

Abstract
Deregulation of the cyclin D‐CDK4/6‐INK4‐RB pathway leading to uncontrolled cell proliferation, is frequently observed in breast cancer. Currently, three selective CDK4/6 inhibitors have been FDA approved: palbociclib, ribociclib and abemaciclib. Despite promising clinical outcomes, intrinsic or acquired resistance to CDK4/6 inhibitors has limited the success of these treatments; therefore, the development of various strategies to overcome this resistance is of great importance. We highlight the various mechanisms that are directly or indirectly responsible for resistance to CDK4/6 inhibitors, categorizing them into two broad groups; cell cycle‐specific mechanisms and cell cycle‐nonspecific mechanisms. Elucidation of the diverse mechanisms through which resistance to CDK4/6 inhibitors occurs, may aid in the design of novel therapeutic strategies to improve patient outcomes. This review summarizes the currently available knowledge regarding mechanisms of resistance to CDK4/6 inhibitors, and possible therapeutic strategies that may overcome this resistance as well.
Keywords: CDK4/6, estrogen receptor‐positive breast cancer, drug resistance
Introduction
Breast cancer is the most common malignancy in women, accounting for approximately 25% of all malignancies worldwide.1 Breast cancer is categorized into three subtypes according to estrogen receptor (ER), progesterone receptor (PR) and HER2 status: hormone receptor (HR)‐positive, HER2‐positive and triple negative subtypes.2 Of these, HR‐positive breast cancers constitute approximately 60%–70%.3 Endocrine therapy is considered to be the mainstay therapy for HR‐positive breast cancer.4 Despite having remarkable improvement in treatment of advanced disease, a large proportion of patients eventually acquire resistance to endocrine therapy.5 Previous studies have suggested multiple mechanisms responsible for endocrine resistance, including ER loss or mutation, alteration of the ER pathway, deregulation of cell cycle signaling molecules, and activation of various escape pathways.6, 7 These limitations regarding endocrine resistance have enabled researchers to identify novel therapeutic targets, such as cyclin‐dependent kinase (CDK) 4/6 inhibitors.
CDK4/6 kinases associate with cyclin D proteins during transition from G1 to S phase of the cell cycle. The cyclin D‐CDK4/6 complex phosphorylates retinoblastoma proteins (RB) and dissociates them from the E2F transcription factors, which are ultimately responsible for cell cycle progression.8 Various factors including the overexpression of cyclin D, mutation or amplification of CDK4/6 and loss of cyclin D‐CDK4/6 negative regulators, elicit the activity of cyclin D‐CDK4/6, which hyperphosphorylates RB, ultimately leading to uncontrolled cell proliferation.9 Thus, specific targeting of CDK4/6 has garnered special interest as an anticancer therapy.
Currently, three CDK 4/6 inhibitors are in clinical development: palbociclib, ribociclib and abemaciclib.10 These three CDK4/6 inhibitors have demonstrated greater efficacy in combination with endocrine therapies, leading to FDA approval.11 When used to treat HR‐positive metastatic breast cancer in postmenopausal women as a first‐line therapy, palbociclib, ribociclib and abemaciclib significantly prolonged progression‐free survival (from 12 to 14 months to ≥25 months) in combination with aromatase inhibitors (PALOMA‐2, MONALEESA‐2 and MONARCH‐3 trials).12, 13, 14 Ribociclib demonstrated similar efficacy in combination with ovarian suppressor and tamoxifen or aromatase inhibitor as a first‐line therapy in premenopausal women (MONALESESA‐7 trial)15 as well. Furthermore, combining fulvestrant with palbociclib, ribociclib, or abemaciclib doubled progression‐free survival compared to fulvestrant alone (PALOMA‐3, MONALEESA‐3 and MONARCH‐2 trials)16, 17, 18 as a second‐line therapy after progression occurred with aromatase inhibitors.
Despite improved disease control that CDK4/6 inhibitors offer to patients with HR‐positive breast cancer, not all patients respond to these drugs and most patients whose tumors respond to CDK4/6 inhibitors eventually develop acquired resistance.19 The early and late adaptation mediated by persistent G1–S‐phase cyclin expression and other bypass signaling limits the effectiveness of CDK4/6 inhibitors.20 In addition, various other mechanisms also exist, which are responsible for intrinsic or acquired resistance to CDK4/6 inhibitors.
This review explores various preclinical biomarkers, which may contribute to intrinsic or acquired resistance to CDK4/6 inhibitors. The main cancer type we will discuss resistance mechanisms in is breast cancer based on current clinical approval status of CDK4/6 inhibitors, despite resistance mechanisms having been studied in several other cancers, too. We will discuss various possible strategies to overcome resistance as well.
Mechanisms of Resistance to CDK4/6 Inhibitors
Cell cycle‐specific mechanisms
The cyclin D‐CDK4/6–INK4–RB pathway is the key regulator of the G1–S transition of the cell cycle.21 Both CDK4 and CDK6 may associate with all three types of cyclin D (cyclin D1, cyclin D2 and cyclin D3), with cyclin D1 being the best characterized.21 Various mitogenic signals activate cyclin D1, which then forms a complex with CDK4/6, thereby phosphorylating RB, and promoting the dissociation of the RB‐E2F complex. Once E2F is released from the complex, it activates genes required for DNA replication, and the cell progresses into the S phase.8, 21 As cyclin D‐CDK4/6 is the key initiator of the G1–S transition, selectively targeting the ATP binding site of CDK4/6 blocks cellular transition from G1 to S phase of the cycle.
The p16INK4A protein, a member of the INK4 family of cell cycle inhibitors, is a tumor suppressor which inhibits cyclin D‐CDK4/6 activity and contributes to G1 arrest by directly binding to CDK4 and inhibiting its catalytic activity.9 In addition, Cip/Kip member proteins, p21and p27 in particular, which inhibit a broader spectrum of cyclin‐CDK complexes, also influence CDK4/6.9, 22, 23 However, specific mutations in the CDKN2A locus, (which encodes p16INK4A) are common in various malignancies, suggesting that the proteins, primarily responsible for inhibiting CDK4/6‐driven signaling, are absent, resulting in aberrant activation of the pathway. This supports the selective inhibition of CDK4/6 as an attractive therapeutic strategy. However, intrinsic or acquired resistance to CDK4/6 inhibitors is an emerging issue, which limits its therapeutic efficacy. Various cell cycle‐specific mechanisms responsible for resistance to CDK4/6 inhibitors are summarized below (Fig. 1).
Figure 1.
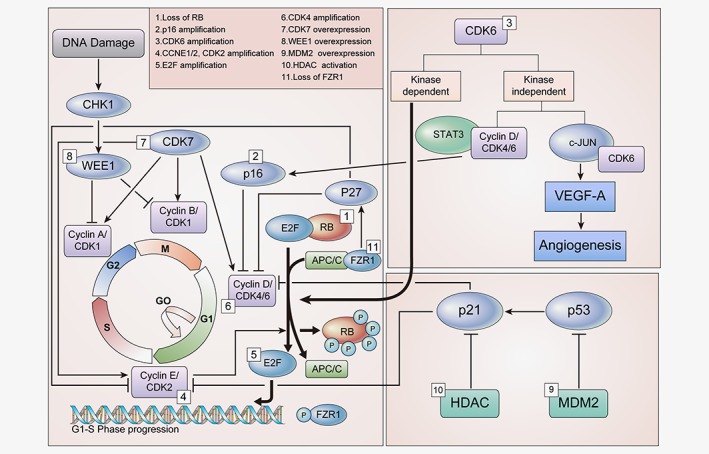
Cell cycle‐specific mechanisms for the resistance to CDK4/6 inhibitors. Multiple factors involved in the regulation of cell cycle are associated with resistance to CDK4/6 inhibitors. Loss of drug target genes, such as RB and FZR1, as well as overexpression of various genes which are directly or indirectly involved in the progression of cell cycle, as shown in Figure 1, are responsible for resistance to CDK4/6 inhibitors. Abbreviations: CDK, Cyclin dependent kinase; RB, Retinoblastoma protein; CHK1, Checkpoint kinase 1; STAT3, Signal transducer and activator of transcription 3; VEGF‐A, Vascular endothelial growth factor A; HDAC, Histone deacetylases; MDM2, Mouse double minute 2 homolog. [Color figure can be viewed at wileyonlinelibrary.com]
Loss of RB
The tumor suppressor RB is the aforementioned key checkpoint in the cell cycle. As the primary target of CDK4/6 inhibitors, RB was considered to be one of the most important biomarkers of sensitivity to therapy.24, 25, 26 In this scenario, loss of RB is the evident cause of resistance to CDK4/6 inhibitors,24 and various preclinical studies have supported this hypothesis.20, 27, 28 In addition, some preclinical and clinical studies have also reported that mutations in RB are responsible for the resistance.29, 30 A study using glioblastoma xenograft cells, a missense mutation in exon 2 of RB(A193T) resulted in resistance to CDK4/6 inhibitors.29 Despite the loss of RB, constitutive progression of the cell cycle continues via the activation of other cell cycle machinery, including such as E2F and the cyclinE‐CDK2 axis, demonstrating the loss of dependence on CDK4/6 for progression from G1 to S phase.10, 19 Therefore, when RB has been lost, inhibition of the cyclin E‐CDK2 axis in combination with CDK4/6 inhibitors may be effective in overcoming resistance to CDK4/6 inhibitors.
p16 amplification
The p16, a member of INK4 family and a natural inhibitor of CDK4, is an important tumor suppressor involved in the regulation of cell cycle.31 Naturally, p16 works as a tumor suppressor in the presence of functional RB because CDK4/6 (a target of p16) requires RB for its own kinase activity.32 Overexpression of p16 occurs during oncogenic stress, with or without the loss of RB.33, 34 Loss of RB with concurrent p16 overexpression resulted in failure to respond to CDK4/6 inhibitors because of the absence of RB function.35 Alternatively, p16 overexpression in the presence of functional RB, also confers resistance to CDK4/6 inhibitors as a result of diminished CDK4, indicating depletion of a target of CDK4/6 inhibitors. Although the loss of RB and p16 overexpression seems to occur consequently together, further studies revealing the mechanistic association of RB loss and p16 overexpression might be beneficial in designing the strategies to overcome acquired resistance to CDK4/6 inhibitors.
CDK6 amplification
In addition to CDK4, CDK6 also plays an important role in the progression of cell cycle from G1 to S phase, as mentioned above. Function of CDK6 is mainly kinase‐dependent. However, CDK6 functions partly kinase‐independently, too.36 CDK6 has a role in transcriptional regulation independently of its protein kinase activity;37 that is, CDK6 has been reported to upregulate the transcription of p16 in the presence of STAT3 and cyclin D. In other way, CDK6 upregulates VEGF‐A in concert with c‐Jun,36 inducing tortuous angiogenesis, which generally promotes cancer progression and drug resistance.38 In some studies, CDK6 overexpression was reported to promote resistance to CDK4/6 inhibitors in preclinical models.39 Possible mechanisms how CDK6 amplification confers resistance to CDK4/6 inhibitor might be due to kinase‐independent function of CDK6, which involves VEGF‐A or p16.
CCNE1/2, CDK2 amplification
Cyclin E‐CDK2 is also known to play a key role in the progression of cell cycle from G1 to S phase. Cyclin E‐CDK2 phosphorylates RB, allowing the release of E2F and promoting entry into the S phase.40 Overexpression of CCNE1, which encodes cyclin E, is a well‐accepted mechanism for resistance to CDK4/6 inhibitors, as demonstrated by previous studies.19, 20, 28, 41
Some of these studies have emphasized that CDK4/6 inhibitor‐resistant cells have lost their dependence on cyclin D1‐CDK4/6 signaling, instead using the bypass signaling pathway. Several bypass mechanisms are described elsewhere in this review. For instance, previous studies have mentioned that, cells may follow the MAPK‐AKT signaling cascade as a bypass to compensate for CDK4/6 inhibition.20, 28, 42 This bypass mechanism is described in detail here, in the section entitled “PI3K/AKT/mTOR pathway.”42 Activation of the cyclin E‐CDK2 pathway is also an important bypass mechanism; in one study, CDK2 inhibitors effectively decreased the growth of cells overexpressing cyclin E1.43 Taken together, inhibiting cyclin E‐CDK2 as well as upstream targets such as PI3K/AKT/mTOR in combination with inhibiting CDK4/6 may also be a successful strategy to overcome resistance.
E2F amplification
E2F is a downstream transcription factor of RB. The RB‐E2F complex plays an important role in the regulation of cell cycle progression from G1 to S phase.8, 21 Phosphorylation of RB by cyclin D‐CDK4/6 releases E2F, leading to the transcription of proteins, including cyclin E, required for cell cycle progression. The cyclin E‐CDK2 complex also phosphorylates RB, releasing E2F and promoting entry into S phase, as mentioned earlier. Loss of RB is correlated with the increased expression of E2F, resulting in the constitutive activation of its downstream target proteins.19, 44 Moreover, E2F has also been shown to upregulate AKT signaling via Gab2.45 Above all, the overexpression of E2F causes the cell to circumvent CDK4/6 inhibition and rely upon signaling pathways other than the cyclin D‐CDK4/6 axis for cell cycle progression.46 Further studies are required to explore the detailed mechanism of this escape pathway. Moreover, inhibition of proteins downstream of E2F, in concert with CDK4/6 inhibition, may increase the efficacy of CDK4/6 inhibitors, overcoming resistance.
CDK4 amplification
CDK4 is one component of the cyclin D‐CDK4/6‐RB pathway. Various mechanisms, such as gene amplification, mutations and epigenetic alterations, serve to activate the cyclin D‐CDK4/6–RB pathway.9 Overexpression of CDK4, which has been described in several cancers, may limit the efficacy of CDK4/6 inhibitors.29, 47, 48 A study in alveolar rhabdomyosarcoma cells Rh 28 and Rh 41 demonstrated the decreased activity of CDK4/6 inhibitors in cells overexpressing CDK4.48 In addition, glioma cells overexpressing CDK4 were completely resistant to CDK4/6 inhibitors.29 Further investigations are needed to confirm whether such a pattern of CDK4 expression associated with resistance to CDK4/6 inhibitors is confined to specific cancer types or is similar in other cancer types as well.
CDK7 overexpression
CDK7 is a cell cycle regulator. In addition, it also acts as a transcription factor, after complexation with cyclin H and MAT1.49, 50 Increased expression of CDK7 is reported to confer resistance to CDK4/6 inhibitors.51 It acts as a CDK‐activating kinase (CAK) and is involved in the G2/M phase by maintaining CDK1 and CDK2 activity.49 Reportedly, CDK7 has CAK activity toward CDK4 and CDK6, which may play a role in mitogen signaling during G1 phase progression.49 Though CDK7 appears to be involved in the G1 phase of the cell cycle, it remains unclear whether it induces resistance to CDK4/6 inhibitors. Further studies are warranted to reveal the detailed underlying mechanism of CDK7’s role in G1 phase progression, and to determine whether, the combined inhibition of CDK7 and CDK4/6 could overcome resistance to CDK4/6 inhibitors.
WEE1 overexpression
WEE1 plays an important role in the G2/M checkpoint. It inhibits the entry of DNA‐damaged cells into mitosis in coordination with CDK1.52 Though the involvement of WEE1 in inducing resistance to CDK4/6 inhibitors is unknown, inhibition of WEE1 has been shown to increase sensitivity to CDK4/6 inhibitors in resistant cells.51 As WEE1 is associated with a resistant phenotype in preclinical models,51 targeting the G2/M phase via the inhibition of WEE1 in combination with CDK4/6 inhibition could be a therapeutic option in overcoming resistance.
MDM2 overexpression
Mouse double minute 2 homolog (MDM2) is a protein that negatively regulates p53 activity, in destabilizing and inhibiting cellular senescence.53 Approximately 20%–30% of breast cancer patients show overexpression of MDM2,54 and this overexpression contributes particularly to the progression of HR‐positive breast cancer.55 It is reported that CDK4/6 inhibitor‐resistant cells have disrupted senescence pathways and insensitivity to the induction of senescence.56 Therefore, interruption of the senescence pathway by MDM2 in a p53‐dependent manner may cause resistance to CDK4/6 inhibitors. MDM2 inhibitors activate p53 by disrupting the MDM2‐p53 complex.57 A combination of palbociclib with an MDM2 inhibitor (RG7388) produced a synergistic anticancer effect in human liposarcoma.57 In addition, another MDM2 inhibitor (CGM097) has shown synergistic effects in combination with CDK4/6 inhibitors or fulvestrant, abrogating cells that are resistant to CDK4/6 inhibitors, as well as those resistant to endocrine therapy in vitro and in vivo.56 These studies highlight the importance of MDM2 in overcoming resistance to CDK4/6 inhibitors.
HDAC activation
Histone deacetylases (HDACs) remove the acetyl group from the ε‐N‐acetyl lysins of histones and play an important role in gene regulation. HDACs suppress the natural CDK inhibitor, p21,58 which interacts with cyclin D throughout the cell cycle, and they associate with cyclin A or cyclin B in the later part of the cell cycle at the G2/M phase.59, 60 Although the involvement of HDAC in resistance to CDK4/6 inhibitors is currently unknown, inhibition of HDAC may increase the efficacy of CDK4/6 inhibitors in CDK4/6 inhibitor‐resistant cells by activating p21, resulting in cell cycle arrest at the G1 and G2/M phases, as demonstrated in CDK4/6 inhibitor‐sensitive cells.61 In other way, HDAC inhibition was reported to induce proapototic proteins such as Noxa and Bim resulting in apoptosis, that is independent of p21, and providing a synergistic effect with CDK4/6 inhibitors in ER‐positive breast cancer cells.61 This supports the hypothesis that HDAC inhibition may enhance the activity of CDK4/6 inhibitors through a non‐overlapping mechanism, suggesting combining HDAC inhibitors with CDK4/6 inhibitors might be beneficial in resistant cases of CDK4/6 inhibitors.
Loss of FZR1
The ubiquitin (Ub) ligase APC/C, which is activated via the co‐activator FZR1, interacts with RB during the G1 phase of cell cycle.62 More notably, APC/CFZR1 complex degrades S‐phase kinase associated protein 2 (SKP2), which inhibits p27, natural CDK inhibitors, resulting in decreased CDK2, CDK4 and CDK6.63 Accordingly, the loss of FZR1 results in uncontrolled cell cycle progression from G1 to S phase. In addition, it is noted that FZR1 serves as a substrate of cyclin D‐CDK4/6, similar to RB and phosphorylated FZR1 loses its function to activate APC/C.64 Furthermore, simultaneous knockdown of both genes was shown to bypass the requirement of cyclin D‐CDK4/6 for the progression of cell cycle.64 Therefore, in addition to RB, FZR1 status may also be correlated with resistance to CDK4/6 inhibitors. Though the detailed mechanism remains unknown, the loss of FZR1 may correspond with the loss of RB to confer resistance to CDK4/6 inhibitors. Further studies are required to explore the mechanism and to overcome resistance to CDK4/6 inhibitors associated with the loss of FZR1.
Cell cycle‐nonspecific mechanisms
Various cell cycle‐nonspecific mechanisms which may confer resistance to CDK4/6 inhibitors are depicted in Figure 2 a and b.
Figure 2.
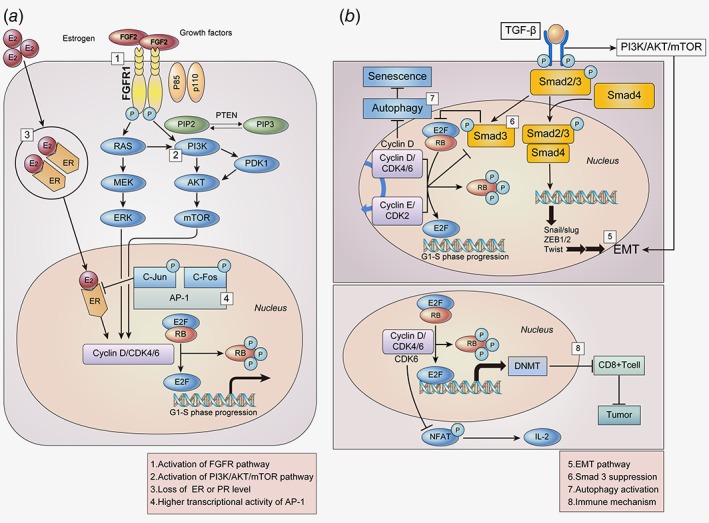
Cell cycle‐non specific mechanisms for the resistance to CDK4/6 inhibitors. (a) Overexpression of various factors which are upstream of the cell cycle, such as FGFR, PI3K/AKT/mTOR, and AP‐1, act as bypass pathways for the progression of the cell cycle, resulting in decreased efficacy of CDK4/6 inhibitors. Loss of ER dependence also drives cells to escape CDK4/6 inhibition. (b) TGF‐β induces the expression of several transcription factors involved in EMT via Smad and the PI3K/AKT/mTOR pathway. The cyclin‐CDK complex phosphorylates and suppresses Smad 3, recovering cell cycle arrest. In addition, inhibition of cyclin D activates autophagy, leading to the reversal of cell cycle arrest mediated by CDK4/6 inhibitors. CDK4/6 inhibitors may activate various immune‐related genes, which may also play a role in the development of resistance. Abbreviations: FGF2, fibroblast growth factor 2; FGFR1, fibroblast growth factor receptor 1; ER, estrogen receptor; TGF‐β, transforming growth factor β; MEK, Mitogen‐activated protein kinase; ERK, extracellular signal‐regulated kinases; PI3K, phosphatidylinositide 3‐kinases; mTOR, mammalian target of rapamycin; PDK1, 3‐phosphoinositide‐dependent protein kinase‐1; AP‐1, Activator protein 1; EMT, epithelial‐mesenchymal transition; NFAT, Nuclear factor of activated T‐cells; IL‐2, Interleukin‐2; DNMT, DNA methyltransferase. [Color figure can be viewed at wileyonlinelibrary.com]
Activation of the FGFR pathway
The fibroblast growth factor receptor (FGFR) signaling pathway is involved in key biological processes such as proliferation, differentiation and cell survival.65 The FGFR pathway is frequently activated in several types of cancer, including breast cancer.66, 67 Of the five FGFRs, FGFR 1–4 have been reported to play an important role in cancer progression.68 Furthermore, FGFR1 and FGFR2 also appear to be associated with resistance to CDK4/6 inhibitors, as well as with endocrine resistance.68, 69 Mechanistic investigation showed that FGFR1 amplification activated the PI3K/AKT and RAS/MEK/ERK signaling pathways in endocrine‐resistant breast cancer cells.70 Preclinically forced overexpression of FGFR1 induced resistance to a combination of fulvestrant and palbociclib, as well as fulvestrant alone.71 The FGFR pathway is primarily activated via FGF2 amplification, rather than FGF3 and FGF4,65 and FGFR1 signaling via FGF2 promotes endocrine resistance.70 Accordingly, silencing FGFR1 prevented FGF2‐mediated endocrine resistance in preclinical studies.70 In addition, a recent study reported that an FGFR2‐activating mutation may also contribute to the development of resistance to CDK4/6 inhibitors, as well as selective estrogen receptor degraders.69 Therefore, the combined inhibition of CDK4/6 and FGFR pathway may be a viable option to overcome resistance to CDK4/6 inhibitors.
Activation of the PI3K/AKT/mTOR pathway
The PI3K/AKT/mTOR signaling pathway is activated in approximately 30%–40% of breast cancers, particularly in the HR‐positive subtype.20, 72 Aberration of this pathway is known to be a crucial factor in resistance to endocrine therapy. Furthermore correlation of the PIK3/AKT/mTOR pathway with resistance to CDK4/6 inhibitors has also been reported recently.20, 42, 73, 74 For instance, CDK4/6 inhibitor‐resistant breast cancer cells were more dependent on PI3K/AKT/mTOR signaling than ER signaling.73 CDK4/6 inhibitors activated the PI3K/AKT pathway via the phosphorylation of S477/T479 of AKT by PDK1 in ribociclib‐resistant breast cancer cells.42 Notably, herein, S477/T479 of p‐AKT is specifically a CDK2‐dependent phosphorylation site. Reactivation of phospho‐RB and E2F, which was noted in CDK4/6 inhibitor‐resistant cell lines, may occur via pathways other than the CDK pathway, such as the mTOR pathway. This may be inferred from preclinical results, wherein mTORC1/2 inhibition suppressed phosphor‐RB and E2F in CDK4/6 inhibitor‐resistant cells, and consequently restored sensitivity to CDK4/6 inhibitors.75 Another recent study suggests that PI3K inhibitors may decrease the cyclin D1 expression and prevent early adaptations to CDK4/6 inhibition.20 Likewise, in CDK4/6 inhibitor‐sensitive preclinical model, complete tumor regression was also observed after combined inhibition of CDK4/6 and PI3K compared to single‐agent treatment.20 These results suggest that the inhibition of the PI3K/AKT/mTOR pathway in combination with CDK4/6 inhibitors may have potential therapeutic benefits in overcoming resistance to CDK4/6 inhibitors as well as in augmenting anticancer activity in CDK4/6 inhibitor‐sensitive setting.
Loss of ER or PR expression
A major driver of cyclin D‐CDK4/6 activity in breast cancer cells is hormone‐mediated activation of the ER.76 Loss of ER/PR expression has been observed in an abemaciclib‐resistant preclinical model.39 Furthermore, in a small number of patient series in which paired biopsies were performed during pre‐CDK4/6 inhibitor treatment and post‐progression,39 three out of seven patients had lost expression of ER or PR. These data suggested that a subset of patients who develop resistance to CDK4/6 inhibitors may be associated with tumoral changes in ER/PR levels. That is, loss of ER dependence may drive cells to escape CDK4/6 inhibition. Therapeutic approaches similar to those that have been successful in HR‐negative subtypes may be required for the treatment of CDK4/6 inhibitor‐resistant patients who have lost ER or PR expression.
Higher transcriptional activity of AP‐1
AP‐1 is a transcription factor that regulates a wide variety of genes, including cyclin D1.77 AP‐1 family is composed of homodimers and heterodimers of the Jun, Fos, activation transcription factor (ATF) and transcription factor MAF sub‐families.77, 78 Approximately 20%–40% of human breast tumors have high levels of activated c‐Jun,79 which is known to interact with the ER and inhibit its activity.80 There are some reports that overexpression of AP‐1 proteins, including c‐Jun and c‐Fos, may account for resistance to CDK4/6 inhibitors, as well as to endocrine therapy. For example, overexpression of c‐Jun in the MCF7 breast cancer cell line was linked with resistance to antiestrogen therapy.80 In addition, higher transcriptional activity of AP‐1 and increased c‐Fos levels were observed in MCF7 cells having acquired resistance to palbociclib and tamoxifen.81 Mechanistically, it is not understood why the overexpression of AP‐1 is correlated with resistance to CDK4/6 inhibitors; however, a plausible explanation is that the suppression of ER by c‐Jun80 may drive cells to escape CDK4/6 inhibition due to the loss of ER dependence. In addition, cyclin D1 overexpression transcribed by c‐Jun may also explain the mechanism.77 Blockade of AP‐1 through the downregulation of c‐Jun via genetic modification synergistically inhibited breast cancer cell growth when applied in combination with palbociclib.81 Further, blockade of AP‐1 in combination with palbociclib and fulvestrant was more effective in inhibiting cell growth than dual‐ or mono‐treatment. Various natural products, bioactive phytochemicals, and small molecules targeting AP‐1 are currently under development.82 However, only one selective c‐Fos/AP‐1 inhibitor (T‐5224), has progressed to phase II clinical trials.78 Combining such AP‐1 specific inhibitors with CDK4/6 inhibitors may have a synergistic effect in overcoming the acquired resistance to CDK4/6 inhibitors.
EMT pathway
The transdifferentiation of cells from epithelial to mesenchymal is known as epithelial–mesenchymal transition (EMT). EMT is an essential phenomenon for tissue morphogenesis in multicellular organisms; however, it promotes invasion and metastasis of cancer cells.83 In addition, the involvement of EMT in anticancer drug resistance has been suggested by many previous studies.84, 85, 86 Furthermore, there are some evidences that EMT may be correlated with resistance to CDK4/6 inhibitors. It was reported that the inhibition of CDK4/6, induced EMT via the activation of TGF‐β.87, 88 In another preclinical study, EMT‐regulating genes were reported to be differentially expressed in CDK4/6 inhibitor‐resistant breast cancer.89 TGF‐β phosphorylates and activates Smad2 and Smad3, which then form a complex with Smad4, leading to EMT via the activation of EMT transcription factors.90, 91 Additionally, TGF‐β also induces EMT via PI3K/AKT/mTOR signaling, which is independent of the Smad pathway.85, 92 Therefore, the inhibition of TGF‐β or EMT in combination with CDK4/6 inhibitors may overcome the resistance to CDK4/6 inhibitors.
Smad 3 suppression
Smad3 is a component of the TGF‐β signaling pathway, having antiproliferative effects that contribute to G1 cell cycle arrest.93 From this perspective, it was demonstrated that the suppression of Smad3 was involved in mechanisms responsible for resistance to certain anticancer drugs, such as trastzumab.94 Furthermore, some evidences suggested that Smad3 may be correlated with resistance to CDK4/6 inhibitors. Mechanistically, Smad3 was reported to be suppressed through phosphorylation by the cyclin E‐CDK2 or cyclin D1‐CDK4/6 complexes.93 This suppression of Smad3 released the RB‐E2F blockade induced by of Smad3,95 and finally recovered cell cycle arrest in breast cancer cells.93 The link between the cyclin E‐CDK2 complex and Smad3 was demonstrated in a trastzumab‐resistant preclinical model; CDK2 inhibition or transfection of trastzumab‐resistant breast cancer cells with a Smad3 construct containing inhibitory mutations in CDK2 phosphorylation sites led to decreased phosphorylation of Smad3, resulting in decreased proliferation of trastzumab‐resistant cells.94 Accordingly, based on the activation of the cyclin E‐CDK2 axis observed in CDK4/6 inhibitor‐resistant models,20, 28, 41, 42, 51 resistance to CDK4/6 inhibitors may result from suppression of Smad3 by the activated cyclin E‐CDK2 axis. Therefore, combined inhibition of CDK2 and CDK4/6 may be a promising strategy in this setting. However, augmenting Smad3 should not be overlooked as a means of overcoming resistance to CDK4/6 inhibitors.
Autophagy activation
Autophagy is thought to be a mechanism of stress tolerance and survival in cancer cells.96 During cell cycle arrest, stress response activates autophagy, which can then degrade ROS and mediate the reversal of G1 arrest and senescence.96 In preclinical studies, autophagy inhibition has been shown to augment the efficacy of many anticancer drugs.96, 97 There are also some evidences that suggests that autophagy accounts, partly, for imparting resistance to CDK4/6 inhibitors.96 In addition, CDK4/6 inhibition activated autophagy, maybe through the inhibition of cyclin D1, which was reported to suppress autophagy in mammary epithelial cells, or as a stress response.96 Accordingly, cell cycle arrest mediated by CDK4/6 inhibitors was reversed by autophagy.96 This phenomenon may support the hypothesis that the activation of autophagy is involved in resistance to CDK4/6 inhibitors. Preclinically, combined inhibition of CDK4/6 and autophagy synergistically increased senescence and sustained growth inhibition.96 The inhibition of autophagy enhanced the efficacy of CDK4/6 inhibitors, and may be help overcome resistance to CDK4/6 inhibitors.
Immune mechanisms
Immune checkpoint blockades in cancer have demonstrated promising efficacy in recent years.98 Various targeted therapies, such as gefitinib, erlotinib, cetuximab and axitinib, are also known to modulate immune responses.99, 100, 101, 102 On the other hand, immune‐related pathways are correlated with the emergence of resistance to various anticancer drugs.89, 102 Moreover, immune‐related pathways, such as those of IFN‐α and IFN‐β, were reported to be enriched in CDK4/6 inhibitor‐resistant breast cancer cells.89 In one preclinical study, CDK4/6 inhibitors were reported to promote anti‐tumor immunity,103 similar to other targeted therapies.104. Mechanistically, CDK4/6 inhibitors reduced the activity of DNA methyltransferase, an E2F target protein which promotes cytotoxic T cell‐mediated tumor inhibition.103 In addition, CDK6 phosphorylated nuclear factor of activated T cell 4 (NFAT4) and suppressed its activity, resulting in reduced IL2 levels, whereas CDK4/6 inhibitors increased IL2 levels by dephosphorylating NFAT4 and enhancing its activity.105 Taken together, CDK4/6 inhibition potentiates anti‐tumor immunity and enhances the response to PD‐1 blockade, providing a rationale for new anti‐cancer therapeutic strategies combining CDK4/6 inhibitors with immunotherapies. Therefore, although it is yet to be preclinically verified, we could hypothesize that combinatorial strategies might have some role in overcoming resistance to CDK4/6 inhibitors.
Potential Biomarkers Tested in Clinical Studies
Based on preclinical observations, potential biomarker analyses were performed using biosamples collected from patients enrolled in palbociclib clinical trials. Five hundred sixty‐six tumor samples from 666 participants enrolled in the PALOMA‐2 trial, in which palbociclib plus letrozole were compared to letrozole alone as a first‐line therapy, were immunohistochemically evaluated for ER, RB, p16, cyclin D1 and Ki‐67.106 The ER‐negative subgroup was not expected to benefit from palbociclib but, unexpectedly, this subgroup (n = 62) showed similar benefits from palbociclib as the ER‐positive subgroup (n = 499) by central confirmation of ER status. Although it was thought that ER‐negative subgroup herein derived benefits from palbociclib because of their PR‐positive status, it was not proved. Although the loss of RB is considered the evident resistant mechanism of CDK4/6 inhibitors, as mentioned earlier, this could not be substantiated because only a small number of patients were RB negative (n = 51). None of the other markers were correlated with sensitivity or resistance to palbociclib. In other biomarker studies using liquid biopsies from the PALOMA‐3 trial, in which palbociclib plus fulvestrant was compared to fulvestrant alone as a second‐line therapy, palbociclib was similarly beneficial, irrespective of mutations in PIK3CA16 or ESR1 mutation.107
Conclusions
CDK4/6 inhibitors are currently the prevailing standard therapy in combination with endocrine therapy in HR‐positive metastatic breast cancer. However, issues resulting from resistance to CDK4/6 inhibitors are emerging. Evidence collected from preclinical studies has suggested that various mechanisms may contribute to intrinsic or acquired resistance to CDK4/6 inhibitors. However, none of the potential resistance mechanisms which were preclinically demonstrated could be confirmed in clinical studies. Therefore, further investigations are warranted for both mechanistic and clinical validation in order to define more precise mechanisms of resistance to CDK4/6 inhibitors, and to develop successful therapeutic strategies to overcome resistance.
Conflicts of interest: The authors declare no conflicts of interest.
References
- 1. Ghoncheh M, Pournamdar Z, Salehiniya H. Incidence and mortality and epidemiology of breast cancer in the world. Asian Pac J Cancer Prev 2016;17:43–6. [DOI] [PubMed] [Google Scholar]
- 2. Parise CA, Caggiano V. Breast cancer survival defined by the ER/PR/HER2 subtypes and a surrogate classification according to tumor grade and immunohistochemical biomarkers. J Cancer Epidemiol 2014;2014:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hart CD, Migliaccio I, Malorni L, et al. Challenges in the management of advanced, ER‐positive, HER2‐negative breast cancer. Nat Rev Clin Oncol 2015;12:541–52. [DOI] [PubMed] [Google Scholar]
- 4. Flaum LE, Gradishar WJ. Advances in endocrine therapy for postmenopausal metastatic breast cancer. Cancer Treat Res. 2018;173:141–54. [DOI] [PubMed] [Google Scholar]
- 5. Hoffmann J, Bohlmann R, Heinrich N, et al. Characterization of new estrogen receptor destabilizing compounds: effects on estrogen‐sensitive and tamoxifen‐resistant breast cancer. J Natl Cancer Inst 2004;96:210–8. [DOI] [PubMed] [Google Scholar]
- 6. Osborne CK, Schiff R. Mechanisms of endocrine resistance in breast cancer. Annu Rev Med 2011;62:233–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kuukasjärvi T, Kononen J, Helin H, et al. Loss of estrogen receptor in recurrent breast cancer is associated with poor response to endocrine therapy. J Clin Oncol 1996;14:2584–9. [DOI] [PubMed] [Google Scholar]
- 8. Clark AS, Karasic TB, Demichele A, et al. Palbociclib (PD0332991)—a selective and potent cyclin‐dependent kinase inhibitor: a review of pharmacodynamics and clinical development. JAMA Oncol 2016;2:253–60. [DOI] [PubMed] [Google Scholar]
- 9. Hamilton E, Infante JR. Targeting CDK4/6 in patients with cancer. Cancer Treat Rev 2016;45:129–38. [DOI] [PubMed] [Google Scholar]
- 10. Finn RS, Crown JP, Lang I, et al. The cyclin‐dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first‐line treatment of oestrogen receptor‐positive, HER2‐negative, advanced breast cancer (PALOMA‐1/TRIO‐18): a randomised phase 2 study. Lancet Oncol 2015;16:25–35. [DOI] [PubMed] [Google Scholar]
- 11. Corona SP, Generali D. Abemaciclib: a CDK4/6 inhibitor for the treatment of HR+/HeR2− advanced breast cancer. Drug Des Devel Ther 2018;12:321–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Finn RS, Martin M, Rugo HS, et al. Palbociclib and letrozole in advanced breast cancer. N Engl J Med 2016;375:1925–36. [DOI] [PubMed] [Google Scholar]
- 13. Hortobagyi GN, Stemmer SM, Burris HA, et al. Ribociclib as first‐line therapy for HR‐positive, advanced breast cancer. N Engl J Med 2016;375:1738–48. [DOI] [PubMed] [Google Scholar]
- 14. Goetz MP, Toi M, Campone M, et al. MONARCH 3: abemaciclib as initial therapy for advanced breast cancer. J Clin Oncol 2017;35:3638–46. [DOI] [PubMed] [Google Scholar]
- 15. Tripathy D, Im S‐A, Colleoni M, et al. Ribociclib plus endocrine therapy for premenopausal women with hormone‐receptor‐positive, advanced breast cancer (MONALEESA‐7): a randomised phase 3 trial. Lancet Oncol 2018;19:904–15. [DOI] [PubMed] [Google Scholar]
- 16. Cristofanilli M, Turner NC, Bondarenko I, et al. Fulvestrant plus palbociclib versus fulvestrant plus placebo for treatment of hormone‐receptor‐positive, HER2‐negative metastatic breast cancer that progressed on previous endocrine therapy (PALOMA‐3): final analysis of the multicentre, double‐blind, phase 3 randomised controlled trial. Lancet Oncol 2016;17:425–39. [DOI] [PubMed] [Google Scholar]
- 17. Sledge GW Jr, Toi M, Neven P, et al. MONARCH 2: abemaciclib in combination with fulvestrant in women with HR+/HER2− advanced breast cancer who had progressed while receiving endocrine therapy. J Clin Oncol 2017;35:2875–84. [DOI] [PubMed] [Google Scholar]
- 18. Dennis JS, Patrick N, Stephen KLC, et al. Ribociclib (RIB) + fulvestrant (FUL) in postmenopausal women with hormone receptor‐positive (HR+), HER2‐negative (HER2–) advanced breast cancer (ABC): results from MONALEESA‐3. J Clin Oncol 2018;36:2465–72.29860922 [Google Scholar]
- 19. Konecny GE, Winterhoff B, Kolarova T, et al. Expression of p16 and retinoblastoma determines response to CDK 4/6 inhibition in ovarian cancer. Clin Cancer Res 2011;17:1591–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Herrera‐Abreu MT, Palafox M, Asghar U, et al. Early adaptation and acquired resistance to CDK4/6 inhibition in estrogen receptor–positive breast cancer. Cancer Res 2016;76:2301–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Diehl JA. Cycling to cancer with cyclin D1. Cancer Biol Ther 2002;1:226–31. [DOI] [PubMed] [Google Scholar]
- 22. Paternot S, Bockstaele L, Bisteau X, et al. Rb inactivation in cell cycle and cancer: the puzzle of highly regulated activating phosphorylation of CDK4 versus constitutively active CDK‐activating kinase. Cell Cycle 2010;9:689–99. [DOI] [PubMed] [Google Scholar]
- 23. Denicourt C, Dowdy SF. Cip/kip proteins: more than just CDKs inhibitors. Genes Dev 2004;18:851–5. [DOI] [PubMed] [Google Scholar]
- 24. O'Leary B, Finn RS, Turner NC. Treating cancer with selective CDK4/6 inhibitors. Nat Rev Clin Oncol 2016;13:417–30. [DOI] [PubMed] [Google Scholar]
- 25. Wiedemeyer WR, Dunn IF, Quayle SN, et al. Pattern of retinoblastoma pathway inactivation dictates response to CDK4/6 inhibition in GBM. Proc Natl Acad Sci 2010;107:11501–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Guarducci C, Bonechi M, Boccalini G, et al. Mechanisms of resistance to CDK4/6 inhibitors in breast cancer and potential biomarkers of response. Breast Care 2017;12:304–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Malorni L, Piazza S, Ciani Y, et al. A gene expression signature of retinoblastoma loss‐of‐function is a predictive biomarker of resistance to palbociclib in breast cancer cell lines and is prognostic in patients with ER positive early breast cancer. Oncotarget 2016;7:68012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Taylor‐Harding B, Aspuria P‐J, Agadjanian H, et al. Cyclin E1 and RTK/RAS signaling drive CDK inhibitor resistance via activation of E2F and ETS. Oncotarget 2015;6:696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cen L, Carlson BL, Schroeder MA, et al. p16‐Cdk4‐Rb axis controls sensitivity to a cyclin‐dependent kinase inhibitor PD0332991 in glioblastoma xenograft cells. Neuro Oncol 2012;14:870–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Condorelli R, Spring L, O'Shaughnessy J, et al. Polyclonal RB1 mutations and acquired resistance to CDK 4/6 inhibitors in patients with metastatic breast cancer. Ann Oncol 2017;29:640–5. [DOI] [PubMed] [Google Scholar]
- 31. Serrano M, Hannon GJ, Beach D. A new regulatory motif in cell‐cycle control causing specific inhibition of cyclin D/CDK4. Nature 1993;366:704–7. [DOI] [PubMed] [Google Scholar]
- 32. Medema RH, Herrera RE, Lam F, et al. Growth suppression by p16ink4 requires functional retinoblastoma protein. Proc Natl Acad Sci 1995;92:6289–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Witkiewicz AK, Knudsen KE, Dicker AP, et al. The meaning of p16(ink4a) expression in tumors: functional significance, clinical associations and future developments. Cell Cycle 2011;10:2497–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liu Y, Zhong X, Wan S, et al. p16(INK4a) expression in retinoblastoma: a marker of differentiation grade. Diagn Pathol 2014;9:180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dean JL, McClendon AK, Hickey TE, et al. Therapeutic response to CDK4/6 inhibition in breast cancer defined by ex vivo analyses of human tumors. Cell Cycle 2012;11:2756–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kollmann K, Heller G, Schneckenleithner C, et al. A kinase‐independent function of CDK6 links the cell cycle to tumor angiogenesis. Cancer Cell 2013;24:167–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tigan AS, Bellutti F, Kollmann K, et al. CDK6‐a review of the past and a glimpse into the future: from cell‐cycle control to transcriptional regulation. Oncogene 2016;35:3083–91. [DOI] [PubMed] [Google Scholar]
- 38. Gacche RN, Assaraf YG. Redundant Angiogenic signaling and tumor drug resistance. Drug Resist Updat 2018;36:47–76. [DOI] [PubMed] [Google Scholar]
- 39. Yang C, Li Z, Bhatt T, et al. Acquired CDK6 amplification promotes breast cancer resistance to CDK4/6 inhibitors and loss of ER signaling and dependence. Oncogene 2017;36:2255–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gladden AB, Diehl JA. Cell cycle progression without cyclin E/CDK2: breaking down the walls of dogma. Cancer Cell 2003;4:160–2. [DOI] [PubMed] [Google Scholar]
- 41. Franco J, Witkiewicz AK, Knudsen ES. CDK4/6 inhibitors have potent activity in combination with pathway selective therapeutic agents in models of pancreatic cancer. Oncotarget 2014;5:6512–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jansen VM, Bhola NE, Bauer JA, et al. Kinome‐wide RNA interference screen reveals a role for PDK1 in acquired resistance to CDK4/6 inhibition in ER‐positive breast cancer. Cancer Res 2017;77:2488–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Etemadmoghadam D, Au‐Yeung G, Wall M, et al. Resistance to CDK2 inhibitors is associated with selection of polyploid cells in CCNE1‐amplified ovarian cancer. Clin Cancer Res 2013;19:5960–71. [DOI] [PubMed] [Google Scholar]
- 44. Finn RS, Dering J, Conklin D, et al. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor‐positive human breast cancer cell lines in vitro. Breast Cancer Res 2009;11:R77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chaussepied M, Ginsberg D. Transcriptional regulation of AKT activation by E2F. Mol Cell 2004;16:831–7. [DOI] [PubMed] [Google Scholar]
- 46. Dean J, Thangavel C, McClendon A, et al. Therapeutic CDK4/6 inhibition in breast cancer: key mechanisms of response and failure. Oncogene 2010;29:4018–32. [DOI] [PubMed] [Google Scholar]
- 47. Wu A, Wu B, Guo J, et al. Elevated expression of CDK4 in lung cancer. J Transl Med 2011;9:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Olanich ME, Sun W, Hewitt SM, et al. CDK4 amplification reduces sensitivity to CDK4/6 inhibition in fusion‐positive rhabdomyosarcoma. Clin Cancer Res 2015;21:4947–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Schachter MM, Merrick KA, Larochelle S, et al. A Cdk7‐Cdk4 T‐loop phosphorylation cascade promotes G1 progression. Mol Cell 2013;50:250–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Larochelle S, Pandur J, Fisher RP, et al. Cdk7 is essential for mitosis and for in vivo Cdk‐activating kinase activity. Genes Dev 1998;12:370–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Martin L‐A, Pancholi S, Ribas R, et al. Resistance to palbociclib depends on multiple targetable mechanisms highlighting the potential of drug holidays and drug switching to improve therapeutic outcome. Cancer Res 2017;77:P3‐03‐09. [Google Scholar]
- 52. Matheson CJ, Backos DS, Reigan P. Targeting WEE1 kinase in cancer. Trends Pharmacol Sci 2016;37:872–81. [DOI] [PubMed] [Google Scholar]
- 53. Efeyan A, Ortega‐Molina A, Velasco‐Miguel S, et al. Induction of p53‐dependent senescence by the MDM2 antagonist nutlin‐3a in mouse cells of fibroblast origin. Cancer Res 2007;67:7350–7. [DOI] [PubMed] [Google Scholar]
- 54. Network CGA. Comprehensive molecular portraits of human breast tumours. Nature 2012;490:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kundu N, Brekman A, Kim JY, et al. Estrogen‐activated MDM2 disrupts mammary tissue architecture through a p53‐independent pathway. Oncotarget 2017;8:47916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lim E, Portman N, Alexandrou S, et al. Therapeutic targeting of CDK4/6 inhibitor resistant breast cancer. Cancer Res 2018;78:P4‐04‐12. [Google Scholar]
- 57. Laroche‐Clary A, Chaire V, Algeo M‐P, et al. Combined targeting of MDM2 and CDK4 is synergistic in dedifferentiated liposarcomas. J Hematol Oncol 2017;10:123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Zupkovitz G, Grausenburger R, Brunmeir R, et al. The cyclin‐dependent kinase inhibitor p21 is a crucial target for histone deacetylase 1 as a regulator of cellular proliferation. Mol Cell Biol 2010;30:1171–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Dash BC, El‐Deiry WS. Phosphorylation of p21 in G2/M promotes cyclin B‐Cdc2 kinase activity. Mol Cell Biol 2005;25:3364–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Lane AA, Chabner BA. Histone deacetylase inhibitors in cancer therapy. J Clin Oncol 2009;27:5459–68. [DOI] [PubMed] [Google Scholar]
- 61. Lee J, Lim B, Pearson T, et al. The synergistic antitumor activity of entinostat (MS‐275) in combination with palbociclib (PD 0332991) in estrogen receptor‐positive and triple‐negative breast cancer. Cancer Res 2018;78:P5‐21‐15‐P5‐21‐15. [Google Scholar]
- 62. Ramanujan A, Tiwari S. APC/C and retinoblastoma interaction: cross‐talk of retinoblastoma protein with the ubiquitin proteasome pathway. Biosci Rep 2016;36:e00377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Fujita T, Liu W, Doihara H, et al. Regulation of Skp2‐p27 axis by the Cdh1/anaphase‐promoting complex pathway in colorectal tumorigenesis. Am J Pathol 2008;173:217–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ruijtenberg S, Bouchet BP, Cristobal A, et al. Rb and FZR1/Cdh1 determine CDK4/6‐cyclin D requirement in C. elegans and human cancer cells. Nat Commun 2015;6:5906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Turner N, Grose R. Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer 2010;10:116–29. [DOI] [PubMed] [Google Scholar]
- 66. Chae YK, Pai SG, Sun P, et al. Fibroblast growth factor receptor (FGFR) as a therapeutic target in lung and head and neck cancer. Am J Hematol Oncol® 2016;12:13–19. [Google Scholar]
- 67. Sahores A, May M, Sequeira G, et al. Targeting FGFR with BGJ398 in breast cancer: effect on tumor growth and metastasis. Curr Cancer Drug Targets 2018;18(10):979–87. [DOI] [PubMed] [Google Scholar]
- 68. Brooks AN, Kilgour E, Smith PD. Molecular pathways: fibroblast growth factor signaling: a new therapeutic opportunity in cancer. Clin Cancer Res 2012;18:1855–62. [DOI] [PubMed] [Google Scholar]
- 69. Mao P, Kusiel J, Cohen O, et al. The role of FGF/FGFR axis in resistance to SERDs and CDK4/6 inhibitors in ER+ breast cancer. Cancer Res 2018;78:PD4‐01. [Google Scholar]
- 70. Turner N, Pearson A, Sharpe R, et al. FGFR1 amplification drives endocrine therapy resistance and is a therapeutic target in breast cancer. Cancer Res 2010;70:2085–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Luigi F, Yao L, Valerie MJ, et al. Gain‐of‐function kinase library screen identifies FGFR1 amplification as a mechanism of resistance to antiestrogens and CDK4/6 inhibitors in ER+ breast cancer. Cancer Res 2017;77:SABCS GS6‐05. [Google Scholar]
- 72. Takeshita T, Yamamoto Y, Yamamoto‐Ibusuki M, et al. Clinical significance of plasma cell‐free DNA mutations in PIK3CA, AKT1, and ESR1 gene according to treatment lines in ER‐positive breast cancer. Mol Cancer 2018;17:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Iida M, Nakamura M, Tokuda E, et al. CDK6 might be a key factor for efficacy of CDK4/6 inhibitor and the hormone sensitivity following acquired resistance Cancer Res 2018;78, Abstract 3541: Epigenetic regulation of KPC1 ubiquitin ligase has a regulatory role on the NF‐κB pathway in metastatic melanoma, 3541.
- 74. O'Brien T, Xiao Y, Ong C, et al. L F. identification of preclinical mechanisms driving acquired resistance to selective ERa degraders (SERDs), CDK4/6 inhibitors, or to combinations of both agents. Cancer Res 2017;77:P3‐04‐24. [Google Scholar]
- 75. Michaloglou C, Crafter C, Siersbaek R, et al. Combined inhibition of mTOR and CDK4/6 is required for optimal blockade of E2F function and long‐term growth inhibition in estrogen receptor–positive breast cancer. Mol Cancer Ther 2018;17:908–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Finn RS, Aleshin A, Slamon DJ. Targeting the cyclin‐dependent kinases (CDK) 4/6 in estrogen receptor‐positive breast cancers. Breast Cancer Res 2016;18:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Shaulian E, Karin M. AP‐1 in cell proliferation and survival. Oncogene 2001;20:2390–400. [DOI] [PubMed] [Google Scholar]
- 78. Ye N, Ding Y, Wild C, et al. Small molecule inhibitors targeting activator protein 1 (AP‐1) miniperspective. J Med Chem 2014;57:6930–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Shen Q, Uray IP, Li Y, et al. Targeting the activator protein 1 transcription factor for the prevention of estrogen receptor–negative mammary tumors. Cancer Prev Res 2008;1:45–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Smith LM, Wise SC, Hendricks DT, et al. cJun overexpression in MCF‐7 breast cancer cells produces a tumorigenic, invasive and hormone resistant phenotype. Oncogene 1999;18:6063–70. [DOI] [PubMed] [Google Scholar]
- 81. De Angelis C, Nardone A, Cataldo ML, et al. AP‐1 as a potential mediator of resistance to the cyclin‐dependent kinase (CDK) 4/6‐inhibitor palbociclib in ER‐positive endocrine‐resistant breast cancer. Cancer Res 2018;78:P4‐03‐05. [Google Scholar]
- 82. Tewari D, Nabavi SF, Nabavi SM, et al. Targeting activator protein 1 signaling pathway by bioactive natural agents: possible therapeutic strategy for cancer prevention and intervention. Pharmacol Res 2018;128:366–75. [DOI] [PubMed] [Google Scholar]
- 83. Singh A, Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene 2010;29:4741–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Polyak K, Weinberg RA. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer 2009;9:265–73. [DOI] [PubMed] [Google Scholar]
- 85. Du B, Shim JS. Targeting epithelial–mesenchymal transition (EMT) to overcome drug resistance in cancer. Molecules 2016;21:965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Mani SA, Guo W, Liao M‐J, et al. The epithelial‐mesenchymal transition generates cells with properties of stem cells. Cell 2008;133:704–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Tobin NP, Sims AH, Lundgren KL, et al. Cyclin D1, Id1 and EMT in breast cancer. BMC Cancer 2011;11:417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Liu F, Korc M. Cdk4/6 inhibition induces epithelial–mesenchymal transition and enhances invasiveness in pancreatic cancer cells. Mol Cancer Ther 2012;11:2138–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Smruthi V, Iman D, Jason PWC, et al. Characterizing acquired resistance to palbociclib in breast cancer. Cancer Res 2017;77:AM2017‐60. [Google Scholar]
- 90. Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial–mesenchymal transition. Nat Rev Mol Cell Biol 2014;15:178–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Yaswen P. Reinforcing targeted therapeutics with phenotypic stability factors. Cell Cycle 2014;13:3818–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Moustakas A, Heldin C‐H. Non‐Smad TGF‐β signals. J Cell Sci 2005;118:3573–84. [DOI] [PubMed] [Google Scholar]
- 93. Zelivianski S, Cooley A, Kall R, et al. CDK4‐mediated phosphorylation inhibits Smad3 activity in cyclin D overexpressing breast cancer cells. Mol Cancer Res 2010;8:1375–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Decker JT, Wan L, Shea LD, et al. Cyclin E affects Smad3 pathway in trastuzumab resistant HER2+ breast cancer. Cancer Res 2018;78:P4‐03‐16. [Google Scholar]
- 95. Yang J, Song K, Krebs TL, et al. Rb/E2F4 and Smad2/3 link survivin to TGF‐β‐induced apoptosis and tumor progression. Oncogene 2008;27:5326–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Vijayaraghavan S, Karakas C, Doostan I, et al. CDK4/6 and autophagy inhibitors synergistically induce senescence in Rb positive cytoplasmic cyclin E negative cancers. Nat Commun 2017;8:15916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Chittaranjan S, Bortnik S, Dragowska WH, et al. Autophagy inhibition augments the anticancer effects of epirubicin treatment in anthracycline‐sensitive and‐resistant triple‐negative breast cancer. Clin Cancer Res 2014;20:3159–73. [DOI] [PubMed] [Google Scholar]
- 98. McArthur HL. Checkpoint inhibitors in breast cancer: hype or promise? Clin Adv Hematol Oncol H&O 2016;14:392. [PubMed] [Google Scholar]
- 99. Correale P, Botta C, Cusi M, et al. Cetuximab±chemotherapy enhances dendritic cell‐mediated phagocytosis of colon cancer cells and ignites a highly efficient colon cancer antigen‐specific cytotoxic T‐cell response in vitro. Int J Cancer 2012;130:1577–89. [DOI] [PubMed] [Google Scholar]
- 100. Läubli H, Müller P, D'Amico L, et al. The multi‐receptor inhibitor axitinib reverses tumor‐induced immunosuppression and potentiates treatment with immune‐modulatory antibodies in preclinical murine models. Cancer Immunol Immunother 2018;67:815–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Sheng J, Fang W, Liu X, et al. Impact of gefitinib in early stage treatment on circulating cytokines and lymphocytes for patients with advanced non‐small cell lung cancer. OncoTargets Therapy 2017;10:1101–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Demuth C, Andersen MN, Jakobsen KR, et al. Increased PD‐L1 expression in erlotinib‐resistant NSCLC cells with MET gene amplification is reversed upon MET‐TKI treatment. Oncotarget 2017;8:68221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Goel S, Decristo MJ, Watt AC, et al. CDK4/6 inhibition triggers anti‐tumour immunity. Nature 2017;548:471–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Kersh AE, Ng S, Chang YM, et al. Targeted therapies: immunologic effects and potential applications outside of cancer. J Clin Pharmacol 2018;58:7–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Deng J, Wang ES, Jenkins RW, et al. CDK4/6 inhibition augments antitumor immunity by enhancing T‐cell activation. Cancer Discov 2018;8:216–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Finn R, Jiang Y, Rugo H, et al. Biomarker analyses from the phase 3 PALOMA‐2 trial of palbociclib (P) with letrozole (L) compared with placebo (PLB) plus L in postmenopausal women with ER+/HER2–advanced breast cancer (ABC). Ann Oncol 2016;27:1–36. [Google Scholar]
- 107. Fribbens C, O'Leary B, Kilburn L, et al. Plasma ESR1 mutations and the treatment of estrogen receptor–positive advanced breast cancer. J Clin Oncol 2016;34:2961–8. [DOI] [PubMed] [Google Scholar]