

Abstract
Pancreatic ductal adenocarcinoma (PDAC) is the most common malignancy of the pancreas and has one of the highest mortality rates of any cancer type with a 5‐year survival rate of <5%. Recent studies of PDAC have provided several transcriptomic classifications based on separate analyses of individual patient cohorts. There is a need to provide a unified transcriptomic PDAC classification driven by therapeutically relevant biologic rationale to inform future treatment strategies. Here, we used an integrative meta‐analysis of 353 patients from four different studies to derive a PDAC classification based on immunologic parameters. This consensus clustering approach indicated transcriptomic signatures based on immune infiltrate classified as adaptive, innate and immune‐exclusion subtypes. This reveals the existence of microenvironmental interpatient heterogeneity within PDAC and could serve to drive novel therapeutic strategies in PDAC including immune modulation approaches to treating this disease.
Keywords: pancreatic cancer, tumour microenvironment, T cells, adaptive immunity, innate immunity, subtypes
Short abstract
What's new?
While several transcriptomic classifications of pancreatic adenocarcinoma (PDAC) have been proposed, a unified classification would be valuable to inform future treatment strategies. Through an integrative meta‐analysis of 353 patients from four different studies, the authors found that the greatest prognostic value in independent cohorts could be achieved through stratification by gene expression signatures associated with tumour‐infiltrating immune cells across different pancreatic cancer subtypes. Recognising the existence of different tumour escape mechanisms (and indeed phenotypes) in pancreatic cancer may guide immunotherapeutic treatment plans and improve patient stratification for maximization of therapeutics.
Introduction
Despite on going improved understanding of the genetics and molecular biology of Pancreatic Ductal Adenocarcinoma (PDAC), prognosis remains strikingly poor with five‐year survival rate <5%.1 Improving outcomes may be achieved through accurate subtyping upon detection to better tailor therapeutic strategies.
Using transcriptomics is an attractive option to sub classify cancers. For example, most notably researchers in colorectal cancer have developed a consensus classification has four subtypes with biological and therapeutic relevance.2 Gene expression profiles of tumours are a combined readout of the tumour's genetic and epigenetic status, as well as the composition of other cells in the microenvironment that are sampled in the biopsy. The microenvironment includes tumour stroma, host connective tissue cells that change in response to signals from the tumour cells, and innate and adaptive immune system cells.3, 4
Several recent studies have described the transcriptomic landscape of PDAC and identified different subtypes with different clinical outcomes and drug sensitivities.5, 6, 7, 8 Table 1 summarises several notable studies. Collisson et al. proposed the first classification in 2011 by using unsupervised clustering methods on transcriptomic data.5 This had identified three subtypes: classical, quasimesenchymal and exocrine‐like, which have different prognoses and different responses to treatment. This classification has subsequently been challenged and revised, with newer classifications being put forward.6, 7, 8 Moffitt et al. identified 4 groups: 2 stroma and 2 non‐stroma tumour groups.6 Moffitt subtyped PDAC ‘classical’ and ‘basal‐like’ based on the pancreatic tumour cells and ‘normal stroma’ and ‘activated stroma’ based on the virtually micro‐dissected stroma cell population. Bailey et al. demonstrated 4 subtypes with different characterisations and different prognoses: ‘squamous’, ‘pancreatic progenitor’, ‘immunogenic’ and ‘aberrantly differentiated endocrine exocrine’ (ADEX).7 Sivakumar et al. used a master regulator approach based on the transcriptional effects of oncogenic KRAS to determine 3 subtypes that showed different prognoses, characteristics and treatment strategies.8
Table 1.
Pancreatic ductal carcinoma and cancer subtyping signatures and studies used in this study
| Technique of discovery | Sub‐type | Prognosis | Composition | Therapeutics | |
|---|---|---|---|---|---|
| Collisson | Non‐negative matrix factorization (NMF) analysis with consensus clustering | Classical | Good | N/A | Erlotinib |
| Exocrine | Average | N/A | Non‐proposed | ||
| Quasi‐mesenchymal | Poor | N/A | Gemcitabine | ||
| Moffitt | Non‐negative matrix factorization (NMF) and digital separation | Tumour: classical | Average (median survival 19 months) | N/A | N/A |
| Tumour: basal‐like | Poor (median survival 11 months) | N/A | Better response to adjuvant therapy | ||
| Stroma: normal | Good (median survival 20 months) | Stellate cells | N/A | ||
| Stroma: activated | Poor (median survival 15 months) | Macrophages | N/A | ||
| Bailey | Unsupervised clustering | Squamous | Poor | Macrophages | N/A |
| Pancreatic Progenitor | N/A | N/A | N/A | ||
| Immunogenic | N/A | B cells and T cells | Checkpoint blockade | ||
| ADEX | N/A | ||||
| Sivakumar | Master regulator Analysis | Notch | Good | T cell infiltration | Immunotherapy |
| Cell cycle | Average | N/A | Adjuvant therapy | ||
| Hedgehog | Poor | Macrophages | N/A |
Other studies that have derived prognostically significant transcriptional classifiers include Birnbaum et al. who performed a previous meta‐analysis of the Collisson, Moffitt and Bailey classifications showing concordance between the Collisson and Moffitt tumour gene lists and concordance between the Moffitt stroma gene lists and Bailey classification.9 Further transcriptome analysis from Mao et al. in a small cohort of 10 patients has shown there is a differential transcriptome in pancreatic cancer.10 A smaller gene expression study showed there were three subtypes using a gene expression microarray that has prognostically significant groups.11 Another smaller master regulator study by Janky et al. demonstrates there are four prognostically significant subtypes.12 Further studies by Haider et al., 13 Kirby et al.14 and Stratford et al.15 all elucidated prognostic transcriptomic signatures of 36, 19 and 6 genes, respectively. Donahue et al. using various cohorts performed an integrative analysis that identified 171 genes that elicited another prognostic signature.16 A study by Connor et al. used mutational signatures to derive four subtypes: age related, double‐strand break repair, mismatch repair, and one with unknown aetiology with the double‐strand break repair and mismatch repair groups were associated with CD8 T cells activation and increased T regs.17 In two papers by Witkiewicz and colleagues, a relationship between the immune status and genomics was described and classified PDAC into four subtypes: mutationally cold, hot, mutationally active and cold with cold having the best prognosis.18, 19
These studies suggest that it is possible to categorise PDAC through transcriptomics, mutational signatures or immunological status. However, this has not become the accepted clinical approach because of problems inherent with the approach – too many non‐overlapping signatures, inadequate clinical relevance and poor mechanistic underpinning. It would thus be valuable to consolidate different stratification schemes into a novel classification of pancreatic cancer and develop reliable and robust biomarkers to better predict outcomes and rationalise therapeutic strategies.
In response to this unmet need, we have developed an immunological classification scheme from four notable transcriptomic PDAC sub typing studies (Table 1).5, 6, 7, 8 PDAC has previously been shown to have different immune populations in its microenvironment.20 In light of the tumour having a complex immune composition, the tumour microenvironment can be immunosuppressive or tolerant and this can be due to immune dysfunction.21, 22 However, immunotherapy approaches have not yet had an impact on pancreatic cancer survival.23, 24 Our meta‐analysis reveals that the greatest prognostic value in independent cohorts could be achieved through stratification by gene expression signatures associated with tumour infiltrating immune cells across different PDAC subtypes. Recognising the existence of different tumour escape mechanisms (and indeed phenotypes) in PDAC may guide distinct immunotherapeutic treatment plans and improve patient stratification for maximisation of therapeutic effect.
Methods
Data download: gene expression profiles, clinical and mutations datasets
The PACA‐AU gene expression data (n = 269) plus clinical and mutational profiles were obtained from ICGC data portal (https://dcc.icgc.org/releases/release_24/Projects/PACA-AU). TCGA raw counts files for RNA‐seq data (n = 177) were obtained from the GDC portal (https://portal.gdc.cancer.gov/). Clinical metadata and Mutation annotation files (MAF) for the TCGA cohort was obtained from the GDC legacy archive (https://portal.gdc.cancer.gov/legacy-archive/) with the UUIDs: a9f29dc4‐6a6a‐42f3‐b06d‐9e6ded926b55 (clinical metadata) and faf50bd9‐bfc8‐4dfa‐b0ca‐9184e44fb07f (MAF file). The UNC gene expression data (n = 132) plus clinical profiles were obtained from gene expression omnibus (GEO) archive under the accession number GSE21501. Out of 132, in the UNC cohort, 30 were excluded due to unavailability of survival time in the clinical table.
Processing of gene expression data
Batch effects were removed by applying the ComBat algorithm.25 TCGA RNA‐seq data was transformed with the variance‐stabilising transformation method26 prior to removing batch effects. Batch IDs for the TCGA cohorts were obtained from the sample barcode, namely the ‘plate’ id as described in https://wiki.nci.nih.gov/display/TCGA/TCGA+Barcode.
Classification of PACA‐AU and TCGA cohorts according to five previous classification schemes
The R package ConsensusClusterPlus27 was employed to subtype PDAC samples according to the expression signatures defined in Moffitt et al.6 and Collisson et al.5 (Supporting Information Fig. S1). The number of clusters was confirmed by examining cumulative distribution function (CDF). We confirmed the existence of well‐separated clusters for Moffitt et al. classification based on tumour (two clusters: basal‐like and classical) and stroma signatures (2 clusters: stroma and activated stroma). For the Collison et al. classification we confirmed the existence of evident 3 clusters (classical, exocrine‐line and quasimesenchymal). Bailey et al. cluster labels were directly downloaded from (PACA‐AU cohort)7 and TCGA cohort).28 Sivakumar et al. cluster labels were directly downloaded from.8
Identification of immunophenotypes
To identify a meaningful agreement of the multiple clustering labels we used consensus clustering and the partition around medoids (PAM) algorithm to cluster the PACA‐AU and TCGA cohorts according the similarity of their labels. For the PACA‐AU cohort, we only included tumours that were classified as ‘Pancreatic Ductal Adenocarcinoma’ according to the tumour histological type classifications and were also labelled by all 5 classification schemes, which corresponded to 204 PDAC tumours. For the TCGA cohort we included only the filtered PDAC cases according to Raphael et al., this corresponded to 149 TCGA tumours.28 The Hamming distance was used as a measure of similarity between PDAC tumours. The robustness of sample classification was analysed by examining cumulative distribution function (CDF) of the proportion of times in which 2 samples are clustered together across the resampling iterations (1,000×).29 For varying number of clusters (K = 2 to K = 7), we examined the area under the curve of the consensus distribution function (CDF) plot (Supporting Information Figs. S2 and S3) and identified three robust novel subtypes in both cohorts.
Survival analysis
Multivariate Cox regression, log‐rank test and Kaplan–Meier estimators were implemented using the R package survival. For the PACA‐AU cohort (Fig. 2c), we adjusted the survival differences for age, gender, and tumour stage and tumour grade. For the TCGA cohort (Fig. 2e), we adjusted for age, gender, tumour stage, tumour grade and targeted therapy indicator (yes/no). Tumour stage refers to the TNM Staging System based on the extent of the tumour (T). The targeted therapy indicator refers to whether the patient had adjuvant and/or postoperative pharmaceutical therapy. More specific information about the treatment regime was not available. The correlation between immune cell scores and survival (Fig. 3c) was performed using a multivariate cox regression model adjusting for age, gender, tumour grade and stage (PACA‐AU and TCGA cohorts) and adjusting for tumour stage (TMN T stage) for the UNC cohort. For the TCGA cohort we only included cases with no targeted therapy.
Analysis of immune infiltrates and tumour purity
Two main methodologies were used to identify immune cell types enriched in the tumour microenvironment. For three of the gene signatures30, 31, 32 we used single‐sample gene set enrichment analysis (ssGSEA) method implemented in the GSVA R package.33 For the Aran gene signatures we used the xCell method implemented in R.34 The xCell method also relies on ssGSEA analysis but contains an additional step, which uses a reference matrix of ‘spillovers’ between cell types. The ‘spillover’ step is thought to better eliminate dependencies between closely related cell types (e.g. such as between CD8+ T‐cells and NK cells). Log2(TPM + 1) gene expression levels from the TCGA cohort were used to estimate the enrichment with the ssGSEA method, and TPM levels were used with the xCell method. Normalised microarray gene expression levels (Illumina Expression BeadChIPs) from the PACA‐AU cohort were used directly in both methods.
ESTIMATE was used to gauge the degree of leukocyte infiltration, stromal content and tumour purity.3 Data was summarised per PDAC subtypes in the PACA‐AU and TCGA cohorts (Fig. 5e; Supporting Information Fig. S5).
Histopathology analysis
TCGA H&E Slides were viewed on the NCI data portal https://portal.gdc.cancer.gov/. Two pathologists reviewed the slides and assessment of tumour cellularity and dominant immune cell type (lymphocyte versus neutrophils) were made.
Mutation analysis
Mutational signature analysis was performed using the DeconstructSigsR Package.35 We determined the contribution of 30signatures defined in COSMIC (http://cancer.sanger.ac.uk/signatures/)to explain each pancreatic cancer mutational profile. Normalisation was relative to the number of times each trinucleotide context is observed in the exome. The output was aset of weights specifying the estimated contribution of each of the 30known signatures to the mutation profile. MAF files were parsed with maftools36 (Supporting Information Fig. S7). Number of neoantigens per tumour in the TCGA cohort was retrieved from The Cancer Immunome Atlas database (https://tcia.at/home).31
Statistical analysis
All statistical analyses: Fisher's exact test, Chi‐square test, Wilcoxon rank sum test, hypergeometric test, and hierarchical clustering, were performed using R.37 Multiple test correction was performed using the R function p.adjust and the Benjamini and Hochberg (FDR) method. Jaccard coefficients were computed using the R package rules. Differential expression analysis between the groups was carried out using the Welch's test implementation in R and by comparing each group against all others. Welch's test is a variant of the classical Student test, whose goal is to test the equality between two means taking assuming different variances between two groups. When necessary, Ensembl or Entrez IDs were mapped to human HUGO identifiers using Ensembl version 89 biomart (http://www.ensembl.org/biomart). R code, sample IDs and final sample cluster memberships for the TCGA and PACA‐AU cohorts are provided as a Supporting Information file (Additional File 1) and in Supporting Information Table S1.
Results
PDAC meta‐analysis
We used published exemplar gene signature for data from Collison et al. and Moffitt et al. to cluster 242 PDAC primary tumour cases from the PACA‐AU cohort (Supporting Information Fig. S1). We further obtained and applied clustering labels identified by Bailey et al. and Sivakumar et al. also from the PACA‐AU cohort. Figure 1 a summarises the workflow of our analysis.
Figure 1.
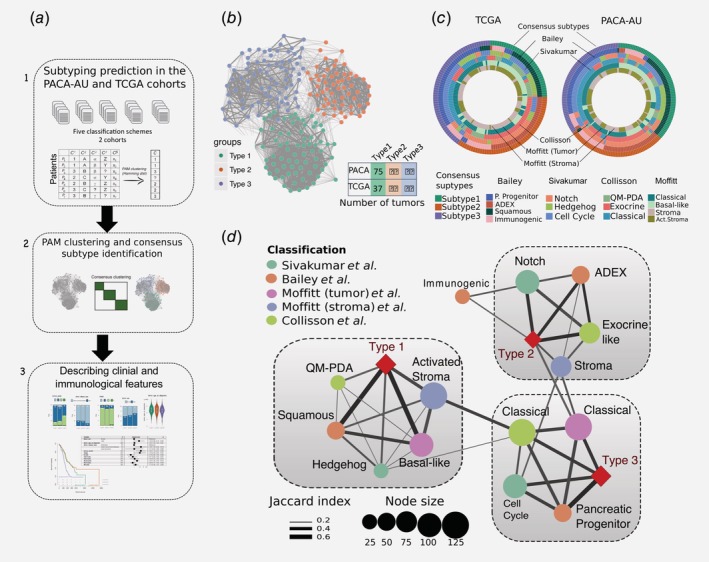
Identification of the consensus subtypes of PDAC. (a) Analytical workflow of the PDAC subtyping: (1) subtype classification using methodology from five different classification schemes; (2) concordance analysis of the five subtyping labels and application of PAM clustering algorithm to identify consensus clusters; (3) analysis of clinicopathological and immunophenotypes of PDAC consensus subtypes and identification of immune cell signatures with prognostic value in independent PDAC cohorts. (b) Patient similarity network. Each node represents a single patient sample in the PACA‐AU cohort (n = 204). Network edges correspond to highly concordant (at least 5 of 6) subtyping calls between samples. Nodes are coloured according to the three clusters identified from the PAM consensus clustering algorithm. (c) Circular heatmap representing sample overlap for consensus PDAC subtypes and mRNA subtypes from Bailey et al., Sivakumar et al., Collisson et al., or Moffitt et al. (from inside to outside, respectively). Significance of sample overlap was assessed with the hypergeometric test, adjusted p values for each pairwise comparison are depicted in Supporting Information Figure S4. (d) Association of consensus PDAC subtypes identified by PAM clustering (red refers to this study: Type 1, Type 2, Type 3) with tumour labels across five classification systems. Each node corresponds to a single subtype (circles are coloured according to classification study; red diamonds correspond to the consensus subtypes). Edge width corresponds to the overlap between labels assessed by the Jaccard coefficient in the PACA‐AU cohort, only significant edges are depicted (hypergeometric P ≤ 0.05. The three grey rectangles delineate clusters of tumour labels that overlap the three PDAC consensus groups (Type 1–3) with FDR adjusted P ≤ 0.05. [Color figure can be viewed at wileyonlinelibrary.com]
To identify a meaningful accordance of the multiple clustering labels we used consensus clustering and the partition around medoids (PAM) algorithm to cluster the 204 PDAC tumours according to the similarity of their labels and identified three robust novel subtypes (Fig. 1 b; Supporting Information Fig. S2). Visualisation of the similarity network between tumour labels revealed that consensus samples remained grouped between the three large primary network hubs (Fig. 1 b). We used 150 TCGA samples that were previously classified to perform the same classification using the PAM algorithm in an independent cohort (Supporting Information Fig. S3). Notably, the same three subtypes were identified in the TCGA cohort (Fig. 1 c) with the three main subtypes showing a stable and consistent pattern of co‐associated labels (Fig. 1 d; Supporting Information Fig. S4). The subtype 1 cluster overlapped between Moffitt basal‐like, Bailey squamous, Collisson QM‐PDA and Moffitt activated stroma subtypes (Fig. 1 d). The subtype 2 cluster consisted of a statistically significant overlap between the Bailey ADEX, Collisson exocrine‐like, Sivakumar Notch and Moffitt normal‐stroma subtypes. Finally, the subtype 3 cluster overlapped with the classical subtypes identified by Moffitt and Collison studies and cell‐cycle group identified by Sivakumar et al. (overlaps were considered significant at hypergeometric FDR adjusted p ≤ 0.05 in both cohorts; Fig. 1 d).
Clinicopathological characteristics of PDAC subtypes
Clinical features, including age, gender, tumour grade and TNM, were statistically compared between the three clusters. Two‐way contingency table analysis showed significant association between grade and cluster subtype, with subtype 1 tumours more likely to be grade 3 (Chi‐square test p = 0.002 PACA‐AU cohort; Fig. 2 a) and subtype 2 tumours more likely to be grade 1 (Chi‐square test p = 0.033 for the TCGA cohort; Fig. 2 b). Tumour stage and gender did not correlate with the subtypes in any of the cohorts. Similarly, the average age of diagnosis was not significantly associated to any cluster subtype (Figs. 2 a and 2b; Supporting Information Table S2).
Figure 2.
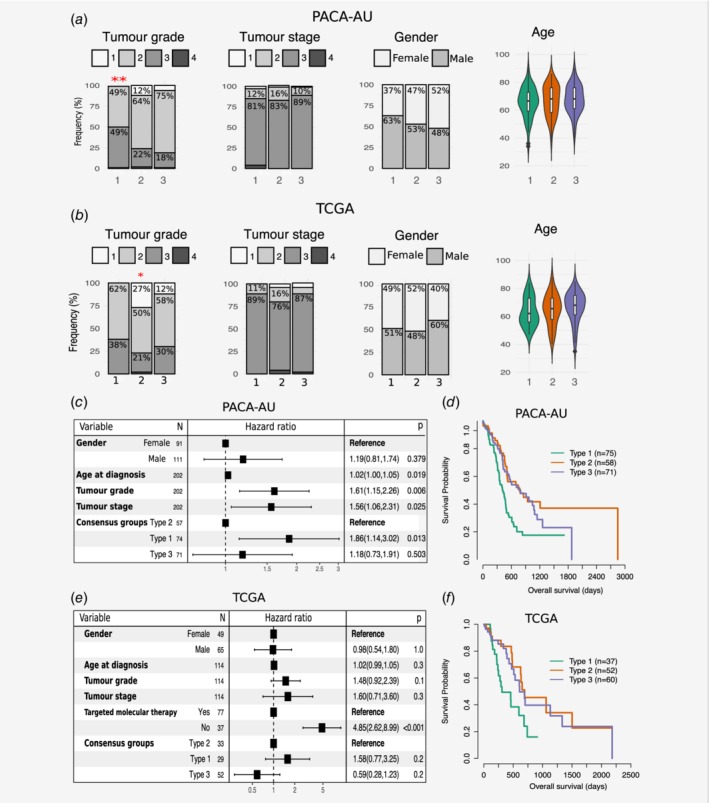
Clinicopathological and prognostic associations of consensus PDAC subtype groups. (a, b) Clinicopathological and prognostic associations of consensus PDAC subtype groups in the PACA‐AU (a) and TCGA (b) cohorts. Distribution of histopathological grade;, TNM system tumour stage at diagnosis, gender, and age at diagnosis, across the three consensus subtypes, represented by the coloured violin plots subtype 1 (green); subtype 2, (orange); subtype 3 (violet). Red asterisks represent significant codes for Chi‐square test p values: p < 0.01 ‘**’; p < 0.05 ‘*’. (c, e) Multivariate Cox proportional hazards regression analysis for the PACA‐AU (c) and TCGA (e) cohorts, with covariates including patient age at diagnosis, tumour stage (TMN system) and tumour grade. Squares represent the hazard ratio (HR) and the horizontal bars extend from the lower limit to the upper limit of the 95% confidence interval of the estimate of the hazard ratio. The plot also shows the number of considered events (N) and p values (p) for the interaction between survival and any covariate. Detailed statistics are in Supporting Information Tables S1 and S2. CI, confidence interval; HR, hazards ratio; p, Wald test p value. (d, f) Prognostic value of subtype 1, 2 and 3 PDAC groups in the PACA‐AU (d) and the TCGA (f) cohorts with Kaplan–Meier overall survival analysis. [Color figure can be viewed at wileyonlinelibrary.com]
To determine whether patients belonging to a certain PDAC cluster have different outcomes, we performed a Cox proportional hazards analysis. We observed differences in prognosis of different PDAC subtypes, with subtype 1 tumours associated to worst overall survival and higher HR (Figs. 2 c–2f; Supporting Information Table S3) in multivariable analyses, after adjustment for several clinicopathological features, including age, gender, tumour stage and grade. This difference was only statistically significant in the PACA‐AU cohort (subtype 1 vs. subtype 2: p = 1.3 × 10−2 HR = 1.9; 95% CI = [1.1–3.0]) and not in the TCGA cohort (subtype 1 vs. subtype 2: p = 0.2; HR = 1.6; CI = [0.8–3.2]). However, we noticed a strong influence (p = 5.35 × 10−7; HR = 4.8; CI = [2.6–9.0]) of ‘targeted therapy’ variable in overall survival (Fig. 2 e) and this might explain our inability to completely reproduce survival results observed in the PACA‐AU cohort. Differential prognosis associated with the stroma type were also evident, as Moffitt classical tumour subtypes with normal stroma (enriched in subtype 2) had the best prognosis, while Moffitt basal‐like tumour subtypes with activated stroma (enriched in subtype 1) had the worse prognosis. Patients in the Sivakumar Notch group (enriched in subtype 2) also displayed the best overall survival rates.8
Immuno‐phenotypes of pancreatic adenocarcinoma
Analysis of the tumour microenvironment has revealed that populations of tumour infiltrating immune cells have significant prognostic value in a variety of solid tumours.3, 38, 39 Analysis in melanoma, breast cancer and colorectal cancer has shown that tumour progression is characterised by distinct immune patterns40, 41, 42, 43 and that assessment of this ‘immunophenotype’ may predict patients prognosis better than beyond that predicted by traditional staging.44
To further explore the composition of different immune infiltrates in the different tumour microenvironments of PDAC cancers, we used single sample gene set enrichment analysis (ssGSEA) to score each tumour based on gene signatures representative of different cell types.45 To test the consistency of the results, we performed the analysis in the PACA‐AU and the TCGA cohorts. We used four published signatures from independent studies30, 31, 32, 34 and focused on 7 immune cell types (NK cells, Neutrophils, Macrophages, Dendritic cells, CD4+ T‐cells, CD8+ T‐cells and B‐cells). We observed that immune signatures are differentially enriched in the tumour microenvironment of PDAC subtypes (Fig. 3 a).
Figure 3.
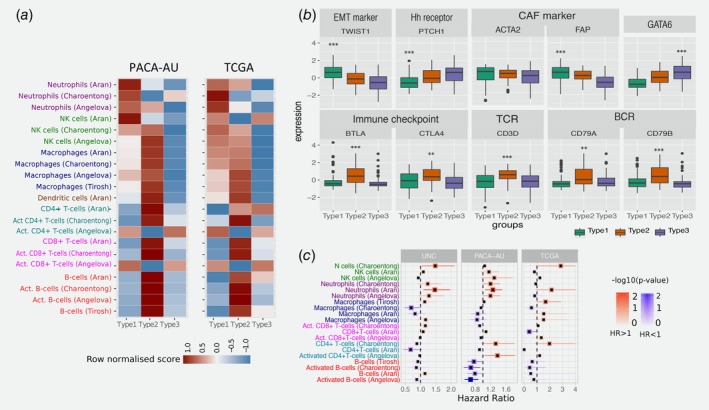
Tumour‐infiltrating subpopulations of immune cells are associated with distinct PDAC subtypes. (a) Heat map of row scaled immune infiltrated scores per immune cell type. Angelova, Charoentong and Tirosh represent single‐sample GSEA scores of signatures for cell types from the corresponding manuscripts. Aran is the inferences produced using xCell algorithm.21 (b) Boxplots showing markers of special interest in PDAC subtypes differentially expressed between groups from the PACA‐AU cohort. Similar trends were observed in the TCGA cohort (Supporting Information Fig. S5). EMT – Epithelial‐mesenchymal transition; Eh – Hedgehog; CAF – Cancer Associated Fibroblasts; TCR – T‐cell receptor; BCR – B‐cell receptor. Asterisks represent significant codes for Welch's t‐test FDR adjusted p values: p < 0.01 ‘**’; p < 0.001 ‘***’. Welch's t‐test was used to test for differential expression of a particular gene in a given subtype compared to all other subtypes. (c) Correlation of tumour infiltrating immune cells with patient overall survival. For three independent cohorts (PACA‐AU, TCGA and UNC), a multivariate Cox proportional hazards regression analysis was performed, with covariates including the enrichment scores of six immune cell types, and when available, patient gender, age at diagnosis, tumour stage and tumour grade. Squares represent the hazard ratio (HR) and the horizontal bars extend from the lower limit to the upper limit of the 95% confidence interval of the estimated of the hazard ratio. The colour scale reflects –log10(p value) and is shown in blue for HR < 1 (good prognosis) and in red for HR > 1 (bad prognosis). [Color figure can be viewed at wileyonlinelibrary.com]
Subtype 1: ‘innate immune’
Our subtype 1 PDAC group showed an enrichment of Natural Killer (NK) cells and neutrophils, and an exclusion of other tumour infiltrated lymphocytes such as activated CD4+ T cells, CD8+ T cells and activated B‐cells. Activation of primary drivers of EMT such as transforming growth factor‐β (TGFβ)7 and the Twist1 gene and de‐regulation of developmental signalling pathways such as Hedgehog (Hh) and Wnt–β‐catenin signalling6, 8 have also been associated to tumours in this category by previous studies. We confirm such findings by showing marked up‐regulation of Twist1 (EMT markers), and down‐regulation of Ptch1 in samples of this subgroup (Fig. 3 b; Welch's t‐test FDR adjusted p = 1.73 × 10−6 and 5.47 × 10−10 for Ptch1 and Twist1, respectively). Ptch1 is a receptor for Hedgehog (Hh) ligands and a tumour suppressor in the Hh pathway. Differential gene expression analysis also showed several de‐regulated pathways related to ‘extracellular matrix organization’, ‘cell adhesion’ and ‘developmental processes’ (Supporting Information Table S3). The Wnt–β‐catenin developmental pathway signalling, which was found to be up‐regulated this PDAC group,8 is known to correlate with T‐cell exclusion across solid tumours.46, 47 This relationship was then validated recently in a clinical setting making the therapeutic strategy of beta‐catenin inhibitors with immunotherapy a potential strategy for T‐cell deficient tumours.48 Bailey et al. also identified gene programmes that included inflammation, hypoxia response and autophagy, and associated to the ‘squamous’ tumours, which are enriched in this group.
Type 1 tumours were associated with the worse survival (Figs. 2 d and 2f). Therefore, the desmoplastic stromal compartment and its interactions with tumour cells have clearly important roles in the poor outcomes for this group of PDAC tumours. This is a very difficult topic to dissect though as there is evidence to demonstrate that the stroma can both restrain and promote the tumour.49
We performed multivariable survival analysis adjusting for clinicopathological features, including age, gender and tumour stage when available, and identified a negative relationship between neutrophil enrichment scores and survival in PDAC (Fig. 3 c). We validate this prediction using three cohorts: PACA‐AU, TCGA and an additional cohort of 102 PDAC tumour samples obtained from the University of North Carolina (UNC). The results of the enrichment of the immune infiltrates showed associations of neutrophils with survival in independent cohorts (Fig. 3 c) highlighting their potential as clinical biomarkers and therapeutic targets.
Subtype 2: ‘T cell dominant’
The second PDAC cluster is characterised by PDAC tumours that displayed enrichment of many tumour infiltrating immune subpopulations related to adaptive immunity including activated CD8+ and CD4+ T‐cells, and B‐cells (Fig. 3 a). Subtype 2 samples exhibited a gene expression profile compatible with increased expression of genes associated with an ‘immune response’, ‘positive regulation of immune system process’ and ‘cell activation’ (Supporting Information Table S4). This immune subtype of PDAC is characterised by marked upregulation of genes known to play roles in immune checkpoint inhibitions (e.g. CTLA4 and BTLA)50; B‐cell receptor and T‐cell receptor genes (e.g. CD3D, CD79A and CD79B) (Fig. 3 b and Supporting Information Fig. S5; Welch's t‐test FDR adjusted p << 0.01 in both cohorts). ‘Normal stroma’, ‘Exocrine‐like’, ‘ADEX’ and ‘Notch’ PDAC tumour samples are also over‐represented in this cluster (Fig. 1 b). Sivakumar et al. also observed enrichment for T cell–related pathways, such as those pertaining to T cell activation, proliferation, and differentiation, adaptive immune response and a significant prevalence of infiltrating CD8+ T cells in tumour samples enriched in this group.
Subtype 2 is the most immunogenic subtype with significantly better survival when compared to samples of subtype 1. It has been shown before that higher levels of CD8+ T cell infiltration correlate with a better survival with Tumeh et al. showing that CD8 T cell infiltration is needed for PD‐1 therapy to work.20, 51, 52 Together these findings indicate that this subtype is potentially amenable to therapy based on immune‐check point inhibitors.
Subtype 3: ‘tumour dominant’
Subtype 3 tumours exhibit lower enrichment scores for immune signatures, which suggests a lack of tumour‐infiltrating lymphocytes in the microenvironment (Fig. 3 a). This group is enriched for PDAC tumours that have been characterised by high expression of adhesion‐associated and epithelial genes5 and genes with distinct roles in the control of cell‐cycle, essential mitotic checkpoint functions, chromosomal stability, and DNA repair.8 We confirmed that GATA6 gene expression is high in the subtype 3 group and low expression in subtype 1 (Fig. 3 b and Supporting Information Fig. S5; Welch's t‐test FDR adjusted p = 1.54 × 10−6 and p = 2.71 × 10−4 for PACA‐AU and TCGA cohorts, respectively), which is expected given the overlap between subtype 3 and classical subgroups defined by Collison et al. and Moffitt et al. Similarly, it has been recently shown that GATA6 expression inhibits the epithelial–mesenchymal transition (EMT) in vitro and cell dissemination in vivo and is associated to suppression of basal‐like (like the one activated in subtype 1) molecular phenotype in PDAC tumours.53 Collison et al. compared PDA cell lines representative of the classical and QM‐PDA subtypes and described that the classical PDA cell lines are enriched in a KRAS‐addiction gene expression signature and more dependent on KRAS than QM‐PDA lines.5 We found a strong enrichment for multiple metabolism signatures (Supporting Information Table S3) in this group indicating prominent metabolic adaptation.
Histopathology analysis of immunophenotypes
We have established clear transcriptional immunophenotypes though this analysis cannot be provided routinely in the clinic. Haematoxylin and Eosin (H&E) staining of the pathology of the specimen can provide a robust and quick assessment of the transcriptional status of the tumour. H&E sections of the cases in the TCGA were provided by TCGA so we were able to assess the relationship of transcriptional immunophenotype to the pathology. We were able to classify each PDAC sample from the TCGA cohort according to their predominant cell type. 149 H&E slides were reviewed in relation to the 149 transcriptomes we analysed from TCGA. We considered three classes: lymphocytes (T cells); neutrophils; or ‘none’ for when no particular immune cell was found. We found that there was no significant difference in the number of neutrophil‐predominant cases in the three immunophenotypes (27% in subtype 1, 18% in subtype 2, and 18% in subtype 3; Fisher test p = 0.15). In the first immunophenotype, there was a larger proportion of cases classified as ‘none’ (21% in subtype 1 vs. 4% and 10% in subtype 2 and subtype 3; Fisher test p = 0.03; Fig. 4 a) and a smaller proportion of samples with lymphocytes as their predominant immune cell type (51% in subtype 1, 77% in subtype 2, 78% in subtype 3; Fisher test p = 0.01; Fig. 4 a). Subtype 1 is the innate subtype so even though on pathology, assessment of macrophages could not be provided, it was clear that there was a smaller proportion of lymphocyte‐predominant samples on histopathology, while in both subtypes 2 and 3, there was an association with lymphocytes.
Figure 4.
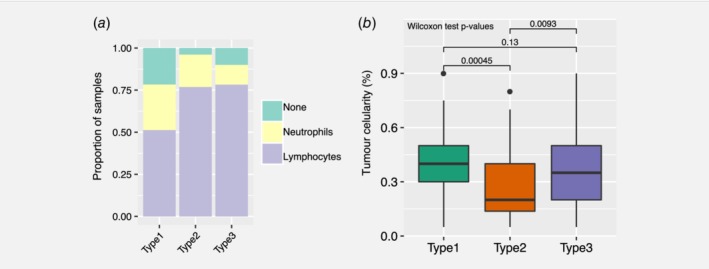
Histopathology in the TCGA cohort. (a) Pathologist assessment of dominant immune cell presence and (b) tumour surface area across the three PDAC consensus subtypes. [Color figure can be viewed at wileyonlinelibrary.com]
While reviewing all 149 available TCGA slides to classify the immunophenotypes; the area percentage of tumour in the slide was estimated concurrently. Furthermore, tumour cellularity was significantly different between subtype 2 and subtype 3; median tumour cellularity was 0.2 (range: 0.05–0.8) and 0.35 (range: 0.05–0.9) in subtype 2 and subtype 3, respectively (Wilcoxon rank sum test p = 9.3 × 10−3, Fig. 4 b). Similarly, the number of cases classified as lymphocyte‐predominant was different between subtype 2 (40 out of 52; 77% in subtype 2) and subtype 3 (47 out of 60; 78% in subtype 3). The ‘type 3′ tumours which we originally called ‘immune exclusion’ has a much higher tumour cellularity compared to ‘type 2′. This could be due to the exact number of immune cells present and one of the limitations of the study is that we do not have a ratio of immune cells to tumour.
Mutational signature analysis
Multiple mutation signatures have been established through pan‐cancer analysis of cancer genomes.54 These have established mutational signatures for pancreatic cancer including a signature associated with failure of double strand break repair by homologous recombination (Signature 3).54 Using a panel of thirty mutational signatures from COSMIC, we established the contribution of each signature to the cohort of pancreatic cancers and sought to identify if any signature that is enriched for the immunophenotypes we have described in this manuscript. We found three major contributory signatures in these pancreatic cancers (Fig. 5 a and Supporting Information Fig. S6) but none of these were enriched for a particular subtype. These were signatures 1, 6 and 15.54 Signature 1 correlates with the ageing process and 6 and 15 are tied to DNA repair. The signature analysis does not help explain the difference in subtypes but does reinforce that certain cases of pancreatic cancer may benefit from treatment with therapeutics against DNA repair.
Figure 5.
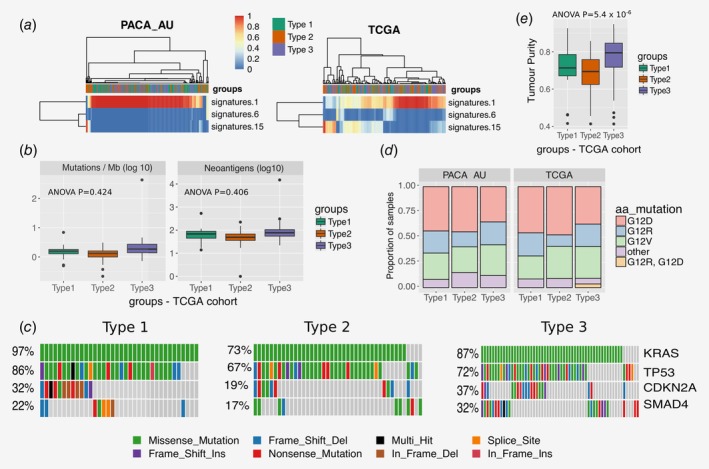
Mutation analysis. (a) Heatmap showing the relative contribution of mutation signatures 1, 6 and 15. (b) Tumour mutational burden and neoantigen load for different tumour subtypes of the TCGA cohort (c) OncoPrint displaying frequency of mutated genes in different PDAC subtypes of the TCGA cohort (d) Distribution of activating KRAS mutations across the three PDAC subtypes in the TCGA and PACA‐AU cohorts (e) Tumour purity (ESTIMATE scores) across subtypes of the TCGA cohort. [Color figure can be viewed at wileyonlinelibrary.com]
Mutational burden
Evidence suggests that the degree of mutagenesis within a tumour may represent a biomarker for response to immunotherapy. It is thought that highly mutated tumours are more likely to harbour neo‐antigens, which make them targets for adaptive immunity. Tumour mutation burden has been shown, in several tumour types, to correlate with patient response to both CTLA‐4 and PD‐1 inhibition.55, 56 We did not find any difference in tumour mutational burden (number of mutations / Mb) and neoantigens between the three subtypes (Fig. 5 b). The most frequently mutated pancreatic cancer genes (KRAS, CDKN2A, SMAD4 and TP53) are equally distributed across the three subtypes (Fig. 5 c), except for KRAS and SMAD4, which are observed in lower frequency in subtype 2 (73 and 17%, respectively) when compared to subtype 1 (KRAS: 97%; Fisher exact test p = 0.003) and subtype 3 (SMAD4: 32%; Fisher exact test p = 0.03). We also analysed the status of the activating KRAS mutation, namely G12D, G12R, G12 V and other (A11T, G12A, G12C, G12 L, G12S, G13C, G13P, GQ60GK, Q61H and Q61R) and verified that KRAS mutations are spread out equally over the three subtypes in both cohorts (Fisher exact test p = 0.76 and p = 0.65 for PACA‐AU and TCGA, respectively; Fig. 5 d). In summary, smaller frequency of mutations of key pancreatic cancer driver mutations are observed in the subtype 2 PDAC subtypes. However these results are inconclusive because it is highly possible that the level of purity across tumour samples affects the interpretation of genomic analyses.57 In fact, in agreement with the tumour cellularity observations in Figure 4 b, we observed striking differences in estimated promotion of tumour cells in the samples between PDAC subtypes (ANOVA p = 5.4 × 10−6), in particular subtype 2 displayed the lowest purity (Fig. 5 e; Supporting Information Fig. S8). This variability confounds the interpretation of genomic analysis results when comparing between PDAC subtypes.
Discussion
In this study, we propose a novel immune clustering of PDAC into three major subtypes that have different molecular and clinical characteristics and may respond differently to selected therapies. We suggest that PDAC subtypes should be considered when stroma and immune modulating therapies are studied in the future. The identified immunophenotypes applies to tumour samples from four independent cohorts, demonstrating the robust nature of this new subtype classification in PDAC. It is important to notice the strong presence of stromal components in subtypes 1 and 2, but also distinct prognosis for each group. However, while the subtype 1 group of tumours is characterised by a reactive desmoplastic stroma and an inflammatory microenvironment with possible epithelial‐to‐mesenchymal transition (EMT) events; the stromal compartment in subtype 2 is enriched in infiltrated CD8+ and CD4+ T‐cells.
Subtype 1 suggests that the functional role of EMT regulators and innate immune cells in immune evasion is complex. The biological link between the inflamed immune subtype and EMT is consistent with the finding that the stroma of subtype 1 tumours is infiltrated not only with innate immune cells, but also markers typically found in activated cancer‐associated‐fibroblasts such as FAP which is significantly over‐expressed in subtype 1 tumours in both cohorts (Welch's t‐test FDR adjusted p PACA‐AU = 1.04 × 10−5 and p TCGA = 6.65 × 10−8; Fig. 3 b) and ACTA2 in the TCGA cohort (Welch's t‐test FDR adjusted p = 7.3 × 10−4; Supporting Information Fig. S5). In addition, it suggests that the worse outcomes seen in the subtype 1 may be partially linked to a pro‐metastatic immune evasive microenvironment. Tumour samples in this group have been characterised by infiltration of desmoplastic stroma (‘activated‐stroma’ such as in Moffitt et al.) and high expression of mesenchyme associated genes (‘basal‐like’ such as in Collison et al.). These results corroborate initial findings by Guerra et al., and others that inflammation increases both EMT and cancer cell invasion21, 22 and that the presence of IL‐6 pro‐inflammatory marker in the serum of patients with pancreatic cancer has been associated with worse survival.23 A better definition of the tumour‐expressed ligands recognised by these myeloid cell subsets and their role in driving tumour progression and anti‐tumour immunity will facilitate more detailed functional analyses and identify possibilities for therapeutic intervention. We had demonstrated on H&Es provided to us that the presence of lymphocytes is the major phenotypic feature, if there are no T cells in the tumour the patient does poorly.
Another challenge that arises is the definition of better preclinical models that recapitulate these subtypes. The interplay between the stroma and the immune components are difficult to model. Previous research suggests that traditional pancreatic cancer cell lines from the Broad Institute Cancer Cell Line Encyclopaedia only recapitulate the two of the three subtypes and do not represent the immunogenic lines5, 8 and PDX models are not able to recapitulate the ‘normal‐like’ subtype.6
Current tumour models such as the KPC mice do not accurately reflect variations in sub‐types of pancreatic cancer and there is generally difficulty at obtaining high quality primary samples. Less then 20% of patients undergo a resection and most of the tumour is infiltrated with a desmoplastic stromal reaction that is composed of collagen, fibroblasts and immune cells. Studies so far have tried to enrich as best as possible the tumour compartment including the ones we have used.
Several lines of evidence suggest differential drug response sensitivity between the different subtypes. Moffitt et al. has shown that patients with ‘basal‐like’ (subtype 1) tumours showed a strong trend toward better response to adjuvant therapy when compared to patients from the ‘classical’ (subtype 3) subtype group6 and that QM‐PDA cell lines were, on average, more sensitive to gemcitabine and less sensitive to erlotinib than the classical cell lines.6 Additionally, it has been shown that patients with GATA6high PDAC tumours have better response to 5‐FU when compared to ‘basal‐like’ GATA6low patients.53 Together these results suggest that KRAS‐directed therapies or therapies targeting growth pathways such as EGFR‐targeted therapy might be best deployed in this subtype 3 classical PDAC subtype when compared to the other PDAC subtypes. The regulatory contribution of the immune system should be assessed more thoroughly in human PDAC cancer to guide new therapeutic interventions tailored to patients with different tumour subtypes.
We think that the pancreatic cancer subtypes identified in our study provide further insights necessary for the understanding of the immunogenicity of PDAC. Being able to determine which PDAC cases are immunogenic or not and what other characteristics they may have, could facilitate development of potential strategies to remove immune suppression. Following on from our current analysis, future work should characterise the immune cell populations in these tumours including their activation status and spatial relationship with the tumour cells. Such investigations required prospective collection of appropriate material from a sufficiently large patients cohort using techniques such as single cell sequencing, CyTOF and multiplexed immunofluorescence imaging with advanced image analysis technology.
Additional work needs to be performed to assess the degree of responsiveness of those infiltrating lymphocytes when the suppression is removed. Our work has already alluded to suppression with macrophages and neutrophils and it will be worth characterising the nature of Tregs in the tumour in subsequent studies as a further immunosuppressive mechanism. An additional issue is that even if we removed the immunosuppression with therapeutics; as the mutational burden of the tumour is low, there is a lower chance a neoantigen will be created that will be recognised by T‐cells. Further strategies will have to be investigated in creating neoantigens such as using radiation therapy or using chemotherapy or to use adoptive T cell strategies to help fight the cancer. Understanding PDAC subtypes could be used improve better patient stratification for clinical drug trial enrichment schemes to better select patients to make detection of a treatment effect more likely. However, more detailed immune characterisation of PDAC tumours is needed before instigating novel therapeutic strategies.
Supporting information
Appendix S1: Supplementary Data.
Figure S1 Clustering of the PACA‐AU cohort according to signatures from Collisson et al. 2011, and Moffitt et al. 2015 (A) Consensus‐clustered heat map of normalised data from PACA‐AU using the gene sets from Collisson et al. 2011 classified as exocrine‐like (green), classical (yellow) and quasimesenchymal (blue) (B) Consensus‐clustered heat map of normalised data from PACA‐AU using the gene sets from the ‘stroma signature’ from Moffitt et al. 2015. (C) Consensus‐clustered heat map of normalised data from PACA‐AU using the gene sets from the ‘tumour signature’ from Moffitt et al. 2015 (E) Consensus‐clustered heat map of normalised data from TCGA using the gene sets from the ‘stroma signature’ from Moffitt et al. 2015.
Figure S2 Partition around medoids (PAM) consensus clustering of PDAC subtypes for the PACA‐AU cohort. (A) Consensus matrix obtained for K = 2 to K = 7 solutions. The consensus matrices display the robustness of sample classification by representing the proportion of times in which 2 samples are clustered together across the resampling iterations (×1,000). Groups of tumours that frequently cluster with one another are represented by darker shades of blue. (B) CDF plot depicting the cumulative distribution functions of the consensus matrix for each k (indicated by colours), estimated by a histogram of 100 bins. (C) Plot showing the relative change in area (Δ) under the CDF plot. The point at which the area under the curve ceases to show marked increases with additional k indicates the ideal number of clusters. Comparing k and k − 1, the optimal cluster number is 3 at which there is no appreciably change in Δ with increasing k.
Figure S3 Partition around medoids (PAM) consensus clustering of PDAC subtypes for the TCGA cohort. (A) Consensus matrix obtained for K = 2 to K = 7 solutions. The consensus matrices display the robustness of sample classification by representing the proportion of times in which 2 samples are clustered together across the resampling iterations (×1,000). Groups of tumours that frequently cluster with one another are represented by darker shades of blue. (B) CDF plot depicting the cumulative distribution functions of the consensus matrix for each k (indicated by colours), estimated by a histogram of 100 bins. (C) Plot showing the relative change in area (Δ) under the CDF plot. The point at which the area under the curve ceases to show marked increases with additional k indicates the ideal number of clusters. Comparing k and k − 1, the optimal cluster number is 3 at which there is no appreciably change in Δ with increasing k.
Figure S4 PDAC subtype classifications across the three consensus groups for the five classification systems. The significance of the overlaps between labels was tested using the hypergeometric distribution. The numbers on the right of the bars (and x‐axis) show the number of overlaps between labels of each classification scheme (y‐axis) and the consensus subtypes (Type 1, Type 2 and Type 3). The bars were ordered and coloured according to the FDR adjusted hypergeometric p‐values.
Figure S5 Boxplots showing markers of special interest in PDAC subtypes differentially expressed between groups from the TCGA cohort. EMT – Epithelial‐mesenchymal transition; Eh – Hedgehog; CAF – Cancer Associated Fibroblasts; TCR – T‐cell receptor; BCR – B‐cell receptor. Asterisks represent significant codes for Welch's T test FDR adjusted p‐values: p < 0.01 ‘**’; p < 0.001 ‘***’. Welch's T test was used to test for differential expression of a particular gene in a given subtype compared to all other subtypes.
Figure S6 Contribution of mutational signatures from Alexandrov et al. Signature‐1. Signature_1 is a result of elevated rate of spontaneous deamination of 5‐methyl‐cytosine, resulting in C > T transitions and is a most common process in PDAC samples. Hierarchical clustering by the proportion of contribution to each signature revealed 2 major groups: a group characterised by signature 1 (which is attributed to mutational processes accumulated over cell divisions) and a group dominated by signatures 6 and 15 (which are attributed to defects in DNA mismatch repair). Colours on the top of the heatmap show the overlap with the RNA subtypes classified according to Sivakumar, Bailey, Moffitt and Collison classification schemes. There is no clear overlap with the RNA subtypes.
Figure S7 Summary of mutations identified in the TCGA cohort. The maftools summary plots display the number of variants in each sample as a stacked barplot and variant types as a boxplot summarised by Variant_Classification field of a MAF file.
Figure S8 Boxplots from the ESTIMATE analysis showing the variation in the stromal, immune content, and tumour purity between the three PDAC consensus subtypes for both the PACA‐AU and TCGA cohorts.
Supplementary Table 1 Clinicopathological associations of PDAC cluster subtypes in the PACA‐AU and the TCGA cohorts. For categorical variables, the Chi‐square test was used to compare counts in each group with the total counts. For continuous variables the t‐test was used to compare the mean in each group against all patients in the corresponding cohort.
Supplementary Table 2 Prognostic value of type 1, 2 and 3 PDAC groups in the PACA‐AU and TCGA cohorts obtained from multivariate Cox proportional hazards regression analysis. Tables show detailed statistics obtained from the models. Significant codes: 0 ‘***’ 0.001 ‘**’ 0.01 ‘*’ 0.05 ‘.’ 0.1 “1.
Supplementary Table 3 Top enriched Gene Ontology (GO) Process terms in PDAC consensus subtypes of PACA‐AU cohort obtained by ranked gene list analysis using the Gene Ontology enRIchment anaLysis and visualisation (GOrilla) tool. The table shows the GO terms enriched in the list of differentially expressed genes obtained by comparing the three consensus PDAC sutbypes. ‘P’ is the enrichment p‐value computed according to the mHG. ‘FDR’ is the correction of the above p‐value for multiple testing using the Benjamini and Hochberg (1995) method. Enrichment (N, B, n, b) is defined as follows:
N ‐ is the total number of genes; B ‐ is the total number of genes associated with a specific GO term; n ‐ is the number of genes in the top of the user's input list or in the target set when appropriate; b ‐ is the number of genes in the intersection. Enrichment = (b/n) / (B/N).
Acknowledgements
This study was supported by the NIHR Oxford Biomedical Research Centre. CY is supported by a UK Medical Research Council Research Grant (MR/P02646X/1). MLD is funded by Wellcome Trust grant 100262Z/12/Z. SS is funded by a NIHR Academic Clinical Lecturership.
Ines de Santiago's current address is: SBG Discovery UK Ltd., 101 Euston Road, London NW1 2RA, United Kingdom
Contributor Information
Ines de Santiago, Email: ines.desantiago@cruk.cam.ac.uk.
Shivan Sivakumar, Email: shivan.sivakumar@oncology.ox.ac.uk.
References
- 1. Ilic M, Ilic I. Epidemiology of pancreatic cancer. World J Gastroenterol 2016;22:9694–705. 10.3748/wjg.v22.i44.9694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Guinney J, Dienstmann R, Wang X, et al. The consensus molecular subtypes of colorectal cancer. Nat Med 2015;21:1350–6. 10.1038/nm.3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yoshihara K, Shahmoradgoli M, Martinez E, et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun 2013;4:2612 10.1038/ncomms3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Newman AM, Liu CL, Green MR, et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods 2015;12:453–7. 10.1038/nmeth.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Collisson EA, Sadanandam A, Olson P, et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat Med 2011;17:500–3. 10.1038/nm.2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Moffitt RA, Marayati R, Flate EL, et al. Virtual microdissection identifies distinct tumor‐ and stroma‐specific subtypes of pancreatic ductal adenocarcinoma. Nat Genet 2015;47:1168–78. 10.1038/ng.3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bailey P, Chang DK, Nones K, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016;531:47–52. 10.1038/nature16965. [DOI] [PubMed] [Google Scholar]
- 8. Sivakumar S, de Santiago I, Chlon L, et al. Master regulators of oncogenic KRAS response in pancreatic cancer: an integrative Network biology analysis. PLoS Med 2017;14:e1002223 10.1371/journal.pmed.1002223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Birnbaum DJ, Finetti P, Birnbaum D, et al. Validation and comparison of the molecular classifications of pancreatic carcinomas. Mol Cancer 2017;16:168 10.1186/s12943-017-0739-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mao Y, Shen J, Lu Y, et al. RNA sequencing analyses reveal novel differentially expressed genes and pathways in pancreatic cancer. Oncotarget 2017;8:42537–47. 10.18632/oncotarget.16451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kim S, Kang M, Lee S, et al. Identifying molecular subtypes related to clinicopathologic factors in pancreatic cancer. Biomed Eng Online 2014;13(Suppl 2):S5 10.1186/1475-925X-13-S2-S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Janky R, Binda MM, Allemeersch J, et al. Prognostic relevance of molecular subtypes and master regulators in pancreatic ductal adenocarcinoma. BMC Cancer 2016;16:632 10.1186/s12885-016-2540-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Haider S, Wang J, Nagano A, et al. A multi‐gene signature predicts outcome in patients with pancreatic ductal adenocarcinoma. Genome Med 2014;6:105 10.1186/s13073-014-0105-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kirby MK, Ramaker RC, Gertz J, et al. RNA sequencing of pancreatic adenocarcinoma tumors yields novel expression patterns associated with long‐term survival and reveals a role for ANGPTL4. Mol Oncol 2016;10:1169–82. 10.1016/j.molonc.2016.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Stratford JK, Bentrem DJ, Anderson JM, et al. A six‐gene signature predicts survival of patients with localized pancreatic ductal adenocarcinoma. PLoS Med 2010;7:e1000307. 10.1371/journal.pmed.1000307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Donahue TR, Tran LM, Hill R, et al. Integrative survival‐based molecular profiling of human pancreatic cancer. Clin Cancer Res 2012;18:1352–63. 10.1158/1078-0432.CCR-11-1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Connor AA, Denroche RE, Jang GH, et al. Association of distinct mutational signatures with correlates of increased immune activity in pancreatic ductal adenocarcinoma. JAMA Oncol 2017;3:774–83. 10.1001/jamaoncol.2016.3916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hutcheson J, Balaji U, Porembka MR, et al. Immunologic and metabolic features of pancreatic ductal adenocarcinoma define prognostic subtypes of disease. Clin Cancer Res 2016;22:3606–17. 10.1158/1078-0432.CCR-15-1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Knudsen ES, Vail P, Balaji U, et al. Stratification of pancreatic ductal adenocarcinoma: combinatorial genetic, stromal, and immunologic markers. Clin Cancer Res 2017;23:4429–40. 10.1158/1078-0432.CCR-17-0162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ino Y, Yamazaki‐Itoh R, Shimada K, et al. Immune cell infiltration as an indicator of the immune microenvironment of pancreatic cancer. Br J Cancer 2013;108:914–23. 10.1038/bjc.2013.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sinha P, Clements VK, Ostrand‐Rosenberg S. Interleukin‐13‐regulated M2 macrophages in combination with myeloid suppressor cells block immune surveillance against metastasis. Cancer Res 2005;65:11743–51. 10.1158/0008-5472.CAN-05-0045. [DOI] [PubMed] [Google Scholar]
- 22. Chen ML, Pittet MJ, Gorelik L, et al. Regulatory T cells suppress tumor‐specific CD8 T cell cytotoxicity through TGF‐beta signals in vivo. Proc Natl Acad Sci U S A 2005;102:419–24. 10.1073/pnas.0408197102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Royal RE, Levy C, Turner K, et al. Phase 2 trial of single agent Ipilimumab (anti‐CTLA‐4) for locally advanced or metastatic pancreatic adenocarcinoma. J Immunother 2010;33:828–33. 10.1097/CJI.0b013e3181eec14c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Brahmer JR, Tykodi SS, Chow LQ, et al. Safety and activity of anti‐PD‐L1 antibody in patients with advanced cancer. N Engl J Med 2012;366:2455–65. 10.1056/NEJMoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 2007;8:118–27. 10.1093/biostatistics/kxj037. [DOI] [PubMed] [Google Scholar]
- 26. Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol 2010;11:R106 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wilkerson MD, Hayes DN. ConsensusClusterPlus: a class discovery tool with confidence assessments and item tracking. Bioinformatics 2010;26:1572–3. 10.1093/bioinformatics/btq170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Network CGAR, Weinstein JN, Colisson EA, et al. The cancer genome atlas pan‐cancer analysis project. Nat Genet 2013;45:1113–20. 10.1038/ng.2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Monti S, Tamayo P, Mesirov J, et al. Consensus clustering: a resampling‐based method for class discovery and visualization of gene expression microarray data. Mach Learn 2003;52:91–118. 10.1023/A:1023949509487. [DOI] [Google Scholar]
- 30. Angelova M, Charoentong P, Hackl H, et al. Characterization of the immunophenotypes and antigenomes of colorectal cancers reveals distinct tumor escape mechanisms and novel targets for immunotherapy. Genome Biol 2015;16:64 10.1186/s13059-015-0620-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Charoentong P, Finotello F, Angelova M, et al. Pan‐cancer immunogenomic analyses reveal genotype‐immunophenotype relationships and predictors of response to checkpoint blockade. Cell Rep 2017;18:248–62. 10.1016/j.celrep.2016.12.019. [DOI] [PubMed] [Google Scholar]
- 32. Tirosh I, Izar B, Prakadan SM, et al. Dissecting the multicellular ecosystem of metastatic melanoma by single‐cell RNA‐seq. Science 2016;352:189–96. 10.1126/science.aad0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hanzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA‐seq data. BMC Bioinf 2013;14:7 10.1186/1471-2105-14-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Aran D, Hu Z, and Butte AJ. xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol 2017;18:220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rosenthal R, McGranahan N, Herrero J, et al. DeconstructSigs: delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol 2016;17:31 10.1186/s13059-016-0893-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mayakonda A, Koeffler HP. Maftools: Efficient analysis, visualization and summarization of MAF files from large‐scale cohort based cancer studies. bioRxiv 2016. 10.1101/052662. [DOI] [Google Scholar]
- 37. Team RC . R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing, 2014. [Google Scholar]
- 38. Fridman WH, Pages F, Sautes‐Fridman C, et al. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer 2012;12:298–306. 10.1038/nrc3245. [DOI] [PubMed] [Google Scholar]
- 39. Galon J, Mlecnik B, Marliot F, et al. Validation of the Immunoscore (IM) as a prognostic marker in stage I/II/III colon cancer: results of a worldwide consortium‐based analysis of 1,336 patients. J Clin Oncol 2016;34:3500. [Google Scholar]
- 40. Bindea G, Mlecnik B, Tosolini M, et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity 2013;39:782–95. 10.1016/j.immuni.2013.10.003. [DOI] [PubMed] [Google Scholar]
- 41. Natrajan R, Sailem H, Mardakheh FK, et al. Microenvironmental heterogeneity parallels breast cancer progression: a histology‐genomic integration analysis. PLoS Med 2016;13:e1001961 10.1371/journal.pmed.1001961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ali HR, Chlon L, Pharoah PD, et al. Patterns of immune infiltration in breast cancer and their clinical implications: a gene‐expression‐based retrospective study. PLoS Med 2016;13:e1002194. 10.1371/journal.pmed.1002194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rosenberg SA, Packard BS, Aebersold PM, et al. Use of tumor‐infiltrating lymphocytes and interleukin‐2 in the immunotherapy of patients with metastatic melanoma. A preliminary report. N Engl J Med 1988;319:1676–80. 10.1056/NEJM198812223192527. [DOI] [PubMed] [Google Scholar]
- 44. Mlecnik B, Tosolini M, Kirilovsky A, et al. Histopathologic‐based prognostic factors of colorectal cancers are associated with the state of the local immune reaction. J Clin Oncol 2011;29:610–8. 10.1200/JCO.2010.30.5425. [DOI] [PubMed] [Google Scholar]
- 45. Barbie DA, Tamayo P, Boehm JS, et al. Systematic RNA interference reveals that oncogenic KRAS‐driven cancers require TBK1. Nature 2009;462:108–12. 10.1038/nature08460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Spranger S, Bao R, Gajewski TF. Melanoma‐intrinsic beta‐catenin signalling prevents anti‐tumour immunity. Nature 2015;523:231–5. 10.1038/nature14404. [DOI] [PubMed] [Google Scholar]
- 47. Ding Y, Shen S, Lino AC, et al. Beta‐catenin stabilization extends regulatory T cell survival and induces anergy in nonregulatory T cells. Nat Med 2008;14:162–9. 10.1038/nm1707. [DOI] [PubMed] [Google Scholar]
- 48. Luke JJ, Bao R, Spranger S, et al. Correlation of WNT/β‐catenin pathway activation with immune exclusion across most human cancers. J Clin Oncol 2016;34:3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Vennin C, Murphy KJ, Morton JP, et al. Reshaping the tumor Stroma for treatment of pancreatic cancer. Gastroenterology 2018;154:820–38. 10.1053/j.gastro.2017.11.280. [DOI] [PubMed] [Google Scholar]
- 50. Turnis ME, Andrews LP, Vignali DA. Inhibitory receptors as targets for cancer immunotherapy. Eur J Immunol 2015;45:1892–905. 10.1002/eji.201344413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ene‐Obong A, Clear AJ, Watt J, et al. Activated pancreatic stellate cells sequester CD8+ T cells to reduce their infiltration of the juxtatumoral compartment of pancreatic ductal adenocarcinoma. Gastroenterology 2013;145:1121–32. 10.1053/j.gastro.2013.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tumeh PC, Harview CL, Yearley JH, et al. PD‐1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014;515:568–71. 10.1038/nature13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Martinelli P, Carrillo‐de Santa Pau E, Cox T, et al. GATA6 regulates EMT and tumour dissemination, and is a marker of response to adjuvant chemotherapy in pancreatic cancer. Gut 2017;66:1665–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Alexandrov LB, Nik‐Zainal S, Wedge DC, et al. Signatures of mutational processes in human cancer. Nature 2013;500:415–21. 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Rizvi NA, Hellmann MD, Snyder A, et al. Cancer immunology. Mutational landscape determines sensitivity to PD‐1 blockade in non‐small cell lung cancer. Science 2015;348:124–8. 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Snyder A, Makarov V, Merghoub T, et al. Genetic basis for clinical response to CTLA‐4 blockade in melanoma. N Engl J Med 2014;371:2189–99. 10.1056/NEJMoa1406498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Aran D, Sirota M, Butte AJ. Systematic pan‐cancer analysis of tumour purity. Nat Commun 2015;6:8971 10.1038/ncomms9971. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supplementary Data.
Figure S1 Clustering of the PACA‐AU cohort according to signatures from Collisson et al. 2011, and Moffitt et al. 2015 (A) Consensus‐clustered heat map of normalised data from PACA‐AU using the gene sets from Collisson et al. 2011 classified as exocrine‐like (green), classical (yellow) and quasimesenchymal (blue) (B) Consensus‐clustered heat map of normalised data from PACA‐AU using the gene sets from the ‘stroma signature’ from Moffitt et al. 2015. (C) Consensus‐clustered heat map of normalised data from PACA‐AU using the gene sets from the ‘tumour signature’ from Moffitt et al. 2015 (E) Consensus‐clustered heat map of normalised data from TCGA using the gene sets from the ‘stroma signature’ from Moffitt et al. 2015.
Figure S2 Partition around medoids (PAM) consensus clustering of PDAC subtypes for the PACA‐AU cohort. (A) Consensus matrix obtained for K = 2 to K = 7 solutions. The consensus matrices display the robustness of sample classification by representing the proportion of times in which 2 samples are clustered together across the resampling iterations (×1,000). Groups of tumours that frequently cluster with one another are represented by darker shades of blue. (B) CDF plot depicting the cumulative distribution functions of the consensus matrix for each k (indicated by colours), estimated by a histogram of 100 bins. (C) Plot showing the relative change in area (Δ) under the CDF plot. The point at which the area under the curve ceases to show marked increases with additional k indicates the ideal number of clusters. Comparing k and k − 1, the optimal cluster number is 3 at which there is no appreciably change in Δ with increasing k.
Figure S3 Partition around medoids (PAM) consensus clustering of PDAC subtypes for the TCGA cohort. (A) Consensus matrix obtained for K = 2 to K = 7 solutions. The consensus matrices display the robustness of sample classification by representing the proportion of times in which 2 samples are clustered together across the resampling iterations (×1,000). Groups of tumours that frequently cluster with one another are represented by darker shades of blue. (B) CDF plot depicting the cumulative distribution functions of the consensus matrix for each k (indicated by colours), estimated by a histogram of 100 bins. (C) Plot showing the relative change in area (Δ) under the CDF plot. The point at which the area under the curve ceases to show marked increases with additional k indicates the ideal number of clusters. Comparing k and k − 1, the optimal cluster number is 3 at which there is no appreciably change in Δ with increasing k.
Figure S4 PDAC subtype classifications across the three consensus groups for the five classification systems. The significance of the overlaps between labels was tested using the hypergeometric distribution. The numbers on the right of the bars (and x‐axis) show the number of overlaps between labels of each classification scheme (y‐axis) and the consensus subtypes (Type 1, Type 2 and Type 3). The bars were ordered and coloured according to the FDR adjusted hypergeometric p‐values.
Figure S5 Boxplots showing markers of special interest in PDAC subtypes differentially expressed between groups from the TCGA cohort. EMT – Epithelial‐mesenchymal transition; Eh – Hedgehog; CAF – Cancer Associated Fibroblasts; TCR – T‐cell receptor; BCR – B‐cell receptor. Asterisks represent significant codes for Welch's T test FDR adjusted p‐values: p < 0.01 ‘**’; p < 0.001 ‘***’. Welch's T test was used to test for differential expression of a particular gene in a given subtype compared to all other subtypes.
Figure S6 Contribution of mutational signatures from Alexandrov et al. Signature‐1. Signature_1 is a result of elevated rate of spontaneous deamination of 5‐methyl‐cytosine, resulting in C > T transitions and is a most common process in PDAC samples. Hierarchical clustering by the proportion of contribution to each signature revealed 2 major groups: a group characterised by signature 1 (which is attributed to mutational processes accumulated over cell divisions) and a group dominated by signatures 6 and 15 (which are attributed to defects in DNA mismatch repair). Colours on the top of the heatmap show the overlap with the RNA subtypes classified according to Sivakumar, Bailey, Moffitt and Collison classification schemes. There is no clear overlap with the RNA subtypes.
Figure S7 Summary of mutations identified in the TCGA cohort. The maftools summary plots display the number of variants in each sample as a stacked barplot and variant types as a boxplot summarised by Variant_Classification field of a MAF file.
Figure S8 Boxplots from the ESTIMATE analysis showing the variation in the stromal, immune content, and tumour purity between the three PDAC consensus subtypes for both the PACA‐AU and TCGA cohorts.
Supplementary Table 1 Clinicopathological associations of PDAC cluster subtypes in the PACA‐AU and the TCGA cohorts. For categorical variables, the Chi‐square test was used to compare counts in each group with the total counts. For continuous variables the t‐test was used to compare the mean in each group against all patients in the corresponding cohort.
Supplementary Table 2 Prognostic value of type 1, 2 and 3 PDAC groups in the PACA‐AU and TCGA cohorts obtained from multivariate Cox proportional hazards regression analysis. Tables show detailed statistics obtained from the models. Significant codes: 0 ‘***’ 0.001 ‘**’ 0.01 ‘*’ 0.05 ‘.’ 0.1 “1.
Supplementary Table 3 Top enriched Gene Ontology (GO) Process terms in PDAC consensus subtypes of PACA‐AU cohort obtained by ranked gene list analysis using the Gene Ontology enRIchment anaLysis and visualisation (GOrilla) tool. The table shows the GO terms enriched in the list of differentially expressed genes obtained by comparing the three consensus PDAC sutbypes. ‘P’ is the enrichment p‐value computed according to the mHG. ‘FDR’ is the correction of the above p‐value for multiple testing using the Benjamini and Hochberg (1995) method. Enrichment (N, B, n, b) is defined as follows:
N ‐ is the total number of genes; B ‐ is the total number of genes associated with a specific GO term; n ‐ is the number of genes in the top of the user's input list or in the target set when appropriate; b ‐ is the number of genes in the intersection. Enrichment = (b/n) / (B/N).