found a dramatic reduced GSH/GSSG ratio. These findings thus further reinforce the evidence of a higher oxidative stress status in GATA‐1FL cells and of a stronger antioxidant capacity in GATA‐1S cells according to what we had previously hypothesized on the basis of the cytoplasmatic ROS levels detected in these cells (Figure 2a).
3.6. Differential response to quercetin treatment in cells expressing GATA‐1 isoforms
The experimental evidence of different oxidative stress conditions in cells expressing GATA‐1 isoforms prompted us to investigate the relationship between variations in viability and apoptosis and the redox state in these cells. To this aim, cells were treated with the flavonoid quercetin whose dose‐ and time‐dependent anti‐ or pro‐oxidant effects have been extensively investigated in several cell lines, including K562 (Akan & Garip, 2013; Brisdelli, Coccia, Cinque, Cifone & Bozzi, 2007; Ferraresi et al., 2005; Ghibellini, Pinti, Nasi, De Biasi, Roat, Bertoncelli & Cossarizza, 2010).
Time‐course and dose‐response experiments were performed in K562 cells treated with 5, 10, 25, 50, 100, and 150 μM quercetin for 3 and 24 hr and cell viability was subsequently assessed by the MTT assay (Figure 6a). Based on these data, the most effective treatments at 50, 100, and 150 μM were chosen for further experiments. Interestingly, opposite effects on cell viability were found in GATA‐1FL and GATA‐1S cells at 3 and 24 hr of treatment. Indeed, after the short‐term exposure (3 hr), a cell viability dose‐dependent increase was found both in GATA‐1FL and GATA‐1S cells, although with some differences between the two cell types. More in detail, in agreement with results reported in Figure 4a, at the lower dose of quercetin, GATA‐1S cells displayed a higher rate of cell viability compared to GATA‐1FL cells whereas, on the contrary, exposure at the higher dose was able to reverse this effect in favor of GATA‐1FL cells (Figure 6b‐i). On the other side, after the long‐term treatment (24 hr) all cell types displayed a dose‐dependent reduction of cell viability, although, noteworthy, this phenomenon was markedly more substantial in GATA‐1S than in GATA‐1FL cells (Figure 6b‐ii).
Figure 6.
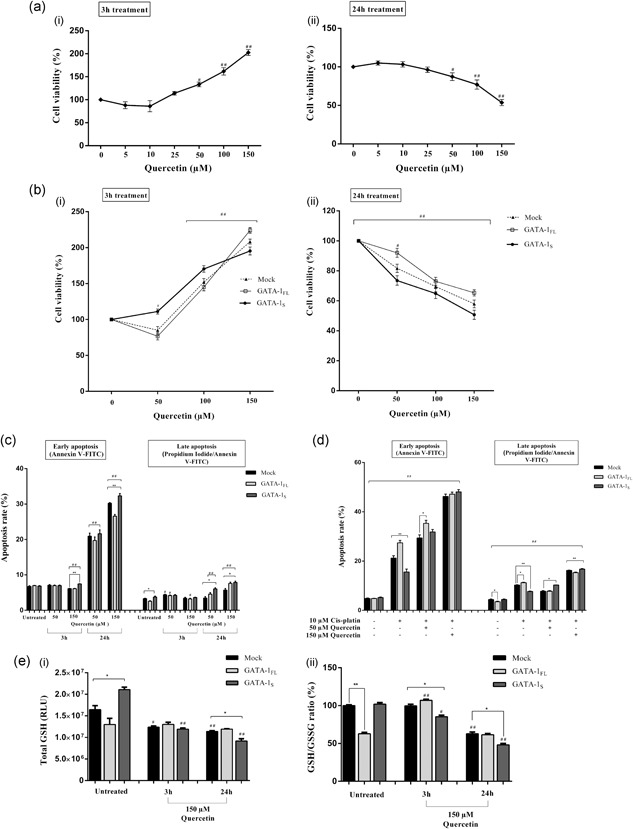
Evaluation of differential response to quercetin treatment in cells expressing GATA‐1 isoforms. (a) Time‐course and dose‐response to 5, 10, 25, 50, 100, 150 μM of quercetin exposure in K562 cells. Cell viability was assessed by the MTT assay after 3 hr (i) and 24 hr exposure (ii). (b) Cell viability after exposure to 50, 100, and 150 μM quercetin for 3 hr (i) and 24 hr (ii) in mock control and in cells overexpressing GATA‐1 isoforms 48 hr after transfection. (c) Early and late apoptosis rate in mock control and in cells overexpressing GATA‐1 isoforms. Forty‐eight hours after transfection, cells were treated for 3 hr and 24 hr with 50 and 150 μM quercetin or with vehicle control (DMSO + PBS); (d) Early and late apoptosis rate in mock control and in cells overexpressing GATA‐1 isoforms after co‐treatment with quercetin and cisplatin. The untreated control group was incubated with vehicle control (DMSO+PBS) under the same conditions. All data represent the mean ± SD of three independent experiments. (e) (i) Total glutathione levels (GSH+GSSG) detected after exposure to 150 μM quercetin or to vehicle control (DMSO+PBS) for 3 hr and 24 hr in K562 cells 48 hr after transfection. Results represent the net luminescence (in RLU) after background subtraction and the mean ± SD of three independent experiments were plotted on the graph; (ii) relative GSH/GSSG ratio for mock control or GATA‐1FL and GATA‐1S cells treated for 3 hr and 24 hr with 150 μM quercetin or vehicle control (DMSO+PBS). Results were obtained using the following formula: (Net transfected cells total glutathione RLU − Net transfected cells GSSG RLU)/(Net transfected cells GSSG RLU/2). Mean ± SD of three independent experiments were plotted on the graph as relative percentage versus mock of the untreated control group. All data were analyzed for statistical significance by one‐way ANOVA, followed by Dunnett's multiple comparison test where appropriate. Differences were considered significant when p < 0.05 and highly significant when p < 0.0001. # p < 0.05, ## p < 0.0001 versus untreated control group, *p < 0.05, ** p < 0.0001 versus mock control. ANOVA: analysis of variance; DMSO: dimethyl sulfoxide; GSH: glutathione; GSSG: oxidized glutathione; PBS: phosphate‐buffered saline; RLU: relative luminescence units; SD: standard deviation
To verify whether variations in cell viability correlate with changes in the apoptotic rate, early and late apoptosis was evaluated by the Annexin V/PI assay in cells exposed to 50 and 150 μM quercetin for 3 and 24 hr. As also expected on the basis of the cell viability data shown in Figure 6b, after 3 hr exposure at the higher dose level, a slight increase in the percentage of apoptotic GATA‐1S cells was found. More interestingly, after 24 hr exposure, at both low and high doses of quercetin, GATA‐1S cells showed an enhanced apoptotic rate with respect to the GATA‐1FL counterpart (Figure 6c). To further confirm the changes in the apoptotic susceptibility induced by quercetin in GATA‐1FL and GATA‐1S cells, apoptosis was also evaluated in cells co‐treated with quercetin and cisplatin. Results clearly confirmed the enhancer activity of quercetin on the apoptotic susceptibility in both cell types. Furthermore, even more importantly, this effect appears to be of greater impact on GATA‐1S cells that showed the most significant dose‐dependent increase with respect to GATA‐1FL cells. Consequently, it is to be emphasized that quercetin is able to revert the resistance to apoptosis shown by GATA‐1S cells when treated with cisplatin alone (Figure 4b,c).
On the basis of these findings, we thus argued that quercetin was able to exert either anti‐ or pro‐oxidant activities, depending on its dose and time of exposure. To clarify this issue, measurements of total GSH and GSH/GSSG ratio were performed in cells treated with the most effective dose of quercetin (150 μM) for 3 and 24 hr. As shown in Figure 6e, a dramatic reduction of total GSH content in GATA‐1S cells was found even after a short‐term exposure to quercetin that is in striking contrast with the unvaried levels detected in GATA‐1FL cells even after 24 hr treatment. Conversely, compared to the untreated samples, the GSH/GSSG ratio resulted to be enhanced in GATA‐1FL cells after 3 hr treatment and progressively decreasing in the other samples (Figure 6e). Our study thus indicates that in our experimental model, during short‐term treatment, quercetin acts as ROS scavenger and protects cells from oxidizer molecules whereas it plays a pro‐oxidant role after a long‐term treatment, as indicated by the variations in GSH levels and in cell viability and apoptosis susceptibility shown by GATA‐1FL and GATA‐1S cells.
3.7. Correlation between GATA‐1 and abnormal expression levels of SDHC
Given the role of the dysregulated activity of complex II in the generation of aberrant levels of superoxide anions (Grimm, 2013; Quinlan et al., 2012; Yankovskaya et al., 2003), we next hypothesized that specific GATA‐1 isoforms could differently influence complex II activity. To this aim, we examined protein levels of each of the SDH components, namely SDHA, SDHB, SDHC, and SDHD, in GATA‐1FL or GATA‐1S cells, respectively. Interestingly, we found variations only in SDHC (Figure 7a,b) that, along with the SDHD subunit, binds cytochrome b‐560 and is required for electron transfer from succinate to the ubiquinone pool (Yankovskaya et al., 2003). However, in spite of apparently increased levels of SDHC in GATA‐1S cells, results revealed a dramatic increased SDHC/mitochondrial mass ratio in GATA‐1FL cells (Figure 7c), a finding that is in agreement with the reduced mitochondrial mass detected in these cells (Figure 2b). Taken as a whole, these data further support our hypothesis that mechanisms triggered by the two GATA‐1 isoforms could involve different structural and functional remodeling of mitochondria.
Figure 7.
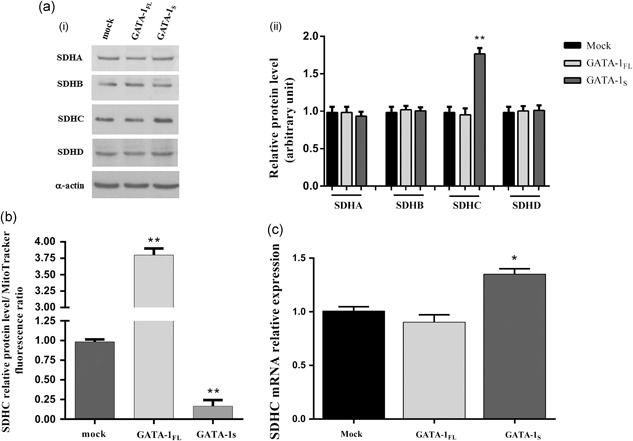
Evaluation of the expression levels of SDH subunits in K562 cells overexpressing GATA‐1 isoforms. (a) Protein levels of SDH complex detected by western blotting in total protein lysates from mock control and from cells overexpressing GATA‐1FL and GATA‐1S, respectively, showing increased levels of SDHC in GATA‐1S cells. (i) Representative western blotting of three independent experiments is shown for each SDH subunit and for α‐actin used as loading control; (ii) densitometric analysis of western blot results. Band intensities from three independent experiments were quantified and normalized to α‐actin. (b) SDHC/mitochondrial mass ratio in mock control and in cells overexpressing GATA‐1 isoforms. (c) Real‐time RT‐PCR quantitative analysis of SDHC mRNA levels in mock control and in cells overexpressing GATA‐1 isoforms. SDHC expression levels were normalized against GAPDH. All data represent the mean ± SD of three independent experiments. Statistical analysis was performed by one‐way ANOVA, followed by Dunnett's multiple comparisons test, where appropriate. Differences were considered significant when p < 0.05 and highly significant when p < 0.0001. # p < 0.05, ## p < 0.0001 versus untreated control group, *p < 0.05, **p < 0.0001 versus mock control. ANOVA: analysis of variance; SD: standard deviation; SDHC: succinate dehydrogenase complex
Given the transcriptional role played by GATA‐1, we next asked whether this factor is directly or indirectly involved in SDHC gene expression. Although evaluation of SDHC mRNA levels confirmed western blot data (Figure 7b,d) analysis of a publically‐available ChIP‐seq data set performed for GATA‐1 binding sites in K562 cells (Snyder, 2012) did not reveal GATA‐1 responsive elements in the SDHC genomic region, thus allowing us to hypothesize that GATA‐1 can indirectly contribute to the modulation of SDHC expression levels by taking part to a multifactorial regulatory network.
3.8. Correlation between SDHC expression levels and redox states of cytochrome b‐560
On the basis of these results, we speculated that different mitochondrial SDHC levels in cells overexpressing specific GATA‐1 isoforms could be related to variations in the redox state of cytochrome b‐560, as also suggested by spectrophotometric data (Figure 1a). To address this hypothesis, we first determined the ratio between the integrated absorbance area of the dip at a wavelength of 560 nm and the integrated absorbance area under the peak at 412 nm, A(560) and A(412). We speculated that since these wavelengths resemble the reduced and oxidized form of cytochrome b‐560, respectively, a depletion of the reduced form of cytochrome b‐560 should result in an absorption decrease causing a dip at 560 nm, whereas an increase of the oxidized form of cytochrome b‐560 leads to an absorption enhancement at the shorter wavelength of 412.5 nm. Accordingly, we chose to use the A(560)/A(412) ratio as an indicator to provide information on the interplay between the reduced and the oxidized form of cytochrome b‐560. In this way, an increase in the A(560)/A(412) ratio indicates enhancement of the oxidized form of cytochrome b‐560, whereas values of the ratio less than unity indicate the prevalence of the reduced form.
The A(560)/A(412) ratio was calculated from the absorbance spectra by integrating the spectral data at the two wavelengths and was plotted in Figure 8 as a function of the relative SHDC/mitochondrial mass ratios obtained from transfected cells with expression vectors for GATA‐1FL or GATA‐1S, respectively, and from mock control.
Figure 8.
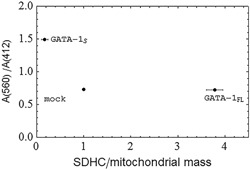
Evaluation of the redox state of cytochrome b‐560 with respect to SDHC levels in K562 cells overexpressing GATA‐1 isoforms. The ratio of the integrated area A (560) under the absorbance dip at 560 nm to the integrated area A (412) under the absorbance peak at 412 nm versus the SDHC levels normalized to the mitochondrial mass. The integrated spectral data are obtained from the absorbance spectra of cells transfected with expression vectors for GATA‐1FL or GATA‐1S, respectively, and with mock control. The positional error bars are the standard deviation of the means (n = 3) of the measurements of the SDHC/mitochondrial mass ratio in cells overexpressing GATA‐1 isoforms. SDHC: succinate dehydrogenase complex
Data shown in Figure 8 indicate the prevalence of the oxidized form of cytochrome b‐560 in GATA‐1S cells as compared with mock or GATA‐1FL cells in which lower A(560)/A(412) ratios were found. Remarkably, according to the model proposed by Quinlan et al. (2012) for superoxide generation by complex II, this result well correlates with the lower mitochondrial superoxide concentration detected in GATA‐1S cells (Figure 2d,e). In fact, it is arguable that the prevalence of the oxidized form of cytochrome b‐560 is related to more efficient electron flow through the respiratory chain that should reduce electron recycling through the heme moiety of cytochrome b‐560 and, consequently, the production of superoxide molecules. This process could eventually contribute to decreasing intracellular oxidative stress, to stimulate cell proliferation and to prevent differentiation (Ye et al., 2015). On the other hand, GATA‐1FL cells show a lower A(560)/A(412) ratio that is indicative of a prevalence of the reduced versus the oxidized form of cytochrome b‐560. This condition is consistent with enhanced electron recycling by complex II and superoxide production and, consequently, with a higher oxidative stress environment in the cell. Data shown in Figure 8 also point out that, despite a similar A(560)/A(412) ratio, GATA‐1FL cells differ from mock cells for the higher SDHC content (as also shown in Figure 7b) that is in agreement with their higher cytosolic ROS levels and mitochondrial superoxide concentration (Figure 2a,d, and e). Therefore, taken as a whole, our data demonstrate that GATA‐1 isoforms differently contribute to the production and compartmentation of ROS species in leukemic cells to control the hematopoietic cell's fate. Furthermore, our study suggests a possible use of the A(560)/A(412) ratio as a molecular biomarker to evaluate the malignant potential of leukemic cells that could be easily measured by spectrophotometric analysis of blood samples.
3.9. Expression levels of SDHC and GATA‐1 isoforms in a patient with AML
Finally, to evaluate possible clinical implications to our findings, GATA‐1 isoforms and SDHC levels were measured in bone marrow specimens of a patient with AML at different stages of the disease. Notably, at diagnosis, during the acute phase of the disease, we found dramatically elevated levels of both GATA‐1S and SDHC that were completely normalized at remission (Figure 9). Interestingly, in these samples, variations in GATA‐1S and SDHC expression levels during the clinical course of the disease showed a similar trend, in contrast with GATA‐1FL that appears to maintain more stable and lower values during the disease, thus further supporting the specific proleukemic role of GATA‐1S in myeloid cells as so far reported (Crispino, 2005; Halsey et al., 2012; Khan et al., 2011). Noteworthy, although it was not possible to evaluate mitochondrial mass and ROS levels in these samples, the elevated SDHC levels detected in AML leukemic cells well correlate with the overexpression of GATA‐1S and are in agreement with the results above reported for K562 cells (Figure 7a).
Figure 9.
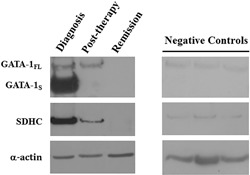
Evaluation of the expression levels of SDHC and GATA‐1 isoforms in a patient with AML. Protein levels of GATA‐1 and SDHC detected by western blotting in total protein lysates from bone marrow biopsies of a patient with AML at different stages of the disease and three healthy negative controls, showing prevalent expression of GATA‐1S isoform and elevated SDHC levels during the acute phase of the disease and their normalization at remission. Representative western blots are shown for GATA‐1, SDHC and α‐actin used as a loading control. AML: acute myeloid leukemia; SDHC: succinate dehydrogenase complex
4. DISCUSSION
Recently we exploited the capabilities of optical techniques to identify novel oncometabolites and biomarkers linked to the expression of specific isoforms of the transcriptional factor GATA‐1. The rationale for using optical techniques is to detect biochemical and morphological features that are concurrent with precancerous conditions because light scattering could be more sensitive to morphological changes than other currently used visualizing techniques (Bellisola & Sorio, 2012). In this study spectral differences at wavelengths of 560 and 412.5 nm were found in K562 cells overexpressing GATA‐1FL (GATA‐1FL cells) or GATA‐1S (GATA‐1S cells), respectively, two GATA‐1 isoforms that play opposite roles in the differentiation and proliferation processes of different hematopoietic lineages (Chlon et al., 2015; Doré & Crispino, 2011; Halsey et al., 2012; Kaneko et al., 2012).
First, we observed that these wavelengths resemble, respectively, the reduced and oxidized form of cytochrome b‐560, the complex II component of the respiratory chain that may be directly involved in electron recycling and superoxide production (Grimm, 2013; Quinlan et al., 2012; Yankovskaya et al., 2003). In this regard, it is noteworthy that, although complex I and III are referred to as chief sites for mitochondrial superoxide production, more recently mitochondrial control of apoptosis and mechanisms of oncogenesis have also been related to complex II whose impaired activity could contribute to generate a higher oxidative stress environment (Di Marcantonio et al., 2018; Farge et al., 2017; Grimm, 2013). According to these observations, several mutations in genes encoding for complex II subunits and for assembly factors have been found in different tumor types, including pheochromocytoma, paraganglioma, gastrointestinal cancer, renal cancer, thyroid cancer, neuroblastoma, and breast cancer (Bardella, Pollard & Tomlinson, 2011; Hensen et al., 2011; Janeway et al., 2011). In addition, the oncogenic activity of these mutations has been associated with a high production of superoxide that may be responsible for the genomic instability of cancer cells (Bardella et al., 2011; Yankovskaya et al., 2003). A simple model for superoxide or hydrogen peroxide production by complex II has recently been proposed by Quinlan et al. (2012). In this model, superoxide is only produced when the flavin is either semi‐reduced (FADH·) or fully reduced (FADH2) and the substrate binding site is empty. In summary, when complex III is inhibited and rapid reoxidation of the ubiquinol (Q) pool is prevented, mitochondrial complex II generates superoxide or hydrogen peroxide, in both forward and reverse reactions. Furthermore, high succinate concentration and mitochondrial inner membrane potential can also induce reverse electron transfer from complex II to complex I, one of the major site of mitochondrial superoxide production (Grimm, 2013; Mailloux, 2015; Wong, Dighe, Mezera, Monternier, & Brand, 2017).
Based on this model and on our preliminary spectra data that were suggestive of different redox states of cytochrome b‐560 in cells overexpressing different GATA‐1 isoforms, we speculated that these isoforms could specifically contribute to inhibit or promote proliferative pathways by differently modulating oxidative stress conditions.
We started to test this hypothesis by investigating the role of GATA‐1 isoforms on ROS production. Variations both in ROS levels and in intracellular compartmentation were found associated with the expression of specific GATA‐1 isoforms: cytosolic ROS resulted to be dramatically increased in GATA‐1FL cells whereas, unexpectedly, increased mitochondrial superoxide levels were found in GATA‐1S cells. Indeed, initially, these data appeared rather incongruent since mitochondrial superoxide is an important source for cytoplasmic ROS (Brand, 2016; Shadel & Horvath, 2015) and, expectedly, a higher superoxide content would correspond to increased cytoplasmic ROS levels. However, an explanation to these apparently abnormal findings was provided when superoxide levels were normalized over the mitochondrial mass. In fact, in this case, according to the differences in mitochondrial mass associated with GATA‐1FL and GATA‐1S expression, the superoxide/mitochondrial mass ratio resulted to be significantly elevated only in GATA‐1FL cells (Figure 2d), thus providing evidence for the higher mitochondrial superoxide concentration in this latter type of cells, a finding that well correlates with the increased cytoplasmic ROS levels detected in these cells with respect to the GATA‐1S counterpart (Figure 2a). On the basis of these findings, we thus speculated that the increased mitochondrial mass in GATA‐1S cells could contribute to modulate redox signaling to promote cell proliferation and resistance to proapoptotic stimuli. Notably, this scenario is also consistent with the different roles played by GATA‐1 isoforms on hematopoietic cells’ fate, in which GATA‐1FL promotes the differentiation process whereas GATA‐1S is mainly involved in the maintenance of the proliferative multipotency (Burda, Laslo & Stopka, 2010; Crispino, 2005; Halsey et al., 2012). On the basis of these observations, we next asked whether the differences in mitochondrial mass found in cells overexpressing GATA‐1 isoforms directly reflected changes in mitochondria size and/or number. To address this issue, mtDNA contents were evaluated along with the levels of canonical mitochondria proteins, including VDAC1, a component of the outer mitochondrial membrane, and the fission or fusion proteins Drp1 and Mfn2. Results clearly allowed us to exclude an increased mitochondria number in GATA‐1S cells, thus confirming that the higher mitochondrial mass detected in these cells can be related to the increased size of these organelles. Also, variations in the protein levels of Drp1 and Mfn2 (Figure 3) are consistent with mechanisms of mitochondrial fusion that could be responsible of the increased mitochondrial mass detected in GATA‐1S cells, thus shedding light on the different roles played by GATA‐1 isoforms in dynamic remodeling of mitochondria.
Therefore, given the relevant role played by ROS in myeloid leukemogenesis, we examined the putative molecular mechanisms underlying the different redox states in cells overexpressing GATA‐1 isoforms. At this regard, we focused our attention on mechanisms involving redox metabolism in mitochondria, that are recognized as the main site for ROS production in the cell, although it cannot be excluded that other organelles or reactions may contribute to modulating redox homeostasis in our cells. We thus asked whether variations in oxidative stress conditions in these cells could be related to differences in their antioxidant capacities since, under normal conditions, ROS are scavenged and converted to nonreactive species by a variety of enzymatic and nonenzymatic antioxidant systems, including superoxide dismutase, peroxidase, catalase, vitamins and reduced GSH. Indeed, in GATA‐1S cells we found a slight increase in the cytoplasmatic SOD1 levels and, conversely, reduced levels of the mitochondrial SOD2 (Figure 5a). These findings are in agreement with the mitochondrial superoxide content detected in these cells (Figure 2b). Importantly, recent literature data report reduced SOD2 expression levels in acute and chronic myeloid leukemia patients (Ciarcia et al., 2010; Girerd et al., 2018; Whang, Xu, Zhang, Li, 2014). Also it is noteworthy that SOD2 plays a crucial role in regulating ROS signaling by controlling the conversion of superoxide anion to other reactive oxygen species that can reach the cytoplasm (Wang, Branicky, Noe, & Hekimi, 2018). Therefore, our findings could provide a possible explanation to the increased mitochondrial superoxide levels and the corresponding reduced cytoplasmatic ROS found in GATA‐1S cells.
Variations were also found in the GSH content, with reduced GSH levels and prevalence of the oxidized form in GATA‐1FL cells and increased GSH levels, mainly in its reduced form, in GATA‐1S cells (Figure 5b). Such findings are consistent with our previous data as they are clearly indicative of a higher oxidative stress status in GATA‐1FL cells and a stronger antioxidant capacity in GATA‐1S cells.
Next, we asked whether changes in cell viability and apoptosis rate detected in these cells were directly related to their redox states. To this aim, cells were treated with the flavonoid quercetin whose dose‐ and time‐dependent anti‐ or pro‐oxidant effects have been extensively investigated in several cell lines, including K562 (Akan & Garip, 2013; Brisdelli, Coccia, Cinque, Cifone & Bozzi, 2007; Ferraresi et al., 2005; Ghibellini, Pinti, Nasi, De Biasi, Roat, Bertoncelli & Cossarizza, 2010). Cell viability and apoptosis rate were evaluated after short‐ and long‐term treatments (3 and 24 hr) with increasing doses of quercetin. GSH measurements were also used to evaluate the pro‐ or antioxidant activity of these treatments (Figure 6e). On the basis of variations in the GSH levels, it is clear that anti‐ or pro‐oxidant activities of quercetin in our cells are time‐ and dose‐dependent: during short‐term exposure quercetin acts as ROS scavenger and protects cells from oxidizer molecules, whereas it plays a pro‐oxidant role at high‐dose and long‐term treatment. In fact, after 3 hr exposure at the lower dose of quercetin, GATA‐1S cells displayed a higher rate of cell viability compared with GATA‐1FL cells, whereas, on the contrary, exposure at the higher dose was able to reverse this effect in favor of GATA‐1FL cells (Figure 6b‐i). Conversely, a long‐term treatment induced a dose‐dependent reduction of cell viability in all cell types, particularly in GATA‐1S (Figure 6b‐ii). Notably, these results are in full agreement with literature data reporting that the anti‐ or pro‐oxidant activity of quercetin particularly depends on its exposure times and on the cell GSH stores, as well as with the evidence that quercetin is able to induce a depletion in the GSH content that is particularly more relevant in cells showing higher GSH levels, as in GATA‐1S cells, where it may exert a more effective pro‐oxidant role (Ferraresi et al., 2005).
According to cell viability data, corresponding variations in the apoptotic rates were found; in particular, after long‐term exposure, GATA‐1S cells showed an enhanced apoptotic rate, in striking contrast with the apoptotic resistance found in these cells after cisplatin treatment (Figures 4b,c, and 6c). To further confirm these changes, apoptosis was also evaluated after cotreatments with quercetin and cisplatin. Besides confirming the enhancer activity of quercetin on the apoptotic susceptibility induced by cisplatin in both cell types, this approach demonstrates the relationship between changes in cell viability and apoptosis rate with the redox states of the cells and, even more importantly, clearly indicates that quercetin is able to revert the resistance to apoptosis shown by GATA‐1S cells when treated with cisplatin alone (Figure 4b–d).
Given the recent role reported for complex II in ROS production (Grimm, 2013; Quinlan et al., 2012) and based on our preliminary data suggesting putative different redox states for cytochrome b‐560, a component of Complex II strictly linked to its subunit C (SDHC) we found interest in investigating the contribution of SDHC to ROS production (Grimm, 2013). We hypothesized that GATA‐1 isoforms may be differently involved in the regulation of SDHC expression levels, according to recent reports indicating that SDHC overexpression can inhibit succinate‐coenzyme Q reductase, without compromising the oxidation activity of succinate to fumarate (SDH; Grimm, 2013). Inhibition of electron transfer to ubiquinone would lead to the reduction of oxygen molecules and, in turn, to the production and accumulation of superoxide anion (Quinlan et al., 2012). Nevertheless, in GATA‐1S cells we found higher levels of SDHC in comparison with GATA‐1FL. However, normalization of SDHC protein levels over the mitochondrial mass revealed an increased SDHC/mitochondrial mass ratio in GATA‐1FL cells with respect to GATA‐1S cells (Figure 4b). According to the role of SDHC overexpression in promoting mitochondrial superoxide production, this data well correlates with the higher mitochondrial concentration of superoxide in GATA‐1FL cells with respect to GATA‐1S cells (Figure 2d,e).
Furthermore, different ROS compartmentations were found related to the expression of the two isoforms of GATA‐1 that could contribute to modulate the redox microenvironment and, eventually, define the cell's fate. According to the role played by mitochondria in the regulation of redox signaling (Sullivan & Chandel, 2014) and to the evidence that the enhanced antioxidant defense and reduced ROS levels in stem cells increase their proliferation rate and contribute to tumor initiation (Diehn et al., 2009; Ishimoto et al., 2011; Janeway et al., 2011; Lartigue et al., 2009), our results allowed us to speculate that GATA‐1FL cells predispose cells to oxidative stress and, consequently, to enhanced susceptibility to apoptosis by increasing cytoplasmatic ROS levels, whereas, on the contrary, GATA‐1S could promote leukemogenesis through reduction of cytoplasmatic ROS levels, inhibition of ROS‐mediated apoptosis and upregulation of prosurvival pathways. Noteworthy, in both cases these effects seem to be mediated via different patterns of mitochondrial remodeling and regulation of respiratory chain activity, thus providing further mechanistic insights into the role of GATA‐1 in onco‐hematological diseases.
Finally, with the aim to explore the potential clinical impact of our findings, we evaluated GATA‐1 isoforms and SDHC levels in bone marrow samples from a patient with AML at different stages of the disease. Our results showed a dramatic increase of GATA‐1S levels at diagnosis that were reduced after chemotherapy and normalized at remission, thus confirming the data obtained in K562 cells as indicative of a proleukemic role of GATA‐1S with respect to its full‐length counterpart. Also, according to our previous results, during the clinical course of the disease, a similar trend was found for SDHC levels. Therefore, although more samples are undoubtedly required to corroborate the clinical relevance of these findings, these preliminary results are in full agreement with our experimental model, therefore providing support to the potential clinical implications of this study and further highlighting the key role played by mitochondria in these pathogenic processes.
5. CONCLUSIONS
A multidisciplinary approach allowed us to shed light on the different effects played by GATA‐1 isoforms in myeloid precursors and to define their potential pathophysiological effects in the leukemogenic process via modulation of ROS production and compartmentation. Although many questions still remain to be addressed, this study highlights a so far unexplored target for intervening mitochondrial ROS‐related processes in leukemogenesis. By providing a better understanding of the source and species of ROS generated by myeloid cells to escape apoptosis and to promote cell proliferation and leukemogenesis, this study promises to pave the way to novel more effective ROS‐based therapies in myeloid leukemia as alternative treatment options particularly in case of a chemotherapy‐resistant disease.
AUTHOR CONTRIBUTIONS
P.R., R.S., S.T., and M.G. designed, performed and analyzed the experiments; F.P. and G.M. performed clinical evaluation of the AML patient and provided bone marrow samples; P.R., S.d.N. and M.G. contributed to the interpretation of data and wrote the manuscript, G.P., P.M., and P.I. contributed to the conception and design of the study and revised the manuscript critically, M.G. coordinated the project. All coauthors approved the final version of the manuscript and declare no conflict of interests.
ACKNOWLEDGMENTS
Authors wish to thank Dr. Sivia Parisi and Dr. Simona Romano for their helpful support in microscopy and cytofluorimetric analysis and are grateful to Prof. Tommaso Russo and Prof. Paola Costanzo for fruitful discussions during the writing of this manuscript. This study was supported by BenTeN (POR Campania FESR 2007/2013 O.O.2.1) and by the Regione Campania “SATIN” grant 2018–2020.
Riccio P, Sessa R, de Nicola S, et al. GATA‐1 isoforms differently contribute to the production and compartmentation of reactive oxygen species in the myeloid leukemia cell line K562. J Cell Physiol. 2019;234:20829–20846. 10.1002/jcp.28688
References
REFERENCES
- Bardella, C. , Pollard, P. J. , & Tomlinson, I. (2011). SDH mutations in cancer. Biochimica et Biophysica Acta, 1807, 1432–1443. 10.1016/j.bbabio.2011.07.003 [DOI] [PubMed] [Google Scholar]
- Bellisola, G. , & Sorio, C. (2012). Infrared spectroscopy, and microscopy in cancer research and diagnosis. American Journal of Cancer Research, 2, 1–21. www.ajcr.us [PMC free article] [PubMed] [Google Scholar]
- Brand, M. D. (2016). Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radicals Biology & Medicine, 100, 14–31. 10.1016/j.freeradbiomed.2016.04.001 [DOI] [PubMed] [Google Scholar]
- Bresnick, E. H. , Katsumura, H. R. , Lee, H. Y. , Johnson, K. D. , & Perkins, A. S. (2012). Master regulatory GATA transcriptional factors: Mechanistic principles and emerging links to hematological malignancies. Nucleic Acids Research, 40, 5819–5831. 10.1093/nar/gks281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burda, P. , Laslo, P. , & Stopka, T. (2010). The role of PU.1 and GATA‐1 transcription factors during normal and leukemogenic hematopoiesis. Leukemia, 24, 1249–1257. 10.1/leu.2010.104 [DOI] [PubMed] [Google Scholar]
- Cantor, A. B. (2016). Megakaryocytic transcription factors in disease and leukemia In Schulze H., & Italiano J. (Eds.), Molecular and cellular biology of platelet formation (pp. pp.61–91). Cham, Switzerland: Springer; 10.1007/978-3-319-39562-3_3. [DOI] [Google Scholar]
- Chlon, T. M. , McNulty, M. , Goldenson, B. , Rosinski, A. , & Crispino, J. D. (2015). Global transcriptome and chromatin occupancy analysis reveal the short isoform of GATA1 is deficient for erythroid specification and gne expression. Haematologica, 100(5), 575–583. 10.3324/haematol.2014.112714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciarcia, R. , d’Angelo, D. , Pacilio, C. , Pagnini, D. , Galdiero, M. , Fiorito, S. , … Giordano, A. (2010). Dysregulated calcium homeostasis and oxidative stress in chronic myeloid leukemia (CML) cells. Journal of Cell Physiology, 224, 443–453. 10.1002/jcp.22140 [DOI] [PubMed] [Google Scholar]
- Ciovacco, W. A. , Raskind, W. H. , & Kacena, M. A. (2008). Human phenotypes associated with GATA‐1 mutations. Gene, 427, 1–6. 10.1016/j.gene.2008.09.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crispino, J. D. (2005). GATA1 in normal and malignant hematopoiesis. Seminars in Cell and Developmental Biology, 16, 137–147. 10.1016/j.semcdb.2004.11.002 [DOI] [PubMed] [Google Scholar]
- Di Marcantonio, D. , Martinez, E. , Sidoli, S. , Vadaketh, J. , Nieborowska‐Skorska, M. , Gupta, A. , … Sykes, S. M. (2018). Protein Kinase C Epsilon is a key regulator of mitochondrial redox homeostasis in acute myeloid leukemia. Clinical Cancer Research, 24, 608–618. 10.1158/1078-0432.CCR-17-2684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diehn, M. , Cho, R. W. , Lobo, N. A. , Kalisky, T. , Dorie, M. J. , Kulp, A. N. , … Clarke, M. F. (2009). Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature, 458, 780–783. 10.1038/nature07733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Döhner, H. , Weisdorf, D. J. , & Bloomfield, C. (2015). Acute myeloid leukemia. The New England Journal of Medicine, 373, 1136–1152. 10.1056/NEJMra1406184 [DOI] [PubMed] [Google Scholar]
- Doi, M. , Takamiya, K. , & Nishimura, M. (1982). Isolation and purification of membrane‐bound cytochrome b‐560 from photosynthetic bacterium Chromatium vinosum . Photosynthesis Research, 3, 131–139. 10.1007/BF00040711 [DOI] [PubMed] [Google Scholar]
- Doré, L. C. , & Crispino, J. D. (2011). Transcription factor networks in erythroid cell and megakaryocyte development. Blood, 118, 231–239. 10.1182/blood-2011-04-285981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farge, T. , Saland, E. , de Toni, F. , Aroua, N. , Hosseini, M. , Perry, R. , … Sarry, J. E. (2017). Chemotherapy‐resistant human acute myeloid leukemia cells are not enriched for leukemic stem cells but require oxidative metabolism. Cancer Discovery, 7, 716–735. 10.1158/2159-8290.CD-16-0441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferraresi, R. , Troiano, L. , Roat, E. , Lugli, E. , Nemes, E. , Nasi, M. , … Cossarizza, A. (2005). Essential requirement of reduced glutathione (GSH) for the anti‐oxidant effect of the flavonoid quercetin. Free Radical Research, 39, 1249–1258. https://doi.org/10.108010715760500306935 [DOI] [PubMed] [Google Scholar]
- Gao, J. , Chen, Y. H. , & Peterson, L. A. (2015). GATA family transcriptional factors: Emerging suspects in hematologic disorders. Experimental Hematology & Oncology, 4, 28 10.1186/s40164-015-0024-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geethakumari, P. R. , Nair, R. G. , Jagannathan, G. , Whitaker‐Menezes, D. , Lin, Z. , Flomenberg, N. , … Martinez‐Outschoorn, U. (2017). Analysis of mitochondrial metabolic profile as a predictor of clinical aggressiveness of acute myeloid leukemia. Blood, 130(Suppl 1), 5072. [Google Scholar]
- Girerd, S. , Tosca, L. , Herault, O. , Vignon, C. , Biard, D. , Aggoune, D. , … Turhan, A. G. (2018). Superoxide dismutase 2 (SOD2) contributes to genetic stability of native and T3151‐mutated BCR‐ABL expressing leukemic cells. Biochemical and Biophysical Research Communications, 498, 715–722. 10.1016/j.bbrc.2018.03.023 [DOI] [PubMed] [Google Scholar]
- Gonçalves, R. P. , Buzhynskyy, N. , Prima, V. , Sturgis, J. N. , & Scheuring, S. (2007). Supramolecular Assembly of VDAC in Native Mitochondrial Outer Membranes. Journal of Molecular Biology, 369(2), 413–418. 10.1016/j.jmb.2007.03.063 [DOI] [PubMed] [Google Scholar]
- Grimm, S. (2013). Respiratory chain complex II as general sensor for apoptosis. Biochimica et Biophysica Acta, 1827, 565–572. 10.1016/j.bbabio.2012.09.009 [DOI] [PubMed] [Google Scholar]
- Halsey, C. , Docherty, M. , McNeill, M. , Gilchrist, D. , Le Brocq, M. , Gibson, B. , & Graham, G. (2012). The GATA1s isoform is normally down‐regulated during terminal haematopoietic differentiation and over‐expression leads to failure to repress MYB, CCND2 and SKI during erythroid differentiation of K562 cells. Journal of Hematology & Oncology, 5, 45 10.1186/1756-8722-5-45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hensen, E. F. , Siemers, M. D. , Jansen, J. C. , Corssmit, E. P. , Romijn, J. A. , Tops, C. M. , … Vriends, A. H. (2011). Mutations in SDHD are the major determinants of the clinical characteristics of Dutch head and neck paraganglioma patients. Clinical Endocrinology, 75, 650–655. 10.1111/j.1365-2265.2011.04097.x [DOI] [PubMed] [Google Scholar]
- Hole, P. S. , Darley, R. L. , & Tonks, A. (2011). Do reactive oxygen species play a role in myeloid leukemias? Blood, 117, 5816–5826. 10.1182/blood-2011-01-326025 [DOI] [PubMed] [Google Scholar]
- Ishimoto, T. , Nagano, O. , Yae, T. , Tamada, M. , Motohara, T. , Oshima, H. , … Saya, H. (2011). CD44 variant regulates redox state in cancer cells by stabilizing the xCT subunit of System xc‐ and thereby promotes tumor growth. Cancer Cell, 19, 387–400. 10.1016/j.ccr.2011.01.038 [DOI] [PubMed] [Google Scholar]
- Janeway, K. A. , Kim, S. Y. , Lodish, M. , Nosé, V. , Rustin, P. , Gaal, J. , … Stratakis, C. A. (2011). Defects in succinate dehydrogenase in gastrointestinal stromal tumors lacking KIT and PDGFRA mutations. Proceedings of the National Academy of Science of the USA, 108, 314–318. 10.1073/pnas.1009199108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko, H. , Kobayashi, E. , Yamamoto, M. , & Shimizu, R. (2012). N‐ and C‐terminal transactivation domains of GATA1 protein coordinate hematopoietic program. The Journal of Biological Chemistry, 287, 21439–21449. 10.1074/jbc.M112.370437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan, I. , Malinge, S. , & Crispino, J. (2011). Myeloid leukemia in down syndrome. Critical Reviews in Oncogenesis, 16, 25–36. 10.1615/CritRevOncog.v16.i1-2.40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuntz, E. M. , Baquero, P. , Michie, A. M. , Dunn, K. , Tardito, S. , Holyoake, T. L. , … Gottlieb, E. (2017). Targeting mitochondrial oxidative phosphorylation eradicates therapy‐resistant chronic myeloid leukemia stem cells. Nature Medicine, 23, 1234–1240. 10.1038/nm.4399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lartigue, L. , Kushnareva, Y. , Seong, Y. , Lin, H. , Faustin, B. , & Newmeyer, D. D. (2009). Caspase‐independent mitochondrial cell death results from loss of respiration, not cytotoxic protein release. Molecular Biology of the Cell, 20, 4871–4884. 10.1091/mbc.e09-07-0649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lentjes, M. H. F. M. , Niessen, H. E. C. , Akiyama, Y. , de Bruine, A. P. , Melotte, V. , & Van Engeland, M. (2016). The emerging role of GATA transcription factors in development and disease. Expert Reviews in Molecular Medicine, 18, e3 10.1017/erm.2016.2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mailloux, R. J. (2015). Teaching the fundamentals of electron transfer reactions in mitochondria and the production and detection of reactive oxygen species. Redox Biology, 4, 381–398. 10.1016/j.redox.2015.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni, H.‐M. , Williams, J. A. , & Ding, W.‐X. (2015). Mitochondrial dynamics and mitochondrial quality control. Redox Biology, 4, 6–13. 10.1016/j.redox.2014.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul, F. , Arkin, Y. , Giladi, A. , Jatin, D. A. , Kenigsberg, E. , Heren‐Shaul, H. , … Amit, I. (2015). Transcriptional heterogeneity and lineage commitment in myeloid precursors. Cell, 7, 1663–1677. 10.1016/j.cell.2015.11.013 [DOI] [PubMed] [Google Scholar]
- Petruzzelli, R. , Gaudino, S. , Amendola, G. , Sessa, R. , Puzone, S. , Di Concilio, R. , … Grosso, M. (2010). Role of the cold shock domain protein A in the transcriptional regulation of HBG expression. The British Journal of Haematology, 150, 689–699. 10.1111/j.1365-2141.2010.08303.x [DOI] [PubMed] [Google Scholar]
- Petruzziello, F. , Sessa, R. , Giovannone, E. D. , Catania, M. , Parasole, R. , Menna, G. , … Grosso, M. (2013). Prognostic significance of GATA‐1 and WT1 levels in pediatric hematological disorders. Leukemia Research, 37(S1), S91–S92. 10.1016/S0145-2126(13)70198-X [DOI] [Google Scholar]
- Quinlan, C. L. , Orr, A. L. , Perevoshchikova, I. V. , Treberg, J. R. , Ackrell, B. A. , & Brand, M. D. (2012). Mitochondrial complex II can generate reactive oxygen species at high rates in both the forward and reverse reactions. The Journal of Biological Chemistry, 287, 27255–27264. 10.1074/jbc.M112.374629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Refinetti, P. , Warren, D. , Morgenthaler, S. , & Ekstrøm, P. O. (2017). Quantifying mitochondrial DNA copy number using robust regression to interpret real time PCR results. BMC Research Notes, 10, 593 10.1186/s13104-017-2913-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosmarin, A. G. , Yang, Z. , & Resendes, K. K. (2005). Transcriptional regulation in myelopoiesis: Hematopoietic cell fate, myeloid differentiation, and leukemogenesis. Experimental Hematology, 33, 131–143. 10.1016/j.exphem.2004.08.015 [DOI] [PubMed] [Google Scholar]
- Sena, L. A. , & Chandel, N. S. (2012). Physiological roles of mitochondrial reactive oxygen species. Molecular Cell, 48, 158–167. 10.1016/j.molcel.2012.09.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shadel, G. S. , & Horvath, T. L. (2015). Mitochondrial ROS signaling in organismal homeostasis. Cell, 163, 560–569. 10.1016/j.cell.2015.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimamoto, T. , Ohyashiki, K. , Ohyashiki, J. H. , Kawakubo, K. , Fujimura, T. , Iwama, H. , … Toyama, K. (1995). The expression pattern of erythrocyte/megakaryocyte‐related transcription factor GATA‐1 and the stem cell leukemia gene correlates with hematopoietic differentiation and is associated with outcome of acute myeloid leukemia. Blood, 86, 3173–3180. [PubMed] [Google Scholar]
- Sies, H. (2015). Oxidative stress: A concept in redox biology and medicine. Redox Biology, 4, 180–183. 10.1016/j.redox.2015.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder, M. (2012). GATA1 ChIP‐seq on human K562 produced by the Snyder lab, Stanford. URL https://www.encodeproject.org/experiments/ENCSR000EWM/
- Sriskanthadevan, S. , Jeyaraju, D. V. , Chung, T. E. , Prabha, S. , Xu, W. , Skrtic, M. , … Schimmer, A. D. (2015). AML cells have low spare reserve capacity in their respiratory chain that renders them susceptible to oxidative metabolic stress. Blood, 125, 2120–2130. 10.1182/blood-2014-08-594408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinmeier, J. , & Dringen, R. (2019). Exposure of cultured astrocytes to menadione triggers rapid radical formation, glutathione oxidation and Mrp1‐mediated export of glutathione disulfide. Neurochemical Research, 1–15. 10.1007/s11064-019-02760-1 [DOI] [PubMed] [Google Scholar]
- Sullivan, L. B. , & Chandel, N. S. (2014). Mitochondrial reactive oxygen species and cancer. Cancer & Metabolism, 2, 17 10.1186/2049-3002-2-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y. , Branicky, R. , Noe, A. , & Hekimi, S. (2018). Superoxide dismutases: Dual roles in controlling ROS damage and regulating ROS signaling. Journal of Cell Biology, 217(6), 1915–1928. 10.1083/jcb.201708007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whang, Y. H. , Xu, X. J. , Zhang, L. F. , & Li, H. L. (2014). Mimic of manganese superoxide dismutase induces apoptosis in human acute myeloid leukemia cells. Leukemia Lymphoma, 55, 1166–1175. 10.3109/10428194.2013.825904 [DOI] [PubMed] [Google Scholar]
- Wong, H. S. , Dighe, P. A. , Mezera, V. , Monternier, P. A. , & Brand, M. D. (2017). Production of superoxide and hydrogen peroxide from specific mitochondrial sites under different bioenergetic conditions. Journal of Biological Chemistry, 292(41), 16804–16809. 10.1074/jbc.R117.789271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wouters, B. J. , & Delwel, R. (2016). Epigenetics and approaches to targeted epigenetic therapy in acute myeloid leukemia. Blood, 127, 42–52. 10.1182/blood-2015-07-604512 [DOI] [PubMed] [Google Scholar]
- Yankovskaya, V. , Horsefield, R. , Törnroth, S. , Luna‐Chavez, C. , Miyoshi, H. , Leger, C. , … Iwata, S. (2003). The architecture of succinate dehydrogenase and reactive oxygen species generation. Science, 299, 700–704. 10.1126/science.1079605 [DOI] [PubMed] [Google Scholar]
- Ye, Z. W. , Zhang, J. , Townsend, D. M. , & Tew, K. D. (2015). Oxidative stress, redox regulation and diseases of cellular differentiation. Biochimica et Biophysica Acta, 1850, 1607–1621. 10.1016/j.bbagen.2014.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, L. , Xu, J. X. , Haley, P. E. , & Yu, C. A. (1987). Properties of bovine heart mitochondrial cytochrome b560 . The Journal of Biological Chemistry, 262, 1137–b1143. [PubMed] [Google Scholar]
- Zelko, I. N. , Mariani, T. J. , & Folz, R. (2002). Superoxide dismutase multigene family: A comparison of the CuZn‐SOD (SOD1), Mn‐SOD (SOD2), and EC‐SOD (SOD3) gene structures, evolution and expression. Free Radical Biology & Medicine, 33, 337–349. 10.1016/S0891-5849(02)00905-X [DOI] [PubMed] [Google Scholar]
- Zhou, F. , Shen, Q. , & Claret, F. X. (2013). Novel roles of reactive oxygen species in the pathogenesis of acute myeloid leukemia. Journal of Leukocyte Biology, 94, 423–429. 10.1189/jlb.0113006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, F. L. , Zhang, W. G. , Wei, Y. C. , Meng, S. , Bai, G. G. , Wang, B. Y. , … Chen, S. P. (2010). Involvement of oxidative stress in the relapse of acute myeloid leukemia. The Journal of Biological Chemistry, 285, 15010–15015. 10.1074/jbc.M110.103713 [DOI] [PMC free article] [PubMed] [Google Scholar]