

Abstract
The aim of this study was to investigate how variability in multiple genes related to pharmacokinetics affects fluvastatin exposure. We determined fluvastatin enantiomer pharmacokinetics and sequenced 379 pharmacokinetic genes in 200 healthy volunteers. CYP2C9*3 associated with significantly increased area under the plasma concentration‐time curve (AUC) of both 3R,5S‐fluvastatin and 3S,5R‐fluvastatin (by 67% and 94% per variant allele copy, P = 3.77 × 10−9 and P = 3.19 × 10−12). In contrast, SLCO1B1 c.521T>C associated with increased AUC of active 3R,5S‐fluvastatin only (by 34% per variant allele copy; P = 8.15 × 10−8). A candidate gene analysis suggested that CYP2C9*2 also affects the AUC of both fluvastatin enantiomers and that SLCO2B1 single‐nucleotide variations may affect the AUC of 3S,5R‐fluvastatin. Thus, SLCO transporters have enantiospecific effects on fluvastatin pharmacokinetics in humans. Genotyping of both CYP2C9 and SLCO1B1 may be useful in predicting fluvastatin efficacy and myotoxicity.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑High interindividual variability exists in the pharmacokinetics of fluvastatin. CYP2C9*3 has previously shown to increase the area under the plasma concentration‐time curve (AUC) of fluvastatin enantiomers, but comprehensive studies evaluating the effects of variability in multiple pharmacokinetic genes on fluvastatin exposure have not been conducted.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑This study investigated how genetic variants in pharmacokinetic genes affect the exposure to fluvastatin enantiomers.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑This study shows that CYP2C9*3 and CYP2C9*2 affect the pharmacokinetics of both fluvastatin enantiomers. SLCO1B1 c.521T>C has an enantiospecific effect on active 3R,5S‐fluvastatin AUC. Furthermore, the results suggest that SLCO2B1 missense variants may affect 3S,5R‐fluvastatin exposure. Based on the results, genotype scores were generated to predict how combinations of CYP2C9 and transporter variants affect fluvastatin enantiomer plasma exposures.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
☑This knowledge might aid in predicting the risk of fluvastatin adverse reactions and thus in individualizing treatment with statins.
Fluvastatin is a 3‐hydroxy‐3‐methylglutaryl‐coenzyme A reductase inhibitor, which is used in the treatment of hypercholesterolemia. It is a racemic mixture of two enantiomers, of which 3R,5S‐fluvastatin is 30 times more active than 3S,5R‐fluvastatin.1 Fluvastatin is extensively metabolized via cytochrome P450 (CYP) 2C9.2, 3 In vitro also CYP3A4, CYP2C8, and possibly CYP2D6 and CYP1A1 contribute to fluvastatin metabolism.2 In addition, fluvastatin is a substrate of several drug transporters, including organic anion transporting polypeptides (OATP) 1B1, 1B3, 2B1, breast cancer resistance protein, multidrug resistance‐associated protein 2, and sodium‐dependent taurocholate cotransporting polypeptide.4, 5, 6, 7, 8
High interindividual variability exists in the pharmacokinetics of fluvastatin. The decreased‐function CYP2C9*3 (c.1075A>C, p.Ile359Leu, rs1057910) allele9, 10 has been associated with markedly increased plasma concentrations of both fluvastatin enantiomers.11 Furthermore, the decreased‐function ABCG2 c.421C>A (p.Gln141Lys, rs2231142) variant12, 13, 14, 15 has been associated with markedly increased plasma concentrations of racemic fluvastatin.14 On the other hand, the decreased‐function SLCO1B1 c.521T>C (p.Val174Ala, rs4149056) variant16, 17, 18, 19, 20, 21, 22, 23 and the ABCB1 c.1236T‐c.2677T‐c.3435T haplotype have not affected fluvastatin pharmacokinetics.21, 24 Comprehensive studies evaluating the effects of variants in multiple genes on fluvastatin pharmacokinetics have not been conducted previously. Therefore, the aim of this study was to investigate how variability in pharmacokinetic genes affects fluvastatin exposure. We determined the pharmacokinetics of 3R,5S‐fluvastatin and 3S,5R‐fluvastatin after a 40 mg oral dose of racemic fluvastatin in 200 healthy volunteers and fully sequenced 379 pharmacokinetic genes using massive parallel sequencing.
Results
Fluvastatin pharmacogenomics
Among the 200 healthy volunteers, the areas under the plasma concentration‐time curve from 0 hour to infinity (AUC0–∞) of 3R,5S‐fluvastatin and 3S,5R‐fluvastatin varied 16‐fold and 19‐fold, respectively (Table S1). A single‐nucleotide variation (SNV), rs77760615, located upstream of CYP2C9 showed the strongest association with the pharmacokinetics of both fluvastatin enantiomers (Table 1, Figure 1). The AUC0–∞ of 3R,5S‐fluvastatin was 70% (P = 2.16 × 10−10) and that of 3S,5R‐fluvastatin was 89% (P = 2.93 × 10−11) larger per copy of the variant allele. After adjusting for this variant, an intronic SLCO1B1 variant, rs58310495, associated with a 34% larger AUC0–∞ of 3R,5S‐fluvastatin per copy of the variant allele (P = 3.07 × 10−10). No other variants remained statistically significantly associated with the AUC0–∞ of 3S,5R‐fluvastatin. The investigated genetic variants had no significant effect on the peak plasma concentrations (Cmax) or the elimination half‐lives (t ½) of fluvastatin enantiomers.
Table 1.
Results of the stepwise forward linear regression analysis of the effects of 46,064 SNVs in 379 genes on fluvastatin pharmacokinetics
| Pharmacokinetic variable | dbSNP ID | Gene | Location | Nucleotide change | MAF | Effecta | P value | |
|---|---|---|---|---|---|---|---|---|
| Average (%) | 90% CI | |||||||
| 3R,5S‐fluvastatin | ||||||||
| AUC0–∞ | 1. rs77760615 | CYP2C9 | Upstream | c.‐5813A>G | 0.072 | 69.7 | 49.0, 93.2 | 2.16 × 10−10 |
| 2. rs58310495 | SLCO1B1 | Intron 10/14 | c.1332‐1091C>T | 0.29 | 34.4 | 24.9, 44.6 | 3.07 × 10−10 | |
| Cmax | — | — | — | — | — | — | — | — |
| t 1/2 | — | — | — | — | — | — | — | — |
| 3S,5R‐fluvastatin | ||||||||
| AUC0–∞ | rs77760615 | CYP2C9 | Upstream | c.‐5813A>G | 0.072 | 88.9 | 62.8, 119.1 | 2.93 × 10−11 |
| Cmax | — | — | — | — | — | — | — | — |
| t 1/2 | — | — | — | — | — | — | — | — |
| Total fluvastatin AUC0–∞ | rs77760615 | CYP2C9 | Upstream | c.‐5813A>G | 0.072 | 81.6 | 57.4, 109.6 | 8.50 × 10−11 |
| 3R,5S/3S,5R AUC0–∞ ratio | 1. rs4149056 | SLCO1B1 | Exon 6/15 | c.521T>C | 0.22 | 23.3 | 19.6, 27.2 | 1.30 × 10−22 |
| 2. rs12367888 | SLCO1B1 | Intron 7/14 | c.728‐2859G>A | 0.16 | −11.6 | −14.7, −8.3 | 5.24 × 10−8 | |
AUC0–∞, area under the plasma concentration‐time curve from 0 hour to infinity; CI, confidence interval; Cmax, peak plasma concentration; dbSNP, National Center for Biotechnology Information Short Genetic Variations database; MAF, minor allele frequency; SNV, single‐nucleotide variation; t ½, elimination half‐life; —, not applicable.
Per variant allele copy.
Figure 1.
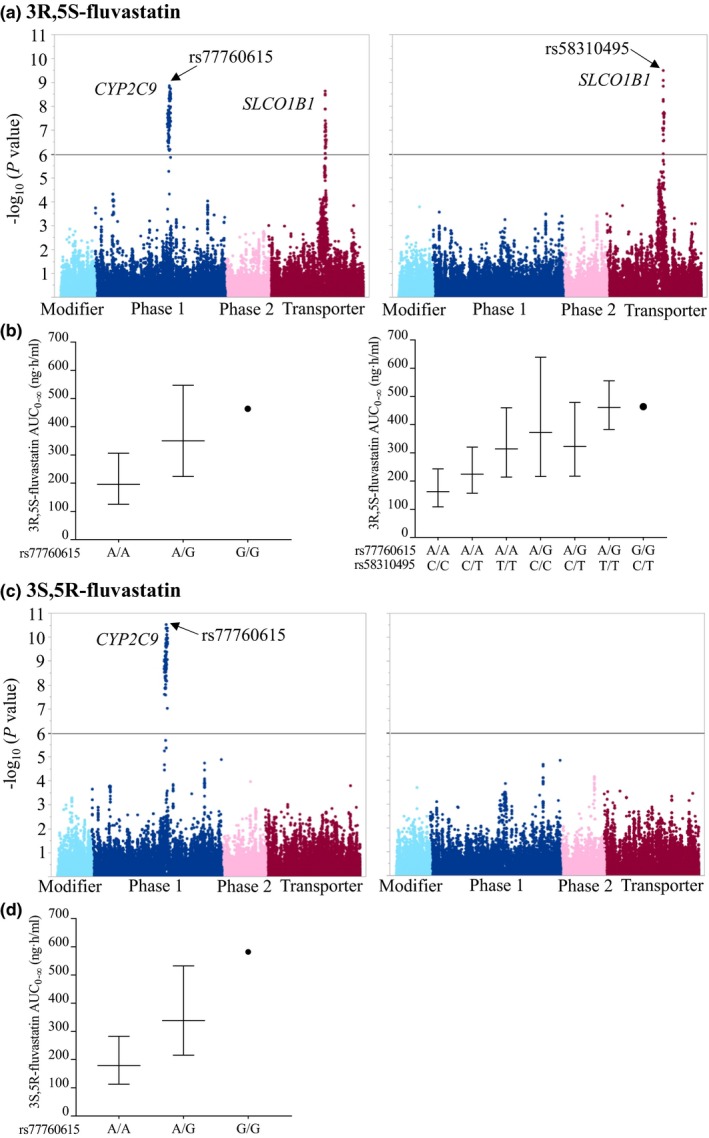
The associations of 46,064 SNVs in 379 pharmacokinetic genes with 3R,5S‐fluvastatin (a) and 3S,5R‐fluvastatin (c) AUC 0–∞, adjusting for BSA (left panel), and BSA and the CYP2C9 rs77760615 SNV (right panel). The y‐axes in (a) and (c) describe the negative logarithm of the P value for each SNV, and the horizontal lines indicate the Bonferroni‐corrected significance level of 1.09 × 10−6. The x‐axes show individual SNVs grouped by protein function. The geometric mean ± geometric standard deviation BSA‐adjusted AUC 0–∞ values grouped by the top associated SNVs are illustrated in (b) and (d). AUC0–∞, area under the plasma concentration‐time curve from 0 hour to infinity; BSA, body surface area; SNV, single‐nucleotide variation. [Colour figure can be viewed at wileyonlinelibrary.com]
In agreement with these results, also the total 3R,5S+3S,5R‐fluvastatin AUC0–∞ associated significantly with the CYP2C9 upstream variant rs77760615 (Table 1). The AUC0–∞ was 82% larger per copy of the variant allele (P = 8.50 × 10−11). After adjusting for this variant, no other variants remained statistically significantly associated with the total fluvastatin AUC0–∞. The investigated variants had no significant effect on the Cmax or t ½ of total fluvastatin.
To further identify the genetic factors that differently affect the two enantiomers, we investigated the associations of the genetic variants with 3R,5S/3S,5R‐fluvastatin AUC0–∞ ratio. The AUC0–∞ ratio was significantly associated with the SLCO1B1 missense SNV c.521T>C (p.V174A, rs4149056). The AUC0–∞ ratio was increased by 23% per copy of the variant allele (P = 1.30 × 10−22). After adjusting for this variant, an intronic triallelic SLCO1B1 SNV rs12367888 remained significantly associated with the AUC0–∞ ratio. The ratio was 12% lower per copy of the rs12367888 A‐allele (P = 5.24 × 10−8).
Linkage disequilibrium and haplotype analysis
In order to identify the causative SNVs underlying the associations of CYP2C9 rs77760615 and SLCO1B1 rs58310495 and rs12367888, we next investigated the linkage disequilibrium of these variants with missense SNVs in the respective genes. The rs77760615 SNV was in a complete linkage disequilibrium (r 2 = 1, P = 7.04 × 10−44) with the c.1075A>C (p.Ile359Leu, rs1057910) missense SNV, defining the CYP2C9*3 allele (Figure 2 a). The SLCO1B1 rs58310495 SNV was strongly linked with the SLCO1B1 missense variants c.521T>C (r 2 = 0.69, P = 5.78 × 10−32) and c.388A>G (p.Asn130Asp, rs2306283; r 2 = 0.41, P = 1.58 × 10−19) (Figure 2 b). Moreover, the A allele of the triallelic rs12367888 SNV was relatively strongly linked with the missense variants c.463C>A (Pro155Thr, rs11045819; r 2 = 0.48, P = 2.95 × 10−22), c.1929A>C (p.Leu643Phe, rs34671512; r 2 = 0.28, P = 1.19 × 10−13), and c.388A>G (r 2 = 0.25, P = 3.42 × 10−12).
Figure 2.
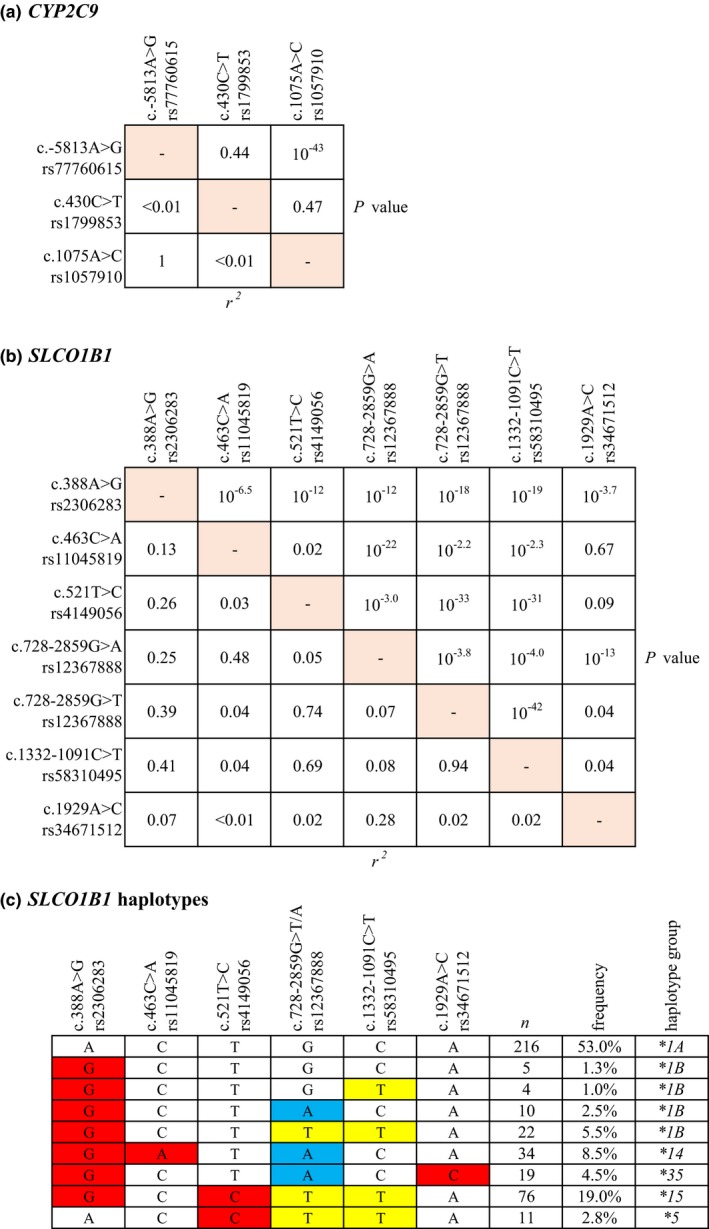
Linkage disequilibrium of (a) CYP2C9 and (b) SLCO1B1 missense and top noncoding SNVs. (c) SLCO1B1 haplotypes (MAF ≥0.01) inferred with missense and top noncoding SLCO1B1 SNVs. Intronic nucleotide changes are depicted in yellow and blue, and missense variations in red. MAF, minor allele frequency; SNV, single‐nucleotide variation. [Colour figure can be viewed at wileyonlinelibrary.com]
Previous studies have suggested that the functional effects of SLCO1B1 SNVs may depend on the combinations of SNVs in the same haplotype.17, 18, 19, 20, 21, 22, 23 We therefore computed SLCO1B1 haplotypes using missense variations and the noncoding rs58310495 and rs12367888 SNVs (Figure 2 c). The rs58310495 variant was found to be present in all SLCO1B1*5 and *15 haplotypes and in 63% of the *1B haplotypes, but not in *14 or *35. The rs12367888 A allele was present in all SLCO1B1*14 and *35 haplotypes, and in 24% of the *1B haplotypes.
Candidate gene analysis
To mitigate the risk of false negatives due to the conservative Bonferroni correction used in the primary analysis, we next carried out a candidate gene analysis without correction for multiple testing, including missense and functional variants with minor allele frequencies of ≥ 0.01 in genes involved in fluvastatin pharmacokinetics (Table S2). The candidate gene analysis focused on AUC0–∞, because it describes total exposure and is likely the best surrogate for drug response. In this analysis, CYP2C9*3 was associated with a 67% (P = 3.77 × 10−9), SLCO1B1 c.521T>C with a 34% (P = 8.15 × 10−8), and CYP2C9*2 (rs1799853, c.430C>T, p.Arg144Cys) with a 23% (P = 0.00213) increased AUC0–∞ of 3R,5S‐fluvastatin per copy of each variant allele (Table 2). The AUC0–∞ of 3S,5R‐fluvastatin was 94% (P = 3.19 × 10−12), 29% (P = 3.94 × 10−4), and 45% (P = 0.0246) larger per copy of CYP2C9*3, CYP2C9*2, and SLCO2B1 c.601G>A (p.Val201Met, rs35199625) variant allele, respectively, and 22% (P = 0.0306) smaller per copy of the SLCO2B1 c.1457C>T (p.Ser486Phe, rs2306168) variant allele. When the candidate gene analysis was performed with SLCO1B1 haplotypes instead of SNVs, the SLCO1B1*15 haplotype was associated with 3R,5S‐fluvastatin AUC0–∞ (Table S3). None of the SLCO1B1 haplotypes associated with 3S,5R‐fluvastatin AUC0–∞.
Table 2.
Results of the candidate gene analysis on 3R,5S‐fluvastatin and 3S,5R‐fluvastatin AUC0–∞, total fluvastatin AUC0–∞, and 3R,5S/3S,5R‐fluvastatin AUC0–∞ ratio
| Pharmacokinetic variable | Covariate/SNV | Effecta | P value | Bonferroni‐adjusted P value | Adjusted R 2 for each step | |
|---|---|---|---|---|---|---|
| Average (%) | 90% CI | |||||
| 3R,5S‐fluvastatin AUC0–∞ | BSA | −19.9 | −23.4, −16.2 | 3.10 × 10−14 | — | 0.24 |
| CYP2C9*3 (rs1057910) | 66.7 | 45.4, 91.0 | 3.77 × 10−9 | 1.01 × 10−7 | 0.36 | |
| SLCO1B1 c.521T>C (rs4149056) | 34.2 | 23.0, 46.4 | 8.15 × 10−8 | 2.20 × 10−6 | 0.44 | |
| CYP2C9*2 (rs1799853) | 22.7 | 10.1, 36.7 | 0.00213 | 0.0575 | 0.46 | |
| 3S,5R‐fluvastatin AUC0–∞ | BSA | −20.7 | −24.4, −16.8 | 1.06 × 10−13 | — | 0.20 |
| CYP2C9*3 (rs1057910) | 93.6 | 67.2, 124.2 | 3.19 × 10−12 | 8.61 × 10−11 | 0.35 | |
| CYP2C9*2 (rs1799853) | 29.1 | 14.9, 45.1 | 3.94 × 10−4 | 0.0106 | 0.38 | |
| SLCO2B1 c.601G>A (rs35199625) | 45.2 | 10.6, 90.7 | 0.0246 | 0.664 | 0.39 | |
| SLCO2B1 c.1457C>T (rs2306168) | −21.7 | −35.0, −5.7 | 0.0306 | 0.825 | 0.40 | |
| Total fluvastatin AUC0–∞ | BSA | −19.7 | −23.3, −16.0 | 1.75 × 10−13 | — | 0.23 |
| CYP2C9*3 (rs1057910) | 75.4 | 52.4, 101.8 | 3.17 × 10−10 | 1.00 × 10−8 | 0.37 | |
| SLCO1B1 c.521T>C (rs4149056) | 20.4 | 10.1, 31.7 | 7.54 × 10−4 | 0.0203 | 0.40 | |
| CYP2C9*2 (rs1799853) | 25.4 | 12.2, 40.2 | 9.63 × 10−4 | 0.0260 | 0.43 | |
| 3R,5S/3S,5R AUC0–∞ ratio | SLCO1B1 c.521T>C (rs4149056) | 28.2 | 23.7, 32.8 | 4.37 × 10−24 | 1.18 × 10−22 | 0.41 |
| SLCO1B1 c.1929A>C (rs34671512) | −15.6 | −20.0, −11.0 | 4.52 × 10−7 | 1.22 × 10−5 | 0.48 | |
| SLCO1B1 c.463C>A (rs11045819) | −10.4 | −14.1, −6.6 | 2.62 × 10−5 | 7.08 × 10−4 | 0.53 | |
| CYP2C9*3 (rs1057910) | −10.7 | −14.7, −6.6 | 4.41 × 10−5 | 0.00119 | 0.55 | |
| SLCO2B1 c.1457C>T (rs2306168) | 8.8 | 2.9, 15.1 | 0.0134 | 0.361 | 0.56 | |
| SLCO2B1 c.935G>A (rs12422149) | −5.1 | −8.5, −1.5 | 0.0221 | 0.597 | 0.57 | |
| SLCO1B3 c.699G>A (rs7311358)b | −4.0 | −7.2, −0.8 | 0.0434 | 1.173 | 0.58 | |
AUC0–∞, area under the plasma concentration‐time curve from 0 hour to infinity; BSA, body surface area; CI, confidence interval; MAF, minor allele frequency; SNV, single nucleotide variation; —, not applicable.
aPer 10% increase in BSA or per variant allele copy. bCompletely linked with SLCO1B3 missense SNV c.334T>G (rs4149117).
The total fluvastatin AUC0–∞ was 75% (P = 3.17 × 10−10), 20% (P = 7.54 × 10−4), and 25% (P = 9.63 × 10−4) larger per copy of the CYP2C9*3, SLCO1B1 c.521T>C, and CYP2C9*2 variant allele, respectively (Table 2). The 3R,5S/3S,5R‐fluvastatin AUC0–∞ ratio showed significant association with variants in the SLCO1B1, CYP2C9, SLCO2B1, and SLCO1B3 genes. Of the SLCO1B1 haplotypes, SLCO1B1*15 was associated with increased total fluvastatin AUC0–∞; SLCO1B1*5 and *15 were associated with increased, and the *14 and *35 haplotypes with decreased, 3R,5S/3S,5R‐fluvastatin AUC0–∞ ratio (Table S3).
To predict the relative effects of combinations of CYP2C9 and SLCO1B1 or SLCO2B1 genotypes on fluvastatin and fluvastatin enantiomer exposures, we calculated genotype scores (GS) using the following equations based on the candidate gene linear regression models:
where n is the number of variant alleles (0, 1, or 2).
The genotype scores predict the fold differences in AUCs between carriers of different genotype combinations and non‐carriers (Table 3, Figure 3). A total of 12%, 2.0%, and 9.5% of the study population had GS predicting more than twofold increased AUCs of 3R,5S‐fluvastatin, 3S,5R‐fluvastatin, and total fluvastatin, respectively.
Table 3.
Pharmacokinetic variables of 3R,5S‐fluvastatin, 3S,5R‐fluvastatin, and total fluvastatin grouped by genotype scoresa
| Cmax (ng/mL) | T max (hour) | AUC0–∞ (ng/hour/mL) | t 1/2 (hour) | |
|---|---|---|---|---|
| 3R,5S‐fluvastatin | ||||
| Genotype score ≥0.80, ≤1.25b | ||||
| Geometric mean (CV%) | 124 (72%) | 1 (0.5–5) | 172 (46%) | 1.9 (33%) |
| Genotype score > 1.25, < 2.00 | ||||
| Geometric mean (CV%) | 183 (65%) | 0.75 (0.5–2) | 249 (37%) | 2.0 (23%) |
| Ratio to control (90%CI) | 1.48 (1.26, 1.73) | — | 1.45 (1.30, 1.61) | 1.05 (0.98, 1.13) |
| P value | 5.37 × 10−5 | 0.547 | 3.02 × 10−8 | 0.241 |
| Genotype score ≥ 2.00 | ||||
| Geometric mean (CV%) | 235 (59%) | 1 (0.5–2) | 386 (49%) | 2.1 (18%) |
| Ratio to control (90% CI) | 1.90 (1.50, 2.40) | — | 2.24 (1.91, 2.63) | 1.10 (0.99, 1.22) |
| P value | 1.03 × 10−5 | 0.400 | 7.53 × 10−15 | 0.141 |
| 3S,5R‐fluvastatin | ||||
| Genotype score ≥0.80, ≤1.25b | ||||
| Geometric mean (CV%) | 145 (78%) | 0.5 (0.5–5) | 175 (45%) | 2.5 (29%) |
| Genotype score < 0.80 | ||||
| Geometric mean (CV%) | 94 (93%) | 1 (0.5–2) | 125 (48%) | 2.3 (18%) |
| Ratio to control (90% CI) | 0.65 (0.44, 0.97) | — | 0.72 (0.55, 0.93) | 0.90 (0.78, 1.04) |
| P value | 0.0740 | 0.088 | 0.0358 | 0.246 |
| Genotype score > 1.25, < 2.00 | ||||
| Geometric mean (CV%) | 187 (78%) | 1 (0.5–2) | 250 (54%) | 2.5 (29%) |
| Ratio to control (90% CI) | 1.30 (1.09, 1.54) | — | 1.43 (1.27, 1.60) | 1.01 (0.95, 1.08) |
| P value | 0.0149 | 0.009 | 9.72 × 10−7 | 0.807 |
| Genotype score ≥ 2.00 | ||||
| Geometric mean (CV%) | 299 (23%) | 1 (0.5–1) | 573 (28%) | 2.4 (20%) |
| Ratio to control (90% CI) | 2.06 (1.16, 3.68) | — | 3.27 (2.23, 4.81) | 0.96 (0.78, 1.19) |
| P value | 0.0396 | 0.349 | 8.65 × 10−7 | 0.762 |
| Total fluvastatin | ||||
| Genotype score ≥0.80, ≤1.25b | ||||
| Geometric mean (CV%) | 286 (75%) | 1 (0.5–5) | 369 (46%) | 2.1 (32%) |
| Genotype score > 1.25, < 2.00 | ||||
| Geometric mean (CV%) | 378 (79%) | 1 (0.5–2) | 543 (47%) | 2.1 (20%) |
| Ratio to control (90% CI) | 1.32 (1.05, 1.67) | — | 1.47 (1.26, 1.71) | 1.01 (0.91, 1.12) |
| P value | 0.0442 | 0.319 | 4.88 × 10−5 | 0.832 |
| Genotype score ≥ 2.00 | ||||
| Geometric mean (CV%) | 481 (50%) | 1 (0.5–1.5) | 763 (47%) | 2.2 (21%) |
| Ratio to control (90% CI) | 1.68 (1.29, 2.19) | — | 2.07 (1.74, 2.47) | 1.08 (0.96, 1.22) |
| P value | 1.26 × 10−3 | 0.368 | 1.05 × 10−10 | 0.263 |
AUC0–∞, area under the plasma concentration‐time curve from 0 hour to infinity; CV, geometric coefficient of variation; Cmax, peak plasma concentration; t ½, elimination half‐life; T max, time to peak plasma concentration. T max data are given as median (range); —, not applicable.
aGenotype scores are the predicted relative effects of combinations of CYP2C9 and SLCO1B1 or SLCO2B1 genotypes on AUC, based on the candidate gene linear regression model. bControl group.
Figure 3.
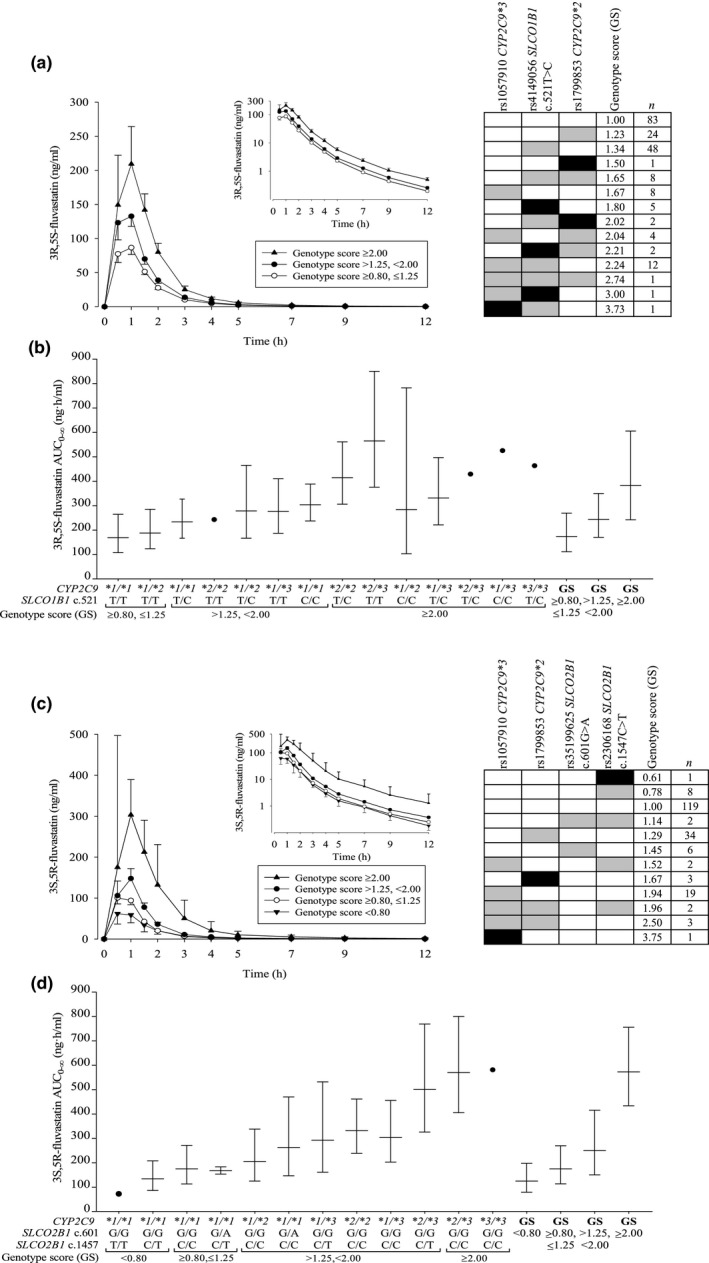
Geometric mean (90% CI) BSA‐adjusted plasma concentrations of (a) 3R,5S‐fluvastatin and (c) 3S,5R‐ fluvastatin after a single 40 mg oral dose of racemic fluvastatin in 200 healthy volunteers with different combinations of CYP2C9 and SLCO1B1 or SLCO2B1 genotypes. The insets depict the same data on a semilogarithmic scale. The volunteers were grouped by genotype scores predicting the fold differences in AUC 0–∞ values between carriers of different genotype combinations and non‐carriers. The right panels in (a) and (c) show the genotype scores for individuals with different genotypes. Reference genotypes are depicted with white, heterozygous with gray, and homozygous variant genotypes with black rectangles. The geometric mean ± geometric standard deviation BSA‐adjusted AUC 0–∞ values grouped by combinations of CYP2C9 and SLCO1B1 or SLCO2B1 genotypes, as well as the genotype scores are illustrated in (b) and (d). AUC0–∞, area under the plasma concentration‐time curve from 0 hour to infinity; BSA, body surface area; CI, confidence interval.
Fluvastatin transport by OATP1B1 and metabolism in vitro
To mechanistically validate the findings, we investigated the transport of fluvastatin enantiomers in OATP1B1‐transfected human embryonic kidney 293 cells, expressing either the reference or the c.521T>C variant SLCO1B1. Both 3R,5S‐fluvastatin and 3S,5R‐fluvastatin were substrates of reference OATP1B1, as indicated by 7.3‐fold and 4.1‐fold higher cellular uptake at 0.5 μM, and 2.5‐fold and 2.5‐fold higher at 4 μM in OATP1B1‐transfected cells than in mock‐transfected cells (P < 0.001). The c.521T>C SNV reduced the uptake of both enantiomers to 7% of the control (P < 10−5) (Figure 4).
Figure 4.
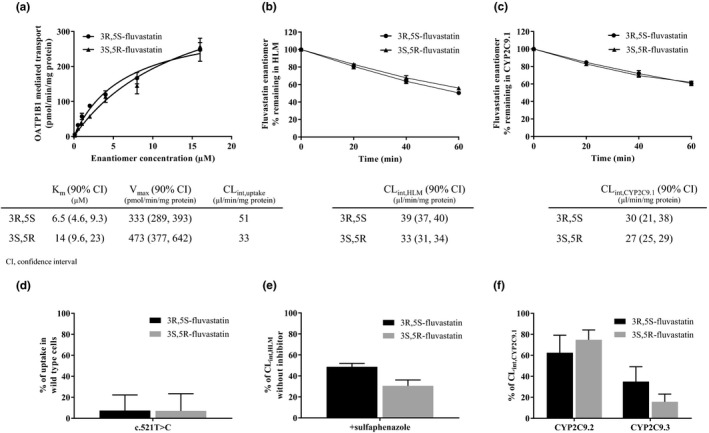
Fluvastatin enantiomer transport by reference OATP1B1 (a), metabolism in HLM (b), and metabolism by reference CYP2C9.1 (c); effects of SLCO1B1 c.521T>C on their transport (d), and sulfaphenazole (e) and CYP2C9.2 and CYP2C9.3 (f) on their metabolism in vitro. Fluvastatin enantiomer concentrations were 0.1–16 μM (a), 0.1 μM (b, c, e, f), and 0.5 μM (d). Sulfaphenazole concentration was 10 μM (e). CLint, intrinsic clearance; HLM, human liver microsomes; Km, Michaelis–Menten constant; Vmax, maximum transport rate.
We also investigated the metabolism of fluvastatin enantiomers by human liver microsomes (HLM), and recombinant CYP2C9.1, CYP2C9.2, and CYP2C9.3 enzymes. The microsomal intrinsic clearance (CLint,HLM) of 3R,5S‐fluvastatin was 39 μL/minute/mg protein and that of 3S,5R‐fluvastatin was 33 μL/minute/mg protein (P = 1.32 × 10−4, Figure 4). The CYP2C9 inhibitor sulfaphenazole25 inhibited the CLint,HLM of 3R,5S‐fluvastatin by 51% (P = 2.14 × 10−9) and that of 3S,5R‐fluvastatin by 69% (P = 1.60 × 10−7). Fluvastatin enantiomers were similarly metabolized by CYP2C9.1 (P = 0.455). The CLint of 3R,5S‐fluvastatin was reduced by 38% (P = 0.0132) and 65% (P = 0.00273), and that of 3S,5R‐fluvastatin by 25% (P = 0.00620) and 84% (P = 4.95 × 10−4) by recombinant CYP2C9.2 and CYP2C9.3, respectively, as compared with CYP2C9.1.
Discussion
These results show that genetic variation in CYP2C9 strongly affects the pharmacokinetics of both fluvastatin enantiomers. SLCO1B1 variants, on the other hand, affect the pharmacokinetics of 3R,5S‐fluvastatin only. Moreover, the candidate gene approach suggests that SLCO2B1 variants may affect the plasma concentrations of 3S,5R‐fluvastatin. Taken together, although the activity of CYP2C9 strongly affects the exposures to both fluvastatin enantiomers, the effects of SLCO transporters are enantiospecific.
The CYP2C9*3 and CYP2C9*2 alleles are known to impair the function of CYP2C9.9, 10, 26 The finding that CYP2C9*3 had a markedly larger effect than CYP2C9*2 in vivo is in line with our in vitro results showing a larger effect of CYP2C9*3 than CYP2C9*2 on fluvastatin metabolism. In a previous study, CYP2C9*3 was associated with increased AUC of both fluvastatin enantiomers, whereas the effect of CYP2C9*2 was not significant.11 However, the number of participants in that study was relatively small. Nevertheless, our results indicate that both CYP2C9*3 and *2 affect the pharmacokinetics of both fluvastatin enantiomers.
The finding that CYP2C9*3 had a larger effect on the AUC of 3S,5R‐fluvastatin than 3R,5S‐fluvastatin could be explained by different contributions of CYP2C9 to the elimination of the enantiomers, enantiomer‐dependent effect of CYP2C9*3, or both. Our in vitro results support both of these hypotheses, as the CYP2C9 inhibitor sulfaphenazole and recombinant CYP2C9.3 reduced the metabolism of 3S,5R‐fluvastatin more than that of 3R,5S‐fluvastatin.
In addition to CYP2C9 variants, an intronic SLCO1B1 rs58310495 SNV was associated with significantly increased AUC of 3R,5S‐fluvastatin. This SNV is strongly linked with the decreased‐function SLCO1B1 c.521T>C variant,16, 17, 18, 19, 20, 21, 22, 23 which suggests that the association is due to c.521T>C. Interestingly, no associations with SLCO1B1 variants were present for 3S,5R‐fluvastatin even in the candidate gene analysis with no correction for multiple testing (effect size 6.5% per copy of the c.521C allele, P = 0.259). Accordingly, the c.521T>C variant was associated with significantly increased 3R,5S‐/3S,5R‐fluvastatin AUC ratio. Thus, our results demonstrate that the effect of the SLCO1B1 genotype on fluvastatin pharmacokinetics is enantiospecific.
In addition to SLCO1B1 c.521T>C, the A allele of a triallelic intronic SLCO1B1 SNV (rs12367888) was associated with significantly decreased 3R,5S/3S,5R‐fluvastatin AUC ratio. Similarly, in the candidate gene analysis, the missense c.463C>A and c.1929A>C SNVs, which are strongly linked with the rs12367888 A allele, significantly decreased the AUC ratio. As the SLCO1B1 genotype has an enantiospecific effect on 3R,5S‐fluvastatin, the decreased AUC ratio is likely due to increased OATP1B1 activity. In line with this, the SLCO1B1*14 (containing c.463C>A) and *35 (containing c.1929A>C) haplotypes have previously been associated with increased hepatic expression of OATP1B127 and increased clearance of the OATP1B1 substrate methotrexate.28 SLCO1B1*14 has also been associated with a decreased AUC of atorvastatin27 and c.463C>A with decreased plasma concentrations of rifampin.29
Although SLCO1B1 c.521T>C markedly raises the plasma concentrations of many statins17, 18, 19, 20, 21, 22, 23 the AUC of total fluvastatin was only nonsignificantly (by 19%) increased in c.521CC homozygotes in a previous study.21 In our candidate gene analysis, the effect of c.521T>C on total fluvastatin was significant and larger than in the previous study. This difference may be explained by the substantially larger sample size in the present study and that the present study also included carriers of SLCO1B1 haplotypes that enhance OATP1B1 activity, e.g., SLCO1B1*14 and *35. In another previous study, individuals homozygous for the ABCG2 missense variant c.421C>A (rs2231142, p.Gln141Lys) had significantly increased plasma concentrations of racemic fluvastatin.14 In the present study, the variant showed no significant association with fluvastatin pharmacokinetics. However, only one participant was homozygous for c.421C>A.
To elucidate the mechanism underlying the enantiospecific effects of SLCO1B1 genotype, we investigated the transport of fluvastatin enantiomers by OATP1B1 in vitro. The enantiomers had less than twofold difference in the in vitro uptake clearance and the c.521T>C variant similarly impaired their uptake. These preliminary in vitro results cannot explain the enantiospecific effects of SLCO1B1 genotype on fluvastatin pharmacokinetics in humans. Therefore, it seems that different contributions of other transporters to the hepatic disposition of the enantiomers is the most likely explanation for the enantiospecific effects of the SLCO1B1 genotype. In addition to OATP1B1, racemic fluvastatin is a substrate of OATP1B3, OATP2B1, and sodium‐dependent taurocholate cotransporting polypeptide, with the highest affinity reported for OATP2B1.4 Our candidate gene analysis suggested that SLCO2B1 missense variants affect 3S,5R‐fluvastatin but not 3R,5S‐fluvastatin AUC. The c.1457C>T SNV, which associated with decreased 3S,5R‐fluvastatin AUC, has previously been associated with decreased fexofenadine and celiprolol AUC.30, 31 In addition to SLCO2B1 SNVs, a missense SNV in SLCO1B3 was associated with a minor effect on the 3R,5S/3S,5R‐fluvastatin AUC ratio. It is noteworthy that the SLCO2B1 and SLCO1B3 associations were significant only when no correction for multiple testing was applied, and therefore the results should be interpreted with caution.
Although statins are generally well tolerated, they can cause muscle toxicity. The clinical spectrum of statin‐induced myotoxicity ranges from mild myopathy to potentially life‐threatening rhabdomyolysis.32 There seem to be no published studies on how the risk of myotoxicity is related to fluvastatin exposure. Because statin‐induced myotoxicity is, however, generally dose‐dependent and concentration‐dependent,32, 33 it is likely that increased systemic exposure to the active 3R,5S‐fluvastatin predisposes to this adverse reaction. Both CYP2C9*3 and *2, as well as SLCO1B1 c.521T>C increase 3R,5S‐fluvastatin AUC. Of these, the CYP2C9*3 allele has previously been associated with fluvastatin‐induced adverse reactions.34 Moreover, the SLCO1B1 c.521T>C SNV increases simvastatin acid AUC more than threefold and associates with a markedly increased risk of simvastatin‐induced myotoxicity.20, 35 As the effect of the c.521T>C SNV on 3R,5S‐fluvastatin AUC is smaller than on simvastatin acid AUC, the increase in myotoxicity risk is also likely smaller. Markedly larger increases in 3R,5S‐fluvastatin AUC are seen in individuals carrying the c.521T>C SNV together with CYP2C9 variants, however. Based on CYP2C9 and SLCO variants, we calculated GS to predict the exposure to fluvastatin. The GS might be useful in predicting the risk of fluvastatin‐induced myotoxicity. For patients with 3R,5S‐fluvastatin GS ≥2.0, it could be advisable to prescribe a lower dose of fluvastatin or consider an alternative statin.
The site of action of statins is within the hepatocytes, and fluvastatin efficacy is therefore related to the hepatocyte exposure to 3R,5S‐fluvastatin.1, 3 The maximum fluvastatin dose (80 mg/day) reduces Low‐density lipoprotein cholesterol on average by 33%,36 which is less than that seen with the maximum doses of rosuvastatin and atorvastatin. As the CYP2C9*3 and *2 variants impair the hepatic metabolism of fluvastatin, they should increase the hepatocyte concentration of 3R,5S‐fluvastatin to at least the same extent as its plasma concentration and therefore enhance its cholesterol‐lowering efficacy. Low‐density lipoprotein cholesterol decreases by 6% for every twofold increase in fluvastatin dose.36 Therefore, individuals with, e.g., the CYP2C9*2/*3 genotype, associated with twofold increased 3R,5S‐fluvastatin AUC (Figure 3), should show at least 6% greater low‐density lipoprotein cholesterol‐lowering efficacy of fluvastatin than individuals with the *1/*1 genotype. In one small study, there was a tendency for greater cholesterol‐lowering effect of fluvastatin in patients with the CYP2C9*1/*3 genotype than in those with the *1/*1 genotype.37 On the other hand, the SLCO1B1 c.521T>C variant reduces the hepatic uptake of 3R,5S‐fluvastatin and might thus reduce fluvastatin efficacy. In a previous study, a trend of impaired fluvastatin efficacy has been reported in c.521C allele carriers.38 Furthermore, the c.463C>A missense SNV, which associates with increased OATP1B1 activity, has been associated with increased cholesterol‐lowering efficacy of fluvastatin.39 Taken together, the CYP2C9*3 and *2 alleles are expected to increase both the efficacy and the risk of myotoxicity of fluvastatin, whereas the SLCO1B1 c.521T>C allele likely increases myotoxicity risk only.
The present study was conducted in Finnish white volunteers. The frequencies of CYP2C9 and SLCO1B1 variants, however, differ between populations. The allele frequencies of CYP2C9*2, *3, and SLCO1B1 c.521T>C are 0.12, 0.056, 0.18 in Europeans, 0.024, 0.013, and 0.019 in Sub‐Saharan Africans, and < 0.001, 0.034, and 0.12 in East Asians, respectively.40, 41 Therefore, the extent to which these variants can explain population variability in fluvastatin pharmacokinetics differs between ethnic groups. Nevertheless, CYP2C9*2, *3, and SLCO1B1 c.521T>C are decreased‐function alleles, as demonstrated both in vitro and in vivo, and individuals carrying these should have similarly increased fluvastatin exposures, irrespective of the ethnic background.
In conclusion, genetic variability in CYP2C9 significantly affects fluvastatin pharmacokinetics. In addition, SLCO1B1 genotype has an enantiospecific effect on active 3R,5S‐fluvastatin pharmacokinetics. However, no obvious difference between the enantiomers was observed in the uptake by OATP1B1 in vitro. The genotype scores may predict how combinations of CYP2C9 and SLCO1B1 variants affect 3R,5S‐fluvastatin exposure and aid in predicting the risk of myotoxicity.
Methods
A total of 200 healthy unrelated Finnish white volunteers participated in the study after giving written informed consent. Their health was confirmed by medical history, clinical examination, and laboratory tests. Participants were not on any continuous medication nor were tobacco smokers. The study was approved by the Coordinating Ethics Committee of the Hospital District of Helsinki and Uusimaa and the Finnish Medicines Agency Fimea. Of the participants, 99 were women and 101 were men. Their mean ± SD age was 23 ± 4 years, height 174 ± 9 cm, weight 70 ± 12 kg, and body mass index 22.8 ± 2.6 kg/m2.
Fluvastatin pharmacokinetics
After fasting overnight, the participants ingested a 40 mg dose of racemic fluvastatin (Lescol capsule, Novartis Finland Oy, Espoo, Finland) with 150 mL of water at 8 am. Standardized meals were served at 4, 7, and 10 hours after fluvastatin ingestion. Timed blood samples (4–9 mL each) were collected to light‐protected ethylenediaminetetraacetic acid tubes prior to and up to 12 hours after fluvastatin administration. Tubes were immediately placed on ice. Plasma was separated within 30 minutes and stored at −70°C until analysis.
The determination of 3R,5S‐fluvastatin and 3S,5R‐fluvastatin in plasma was based on chiral liquid chromatographic‐tandem mass spectrometric (LC‐MS/MS) method employing commercial pure reference compounds (Toronto Research Chemicals, Inc., North York, ON). The analytical system consisted of a Nexera X2 liquid chromatography instrument (Shimadzu, Kyoto, Japan) interfaced with a 5500 Qtrap mass spectrometer (AB Sciex, Toronto, ON, Canada). Chromatographic separation of the enantiomers was achieved on a Lux Cellulose‐1 chiral column (150 × 2.0‐mm internal diameter, 3‐μm particle size; Phenomenex, Torrance, CA). The mobile phase consisted of a mixture of acetonitrile, methanol and water (12:18:20 v/v/v) containing 0.01% formic acid, and the flow rate was 220 μL/minute. The mass spectrometer was operated in a negative electrospray ionization mode, and the targeted mass‐to‐charge ion transition for both fluvastatin enantiomers was 410–348. Deuterium‐labeled fluvastatin was monitored as an internal standard. The lower limits of quantification were 0.25 ng/mL, and the inter day coefficients of variation of the quality control samples were below 7% for both enantiomers. The AUC0–∞, Cmax, and t ½ values were calculated for 3R,5S‐fluvastatin, 3S,5R‐fluvastatin, and total fluvastatin with standard noncompartmental methods using Phoenix WinNonlin, version 6.3 (Certara, Princeton, NJ).
DNA sequencing and genotyping
Genomic DNA was extracted from ethylenediaminetetraacetic acid blood samples using the Maxwell 16 LEV Blood DNA Kit on a Maxwell 16 Research automated nucleic acid extraction system (Promega, Madison, WI). A total of 379 pharmacokinetic genes ± 20 kb were completely sequenced in the study participants using targeted massive parallel sequencing at the Technology Centre at Institute for Molecular Medicine Finland (Helsinki, Finland).42, 43 A total of 46,064 SNVs with minor allele frequency ≥ 0.05 were included in the statistical analysis.
In order to verify genotype calls and to supplement missing data, all study participants were genotyped for the CYP2C9*3, CYP2C9*2, and SLCO1B1 c.521T>C, c.388A>G, and c.463C>A SNVs with TaqMan genotyping assays on a QuantStudio 12K Flex Real‐Time PCR System (Thermo Fisher Scientific, Waltham, MA). Call identity with sequencing data was 100%. TaqMan genotyping was also used to supplement missing data of SLCO1B1 c.1929A>C, SLCO1B3 c.334T>G (p.Ser112Ala, rs4149117), SLCO2B1 c.1457C>T, and SLCO2B1 c.601G>A SNVs. Triallelic rs12367888 genotype calling was verified and missing data supplemented by Sanger sequencing. All participants were genotyped for relevant CYP2D6 alleles with TaqMan genotyping assays.44 The CYP2D6 copy number was determined with a TaqMan copy number assay targeting exon 9. CYP2D6 metabolizer status was inferred from the genotypes using the activity score method.45
In vitro studies with transfected OATP1B1 cells
Recombinant baculoviruses containing reference or c.521T>C variant SLCO1B1 gene or the gene for enhanced yellow fluorescent protein (eYFP) were prepared using a modified Bac‐to‐Bac method.46 Human embryonic kidney 293 cells were seeded on Corning BioCoat Poly‐D‐Lysine 24 well plates (Corning, Woburn, MA) at a density of 250,000 cells per well and transfected with recombinant baculoviruses carrying either reference or variant SLCO1B1 gene or the eYFP gene for the control cells. Sodium butyrate (5 mM) was used to stimulate the expression of the recombinant proteins.
The cellular uptake assays were performed on a heated orbital shaker plate 48 hours post‐transfection. Cell culture medium was first removed and 1 mL of transport buffer (Hank's Balanced Salt Solution; Gibco, Thermo Fisher Scientific; 4.17 mM NaHCO3 and 25 mM HEPES adjusted to pH 7.4 with HCl) was added to wells for 3‐minute preincubation. The buffer was then replaced with 250 μL of test solution, which contained 0.1, 0.5, 1, 2, 4, 8, or 16 μM (n = 2–3) of 3R,5S‐fluvastatin or 3S,5R‐fluvastatin for determining the transport kinetics, or 0.5 μM (n = 6) for testing the effect of c.521T>C. After 1 minute, the test solution was removed, and the wells were washed with ice‐cold transport buffer three times and left to dry. Thereafter, 250 μL of methanol water mixture (3:1) containing deuterium‐labeled fluvastatin was added to lyse the cells. After 30 minutes, the samples were centrifuged and the supernatant was separated for LC‐MS/MS analysis.
Fluvastatin concentrations were measured with the Nexera X2 liquid chromatography‐AB Sciex 5500 Qtrap mass spectrometer. The chromatographic separation was achieved on a reversed phase Kinetex C8 column (75 × 2.1‐mm internal diameter, 2.6‐μm particle size; Phenomenex) with a mobile phase of 0.1% formic acid (A) and acetonitrile (B). The mobile phase flow rate was 300 μL/minute, and a gradient profile was applied as follows: a linear increase from 25% B to 65% B over 2.5 minute, then 0.5 minute at 90% B on hold before a re‐equilibration step to the starting composition. The mass spectrometric detection was performed as described for plasma samples. Dichlorofluorescein (Santa Cruz Biotechnology, Dallas, TX) was used for assay quality control in three wells expressing reference OATP1B1 or eYFP. These wells were lysed with 0.1 M NaOH. Protein concentration was determined by adding 300 μL of Coomassie Plus reagent (Thermo Fisher Scientific) to 10 μL of the quality control cell lysate and measuring absorbance at 595 nm with Varioskan LUX (Thermo Fischer Scientific). The cellular uptake was normalized to the protein content.
The OATP1B1‐mediated uptake was obtained by subtracting the uptake in eYFP‐transfected cells from that in OATP1B1‐transfected cells. Transport kinetics were calculated with GraphPad Prism 7 (GraphPad Software, San Diego, CA) to obtain Michaelis‐Menten constant (K m) and maximum transport rate (V max) values for both fluvastatin enantiomers. The CLint,uptake was calculated as V max/K m.
In vitro studies with HLM and recombinant enzymes
The incubations were performed with pooled HLM (Corning) and recombinant CYP2C9.1, CYP2C9.2, and CYP2C9.3 (EasyCYP, Cypex Ltd., Dundee, UK) enzymes. The incubation mixtures (n = 3 or 6) contained 100 mM phosphate buffer, pH 7.4, 0.3 mg/mL of protein, 1 mM β‐nicotinamide adenine dinucleotide phosphate (β‐NADPH, tetrasodium salt), and 0.1 μM 3R,5S‐fluvastatin or 3S,5R‐fluvastatin in a total volume of 500 μL. Sulfaphenazole concentration was 10 μM in the inhibition experiments. Control incubations were performed in duplicate without the addition of NADPH. The reactions were initiated by the addition of the substrate or the mixture of substrate and inhibitor, and incubated in a shaking ThermoMixer (Eppendorf AG, Hamburg, Germany) at 37 °C. Reactions were stopped by transferring 50 μL samples to 50‐μL acetonitrile‐containing deuterium‐labeled fluvastatin at time points 0, 20, 40, and 60 minutes. After centrifugation, the supernatants were analyzed by using LC‐MS/MS as described for the OATP1B1 uptake experiments. The depletion rate constants (k dep) of the substrates were calculated with the first‐order elimination kinetics equation. CLint in each matrix was obtained by dividing the k dep with protein concentration.
Statistical analysis
The data were analyzed with the statistical programs JMP Genomics 7.0 (SAS Institute, Inc., Cary, NC) and IBM SPSS 22.0 for Windows (Armonk, NY). The pharmacokinetic variables were logarithmically transformed before analysis. Sex, body weight, lean body weight,47 and body surface area48 were tested as demographic covariates for pharmacokinetic data using stepwise linear regression analysis, with P value thresholds of 0.05 for entry and 0.10 for removal. Possible effects of genetic variants on pharmacokinetic variables were investigated using linear regression analysis fixed for significant demographic covariates with a stepwise approach. A Bonferroni‐corrected P value threshold of 1.09 × 10−6 was employed for the 379 genes and thresholds of 0.05 for entry and 0.10 for removal for the candidate gene analysis. Additive coding was employed for genetic variants, and multiallelic variants were expanded. CYP2D6 data were included as the activity scores in the candidate gene analysis. Haplotype computations for SLCO1B1 were performed with PHASE v2.1.1.49, 50 Statistical comparisons of in vitro data were done using independent samples Student's t‐test, with logarithmic transformation as appropriate.
Funding
This study was supported by grants from the European Research Council (Grant agreement 282106), State funding for university‐level health research, and the Sigrid Jusélius Foundation (Helsinki, Finland).
Conflicts of Interest
The authors declared no competing interests for this work.
Author Contributions
P.H. and M.Ni. wrote the manuscript; P.H., A.T., J.T.B., and M.Ni. designed the research; P.H., A.T., M.Ne., W.K., T.T., M.P‐H., H.K., J.T.B., and M.Ni. performed the research; P.H. and M.Ni. analyzed the data.
Supporting information
Table S1. Pharmacokinetic variables of 3R,5S‐fluvastatin, 3S,5R‐fluvastatin, and total fluvastatin in 200 healthy volunteers.
Table S2. SNVs (MAF ≥ 0.01) included in the candidate gene analysis.
Table S3. Results of the candidate gene analysis with SLCO1B1 haplotypes on 3R,5S‐fluvastatin and 3S,5R‐fluvastatin AUC0–∞, total fluvastatin AUC0–∞, and 3R,5S/3S,5R‐fluvastatin AUC0–∞ ratio.
Acknowledgments
The authors thank Katja Halme, Hanna Hyvärinen, Satu Karjalainen, Jouko Laitila, Eija Mäkinen‐Pulli, Raija Nevala, and Lisbet Partanen for skillful assistance in conducting the clinical pharmacokinetic study, and Pekka Ellonen and Maija Lepistö, for the massive parallel sequencing.
References
- 1. Kathawala, F.G. HMG‐CoA reductase inhibitors: an exciting development in the treatment of hyperlipoproteinemia. Med. Res. Rev. 11, 121–146 (1991). [PubMed] [Google Scholar]
- 2. Fischer, V. et al The 3‐hydroxy‐3‐methylglutaryl coenzyme A reductase inhibitor fluvastatin: effect on human cytochrome P‐450 and implications for metabolic drug interactions. Drug Metab. Dispos. 27, 410–416 (1999). [PubMed] [Google Scholar]
- 3. Neuvonen, P.J. , Niemi, M. & Backman, J.T. Drug interactions with lipid‐lowering drugs: mechanisms and clinical relevance. Clin. Pharmacol. Ther. 80, 565–581 (2006). [DOI] [PubMed] [Google Scholar]
- 4. Kopplow, K. , Letschert, K. , König, J. , Walter, B. & Keppler, D. Human hepatobiliary transport of organic anions analyzed by quadruple‐transfected cells. Mol. Pharmacol. 68, 1031–1038 (2005). [DOI] [PubMed] [Google Scholar]
- 5. Hirano, M. , Maeda, K. , Matsushima, S. , Nozaki, Y. , Kusuhara, H. & Sugiyama, Y. Involvement of BCRP (ABCG2) in the biliary excretion of pitavastatin. Mol. Pharmacol. 68, 800–807 (2005). [DOI] [PubMed] [Google Scholar]
- 6. Kalliokoski, A. & Niemi, M. Impact of OATP transporters on pharmacokinetics. Br. J. Pharmacol. 158, 693–705 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Greupink, R. , Dillen, L. , Monshouwer, M. , Huisman, M.T. & Russel, F.G. Interaction of fluvastatin with the liver‐specific Na + ‐dependent taurocholate cotransporting polypeptide (NTCP). Eur. J. Pharm. Sci. 44, 487–496 (2011). [DOI] [PubMed] [Google Scholar]
- 8. Ellis, L.C. , Hawksworth, G.M. & Weaver, R.J. ATP‐dependent transport of statins by human and rat MRP2/Mrp2. Toxicol. Appl. Pharmacol. 269, 187–194 (2013). [DOI] [PubMed] [Google Scholar]
- 9. Haining, R.L. , Hunter, A.P. , Veronese, M.E. , Trager, W.F. & Rettie, A.E. Allelic variants of human cytochrome P450 2C9: baculovirus‐mediated expression, purification, structural characterization, substrate stereoselectivity, and prochiral selectivity of the wild‐type and I359L mutant forms. Arch. Biochem. Biophys. 333, 447–458 (1996). [DOI] [PubMed] [Google Scholar]
- 10. Daly, A.K. , Rettie, A.E. , Fowler, D.M. & Miners, J.O. Pharmacogenomics of CYP2C9: functional and clinical considerations. J. Pers. Med. 8 (2017) 10.3390/jpm8010001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kirchheiner, J. et al Influence of CYP2C9 polymorphisms on the pharmacokinetics and cholesterol‐lowering activity of (‐)‐3S,5R‐fluvastatin and (+)‐3R,5S‐fluvastatin in healthy volunteers. Clin. Pharmacol. Ther. 74, 186–194 (2003). [DOI] [PubMed] [Google Scholar]
- 12. Kondo, C. et al Functional analysis of SNPs variants of BCRP/ABCG2. Pharm. Res. 21, 1895–1903 (2004). [DOI] [PubMed] [Google Scholar]
- 13. Keskitalo, J.E. , Zolk, O. , Fromm, M.F. , Kurkinen, K.J. , Neuvonen, P.J. & Niemi, M. ABCG2 polymorphism markedly affects the pharmacokinetics of atorvastatin and rosuvastatin. Clin. Pharmacol. Ther. 86, 197–203 (2009). [DOI] [PubMed] [Google Scholar]
- 14. Keskitalo, J.E. , Pasanen, M.K. , Neuvonen, P.J. & Niemi, M. Different effects of the ABCG2 c.421C>A SNP on the pharmacokinetics of fluvastatin, pravastatin and simvastatin. Pharmacogenomics 10, 1617–1624 (2009). [DOI] [PubMed] [Google Scholar]
- 15. Giacomini, K.M. et al International Transporter Consortium commentary on clinically important transporter polymorphisms. Clin. Pharmacol. Ther. 94, 23–26 (2013). [DOI] [PubMed] [Google Scholar]
- 16. Tirona, R.G. , Leake, B.F. , Merino, G. & Kim, R.B. Polymorphisms in OATP‐C: identification of multiple allelic variants associated with altered transport activity among European‐ and African‐Americans. J. Biol. Chem. 276, 35669–35675 (2001). [DOI] [PubMed] [Google Scholar]
- 17. Nishizato, Y. et al Polymorphisms of OATP‐C (SLC21A6) and OAT3 (SLC22A8) genes: consequences for pravastatin pharmacokinetics. Clin. Pharmacol. Ther. 73, 554–565 (2003). [DOI] [PubMed] [Google Scholar]
- 18. Niemi, M. et al High plasma pravastatin concentrations are associated with single nucleotide polymorphisms and haplotypes of organic anion transporting polypeptide‐C (OATP‐C, SLCO1B1). Pharmacogenetics 14, 429–440 (2004). [DOI] [PubMed] [Google Scholar]
- 19. Mwinyi, J. , Johne, A. , Bauer, S. , Roots, I. & Gerloff, T. Evidence for inverse effects of OATP‐C (SLC21A6) 5 and 1b haplotypes on pravastatin kinetics. Clin. Pharmacol. Ther. 75, 415–421 (2004). [DOI] [PubMed] [Google Scholar]
- 20. Pasanen, M.K. , Neuvonen, M. , Neuvonen, P.J. & Niemi, M. SLCO1B1 polymorphism markedly affects the pharmacokinetics of simvastatin acid. Pharmacogenet. Genomics 16, 873–879 (2006). [DOI] [PubMed] [Google Scholar]
- 21. Niemi, M. , Pasanen, M.K. & Neuvonen, P.J. SLCO1B1 polymorphism and sex affect the pharmacokinetics of pravastatin but not fluvastatin. Clin. Pharmacol. Ther. 80, 356–366 (2006). [DOI] [PubMed] [Google Scholar]
- 22. Niemi, M. , Pasanen, M.K. & Neuvonen, P.J. Organic anion transporting polypeptide 1B1: a genetically polymorphic transporter of major importance for hepatic drug uptake. Pharmacol. Rev. 63, 157–181 (2011). [DOI] [PubMed] [Google Scholar]
- 23. Tornio, A. , Vakkilainen, J. , Neuvonen, M. , Backman, J.T. , Neuvonen, P.J. & Niemi, M. SLCO1B1 polymorphism markedly affects the pharmacokinetics of lovastatin acid. Pharmacogenet. Genomics 25, 382–387 (2015). [DOI] [PubMed] [Google Scholar]
- 24. Keskitalo, J.E. , Kurkinen, K.J. , Neuvonen, M. , Backman, J.T. , Neuvonen, P.J. & Niemi, M. No significant effect of ABCB1 haplotypes on the pharmacokinetics of fluvastatin, pravastatin, lovastatin, and rosuvastatin. Br. J. Clin. Pharmacol. 68, 207–213 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Baldwin, S.J. , Bloomer, J.C. , Smith, G.J. , Ayrton, A.D. , Clarke, S.E. & Chenery, R.J. Ketoconazole and sulphaphenazole as the respective selective inhibitors of P4503A and 2C9. Xenobiotica 25, 261–270 (1995). [DOI] [PubMed] [Google Scholar]
- 26. Rettie, A.E. , Wienkers, L.C. , Gonzalez, F.J. , Trager, W.F. & Korzekwa, K.R. Impaired (S)‐warfarin metabolism catalysed by the R144C allelic variant of CYP2C9. Pharmacogenetics 4, 39–42 (1994). [DOI] [PubMed] [Google Scholar]
- 27. Nies, A.T. et al Genetics is a major determinant of expression of the human hepatic uptake transporter OATP1B1, but not of OATP1B3 and OATP2B1. Genome Med. 5 (2013) 10.1186/gm405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ramsey, L.B. et al Rare versus common variants in pharmacogenetics: SLCO1B1 variation and methotrexate disposition. Genome Res. 22, 1–8 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Weiner, M. et al Effects of tuberculosis, race, and human gene SLCO1B1 polymorphisms on rifampin concentrations. Antimicrob. Agents Chemother. 54, 4192–4200 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Imanaga, J. et al The effects of the SLCO2B1 c.1457C > T polymorphism and apple juice on the pharmacokinetics of fexofenadine and midazolam in humans. Pharmacogenet. Genomics 21, 84–93 (2011). [DOI] [PubMed] [Google Scholar]
- 31. Ieiri, I. et al Microdosing clinical study: pharmacokinetic, pharmacogenomic (SLCO2B1), and interaction (grapefruit juice) profiles of celiprolol following the oral microdose and therapeutic dose. J. Clin. Pharmacol. 52, 1078–1089 (2012). [DOI] [PubMed] [Google Scholar]
- 32. Alfirevic, A. et al Phenotype standardization for statin‐induced myotoxicity. Clin. Pharmacol. Ther. 96, 470–476 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Thompson, P.D. , Clarkson, P. & Karas, R.H. Statin‐associated myopathy. JAMA 289, 1681–1690 (2003). [DOI] [PubMed] [Google Scholar]
- 34. Miroševic Skvrce, N. , Božina, N. , Zibar, L. , Barišic, I. , Pejnovic, L. & Macolic Šarinic, V. CYP2C9 and ABCG2 polymorphisms as risk factors for developing adverse drug reactions in renal transplant patients taking fluvastatin: a case‐control study. Pharmacogenomics 14, 1419–1431 (2013). [DOI] [PubMed] [Google Scholar]
- 35. SEARCH Collaborative Group SLCO1B1 variants and statin‐induced myopathy–a genomewide study. N. Engl. J. Med. 359, 789–799 (2008). [DOI] [PubMed] [Google Scholar]
- 36. Adams, S.P. , Sekhon, S.S. , Tsang, M. & Wright, J.M. Fluvastatin for lowering lipids. Cochrane Database Syst. Rev. 3, CD012282 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Buzková, H. et al Lipid‐lowering effect of fluvastatin in relation to cytochrome P450 2C9 variant alleles frequently distributed in the Czech population. Med. Sci. Monit. 18, 512–517 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Meyer zu Schwabedissen, H.E. et al Function‐impairing polymorphisms of the hepatic uptake transporter SLCO1B1 modify the therapeutic efficacy of statins in a population‐based cohort. Pharmacogenet. Genomics 25, 8–18 (2015). [DOI] [PubMed] [Google Scholar]
- 39. Couvert, P. et al Association between a frequent allele of the gene encoding OATP1B1 and enhanced LDL‐lowering response to fluvastatin therapy. Pharmacogenomics 9, 1217–1227 (2008). [DOI] [PubMed] [Google Scholar]
- 40. Pasanen, M.K. , Neuvonen, P.J. & Niemi, M. Global analysis of genetic variation in SLCO1B1 . Pharmacogenomics 9, 19–33 (2008). [DOI] [PubMed] [Google Scholar]
- 41. Zhou, Y. , Ingelman‐Sundberg, M. & Lauschke, V.M. Worldwide distribution of cytochrome P450 alleles: a meta‐analysis of population‐scale sequencing projects. Clin. Pharmacol. Ther. 102, 688–700 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hirvensalo, P. et al Comprehensive pharmacogenomic study reveals an important role of UGT1A3 in montelukast pharmacokinetics. Clin. Pharmacol. Ther. 104, 158–168 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sulonen, A.M. et al Comparison of solution‐based exome capture methods for next generation sequencing. Genome Biol. 12, R94 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gonçalves, B.P. et al Age, weight, and CYP2D6 genotype are major determinants of primaquine pharmacokinetics in African children. Antimicrob. Agents Chemother. 61, e02590‐16 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gaedigk, A. , Simon, S.D. , Pearce, R.E. , Bradford, L.D. , Kennedy, M.J. & Leeder, J.S. The CYP2D6 activity score: translating genotype information into a qualitative measure of phenotype. Clin. Pharmacol. Ther. 83, 234–242 (2008). [DOI] [PubMed] [Google Scholar]
- 46. Sjöstedt, N. , van den Heuvel, J.J.M.W. , Koenderink, J.B. & Kidron, H. Transmembrane domain single‐nucleotide polymorphisms impair expression and transport activity of ABC transporter ABCG2. Pharm. Res. 34, 1626–1636 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hallynck, T.H. , Soep, H.H. , Thomis, J.A. , Boelaert, J. , Daneels, R. & Dettli, L. Should clearance be normalised to body surface or to lean body mass? Br. J. Clin. Pharmacol. 11, 523–526 (1981). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Haycock, G.B. , Schwartz, G.J. & Wisotsky, D.H. Geometric method for measuring body surface area: a height‐weight formula validated in infants, children, and adults. J. Pediatr. 93, 62–66 (1978). [DOI] [PubMed] [Google Scholar]
- 49. Stephens, M. , Smith, N.J. & Donnelly, P. A new statistical method for haplotype reconstruction from population data. Am. J. Hum. Genet. 68, 978–989 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Stephens, M. & Donnelly, P. A comparison of bayesian methods for haplotype reconstruction from population genotype data. Am. J. Hum. Genet. 73, 1162–1169 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Pharmacokinetic variables of 3R,5S‐fluvastatin, 3S,5R‐fluvastatin, and total fluvastatin in 200 healthy volunteers.
Table S2. SNVs (MAF ≥ 0.01) included in the candidate gene analysis.
Table S3. Results of the candidate gene analysis with SLCO1B1 haplotypes on 3R,5S‐fluvastatin and 3S,5R‐fluvastatin AUC0–∞, total fluvastatin AUC0–∞, and 3R,5S/3S,5R‐fluvastatin AUC0–∞ ratio.