

Abstract
Gas-phase infrared multiple-photon dissociation (IRMPD) spectra are recorded for the protonated dye molecules indigo and isoindigo by using a quadrupole ion trap (QIT) mass spectrometer coupled to the free electron laser for infrared experiments (FELIX). From their fingerprint IR spectra (600—1800 cm–1) and comparison with quantum-chemical calculations at the density functional level of theory (B3LYP/6-31++G(d,p)), we derive their structures. We focus particularly on the question of whether trans-to-cis isomerization occurs upon protonation and transfer to the gas phase. The trans-configuration is energetically favored in the neutral forms of the dyes in solution and in the gas phase. Instead, the cis-isomer is lower in energy for the protonated forms of both species, but indigo is also notorious for not undergoing double-bond trans-to-cis isomerization, in contrast to many other conjugated systems. The IR spectra suggest that protoisomerization from trans to cis indeed occurs for both dyes. To estimate the extent of isomerization, on-resonance kinetics are measured on diagnostic and common vibrational frequencies to determine the ratio of cis-to-trans isomers. We find ratios of 65–70% cis and 30–35% trans for indigo versus 75–80% cis and 20–25% trans for isoindigo. Transition-state calculations for the isomerization reactions have been carried out, which indeed suggest a lower barrier for protonated isoindigo, qualitatively explaining the more efficient isomerization.
Introduction
Indigo (C16H10N2O2) is a common pigment with a distinctive blue color. Double-bond isomerization1 of indigo and its various derivatives is of the essence because of its potential use as a molecular switch2,3 in the ongoing challenge to harness and exploit the well-defined mechanical properties of molecular compounds and the design of small, molecular-sized devices. Cis–trans isomerization around double bonds in conjugated compounds can be triggered by heat, light, or catalysts such as the addition of protons, transition metal ions, Lewis acids, and so on. Especially light-induced trans–cis photoisomerization of indigo and a variety of its derivatives has been widely studied.1,4−10 As general conclusion from these studies, it was found that although many of the derivatives undergo double-bond isomerization in the excited state, indigo itself does not. The distinctive ingredients inhibiting photoisomerization in indigo have been suggested to be the NH···O=C hydrogen bonds in the trans isomer, efficient excited-state proton transfer, and efficient nonradiative internal conversion quenching the photoisomerization channel.5,6,9,11 Indigo’s resistance to photoisomerization is key to its photostability as a pigment.12
As an alternative to photoisomerization, protoisomerization of indigo, i.e., trans-to-cis isomerization induced by protonation, has also been addressed. Indigo and its various derivatives were characterized experimentally in the condensed phase.13,14 Studies of imine derivatives of indigo in strong acids provide evidence for efficient trans-to-cis isomerization, but this could not be established for indigo itself.13 Theoretical studies of protoisomerization of indigo and some of its imine derivatives considering both the gas-phase and solution conditions indeed predict lower activation energies for the imine derivatives.15
Here we address the question of whether and to what extent isomerization to the cis-configuration occurs for protonated indigo, as well as of isoindigo (Scheme 1), when fully isolated in the gas phase of a mass spectrometer. As neutral species, indigo and isoindigo are characterized by a trans-configuration ground state, where the carbonyl groups on the two subunits are antiparallel to each other. Two NH···O=C hydrogen bonds stabilize the trans-configuration of the indigo molecule with respect to the cis-isomer. The absence of H-bonds in isoindigo brings cis- and trans-configurations closer in energy, although the trans-configuration is still lower in energy (vide infra).
Scheme 1. Schematic Showing Neutral (a) trans-Indigo and (b) Isoindigo.
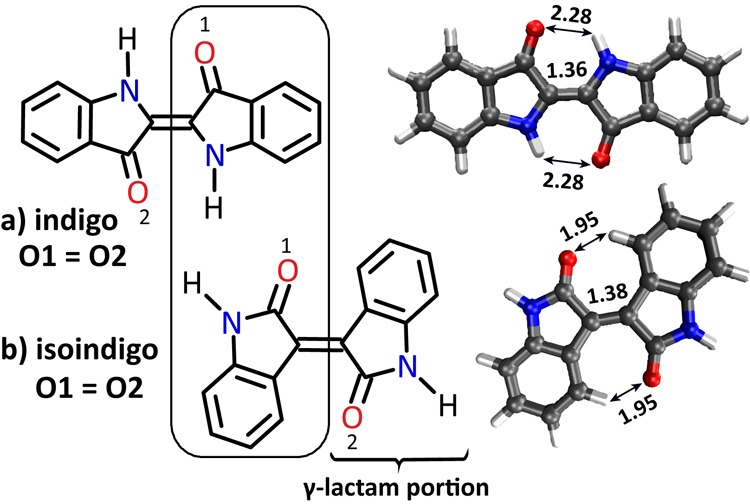
Isoindigo is a structural isomer of indigo and has a 5-membered cyclic amide (γ-lactam) arrangement. Oxygen atoms are identical (O1 = O2) in both isomers. DFT optimized structures are also shown with important bond distances (in Å) indicated.
Of the possible protonation sites (NH nitrogen and C=O oxygen atoms), the oxygen atom is preferred, and it is noted that in indigo as well as in isoindigo both carbonyl oxygens are symmetrically identical. Upon protonation on one of the oxygen atoms, the charge is delocalized over the conjugated system as suggested by the resonance structures shown in Scheme 2. The reduced double-bond character of the central C=C bond suggested by the resonance structures indicates that trans-to-cis isomerization may become more facile. In the cis-configuration, O-protonated indigo can form a proton bridge between the two carbonyl O atoms, providing additional stability to the cis-isomer. On the other hand, the trans-configuration is destabilized upon protonation due to the partial positive charge on the protonated carbonyl O atom, reducing the NH···O=C hydrogen bond strength. Computational investigations have indicated that in its protonated form cis-indigo is indeed lower in energy than trans-indigo, although the barrier to isomerization remains substantial (125 kJ mol–1).15
Scheme 2. Protonation on One of the Two Identical Oxygen Atoms in Indigo Allows for Mesomeric Structures Having a Single Bond Connecting the Two Subunits, Suggesting More Facile Trans-to-Cis Isomerization of the Protonated Species (Similar Resonance Structures Can Be Drawn for Isoindigo).

We investigate the molecular structures of the gaseous protonated dyes using infrared multiple-photon dissociation (IRMPD) spectroscopy16−18 in an ion-trap mass spectrometer coupled to the beamline of our infrared free electron laser FELIX. Gas-phase IR spectra are employed for structural identification by comparison with harmonic frequency calculations at the density functional theory (DFT) level. In addition, we employ wavelength selective IRMPD kinetics to estimate the relative cis- and trans-isomer abundances.
Experimental Section
IRMPD Spectroscopy
Protonated (iso)indigo ions are generated by electrospray ionization (ESI) and stored in a modified 3D quadrupole ion trap (QIT) mass spectrometer (Bruker, AmaZon Speed ETD, Bremen, Germany).19 Solutions containing 1:1 MeOH:H2O and about 1.0 μM of one of the dye molecules and about 0.1% of formic acid (to enhance protonation) are used for ESI. Ions are mass-isolated in the trap and irradiated with two pulses of tunable infrared radiation generated by the FELIX free electron laser (FEL) source.16 In these experiments, the FEL typically produces radiation in the form of 6 μs long macropulses at a 10 Hz repetition rate, which have ∼100 mJ of energy and a bandwidth of about 0.4% of the central frequency. Every macropulse consists of a series of micropulses of a few picoseconds, separated by a 1 ns time interval. Upon resonance of the FEL frequency with a vibrational transition of the stored ion, absorption of multiple photons occurs, aided by intramolecular vibrational redistribution (IVR),20 which raises the ion’s internal energy and eventually results in unimolecular dissociation. IR-frequency-dependent fragment and precursor ion intensities are monitored in the QIT-MS. The mass spectral data are then converted into an IR spectrum of the precursor ion by plotting the dissociation yield21−24 as a function of the FEL frequency:
Each data point in the IRMPD spectrum is obtained from six averaged mass spectra. The yield is linearly corrected for frequency-dependent variations in FEL pulse energy. The FEL frequency is tuned with a 3 cm–1 step size reconstructing the fingerprint IR spectra from 700 to 1800 cm–1. The FEL wavelength is calibrated by using a grating spectrometer.
In addition, isomer selective on-resonance IRMPD kinetics25,26 are measured to estimate the relative abundance of isomers in the ion population, which eventually yields the extent of protoisomerization. This method is described in detail in ref (25). The intensity of the precursor ion (protonated indigo or isoindigo) is measured as a function of the number of FEL pulses at selected IR frequencies. At an IR frequency where both trans- and cis-isomers absorb, all precursor ions should be dissociated if irradiated sufficiently long. As a result of a nonperfect overlap of the laser focus with the ion cloud, a small fraction of precursor ions survive even after 60 laser pulses (2% in the current experiments; see below). In contrast, irradiation at isomer-specific frequencies selectively depletes only the cis or trans isomeric ions. The decay of the ion intensity as a function of the number of laser pulses provides information about the relative abundance of isomers in the ion population.
Theoretical Modeling
For all calculations, the B3LYP/6-31++G(d,p) level of theory8,27−29 is chosen to optimize the geometry and to compute the harmonic frequencies employing Gaussian 09, revision D 01.30 To compare the computed (linear) IR spectra with IRMPD spectra, the calculated harmonic frequencies are scaled by a factor of 0.975, which is considered to be appropriate at this level of theory, to compensate for anharmonicity and basis set incompleteness.31,32 Computed IR spectra are convoluted with a 15 cm–1 full width at half-maximum (fwhm) Gaussian line shape function. The relative Gibbs free energies of the isomers are also considered for comparison. Transition-state (TS) geometries for the trans-to-cis isomerization are computed by using the opt=TS keyword and a single negative frequency is confirmed, corresponding to the torsional motion around the central CC bond of the molecule (vide infra).
Results and Discussion
Resonance Structures of Protonated Indigo and Isoindigo
Before analyzing the IRMPD spectrum, we first consider the consequences of protonation for the resonance structures in indigo and isoindigo, which can be considered as α,β-unsaturated carbonyl compounds. In particular, the carbonyl moieties are conjugated with the C=C bond connecting the two subunits of the dye. Scheme 2 shows that protonation at one of the (identical) carbonyl groups allows one to draw mesomeric structures having a single bond connecting the two subunits, which suggests not only that the charge is well delocalized over the entire molecule but also that the partial single-bond character may enhance trans-to-cis isomerization of the system. The calculations confirm the partial single-bond character of the central CC linkage by a lengthening of this bond of 0.01–0.02 Å as compared to the neutral molecule. Neutral trans-indigo is stabilized by 68 kJ mol–1 due to two N–H···O=C hydrogen bonds with respect to the cis isomer. In the protonated form, cis-indigo is stabilized by a shared-proton hydrogen bonding structure (O–H+···O). The absence of strong H-bonds in neutral isoindigo brings cis- and trans-configurations closer in energy, with trans being stabilized by 31 kJ mol–1.
For both indigo and isoindigo, protonation at one of the carbonyl oxygen atoms is expected to significantly affect the carbonyl stretching frequencies. For our IR spectroscopic investigation, the position of the C=O stretch frequency is therefore of special interest as it is expected to be a sensitive probe of the cis/trans structure of the system. If both trans and cis protonated dyes are present, we expect to observe two distinct C=O stretch frequencies.
Finally, we note that the N-protonated forms of indigo and isoindigo are computed to be +54 and +118 kJ mol–1 higher in energy than the O-protonated forms, respectively, and that their predicted IR spectra do not match with experiment (see the Supporting Information, Figure S1); we shall therefore not further consider these alternative protomers.
Protonated Indigo
Figure 1 shows the IRMPD spectrum of protonated indigo (m/z 263) from 600 to 1850 cm–1. It has been recorded by monitoring IR-induced fragments at m/z 262, 245, 235, 219, 217, 206, and 190 (Figure S2), which is consistent with observed dissociation channels in collision-induced dissociation (CID) mass spectra of protonated indigo.33,34 The dominant experimental IR bands are labeled with their IR frequency for assignment (Figure 1a). Computed linear-IR absorption spectra for cis- (Figure 1b) and trans-isomers (Figure 1c) of protonated indigo are also shown. A quick comparison of the computed and measured spectra shows that (i) in the 600–1550 cm–1 range the cis and trans spectra match roughly equally well to the experimental spectrum and (ii) the most striking differences are in the 1550–1800 cm–1 range. In the experimental spectrum, a low-intensity band is observed centered at 1725 cm–1, which is attributed to stretching of the unprotonated C=O group that is hydrogen bonded with the neighboring N–H group in the trans-isomer. Theory predicts this band with relatively low intensity at 1733 cm–1 in the spectrum of protonated trans-indigo. The observation of this band, though weak, suggests the presence of protonated indigo in its trans isomeric form.
Figure 1.
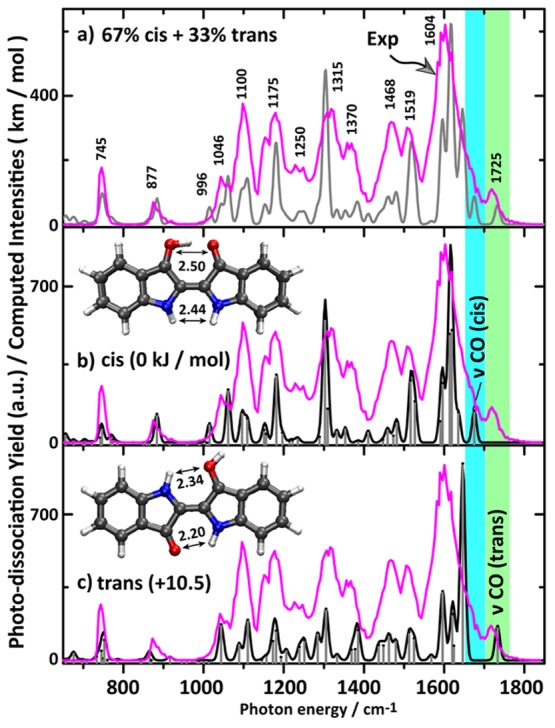
(a) Gas-phase IRMPD spectrum of protonated indigo (magenta trace) with band centers for the main experimental bands indicated. The experimental spectrum is compared with computed spectra for (b) the cis- and (c) trans-isomer. The gray trace in (a) represents the aggregate spectrum of cis- and trans-isomers assuming fractional populations as suggested by the kinetic measurements described in the text. Computed intensities in km mol–1 refer to the stick spectra. The optimized structures along with their relative Gibbs energies are shown. Atomic distances shown are in Å.
DFT predicts a similar C=O stretch band at 1675 cm–1 for the cis-isomer, but experimentally this band is not well resolved. The dominant experimental band is centered at 1604 cm–1, which clearly matches mostly with the cis-isomer due to a set of delocalized vibrations of CC stretching and OH+···O in-plane bending character. In addition, the theoretical bands of the cis-isomer at 1517 and 1527 cm–1 are attributed to C=O stretching of the protonated carbonyl and a ring vibration involving the 5- and 6-membered rings, which is coupled to the OH in-plane bending vibration; the computations match with the experimental band at 1519 cm–1. The experimental band at 1468 cm–1 is due to 6-membered-ring vibrations along with OH in-plane bending modes of the cis-isomer, which is predicted at 1459 cm–1, while the trans-isomer has a band at 1462 cm–1 due to a 6-membered-ring vibration alone. The strong experimental band at 1315 cm–1 also matches with a predicted band for the cis-isomer having mainly ring vibration character combined with CH and NH in-plane bending. Similar vibrations are present in the trans-isomer, but their intensities are not as high. The shoulder at 1370 cm–1 appears to have dominant contributions from the trans-isomer predicted at 1378 and 1385 cm–1 and attributed to ring vibrations coupled with NH and CH in-plane bending. The band at 1250 cm–1 also has more contribution from the trans-isomer showing predicted bands at 1239 and 1251 cm–1. Moreover, the predicted band for the cis-isomer at 1181 cm–1 due to a 6-membered ring vibration matches with the experiment at 1175 cm–1. The trans-isomer contributes with bands at 1181 and 1173 cm–1. The last dominant experimental band at 1100 cm–1 appears to represent contributions from both cis- (1109 cm–1) and trans-isomers (1110 cm–1), involving 6-membered-ring vibrations combined with CH in-plane bending. The predicted band at 1060 cm–1 for the cis-isomer due to a 5-membered-ring vibration coupled to NH in-plane bending matches the experimental band at 1060 cm–1, while the trans-isomer has a similar band at 1043 cm–1 that also matches the experiment at 1046 cm–1.
Apart from these dominant bands, there are relatively low-intensity experimental bands toward the low-frequency region of the IR spectrum. For instance, the OH in-plane bending mode at 1015 cm–1 for the cis-isomer is experimentally confirmed by the shoulder at 1000 cm–1. There is no obvious contribution from the trans-isomer to this band. The cis- and trans-isomers contribute roughly equally to the experimental band at 877 cm–1 due to computed intensities at 884 and 865 cm–1, respectively, with both bands being attributed to in-plane ring vibrations. The low-frequency experimental band at 745 cm–1 is the typical CH out-of-plane bending mode predicted at 752 cm–1 for the cis-isomer and at 746 cm–1 for the trans-isomer.
From this analysis of the spectrum, we conclude that both cis- and trans-species are present in the ion population and therefore that protoisomerization occurs in part for the ion population. From the kinetic analysis below (involving the 1725 cm–1 band diagnostic for the trans-isomer and the 1604 cm–1 band attributed to both isomers), we estimate a 65–70% cis plus 30–35% trans mixture. Combining the two predicted spectra in this ratio gives the gray trace in Figure 1a. The calculations show a H-bond distance in the cis-isomer of 1.48 Å (C=O···H+—O=C) indicative of a strong H-bond. The C=C distance (1.38 Å) is increased slightly by 0.015 Å as compared to neutral (trans) indigo. The increment of the C=C distance for the trans-isomer upon protonation is 0.02 Å.
Protonated Isoindigo
Figure 2 shows the IRMPD spectrum of protonated isoindigo (m/z 263), which has been recorded by monitoring the fragments at m/z 245, 235, 219, 217, and 190 (see Figure S2). Theoretical IR spectra of the cis- and trans-isomers are again overlaid with experiment in panels b and c for comparison. The two highest frequency IR bands in the observed spectrum at 1745 and 1686 cm–1 are assigned as the carbonyl C=O stretch bands of the trans- and cis-isomers, respectively. Hence, we conclude immediately that the IRMPD spectrum suggests again the coexistence of the two isomers. The experimental C=O stretch band for the cis-isomer appears broadened, possibly due to the strong hydrogen bond of the added proton with the unprotonated carbonyl group, forming a shared proton motif (O–H+···O).35−38 DFT calculations predict the carbonyl stretch bands in both isomers to be separated by 65 cm–1, close to the observed splitting. Absolute band positions are calculated at 1765 and 1700 cm–1 for trans and cis, respectively, showing a slight systematic shift. In contrast to protonated indigo, the band for the cis-isomer is well resolved. The trans-isomer of protonated isoindigo is computed to lie 75 kJ mol–1 higher in energy than the cis-isomer so that the presence of any trans-isomers must be due to kinetic trapping and a reasonable barrier between the two isomers. IRMPD kinetics experiments (vide infra) suggest that the trans-isomer contributes 20–25% to the ion population, where the other 75–80% is cis. Thus, isomerization from trans to cis indeed occurs upon protonation for the majority of the isoindigo molecules as evidenced by IRMPD spectroscopy.
Figure 2.
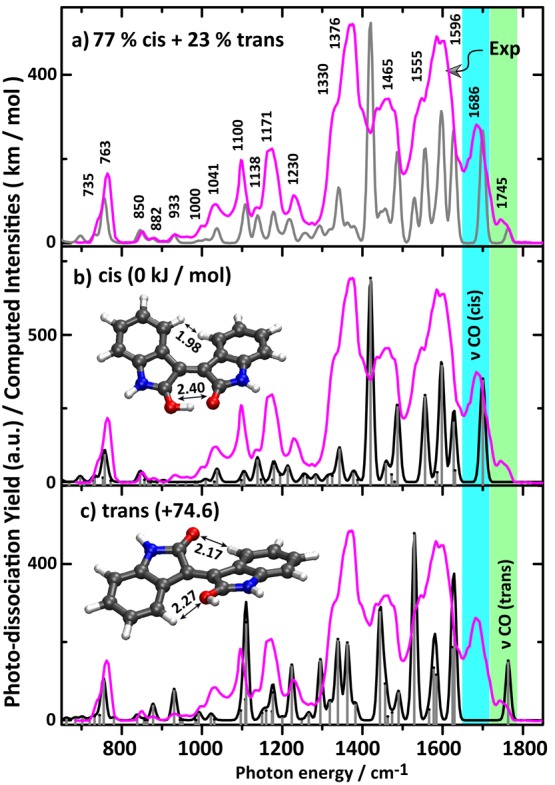
(a) Experimental IRMPD spectrum of protonated isoindigo (magenta trace). (b) Overlay of the experimental spectrum with the calculated IR spectrum for the cis-isomer and (c) with that of the trans-isomer. The gray trace in (a) represents a composite computed spectrum with 77% cis and 23% trans, as suggested by the kinetic measurements described in the text. Optimized structures along with the relative Gibbs energies are shown as well as some relevant atomic distances (in Å).
Upon analysis of the experimental spectrum in detail, the band centered at 1596 cm–1 with a red shoulder at 1555 cm–1 is due to three dominant bands of both cis- and trans-isomers as predicted by theory. The band centered at 1596 cm–1 is mostly due to the cis-isomer, in particular, attributed to the hydrogen-bonded OH in-plane-bending mode. The dominant band predicted for trans at 1529 cm–1 is the C=C stretch of the central CC bond; it is predicted at 1556 cm–1 for cis.
The experimental band at 1465 cm–1 is broadened and perhaps due to an overlap of bands at 1487 and 1419 cm–1 predicted for the cis-isomer and a band predicted at 1444 cm–1 for the trans-isomer. These bands have mainly ring stretching and in-plane CH bending character. The experimental bands at 1376 and 1330 cm–1 form the dominant feature in the experimental spectrum, but they are not well reproduced by the computed spectrum for the cis-isomer, which we believe to be dominant in the ion population. In part, this band may be due to the trans-isomer which has predicted bands at 1362, 1339, and 1295 cm–1 having ring vibration character along with CH in-plane bending. However, the main contribution may be due to the 1419 cm–1 predicted band in the cis-isomer; this band is due to the O–H stretch vibration of the shared-proton (OH+···O), which we suspect to behave particularly anharmonically due to the small OO distance of 2.40 Å in protonated cis-isoindigo. The larger OO distance in protonated cis-indigo of 2.50 Å reduces the anharmonic behavior of this band, placing it closer to 1600 cm–1 and giving a better match with the harmonic calculations. Weaker bands predicted near 1377 and 1342 cm–1 may give further contributions to this strong feature.
Experimental features at 1230, 1171, and 1100 cm–1 are not accurately reproduced but are assumed to be due to the overlap of a large number of weaker bands predicted in this range for both cis- and trans-isomers. They have largely in-plane ring deformation and in-plane CH, NH, and OH bending character. The experimental band 1041 cm–1 is a well-resolved band which appears to be mainly due to the cis-isomer, which features a predicted band at 1038 cm–1 with ring vibration character. The observed spectrum below 1000 cm–1 matches particularly well with that predicted for the cis-isomer, further confirming its dominant contribution to the ion population. The band observed at 850 cm–1 is attributed to a ring-breathing vibration of the cis-isomer, and the intense band at 763 cm–1 is in good agreement with the typical CH out-of-plane bending mode predicted at 758 cm–1 for cis (755 cm–1 for trans). The minor population of trans-isoindigo is evidenced by a weak feature at 933 cm–1 which according to the calculations is exclusively due to the trans-isomer. Overall, the spectrum in this range is well reproduced by the composite spectrum of 77% cis and 23% trans protonated isoindigo.
The H-bond distance in the cis-isomer is about 1.32 Å (C=O···H+–O=C) while both O atoms are 2.40 Å apart, much shorter than in DNA base pairs,39 indicative of a strong H-bond.35−38 The central C=C bond length is 1.39 Å in the cis-isomer, which is similar to the bond length in the trans-isomer but 0.01 Å larger than the neutral.
Determination of Relative Ion Populations by Wavelength-Selective IR-Induced Dissociation Kinetics
IRMPD kinetics are used to probe the relative populations of the cis- and trans-isomers. First, a kinetic measurement on an IR frequency where both cis and trans absorb has been performed to determine the extent of overlap between the laser beam and the ion cloud in the ion trap. The CH out-of-plane bending modes near 750 cm–1 were selected for this experiment (see overlap at this frequency for cis- and trans-isomers in Figures 1 and 2). Figure 3 shows that for both indigo and isoindigo the precursor ion intensities level off after 20 pulses, leaving about 2% of the ions undissociated in the trap. A similar result is obtained for indigo exciting the ions at the most intense band at 1604 cm–1.
Figure 3.

(a) Photofragmentation decay of mass-isolated protonated isoindigo as a function of the number of IR FEL pulses at fixed frequencies diagnostic for the trans-isomer (1745 cm–1, ●) and for the cis-isomer (1686 cm–1, ★, ☆: replicate data). Data points indicated with ▷ were taken with the laser set at 763 cm–1, where both isomers absorb leading to complete depletion of the precursor ion population. (b) Results of similar experiments for protonated indigo, performed at frequencies resonant with trans (1725 cm–1, ●, ○: replicate), and with both cis + trans (1604 cm–1, ★, ☆ replicate, and 745 cm–1, ▷). Each data point is obtained from 10 averaged mass spectra. Solid curves are single-exponential decay fits to a limited range of the experimental data points, except for trans-isoindigo where all data points are included.
Next, kinetic measurements are performed at IR frequencies which exclusively probe the cis- or trans-isomer of each of the molecules, using the C=O stretch bands. For isoindigo, kinetic measurements at 1745 cm–1, probing trans, and at 1686 cm–1, probing cis, are displayed in Figure 3. After irradiation with 40 pulses a steady state appears to be reached, suggesting that about 15% of the ions are trans and 85% cis (taking into account that 2% of the ions cannot be dissociated at all). However, the curve for cis shows a plateau around 10 pulses, indicative of a double-exponential decay likely due to another species dissociating at a slower rate upon irradiation at 1686 cm–1.
In Figure 3, the normalized precursor ion intensity on the y-axis is defined as the precursor intensity divided by the sum of fragment and precursor intensities. All fragment ions identified as belonging to isoindigo (see Figure S2) are included. Plotting the precursor intensity without normalization gives the same curves, albeit more noisy, with the plateau clearly visible. This suggest that the unknown species shares some or all of the same fragment ions. The unknown species appears not to be present in the isoindigo sample as was verified with HPLC measurements.
In a control experiment, ESI of pure 1:1 MeOH:H2O solvent (without isoindigo) and about 0.1% of formic acid shows that a background ion at m/z 263, the mass of isoindigo, is present in our mass spectrometers. The ion intensity is 50–100 times lower than that of isoindigo. We have recorded the IRMPD spectrum of this background ion (shown in Figure S3). The dominant fragment ion is m/z 235, the same as for isoindigo. The spectrum consists of a broad structured band between 650 and 1300 cm–1, and some weak bands between 1300 and 1720 cm–1. Note that our measurements have been repeated over a period of 10 months on two mass spectrometers; the origin of this contamination is currently unknown.
The presence of this background ion can explain the observed curves for isoindigo (Figure 3a). At 763 cm–1, all ions, cis- and trans-indigo, and the background ions, absorb the IR radiation, and within 5–10 laser pulses all ions are dissociated. The background ion does not absorb at 1745 cm–1, the frequency diagnostic for trans-isoindigo, indicating that about 15% of the total ion population is trans-isoindigo. Note that after about two pulses 50% of the trans-isomers have dissociated. At the diagnostic cis-isoindigo frequency (1686 cm–1), the background ion has a weak overlapping absorption band. The observed dissociation yield shows that <3% of the background ions dissociate per IR laser pulse at this frequency. The steep decay of the kinetics curve recorded at 1686 cm–1 (Figure 3a) can then be attributed almost exclusively to cis-isoindigo. This suggest the presence of about 65–70% cis-isoindigo in the ion population, of which 50% have dissociated after 1–2 pulses (as for the trans-isomer). About 20% of the ion population can be attributed to the background ions, which dissociate at a much slower rate, explaining the plateau in the kinetics plot.
These results indicate that about 20–25% of the protonated isoindigo population are in the trans-configuration and 75–80% are cis. In the top panel of Figure 2, the experimental IR spectrum is compared to a composite calculated spectrum based on this cis/trans ratio in the ion population. Note that although 20% of the total ion population is due to the contaminant ions, they have a negligible contribution to the experimental IRMPD spectrum, which was recorded with two pulses of irradiation, thus nearly avoiding any dissociation of the contaminant ions.
For protonated indigo, the experimental situation is different since only the trans-isomer has a well-resolved carbonyl band at 1725 cm–1 (see Figure 1). The kinetics plot recorded at this frequency suggests that about 40% of the ion population is present as trans-indigo. However, the contaminant background ions show a low, but non-negligible, IRMPD intensity (see Figure S3), and they are responsible for the barely visible plateau around 10 laser pulses in the kinetics plot. The plateau suggests that about 20% of the ions are protonated trans-indigo and 20% of the ions are contaminant ions. The kinetics plots recorded at 745 and 1604 cm–1 have contributions from both trans- and cis-indigo and the contaminant ions and cannot be used to estimate the fractional cis-indigo population. Therefore, we tentatively attribute the remaining 60% of the ion population to cis-indigo. These results indicate that for protonated indigo about 30–35% are in the trans configuration and 65–70% are cis. Again, the contaminant ion does not contribute significantly to the IRMPD spectrum of Figure 1, since only a minute fraction undergoes dissociation upon irradiation with two laser pulses.
Transition-State Calculations
Figure 4 shows the results of transition-state (TS) calculations for trans-to-cis isomerization for protonated indigo and isoindigo. A singlet electronic state is considered at the TS, and the C=C bond rotation barrier represents the rate-limiting step; rotation of the OH group is disregarded here. Extensive TS calculations on indigo and some of its derivatives have been reported previously,15 and our theoretical method reproduces these TS geometries. The barrier to rotation about the central C=C bond is estimated to be 141 kJ mol–1 for protonated trans-indigo in the gas phase, while the reported value in solution is 125 kJ mol–1.15
Figure 4.
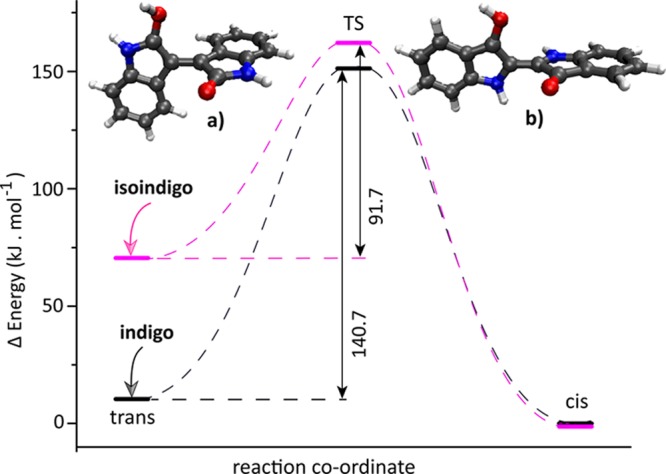
Computed TS barriers for trans-to-cis isomerization and optimized TS geometries of protonated isoindigo (a) and indigo (b). Values given for the TS barriers are in kJ mol–1 relative to the trans-form of the protonated molecules. For the protonated systems, unlike for the neutral molecules, cis is the minimum-energy isomer. Although both cis-isomers are positioned at the same energy in the plot, protonated cis-isoindigo is 46.3 kJ mol–1 more stable than protonated cis-indigo as a consequence of the stronger shared-proton interaction induced by the smaller OO distance (see structures in Figures 1 and 2).
From a thermochemical viewpoint, trans-to-cis protoisomerization can be well-understood from the fact that the energetic ordering of trans- and cis-isomers inverts upon protonation (for both indigo and isoindigo). In their neutral forms, trans-indigo is higher in energy (+22 kJ mol–1) than trans-isoindigo (Scheme 1).
The relative H-bond stabilization of cis- and trans-isomers is altered after protonation, destabilizing the trans-isomer of both systems. Protonated trans-isoindigo destabilizes more, and the geometry also distorts to become nonplanar, whereas indigo retains its planar geometry. In protonated trans-indigo, the H-bond between the nonprotonated C=O and N–H shortens by 0.08 Å, whereas the other H-bond increases by 0.06 Å. On the other hand, in protonated trans-isoindigo, both H-bonds are increased significantly (by 0.22 Å for the nonprotonated C=O and C–H and 0.32 Å for the protonated C=O and C–H) as a consequence of the nonplanar structure (Figure 2). The relatively weak H-bonds and nonplanar geometry result in a significant destabilization but simultaneously in a relatively low trans-to-cis TS barrier of +92 kJ mol–1. On the other hand, destabilization of protonated indigo is limited, and the barrier for trans-to-cis isomerization is significantly higher (+141 kJ mol–1).
In addition, the lone-pair electrons on both nitrogen atoms in indigo may participate in conjugation (delocalization), reinforcing the central C=C bond and hence the planar structure. In isoindigo, this effect is smaller because the nitrogen atoms are further away from the central C=C bond. In this respect, it is also interesting to note that in the γ-lactam arrangement of isoindigo (Scheme 1) the nitrogen lone-pair electrons can participate in resonance with the C=O group, shifting the C=O stretch frequency to higher frequencies than in indigo (1745 cm–1 in protonated isoindigo versus 1725 cm–1 in protonated indigo).
The computed TS geometry of isoindigo has a nearly perpendicular arrangement with a C–C=C–C dihedral angle of 88°; in protonated indigo, the dihedral angle at the TS is about 82°. Despite the high TS barrier, indigo is clearly observed to undergo protoisomerization in our experiments. The conversion from trans to cis involves the breaking of existing H-bonds upon rotation around the central C=C bond as well as rotation of the −O–H group in the required direction to make a new H-bond with the other carbonyl.15 The rate-limiting step is the rotation about the central CC bond, which relies on its reduced double-bond character upon protonation (Scheme 2). Experimentally, we clearly observe protonation-induced double-bond isomerization in both indigo and isoindigo. Moreover, our isomer population analysis indicates that conversion to the cis-isomer is more efficient for isoindigo than for indigo, which is qualitatively corroborated by the difference in computed TS energies for the two systems.
Conclusion
IRMPD spectra of protonated indigo and isoindigo have been measured by using a quadrupole ion trap mass spectrometer coupled to the tunable infrared free electron laser source FELIX. Comparisons of the experimental spectra with DFT computations led to the identification of two coexisting isomers for both dyes: cis being the minimum-energy isomer and trans being the ground state in the neutral molecule. As the starting sample material has the neutral molecule in the trans-form, protoisomerization from trans to cis is confirmed for both species.
To quantify the ratio between the cis- and trans-isomers present in the mixture, on-resonance IRMPD kinetics are performed on diagnostic IR bands. This provides an estimate for the relative abundances of 65–70% cis and 30–35% trans for protonated indigo and 75–80% cis and 20–25% trans for isoindigo. Thus, isoindigo appears to be somewhat more prone to protoisomerization than indigo, which is qualitatively explained by significant differences in the DFT computed isomerization barriers. The remaining fraction of trans-isomers observed in the spectra are likely kinetically trapped.
Acknowledgments
We gratefully acknowledge the Nederlandse Organisatie voor Wetenschappelijk Onderzoek (NWO) for the support of the FELIX Laboratory and the FELIX staff for their assistance. Financial support for this project was provided by NWO Chemical Sciences (CW) under VICI project 724.011.002 and TTW OTP project 15769. We also thank NWO Physical Sciences (EW) and the SurfSARA Supercomputer Centre for providing the computational time and resources (grant 17603).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jpca.9b06858.
Additional figures showing calculated spectra for N-protonated indigo and isoindigo (Figure S1), MS/MS spectra upon collision-induced and IR-induced dissociation (Figure S2), and the IRMPD spectrum of an as yet unknown minor contamination having m/z 263 (Figure S3); optimized geometries for all structures discussed (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Wyman G. M. The Cis-Trans Isomerization of Conjugated Compounds. Chem. Rev. 1955, 55, 625–657. 10.1021/cr50004a001. [DOI] [Google Scholar]
- Feringa B. L. The Art of Building Small: From Molecular Switches to Molecular Motors. J. Org. Chem. 2007, 72, 6635–6652. 10.1021/jo070394d. [DOI] [PubMed] [Google Scholar]
- Glowacki E. D.; Voss G.; Sariciftci N. S. 25th Anniversary Article: Progress in Chemistry and Applications of Functional Indigos for Organic Electronics. Adv. Mater. 2013, 25, 6783–6800. 10.1002/adma.201302652. [DOI] [PubMed] [Google Scholar]
- Wyman G. M.; Brode W. R. The Relation between the Absorption Spectra and the Chemical Constitution of Dyes XXII. Cis-Trans Isomerism in Thioindigo Dyes. J. Am. Chem. Soc. 1951, 73, 1487–1493. 10.1021/ja01148a023. [DOI] [Google Scholar]
- Brode W. R.; Pearson E. G.; Wyman G. M. The Relation between the Absorption Spectra and the Chemical Constitution of Dyes. XXVII. Cis-Trans Isomerism and Hydrogen Bonding in Indigo Dyes. J. Am. Chem. Soc. 1954, 76, 1034–1036. 10.1021/ja01633a033. [DOI] [Google Scholar]
- Haggmark M. R.; Gate G.; Boldissar S.; Berenbeim J.; Sobolewski A. L.; de Vries M. S. Evidence for Competing Proton-Transfer and Hydrogen-Transfer Reactions Inthe S1 State of Indigo. Chem. Phys. 2018, 515, 535–542. 10.1016/j.chemphys.2018.09.027. [DOI] [Google Scholar]
- Brode W. R.; Gould J. H.; Wyman G. M. The Relation between the Absorption Spectra and the Chemical Constitution of Dyes. XXV. Phototropism and Cis-Trans Isomerism in Aromatic Azo Compounds. J. Am. Chem. Soc. 1952, 74, 4641–4646. 10.1021/ja01138a059. [DOI] [Google Scholar]
- Jacquemin D.; Preat J.; Wathelet V.; Perpète E. A. Substitution and Chemical Environment Effects on the Absorption Spectrum of Indigo. J. Chem. Phys. 2006, 124, 074104. 10.1063/1.2166018. [DOI] [PubMed] [Google Scholar]
- Kobayashi T.; Rentzepis P. M. On the Picosecond Kinetics and Photostability of Indigo and 6,6′-Dimethoxyindigo. J. Chem. Phys. 1979, 70, 886–892. 10.1063/1.437479. [DOI] [Google Scholar]
- Giuliano C. R.; Hess L. D.; Margerum J. D. Cis-Trans Isomerization and Pulsed Laser Studies of Substituted Indigo Dyes. J. Am. Chem. Soc. 1968, 90, 587–594. 10.1021/ja01005a600. [DOI] [Google Scholar]
- Moreno M.; Ortiz-Sánchez J. M.; Gelabert R.; Lluch J. M. A Theoretical Study of the Photochemistry of Indigo in Its Neutral and Dianionic (Leucoindigo) Forms. Phys. Chem. Chem. Phys. 2013, 15, 20236–20246. 10.1039/c3cp52763h. [DOI] [PubMed] [Google Scholar]
- Miliani C.; Romani A.; Favaro G. A Spectrophotometric and Fluorometric Study of Some Anthraquinoid and Indigoid Colorants Used in Artistic Paintings. Spectrochim. Acta, Part A 1998, 54, 581–588. 10.1016/S1386-1425(97)00240-0. [DOI] [Google Scholar]
- Nicholls-Allison E. C.; Nawn G.; Patrick B. O.; Hicks R. G. Protoisomerization of Indigo Di-and Monoimines. Chem. Commun. 2015, 51, 12482–12485. 10.1039/C5CC04492H. [DOI] [PubMed] [Google Scholar]
- Brode W. R.; Wyman G. M. The Relation between the Absorption Spectra and the Chemical Constitution of Dyes. XXIV. Absorption Spectra of Some Thioindigo Dyes in Sulfuric Acid. J. Am. Chem. Soc. 1951, 73, 4267–4270. 10.1021/ja01153a070. [DOI] [Google Scholar]
- Hajjar L.; Hicks R. G.; Zeng T. A Computational Study of the Protoisomerization of Indigo and Its Imine Derivatives. J. Phys. Chem. A 2016, 120, 7569–7576. 10.1021/acs.jpca.6b07838. [DOI] [PubMed] [Google Scholar]
- Oomens J.; Sartakov B. G.; Meijer G.; Von Helden G. Gas-Phase Infrared Multiple Photon Dissociation Spectroscopy of Mass-Selected Molecular Ions. Int. J. Mass Spectrom. 2006, 254, 1–19. 10.1016/j.ijms.2006.05.009. [DOI] [Google Scholar]
- Polfer N. C. Infrared Multiple Photon Dissociation Spectroscopy of Trapped Ions. Chem. Soc. Rev. 2011, 40, 2211–2221. 10.1039/c0cs00171f. [DOI] [PubMed] [Google Scholar]
- Rijs A. M.; Oomens J. IR Spectroscopic Techniques to Study Isolated Biomolecules. Top. Curr. Chem. 2014, 364, 1–42. 10.1007/128_2014_621. [DOI] [PubMed] [Google Scholar]
- Martens J.; Berden G.; Gebhardt C. R.; Oomens J. Infrared Ion Spectroscopy in a Modified Quadrupole Ion Trap Mass Spectrometer at the FELIX Free Electron Laser Laboratory. Rev. Sci. Instrum. 2016, 87, 103108. 10.1063/1.4964703. [DOI] [PubMed] [Google Scholar]
- Lehmann K.; Scoles G.; Pate B. Intramolecular Dynamics from Eigenstate-Resolved Infrared Spectra. Annu. Rev. Phys. Chem. 1994, 45, 241–274. 10.1146/annurev.pc.45.100194.001325. [DOI] [Google Scholar]
- Lucas B.; Gregoire G.; Lemaire J.; Maitre P.; Glotin F.; Schermann J.; Desfrancois C. Infrared Multiphoton Dissociation Spectroscopy of Protonated N-Acetyl-Alanine and Alanyl-Histidine. Int. J. Mass Spectrom. 2005, 243, 105–113. 10.1016/j.ijms.2005.01.002. [DOI] [Google Scholar]
- Gao J.; Berden G.; Rodgers M. T.; Oomens J. Interaction of Cu+ with Cytosine and Formation of i-Motif-Like C–M+–C Complexes: Alkali Versus Coinage Metals. Phys. Chem. Chem. Phys. 2016, 18, 7269–7277. 10.1039/C6CP00234J. [DOI] [PubMed] [Google Scholar]
- Prell J. S.; O’Brien J. T.; Williams E. R. IRPD Spectroscopy and Ensemble Measurements: Effects of Different Data Acquisition and Analysis Methods. J. Am. Soc. Mass Spectrom. 2010, 21, 800–809. 10.1016/j.jasms.2010.01.010. [DOI] [PubMed] [Google Scholar]
- Berden G.; Derksen M.; Houthuijs K. J.; Martens J.; Oomens J. An Automatic Variable Laser Attenuator for IRMPD Spectroscopy and Analysis of Power-Dependence in Fragmentation Spectra. Int. J. Mass Spectrom. 2019, 443, 1–8. 10.1016/j.ijms.2019.05.013. [DOI] [Google Scholar]
- Prell J. S.; Chang T. M.; Biles J. A.; Berden G.; Oomens J.; Williams E. R. Isomer Population Analysis of Gaseous Ions from Infrared Multiple Photon Dissociation Kinetics. J. Phys. Chem. A 2011, 115, 2745–2751. 10.1021/jp2004166. [DOI] [PubMed] [Google Scholar]
- Patrick A. L.; Cismesia A. P.; Tesler L. F.; Polfer N. C. Effects of Esi Conditions on Kinetic Trapping of the Solution-Phase Protonation Isomer of p-Aminobenzoic Acid in the Gas Phase. Int. J. Mass Spectrom. 2017, 418, 148–155. 10.1016/j.ijms.2016.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becke A. D. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. 10.1063/1.464913. [DOI] [Google Scholar]
- Becke A. D. Becke’s Three Parameter Hybrid Method Using the LYP Correlation Functional. J. Chem. Phys. 1993, 98, 5648–5652. 10.1063/1.464913. [DOI] [Google Scholar]
- Lee C.; Yang W.; Parr R. G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B: Condens. Matter Mater. Phys. 1988, 37, 785. 10.1103/PhysRevB.37.785. [DOI] [PubMed] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, 2013.
- Laury M. L.; Carlson M. J.; Wilson A. K. Vibrational Frequency Scale Factors for Density Functional Theory and the Polarization Consistent Basis Sets. J. Comput. Chem. 2012, 33, 2380–2387. 10.1002/jcc.23073. [DOI] [PubMed] [Google Scholar]
- Scott A. P.; Radom L. Harmonic Vibrational Frequencies: An Evaluation of Hartree–Fock, Møller–Plesset, Quadratic Configuration Interaction, Density Functional Theory, and Semiempirical Scale Factors. J. Phys. Chem. 1996, 100, 16502–16513. 10.1021/jp960976r. [DOI] [Google Scholar]
- Zou P.; Koh H. L. Determination of Indican, Isatin, Indirubin and Indigotin in Isatis Indigotica by Liquid Chromatography/Electrospray Ionization Tandem Mass Spectrometry. Rapid Commun. Mass Spectrom. 2007, 21, 1239–1246. 10.1002/rcm.2954. [DOI] [PubMed] [Google Scholar]
- Puchalska M.; Połeć-Pawlak K.; Zadrożna I.; Hryszko H.; Jarosz M. Identification of Indigoid Dyes in Natural Organic Pigments Used in Historical Art Objects by High-Performance Liquid Chromatography Coupled to Electrospray Ionization Mass Spectrometry. J. Mass Spectrom. 2004, 39, 1441–1449. 10.1002/jms.728. [DOI] [PubMed] [Google Scholar]
- Baughcum S. L.; Duerst R. W.; Rowe W. F.; Smith Z.; Wilson E. B. Microwave Spectroscopic Study of Malonaldehyde (3-Hydroxy-2-Propenal). 2. Structure, Dipole Moment, and Tunneling. J. Am. Chem. Soc. 1981, 103, 6296–6303. 10.1021/ja00411a005. [DOI] [Google Scholar]
- Ceccarelli C.; Jeffrey G.; Taylor R. A Survey of OH···O Hydrogen Bond Geometries Determined by Neutron Diffraction. J. Mol. Struct. 1981, 70, 255–271. 10.1016/0022-2860(81)80112-3. [DOI] [Google Scholar]
- Ubbelohde A.; Gallagher K. Acid-Base Effects in Hydrogen Bonds in Crystals. Acta Crystallogr. 1955, 8, 71–83. 10.1107/S0365110X55000340. [DOI] [Google Scholar]
- Ubbelohde A.; Woodward I. Structure and Thermal Properties of Crystals, VI. The Role of Hydrogen Bonds in Rochelle Salt. Proc. R. Soc. London A 1946, 185, 448–465. 10.1098/rspa.1946.0030. [DOI] [PubMed] [Google Scholar]
- FonsecaGuerra C.; Bickelhaupt F. M.; Snijders J. G.; Baerends E. J. The Nature of the Hydrogen Bond in DNA Base Pairs: The Role of Charge Transfer and Resonance Assistance. Chem. - Eur. J. 1999, 5, 3581–3594. . [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.