

Abstract
Emicizumab (ACE910) is a bispecific antibody that is a novel, subcutaneously injectable treatment for patients with hemophilia A. This study assessed the relative bioavailability of emicizumab between old and new drug products (DPs) and among 3 commonly used subcutaneous injection sites (abdomen, upper arm, and thigh), together with its absolute bioavailability in healthy volunteers. Forty‐eight healthy volunteers were randomized into 4 groups to receive a single subcutaneous injection of 1 mg/kg with the old or new DP, and another 12 volunteers each received a single, 90‐minute, intravenous infusion of 0.25 mg/kg with the new DP. Similar pharmacokinetic profiles were observed between the DPs, with geometric mean ratios of 1.199 (90% confidence interval [CI] 1.060‐1.355) for the maximum plasma concentration and 1.083 (90% CI 0.920‐1.275) for area under the plasma concentration‐time curve extrapolated to infinity. The geometric mean ratios of maximum plasma concentration and area under the plasma concentration‐time curve extrapolated to infinity for upper arm versus abdomen were 0.823 (90% CI 0.718‐0.943) and 0.926 (90% CI 0.814‐1.053), respectively, and those for thigh versus abdomen were 1.168 (90% CI 1.030‐1.324) and 1.073 (90% CI 0.969‐1.189), respectively. Absolute bioavailability ranged from 80.4% to 93.1%. These results suggested that no emicizumab dose adjustment would be needed when switching the DPs or injecting to different sites interchangeably and that emicizumab injected subcutaneously is highly bioavailable.
Keywords: emicizumab, hemophilia A, bioavailability, monoclonal antibody, drug product bridging strategy
Hemophilia A is a bleeding disorder attributable to a congenital absence or dysfunction of coagulation factor VIII (FVIII) that occurs in approximately 1 in 5000 live‐born males.1 The standard therapy for patients with hemophilia A is categorized into 2 types: on‐demand therapy, for which coagulation factor products are administered when bleeding occurs, and regular replacement therapy (prophylaxis), for which coagulation factor products are administered regularly to prevent the onset of bleeding. Coagulation factor products used for treatment differ between patients without alloantibodies to FVIII (“FVIII inhibitors”), who are treated with plasma‐derived or recombinant FVIII products, and patients with FVIII inhibitors, who are treated with bypassing agents such as recombinant activated coagulation factor VII or activated prothrombin complex concentrate. However, there is a common burden in the treatment for patients both with and without FVIII inhibitors, that is, the necessity of frequent intravenous (IV) infusions due to their short half‐lives (FVIII 8 to 19 hours,2, 3, 4, 5 recombinant activated coagulation factor VII 2.3 to 6.0 hours,6, 7, 8, 9 activated prothrombin complex concentrate 4 to 7 hours10). Given this drawback, new drugs that can be administered subcutaneously with a low frequency of administration are desired.
Emicizumab (ACE910) is a recombinant humanized bispecific monoclonal antibody that bridges activated factor IX (FIXa) and factor X (FX) to restore the function of activated FVIII that is missing in patients with hemophilia A.11, 12, 13 Based on the series of nonclinical investigations,12, 13, 14, 15 the clinical development of emicizumab started from a first‐in‐human, single‐ascending‐dose phase 1 study in Japanese and white healthy volunteers,16 followed by a first‐in‐patient, multiple‐ascending‐dose phase 1 study17 and its long‐term extension phase 1/2 study18 in Japanese patients with severe hemophilia A with or without FVIII inhibitors. In these early‐stage clinical studies, emicizumab demonstrated favorable safety profiles at up to 1 mg/kg as a single subcutaneous (SC) injection in healthy volunteers and up to 3 mg/kg as once‐weekly SC injections in patients with hemophilia A. The absorption of emicizumab after SC injection was gradual and reached the maximum plasma concentration (Cmax) 1 to 2 weeks after administration, and its subsequent elimination appeared monophasic. Emicizumab showed a linear pharmacokinetic (PK) profile from 0.01 mg/kg to 3 mg/kg without affecting the plasma concentrations of the target antigens (FIX and FX), suggesting that target‐mediated drug disposition is negligible for emicizumab. The half‐life (t1/2) of emicizumab was approximately 4 to 5 weeks, which was much longer than those of FVIII products and bypassing agents. The PK profiles in Japanese and white populations were similar. In addition, in the patient study, the efficacy potential of emicizumab was demonstrated by reduction in the individual annualized bleeding rates compared with each patient's own historical data. Emicizumab was given as a SC injection to the abdomen in all of these early‐stage clinical studies. Based on the study data, modeling and simulation analyses were conducted to select the dosing regimens to be tested in phase 3 pivotal studies.19 Consequently, a previously untested dosing regimen of a loading dose of 3 mg/kg once weekly for the first 4 weeks followed by a maintenance dose of 1.5 mg/kg once weekly was selected for the first phase 3 trial enrolling adult and adolescent patients with FVIII inhibitors.20
Emicizumab drug product (DP) was changed from that for early‐stage development to that for late‐stage development, with differences between the old and new DPs in drug substance manufacturing process and drug concentration. In the manufacturing process of the new DP, the purification process was upgraded to decrease the impurities in drug substance, and the cell culture process was also upgraded to improve the manufacturing productivity. As a consequence of these manufacturing process changes, the new DP showed some different pharmaceutical compositions compared with those of the old DP, including a slight change of glycosylation profile such as an increase of high‐mannose form.
In previous publications it was reported that an increased relative abundance ratio of high‐mannose forms in drug substance of monoclonal antibody is associated with higher drug clearance from blood circulation in mice and humans.21, 22 This raised a potential concern of decreased exposure and thus potential decreased efficacy of emicizumab with the new DP. With this potential concern taken into account, together with the International Council of Harmonisation (ICH) Q5E guideline, which states that “Comparability exercises are generally performed to demonstrate that nonclinical and clinical data generated with pre‐change product are applicable to post‐change product in order to facilitate further development and, ultimately, to support the marketing authorisation,”23 conducting a clinical study to compare the PK properties between the old and new DPs before embarking on the late‐stage phase 3 program was considered to be a rapid and conservative DP‐bridging strategy.
In addition, expanding the body locations available for SC injection was considered to be beneficial for patients, particularly in this disease area in which frequent IV infusions are required as the standard treatment. Increase of flexibility in choice of injection sites may lead to improved patient adherence in lifelong treatment and may also increase ease in giving treatment, particularly to children or infants who have difficulty in securing vascular access.
On the basis of these situations taken together, the present study was conducted to investigate whether the PK profile of emicizumab differs between the 2 DPs and among 3 commonly used SC injection sites (abdomen, upper arm, and thigh) and to explore the necessity of dose adjustment when the DPs are switched or when emicizumab is injected into different SC injection sites interchangeably. In addition, the absolute SC bioavailability of emicizumab was investigated in this study.
Methods
This single‐center, open‐label, randomized, parallel‐group study was conducted at CPC Clinic, Medipolis Medical Research Institute (Kagoshima, Japan) between April 2015 and October 2015 in accordance with the Declaration of Helsinki and ICH‐Good Clinical Practice. The study protocol was approved by the institutional review board of the trial site (CPC Clinical Trial Hospital IRB, Kagoshima, Japan), and all subjects gave written informed consent before enrollment. This study was registered at www.clinicaltrials.jp (JapicCTI‐152888).
Subjects
Healthy Japanese male volunteers aged 20 to <45 years with a body mass index of 18.5 to <25.0 kg/m2 were enrolled in this study. Key exclusion criteria were previous or current history of clinically significant allergy, hypersensitivity associated with globulin preparations, thromboembolic diseases, or FVIII activity of ≥120 IU/dL. Subjects were screened for eligibility during a 3‐week period before the administration of emicizumab.
Study Design
Forty‐eight healthy Japanese male volunteers were randomized into 4 SC‐dosing groups (groups A, B, C, and D; N = 12 each). In addition, 12 volunteers were enrolled in a separate IV‐dosing group (group E). The subjects received either of the following emicizumab doses: a single SC injection of 1 mg/kg with the old DP to the abdomen (group A); a single SC injection of 1 mg/kg with the new DP to the abdomen, upper arm, or thigh (groups B, C, and D, respectively); or a single IV infusion of 0.25 mg/kg with the new DP (group E). Because it was the first experience to administer emicizumab intravenously to humans, emicizumab administration in the IV‐dosing group was initiated after all the emicizumab administrations in the SC‐dosing groups had been completed, and thus, the IV‐dosing group was not involved in the randomization (Figure 1). Intravenous infusions were performed over a duration of 90 minutes.
Figure 1.
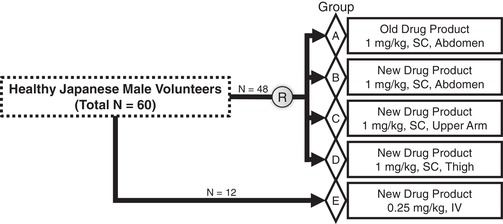
Study design and subject enrollment. Forty‐eight healthy Japanese male volunteers were randomized into 4 subcutaneous dosing groups (groups A, B, C, and D; N = 12 each), and 12 volunteers were enrolled in a separate intravenous dosing group (group E). Drug administration in the intravenous dosing group was initiated after all the drug administrations in the subcutaneous dosing groups had been completed, and thus, the intravenous dosing group was not involved in the randomization. IV indicates intravenous; SC, subcutaneous.
The dose of 1 mg/kg for the SC‐dosing groups was selected based on the following considerations. In the first‐in‐human study of emicizumab, the safety and tolerability of single SC doses up to 1 mg/kg were confirmed in healthy volunteers. Considering a potential risk of hypercoagulability in healthy volunteers who have normal FVIII activity, the dose level to be tested in this study was selected not to exceed the maximum dose tested previously in healthy volunteers. In addition, because PK linearity of emicizumab was confirmed over the dose range of 0.01 to 3 mg/kg in the phase 1‐1/2 studies, and the given doses constituting the first phase 3 dosing regimen (1.5 and 3 mg/kg) were within this dose range, it was supported that the relative bioavailability evaluated at the dose of 1 mg/kg can be extrapolated to the potential therapeutic dosing regimen. The dose of 0.25 mg/kg for the IV‐dosing group was also selected based on PK and safety considerations. Assuming a typical plasma volume for a 70‐kg human of 3 L,24 the plasma emicizumab concentration immediately after a single IV administration of 0.25 mg/kg was predicted to be 5.83 µg/mL, which would not exceed the highest previously experienced exposure level in healthy volunteers (ie, mean Cmax of 5.56 to 5.92 µg/mL after a single SC injection of 1 mg/kg in the first‐in‐human study16). Moreover, the linear PK profile of emicizumab justified the selection of 0.25 mg/kg for the IV‐dosing group to derive the absolute bioavailability, which was to be made by comparing area under the plasma concentration‐time curve extrapolated to infinity (AUCinf) with those from the SC‐dosing groups with normalization by the dose levels of each dosing group.
Subjects were observed for 16 weeks in duration, which was expected to capture ≥80% of AUCinf, until the study end. Blood samples were drawn from each subject before administration and at 8, 24, 48, 96, and 168 hours, 10 days, and 2, 3, 4, 6, 8, 10, 12, 14, and 16 weeks after injection in the SC dosing groups, and used for determination of plasma emicizumab concentrations. In the IV‐dosing group, blood samples were drawn at 1, 4, and 72 hours after the end of infusion in addition to the time points defined for the SC‐dosing groups. For immunogenicity evaluations, blood samples were drawn from each subject before administration and at 8 and 16 weeks after administration in both the SC‐ and IV‐dosing groups. After centrifugation of the blood samples (1700g, 15 minutes, 4°C), plasma was transferred to storage tubes and immediately stored at or below −70°C until the respective assays.
Drug Product
Emicizumab was produced from a Chinese hamster ovary cell line using recombinant DNA technology. The concentrations of emicizumab in the old and new DPs were 80 and 150 mg/mL, respectively.
Outcome Measures
Pharmacokinetics
Plasma emicizumab concentrations were determined by a validated sandwich enzyme‐linked immunosorbent assay as previously described.16 Plasma concentrations of emicizumab's target antigens (ie, FIX and FX, including their activated forms) were determined using the same commercially available enzyme‐linked immunosorbent assay kits as in the previous report.16
Pharmacodynamics
Activated partial thromboplastin time (aPTT) and peak height of activated factor XI–triggered thrombin generation (TG) were measured in plasma both with and without neutralization of endogenous FVIII using the same assay methods as previously described16 with change in anti‐FVIII neutralizing antibodies used; the host animals of the antibodies were changed from mice to monkeys. Endogenous FVIII activity in plasma samples from the healthy subjects was neutralized by addition of a dual anti‐FVIII neutralizing antibody cocktail ex vivo to artificially mimic the FVIII deficiency observed in patients with hemophilia A.
Safety
Safety was assessed by physical examination, adverse events (AEs; categorized by the Medical Dictionary for Regulatory Activities code version 17.1), vital signs, 12‐lead electrocardiogram, and laboratory tests. The severity of AEs was determined by the investigator as mild, moderate, or severe. Laboratory tests consisted of hematology, blood chemistry and coagulation tests, urinalysis, and serum cytokine concentrations. Blood coagulation tests included D‐dimer and prothrombin time international normalized ratio.
Immunogenicity
Antiemicizumab antibodies (antidrug antibodies [ADAs]) in plasma were detected by a validated electrochemiluminescence immunoassay.16 Subjects who had at least 1 ADA‐positive plasma sample at any time point including baseline were defined as ADA‐positive subjects; if not, as ADA‐negative subjects.
Statistical Analyses
The target sample size for this study was set at 60 subjects (12 subjects for each group) to enable investigation of the safety, PK, and pharmacodynamics (PD) of single SC and IV doses of emicizumab. Because demonstration of bioequivalence was not an objective of this study, the target sample size of 12 subjects per group was based on a presumed minimum acceptable number for a bioequivalence study25 rather than based on statistical considerations. Of note, the probability that the point estimates of the geometric mean ratios (GMRs) of Cmax and AUCinf would simultaneously fall within the range of 0.80 to 1.25 with 12 subjects in 2 groups each was estimated to be approximately 92% assuming that the intersubject coefficients of variation for the geometric means of Cmax and AUCinf are 23.6% and 20.5%, respectively, based on the previous study results in healthy Japanese adults,16 and assuming that the expected GMRs between the 2 groups (eg, group A and group B) are 1.00.
Baseline characteristics, PK, PD, and safety data were summarized by dosing group. PK parameters including Cmax, time to reach maximum plasma concentration, area under the plasma concentration‐time curve to the last measurable plasma concentration (AUClast), AUCinf, t1/2, (apparent) total clearance (CL[/F]), volume of distribution during the terminal phase (Vd,z; for the IV‐dosing group only), and volume of distribution at steady state (Vd,ss; for the IV‐dosing group only) were calculated from each subject's emicizumab plasma concentration‐time profile by a noncompartmental analysis method. In addition, the key exposure parameters (ie, Cmax, AUClast, and AUCinf) following a single SC administration of emicizumab were normalized by the actually given dose in mg/kg and then applied to the analysis of variance (ANOVA) to derive the GMRs as the relative bioavailability estimates, and similarly, AUCinf following a single SC or IV administration was normalized and then applied to the ANOVA to derive the GMRs as the absolute bioavailability estimates. Data from ADA‐positive subjects were not included in the ANOVA to avoid the potential bias on the estimates. All the data analyses were performed using SAS software version 9.2 (SAS Institute Inc, Cary, North Carolina).
Results
Baseline Characteristics
A total of 60 healthy Japanese volunteers were enrolled in this study (48 to the 4 SC‐dosing groups and 12 to the IV‐dosing group; N = 12 per group). No subjects were withdrawn, and all subjects completed the study; as a consequence, no subjects were excluded from the data set.
A summary of the subject baseline characteristics by dosing group is shown in Table 1. The mean age, body weight, and body mass index in each group ranged from 25.7 to 30.3 years, 58.7 to 66.2 kg, and 20.6 to 22.3 kg/m2, respectively. There were no imbalances in baseline characteristics among the groups, even including the IV‐dosing group (group E), which was not involved in the randomization.
Table 1.
Baseline Demographic Characteristics of Study Subjects
| Group | |||||
|---|---|---|---|---|---|
| A | B | C | D | E | |
| N | 12 | 12 | 12 | 12 | 12 |
| Age (y) | 29.2 ± 5.17 | 29.4 ± 7.19 | 28.8 ± 6.98 | 25.7 ± 6.23 | 30.3 ± 5.90 |
| Body weight (kg) | 62.4 ± 6.02 | 66.2 ± 9.78 | 63.8 ± 7.61 | 62.3 ± 5.46 | 58.7 ± 5.95 |
| BMI (kg/m2) | 21.6 ± 1.90 | 22.3 ± 1.86 | 21.7 ± 1.99 | 21.5 ± 1.43 | 20.6 ± 1.69 |
Data are presented as mean ± SD. BMI indicates body mass index.
Overall Pharmacokinetics
The time course of plasma emicizumab concentration after a single SC or IV administration is shown in Figure 2. In the SC‐dosing groups, plasma emicizumab concentrations peaked by 2 weeks after SC administration and subsequently decreased, exhibiting a monophasic time course. The PK time‐course profiles of emicizumab were well overlapped between the DPs and among the sites of SC injection. In the IV‐dosing group, plasma emicizumab concentrations decreased exhibiting a biphasic time course after IV administration.
Figure 2.
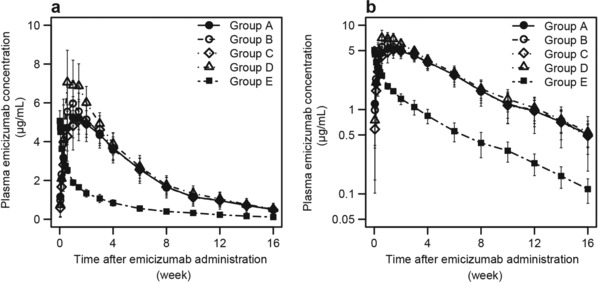
Mean (± SD) time courses of plasma emicizumab concentration following a single subcutaneous injection of 1 mg/kg with the old drug product into the abdomen (group A), a single subcutaneous injection of 1 mg/kg with the new drug product into the abdomen (group B), upper arm (group C), and thigh (group D), and a single intravenous infusion of 0.25 mg/kg with the new drug product (group E). Data below the quantification range were handled as missing in summary statistics calculation. Summary statistics were not calculated when measured values were below the quantification range in the majority of subjects for each group and time point. a, Linear plot. b, Semilogarithmic plot.
The summary statistics of PK parameters of emicizumab following a single SC or IV administration are shown in Table 2. The ranges of mean Cmax, AUClast, and AUCinf observed after a single SC injection of 1 mg/kg were 5.29 to 7.56 µg/mL, 241 to 284 day·µg/mL, and 260 to 307 day·µg/mL, respectively, and the range of median time to reach maximum plasma concentration was 6.97 to 11.4 days. No obvious differences for these PK parameters were observed among the SC‐dosing groups. The mean Cmax, AUClast, and AUCinf observed after a single IV infusion of 0.25 mg/kg were 5.10 µg/mL, 74.7 day·µg/mL, and 79.2 day·µg/mL, respectively, and the mean CL and Vd,ss of emicizumab were 3.26 mL/(day·kg) and 106 mL/kg, respectively. The t1/2 of emicizumab averaged 25.6 to 28.7 days without obvious differences among all of the dosing groups.
Table 2.
Pharmacokinetic Parameters of Emicizumab Following a Single Subcutaneous or Intravenous Administration
| Group | |||||
|---|---|---|---|---|---|
| Parameter | A (SC) | B (SC) | C (SC) | D (SC) | E (IV) |
| N | 12 | 12 | 12 | 12 | 12 |
| Cmax (µg/mL) | 5.40 ± 0.907 | 6.26 ± 1.26 | 5.29 ± 0.960 | 7.56 ± 1.38 | 5.10 ± 0.509 |
| Tmax (d) | 6.97 | 6.97 | 11.4 | 8.47 | 0.104 |
| (3.99‐10.9) | (3.98‐14.0) | (6.97‐21.0) | (3.99‐10.9) | (0.104‐0.396) | |
| AUClast (d·µg/mL) | 247 ± 56.8 | 253 ± 47.7 | 241 ± 40.4 | 284 ± 38.2 | 74.7 ± 10.9 |
| AUCinf (d·µg/mL) | 271 ± 76.2 | 274 ± 53.3 | 260 ± 47.5 | 307 ± 45.6 | 79.2 ± 12.8 |
| t1/2 (d) | 28.7 ± 7.43 | 28.0 ± 5.53 | 25.6 ± 6.97 | 28.7 ± 4.21 | 26.7 ± 6.62 |
| CL/F (mL/[d·kg]) | 3.98 ± 1.19 | 3.84 ± 1.05 | 4.00 ± 0.863 | 3.33 ± 0.503 | … |
| CL (mL/[d·kg]) | … | … | … | … | 3.26 ± 0.681 |
| Vd,z (mL/kg) | … | … | … | … | 120 ± 20.5 |
| Vd,ss (mL/kg) | … | … | … | … | 106 ± 14.8 |
| F (%) | … | 0.868a | 0.804b | 0.931c | … |
| (0.795‐0.948) | (0.712‐0.906) | (0.849‐1.022) | |||
AUCinf indicates area under the plasma concentration‐time curve extrapolated to infinity; AUClast, area under the plasma concentration‐time curve to the last measurable plasma concentration; CL, total clearance; CL/F, apparent total clearance; Cmax, maximum plasma concentration; F, absolute bioavailability; IV, intravenous; SC, subcutaneous; Tmax, time to reach maximum plasma concentration; t1/2, half‐life; Vd,z, volume of distribution during the terminal phase; Vd,ss, volume of distribution at steady state.
Data are presented as mean ± SD for Cmax, AUClast, AUCinf, t1/2, CL/F, CL, Vd,z and Vd,ss; median (range) for Tmax; geometric mean ratio (90% confidence interval) for F.
Subjects who tested positive for antiemicizumab antibodies (1 each in groups B and E) were excluded from the bioavailability estimation (N of subjects included in the analysis = 22).
Subjects who tested positive for antiemicizumab antibodies (2 in group C and 1 in group E) were excluded from the bioavailability estimation (N of subjects included in the analysis = 21).
Subjects who tested positive for antiemicizumab antibodies (1 in group E) were excluded from the bioavailability estimation (N of subjects included in the analysis = 23).
A single SC (1 mg/kg) or IV (0.25 mg/kg) administration of emicizumab did not affect the plasma concentrations of the target antigens (FIX and FX) in any of the dosing groups (data not shown).
Relative and Absolute Bioavailability
The results of the ANOVA on relative and absolute bioavailability are shown in Tables 3 and 2, respectively. A total of 4 subjects who tested positive for ADAs were excluded from this analysis.
Table 3.
Relative Bioavailability of Emicizumab Between Drug Products and Among Sites of Subcutaneous Injection
| Relative Bioavailability (GMR [90% CI]) | |||
|---|---|---|---|
| New vs Old Drug Products | Upper Arm vs Abdomen | Thigh vs Abdomen | |
| (Group B vs Group A) | (Group C vs Group B) | (Group D vs Group B) | |
| Parameter | N = 23 | N = 21 | N = 23 |
| Cmax/Dose | 1.199 (1.060‐1.355) | 0.823 (0.718‐0.943) | 1.168 (1.030‐1.324) |
| AUClast/Dose | 1.085 (0.942‐1.250) | 0.931 (0.824‐1.051) | 1.077 (0.979‐1.184) |
| AUCinf/Dose | 1.083 (0.920‐1.275) | 0.926 (0.814‐1.053) | 1.073 (0.969‐1.189) |
AUCinf indicates area under the plasma concentration‐time curve extrapolated to infinity; AUClast, area under the plasma concentration‐time curve to the last measurable plasma concentration; CI, confidence interval; Cmax, maximum plasma concentration; GMR, geometric mean ratio.
Subjects who tested positive for antiemicizumab antibodies (1 in group B, 2 in group C, and 1 in group E) were excluded from the bioavailability estimation.
The point estimates (90% confidence intervals) of the GMRs of dose‐normalized Cmax, AUClast, and AUCinf for the new DP compared with the old DP were 1.199 (1.060 to 1.355), 1.085 (0.942 to 1.250), and 1.083 (0.920 to 1.275), respectively. Similarly, those for upper arm compared with abdomen were 0.823 (0.718 to 0.943), 0.931 (0.824 to 1.051), and 0.926 (0.814 to 1.053), respectively, and those for thigh compared with abdomen were 1.168 (1.030 to 1.324), 1.077 (0.979 to 1.184), and 1.073 (0.969 to 1.189), respectively. All the point estimates of GMRs were within the standard criterion for bioequivalence (ie, 0.80‐1.25).
The point estimates (90% confidence intervals) of the GMRs of dose‐normalized AUCinf for SC injection to abdomen, upper arm, and thigh compared with IV infusion were 0.868 (0.795 to 0.948), 0.804 (0.712 to 0.906), and 0.931 (0.849 to 1.022), respectively.
Pharmacodynamics
In plasma without ex vivo neutralization of FVIII, aPTT was slightly shortened after emicizumab administration in all the dosing groups (data not shown). In plasma with ex vivo neutralization of FVIII, the shortening of aPTT was more obviously observed after emicizumab administration in all the dosing groups (Figure 3A). Similarly, obvious promotion of TG was observed in plasma with ex vivo neutralization of FVIII after emicizumab administration in all the dosing groups (Figure 3B). The PD profiles of emicizumab following single SC administration were similar between the old and new DPs and among the sites of SC injection of abdomen, upper arm, and thigh.
Figure 3.
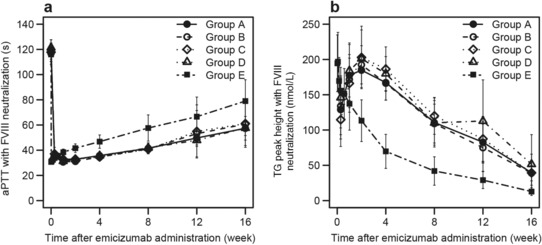
Mean (± SD) time courses of pharmacodynamic responses in ex vivo factor VIII–neutralized plasma following a single subcutaneous injection of 1 mg/kg with the old drug product into the abdomen (group A), a single subcutaneous injection of 1 mg/kg with the new drug product into the abdomen (group B), upper arm (group C), and thigh (group D), and a single intravenous infusion of 0.25 mg/kg with the new drug product (group E). Data below the quantification range were handled as missing in summary statistics calculations. Summary statistics were not calculated when measured values were below the quantification range in the majority of subjects for each group and time point. a, Activated partial thromboplastin time. b, Peak height of activated factor XI–triggered thrombin generation. aPTT indicates activated partial thromboplastin time; FVIII, factor VIII; TG, thrombin generation.
Safety
Twenty‐seven AEs occurred in 22 of 60 subjects; the summary of the incidence of AEs in each group is shown in Table 4. The only AE that occurred in at least 2 subjects in any group was upper respiratory tract infection (group A 3 subjects [25.0%], group B 2 subjects [16.7%], group C 1 subject [8.3%], group D 1 subject [8.3%], and group E 1 subject [8.3%]). All of the upper respiratory tract infections were considered unrelated to emicizumab administration by the investigator.
Table 4.
Adverse Events Reported Following a Single Subcutaneous or Intravenous Administration
| Group | |||||
|---|---|---|---|---|---|
| A | B | C | D | E | |
| Adverse Event | N (%) | N (%) | N (%) | N (%) | N (%) |
| Total number of subjects with at least 1 adverse event | 6 (50.0) | 5 (41.7) | 4 (33.3) | 3 (25.0) | 4 (33.3) |
| Upper respiratory tract infection | 3 (25.0) | 2 (16.7) | 1 (8.3) | 1 (8.3) | 1 (8.3) |
| Acute sinusitis | 0 (0.0) | 1 (8.3) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Acute tonsillitis | 1 (8.3) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Conjunctivitis | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (8.3) | 0 (0.0) |
| Nasopharyngitis | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (8.3) | 0 (0.0) |
| Paronychia | 0 (0.0) | 1 (8.3) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Pharyngitis | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (8.3) | 0 (0.0) |
| Abdominal pain | 1 (8.3) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Aphthous stomatitis | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (8.3) | 0 (0.0) |
| Dental caries | 0 (0.0) | 1 (8.3) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Oropharyngeal pain | 0 (0.0) | 1 (8.3) | 0 (0.0) | 0 (0.0) | 1 (8.3) |
| Punctate keratitis | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (8.3) | 0 (0.0) |
| Facial pain | 1 (8.3) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Venomous sting | 0 (0.0) | 0 (0.0) | 1 (8.3) | 0 (0.0) | 0 (0.0) |
| Blood glucose decreased | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (8.3) |
| Myalgia | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (8.3) |
| Cervicobrachial syndrome | 1 (8.3) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Dermatitis contact | 0 (0.0) | 0 (0.0) | 1 (8.3) | 0 (0.0) | 0 (0.0) |
| Hot flush | 0 (0.0) | 0 (0.0) | 1 (8.3) | 0 (0.0) | 0 (0.0) |
AEs occurring within 24 hours after administration of emicizumab were defined as injection/infusion‐related reactions, which included oropharyngeal pain in 1 subject (group B) and hot flush in 1 subject (group C). Although a causal relationship with emicizumab administration was not ruled out, both of the events were mild in severity and resolved or improved within 3 days without any treatment.
No severe AEs, AEs leading to withdrawal, or serious AEs occurred. In addition, no clinically significant changes in laboratory test values, vital signs, or electrocardiogram parameters were observed in any group (data not shown).
Overall, no significant differences in the safety profile of emicizumab were observed by DP, injection site, or administration route.
Immunogenicity
ADAs were detected in 4 of a total of 60 subjects. Of these subjects, 1 subject tested positive for ADAs before and after emicizumab administration (preexisting ADAs), and 3 subjects tested positive for ADAs only after emicizumab administration (treatment‐induced ADAs). The subjects with treatment‐induced ADAs consisted of 1 of 12 subjects (8.3%) in groups B, C, and E each; therefore, 2 of 36 subjects (5.6%) receiving a single SC injection of the new DP had treatment‐induced ADAs.
The time courses of plasma emicizumab concentration, aPTT, and peak height of TG in FVIII‐neutralized plasma were not different between the subject who tested positive for preexisting ADAs and ADA‐negative subjects. On the other hand, the t1/2 of emicizumab for the subjects who tested positive for treatment‐induced ADAs were shorter than those for ADA‐negative subjects (data not shown). Correspondingly, in FVIII‐neutralized plasma, the effect of emicizumab on both aPTT and peak height of TG dissipated earlier in the subjects with treatment‐induced ADAs (data not shown). No AEs were observed in either ADA‐positive subjects.
Discussion
This study was conducted to compare PK profiles of emicizumab between 2 different DPs used during the clinical development and among 3 different injection sites for which we intended to produce supportive clinical data to expand SC injection sites of emicizumab and thus to enable patients with hemophilia A to have the flexibility in choice of the injection site in their lifelong treatment. In addition, the absolute SC bioavailability of emicizumab was investigated.
The similar PK profiles after a single SC injection of emicizumab 1 mg/kg were confirmed between the old and new DPs and among 3 injection sites of abdomen, upper arm, and thigh, with all the point estimates of GMRs within the standard criterion for bioequivalence (ie, 0.80‐1.25). The PK similarity of emicizumab among the 3 SC injection sites was consistent with a previous investigation with another monoclonal antibody.26 In addition, the PD responses (aPTT and TG) in plasma with ex vivo neutralization of FVIII after a single SC injection of emicizumab 1 mg/kg were similar as well. These results supported the idea that no dose adjustment was needed when the new DP replaced the old one during the clinical development of emicizumab. Moreover, the results indicated that emicizumab can be administered to the abdomen, upper arm, and thigh, interchangeably, without dose adjustment.
Group A reproduced similar PK profiles to those observed in the first‐in‐human study of emicizumab in which the old DP was subcutaneously injected into the skin of the abdomen in healthy volunteers as well. The incidence of AEs did not differ among the SC‐dosing groups, and it was similar to that observed in the first‐in‐human study.16 It was reconfirmed that a single SC injection of emicizumab 1 mg/kg was tolerable in healthy volunteers. These results did not identify any new safety risk with the new DP compared with the old DP.
This study was the first clinical investigation of emicizumab administered intravenously to humans. The observed Cmax after a single IV infusion of emicizumab 0.25 mg/kg with the new DP was similar to that after a single SC injection of emicizumab 1 mg/kg with the new DP as hypothesized before the study initiation. The incidence of AEs did not differ from that in the SC‐dosing groups, and thus, it can be concluded that emicizumab is tolerable up to 0.25 mg/kg when it is administered intravenously in healthy volunteers. The mean CL of emicizumab after IV infusion was 3.26 mL/(day·kg), which suggested that the elimination of emicizumab from the circulating blood is comparable to or even slower than that of other monoclonal antibodies.27 This might be a consequence of a reduced isoelectric point resulting from the amino acid–substituting antibody engineering technology applied.13, 28 The mean Vd,ss of emicizumab was 106 mL/kg, which was approximately 2‐ to 3‐fold the plasma volume of humans but much less than the extracellular fluid volume,24 suggesting that the tissue distribution of emicizumab is limited in humans as commonly seen for monoclonal antibodies.27 In addition, this IV‐dosing experience revealed that the absolute SC bioavailability of emicizumab ranged from 80.4% to 93.1%. This finding indicates a high SC absorbability of emicizumab, which is consistent with the previous nonclinical results and is in line with other monoclonal antibodies.27
Two of 36 subjects who received a single SC injection of emicizumab with the new DP tested positive for treatment‐induced ADAs, and no ADAs were detected in subjects who received a single SC injection of emicizumab with the old DP in this study. On the other hand, in the first‐in‐human study, 1 of a total of 48 subjects who received a single SC injection of emicizumab with the old DP tested positive for treatment‐induced ADAs. Altogether, treatment‐induced ADAs were observed with administration of either the old or the new DP across the 2 studies in healthy subjects, but the overall incidence was within the ADA incidence reported in other monoclonal antibodies.29, 30 The immunogenicity of emicizumab, including the incidence and clinical relevance of ADAs, should be further investigated in patients with hemophilia A who will receive emicizumab repeatedly at higher doses.
The regulatory guidance, stipulating comparability assessments to support manufacturing process changes during the development of a biological product, and some previous reports have suggested a hierarchical risk‐based approach for DP bridging.23, 31, 32, 33, 34 It starts with analytical testing to ensure quality, followed by biological characterization, animal in vivo PK or PK‐PD studies, and finally clinical PK, safety, and/or efficacy studies, if needed. However, such a stepwise approach may not completely remove the uncertainty associated with extrapolation of in vitro and/or animal study findings to humans, may be potentially time consuming where a clinical study is eventually required, and may not be fit for some disease areas, especially in life‐threatening or rare diseases when rapid development of new drugs is desired. In the case of emicizumab, the experimental evidence indicating the lack of need for dose adjustment between the old and new DPs was obtained by conducting this clinical bioavailability study before initiating phase 3 studies. This approach was intended to avoid delaying the development of emicizumab in hemophilia A, a rare disease in which unmet medical need is high, especially in patients with FVIII inhibitors due to suboptimal efficacy of bypassing agents.
Although this study presents a successful bridging case with emicizumab between 2 DPs and among 3 SC injection sites, there are some limitations. The current study was not a statistically powered bioequivalence study, and thus, PK equivalency cannot be concluded. The data were obtained from healthy volunteers who received only a single SC injection or IV infusion at a dose of 1 or 0.25 mg/kg, which is lower than the doses given to patients with hemophilia A in the first phase 3 study.
Based on the results from the early‐stage clinical studies16, 17, 18 and bridging practices from early‐ to late‐stage development (ie, model‐guided phase 3 dose selection19 and this bioavailability study), the phase 3 program of emicizumab is now ongoing, and recently, it was demonstrated that emicizumab prophylaxis significantly reduced the bleeding rate compared with no prophylaxis in patients with hemophilia A with FVIII inhibitors in the first phase 3 study.20
Conclusions
Similar PK and PD profiles of emicizumab between the DPs indicated that no dose adjustment would be needed in switching the DPs during the emicizumab development program. In addition, the study results supported the conclusion that emicizumab can be injected into the abdomen, upper arm, or thigh, interchangeably. Availability of multiple injection sites for SC dosing would allow for increased convenience in treatment and may thus improve patients’ therapeutic adherence. The study results also indicated that emicizumab injected subcutaneously is highly bioavailable.
Acknowledgments
We thank the subjects for their involvement in this study. We also sincerely thank our colleagues at Chugai Pharmaceutical Co, Ltd, especially Ryu Kasai, Mariko Hoshiba, and Isamu Terashima for support in the conduct of the research, and Taku Sakaue for support in planning and conducting the statistical analysis.
Declaration of Conflicting Interests
This study was sponsored by Chugai Pharmaceutical Co, Ltd. N. Kotani, K.Y., N. Kawakami, T.S., and T.K. are employees of Chugai Pharmaceutical Co, Ltd. N. Kawakami, T.S., and T.K. hold stock/stock options in Chugai Pharmaceutical Co, Ltd. K.Y. is an inventor of the patents relating to anti‐FIXa/X bispecific antibodies. H.F. declares no conflict of interest.
References
- 1. Mannucci PM, Tuddenham EG. The hemophilias—from royal genes to gene therapy. N Engl J Med. 2001;344(23):1773–1779. [DOI] [PubMed] [Google Scholar]
- 2. Collins P, Chalmers E, Chowdary P, et al. The use of enhanced half‐life coagulation factor concentrates in routine clinical practice: guidance from UKHCDO. Haemophilia. 2016;22(4):487–498. [DOI] [PubMed] [Google Scholar]
- 3. Collins PW, Fischer K, Morfini M, Blanchette VS, Bjorkman S, International Prophylaxis Study Group Pharmacokinetics Expert Working Group . Implications of coagulation factor VIII and IX pharmacokinetics in the prophylactic treatment of haemophilia. Haemophilia. 2011;17(1):2–10. [DOI] [PubMed] [Google Scholar]
- 4. Manco‐Johnson MJ, Abshire TC, Shapiro AD, et al. Prophylaxis versus episodic treatment to prevent joint disease in boys with severe hemophilia. N Engl J Med. 2007;357(6):535–544. [DOI] [PubMed] [Google Scholar]
- 5. Srivastava A, Brewer AK, Mauser‐Bunschoten EP, et al. Guidelines for the management of hemophilia. Haemophilia. 2013;19(1):e1–e47. [DOI] [PubMed] [Google Scholar]
- 6. Bysted BV, Scharling B, Møller T, Hansen BL. A randomized, double‐blind trial demonstrating bioequivalence of the current recombinant activated factor VII formulation and a new robust 25 degrees C stable formulation. Haemophilia. 2007;13(5):527–532. [DOI] [PubMed] [Google Scholar]
- 7. Fridberg MJ, Hedner U, Roberts HR, Erhardtsen E. A study of the pharmacokinetics and safety of recombinant activated factor VII in healthy Caucasian and Japanese subjects. Blood Coagul Fibrinolysis. 2005;16(4):259‐266. [DOI] [PubMed] [Google Scholar]
- 8. Lindley CM, Sawyer WT, Macik BG, et al. Pharmacokinetics and pharmacodynamics of recombinant factor VIIa. Clin Pharmacol Ther. 1994;55(6):638–648. [DOI] [PubMed] [Google Scholar]
- 9. Villar A, Aronis S, Morfini M, et al. Pharmacokinetics of activated recombinant coagulation factor VII (NovoSeven) in children vs adults with haemophilia A . Haemophilia. 2004;10(4):352–359. [DOI] [PubMed] [Google Scholar]
- 10. Varadi K, Negrier C, Berntorp E, et al. Monitoring the bioavailability of FEIBA with a thrombin generation assay. J Thromb Haemost. 2003;1(11):2374–2380. [DOI] [PubMed] [Google Scholar]
- 11. Kitazawa T, Esaki K, Tachibana T, et al. Factor VIIIa‐mimetic cofactor activity of a bispecific antibody to factors IX/IXa and X/Xa, emicizumab, depends on its ability to bridge the antigens. Thromb Haemost. 2017;117(7):1348–1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kitazawa T, Igawa T, Sampei Z, et al. A bispecific antibody to factors IXa and X restores factor VIII hemostatic activity in a hemophilia A model. Nat Med. 2012;18(10):1570–1574. [DOI] [PubMed] [Google Scholar]
- 13. Sampei Z, Igawa T, Soeda T, et al. Identification and multidimensional optimization of an asymmetric bispecific IgG antibody mimicking the function of factor VIII cofactor activity. PLoS One. 2013;8(2):e57479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Muto A, Yoshihashi K, Takeda M, et al. Anti‐factor IXa/X bispecific antibody (ACE910): hemostatic potency against ongoing bleeds in a hemophilia A model and the possibility of routine supplementation. J Thromb Haemost. 2014;12(2):206–213. [PubMed] [Google Scholar]
- 15. Muto A, Yoshihashi K, Takeda M, et al. Anti‐factor IXa/X bispecific antibody ACE910 prevents joint bleeds in a long‐term primate model of acquired hemophilia A. Blood. 2014;124(20):3165–3171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Uchida N, Sambe T, Yoneyama K, et al. A first‐in‐human phase 1 study of ACE910, a novel factor VIII‐mimetic bispecific antibody, in healthy subjects. Blood. 2016;127(13):1633–1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shima M, Hanabusa H, Taki M, et al. Factor VIII‐mimetic function of humanized bispecific antibody in hemophilia A. N Engl J Med. 2016;374(21):2044‐2053. [DOI] [PubMed] [Google Scholar]
- 18. Shima M, Hanabusa H, Taki M, et al. Long‐term safety and efficacy of emicizumab in a phase 1/2 study in patients with hemophilia A with or without inhibitors. Blood Adv. 2017;1(22):1891–1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yoneyama K, Schmitt C, Kotani N, et al. A pharmacometric approach to substitute for a conventional dose‐finding study in rare diseases: example of phase III dose selection for emicizumab in hemophilia A. Clin Pharmacokinet. 2018;57(9):1123–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Oldenburg J, Mahlangu JN, Kim B, et al. Emicizumab prophylaxis in hemophilia A with inhibitors. N Engl J Med. 2017;377(9):809–818. [DOI] [PubMed] [Google Scholar]
- 21. Goetze AM, Liu YD, Zhang Z, et al. High‐mannose glycans on the Fc region of therapeutic IgG antibodies increase serum clearance in humans. Glycobiology. 2011;21(7):949–959. [DOI] [PubMed] [Google Scholar]
- 22. Yu M, Brown D, Reed C, et al. Production, characterization, and pharmacokinetic properties of antibodies with N‐linked mannose‐5 glycans. MAbs. 2012;4(4):475–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH Harmonized Tripartite Guideline Q5E: Comparability of Biotechnological/Biological Products Subject to Changes in their Manufacturing Process (CPMP/ICH/5721/03) . Geneva: ICH; 2004.
- 24. Davies B, Morris T. Physiological parameters in laboratory animals and humans. Pharm Res. 1993;10(7):1093–1095. [DOI] [PubMed] [Google Scholar]
- 25. The European Agency for the Evaluation of Medicinal Products. Guideline on the Investigation of Bioequivalence (EMEA/CHMP/BMWP/14327/2006) . London: EMA; 2010.
- 26. Xu Z, Wang Q, Zhuang Y, et al. Subcutaneous bioavailability of golimumab at 3 different injection sites in healthy subjects. J Clin Pharmacol. 2010;50(3):276–284. [DOI] [PubMed] [Google Scholar]
- 27. Lobo ED, Hansen RJ, Balthasar JP. Antibody pharmacokinetics and pharmacodynamics. J Pharm Sci. 2004;93(11):2645–2668. [DOI] [PubMed] [Google Scholar]
- 28. Igawa T, Tsunoda H, Tachibana T, et al. Reduced elimination of IgG antibodies by engineering the variable region. Protein Eng Des Sel. 2010;23(5):385–392. [DOI] [PubMed] [Google Scholar]
- 29. Baker MP, Reynolds HM, Lumicisi B, Bryson CJ. Immunogenicity of protein therapeutics: the key causes, consequences and challenges. Self Nonself. 2010;1(4):314–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wang W, Wang EQ, Balthasar JP. Monoclonal antibody pharmacokinetics and pharmacodynamics. Clin Pharmacol Ther. 2008;84(5):548–558. [DOI] [PubMed] [Google Scholar]
- 31. US Department of Health and Human Services Food and Drug Administration. Guidance for Industry (draft): Comparability Protocols—Protein Drug Products and Biological Products—Chemistry, Manufacturing and Controls Information . Rockville, MD: FDA; 2003.
- 32. The European Agency for the Evaluation of Medicinal Products. Guideline on Comparability of Biotechnology‐Derived Medicinal Products After a Change in the Manufacturing Process: Non‐Clinical and Clinical Issues (EMEA/CHMP/BMWP/101695/2006) . London: EMA; 2007.
- 33. Putnam WS, Prabhu S, Zheng Y, Subramanyam M, Wang YM. Pharmacokinetic, pharmacodynamic and immunogenicity comparability assessment strategies for monoclonal antibodies. Trends Biotechnol. 2010;28(10):509–516. [DOI] [PubMed] [Google Scholar]
- 34. Center for Drug Evaluation and Research Center for Biologics Evaluation and Research. FDA Guidance Concerning Demonstration of Comparability of Human Biological Products, Including Therapeutic Biotechnology‐Derived Products . Rockville, MD: CBER; 1996.