

Abstract
Deregulation of cellular metabolism is well established in cancer. The mitochondria are dynamic organelles and act as the center stage for energy metabolism. Central to mitochondrial regulatory network is peroxisome proliferator‐activated receptor γ coactivator 1a (PGC‐1α), which serves as a master regulator of mitochondrial proliferation and metabolism. The activity and stability of PGC‐1α are subject to dynamic and versatile posttranslational modifications including phosphorylation, ubiquitination, methylation and acetylation in response to metabolic stress and other environmental signals. In this review, we describe the structure of PGC‐1α. Then, we discuss recent advances in the posttranslational regulatory machinery of PGC‐1α, which affects its transcriptional activity, stability and organelle localization. Furthermore, we address the important roles of PGC‐1α in tumorigenesis and malignancy. Finally, we also mention the clinical therapeutic potentials of PGC‐1α modulators. A better understanding of the elegant function of PGC‐1α in cancer progression could provide novel insights into therapeutic interventions through the targeting of PGC‐1α signaling.
Keywords: PGC‐1α, posttranslational modifications, cancer metabolism
Introduction
Deregulation of cellular metabolism is well established in many diseases including diabetes, neurodegeneration, cardiovascular disease, muscular dystrophies and cancer.1, 2, 3, 4 The complex regulatory machinery controlling mitochondrial bioenergetic homeostasis represents one of the core contents of metabolism. Central to this regulatory network is the peroxisome proliferator‐activated receptor γ coactivator 1 (PGC‐1) family, which is composed of three members, PGC‐1α, PGC‐1β and PGC‐1‐related coactivator (PRC). A high amino acid sequence homology is present within the amino (N)‐ and carboxy (C)‐terminal ends of the three‐member proteins5, 6 (Fig. 1). PGC‐1α has emerged as a master regulator of mitochondrial biogenesis and energy expenditure and plays a central role in adapted metabolic responses and PGC‐1βis responsible for the maintenance of basal mitochondrial function.2 PGC‐1α and PGC‐1βare highly expressed in oxidative tissues, such as the heart, kidney, muscle, brown adipose tissue (BAT) and brain. In contrast, the tissue‐specific expression level of PRC corresponds with the proliferative status of the cell and is considered to be a regulator of cell growth.5
Figure 1.
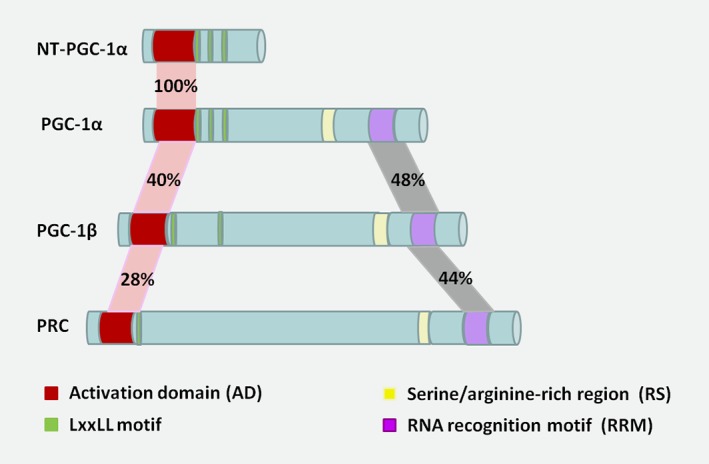
Schematic domain structure of PGC‐1 family and isoforms. Sequence homology of the transcriptional coactivator PGC‐1 family members, including PGC‐1α, PGC‐1β, PRC and a short isoform, NT‐PGC‐1α. PGC‐1β shares sequence identity with PGC‐1α in AD (40%) and RRM (48%), respectively. As to PRC, the homology of each domain is 28 and 44%. As to NT‐PGC‐1α, the homology of AD domain with PGC‐1α is 100%. For each domain structure, the left corresponds to the N‐terminal of a protein, and the right to the C‐terminal. [Color figure can be viewed at wileyonlinelibrary.com]
PGC‐1α is the most well‐studied member of the PGC‐1 family coactivators. PGC‐1α is reportedly induced in adaptive thermogenesis in BAT, gluconeogenesis in liver, fiber‐type switching in muscle, fatty acid oxidation in heart and clock gene expression in liver and muscle.2, 7 Both the expression and activity of PGC‐1α are precisely modulated to maintain its temporal and tissue‐specific functions in response to diverse metabolic demands. Posttranslational modifications of PGC‐1α affect different regions of the protein, allowing for a quick, multifaceted and flexible regulation of PGC‐1α activity that is required for environmental stress responses. Many of these modifications are clearly associated with a specific structural or functional mechanism.8, 9, 10, 11
Here, we describe the structure of PGC‐1α and then we discuss the recent advances in the posttranslational regulatory machinery of PGC‐1α, which affects its transcriptional activity, stability and organelle localization. Furthermore, we address the important roles of PGC‐1α in tumorigenesis and malignancy. Finally, we also provide an overview of the clinical therapeutic potentials of PGC‐1α modulators.
Structure of PGC‐1α
PGC‐1α has a complex structure with distinctive domains that orchestrate its multifaceted biological and physiological functions (Fig. 1). The N‐terminal of PGC‐1α harbors a highly conserved autonomous activation domain (AD) and a leucine‐rich motif (LxxLL, also called NR boxes) that binds to nuclear hormone receptors (NR). The transcriptional AD provides a docking platform for PGC‐1α to recruit coregulators such as histone acetyltransferase protein complexes (CBP/p300) and steroid receptor coactivator 1 (SRC‐1). This implies that PGC‐1α is a typical coactivator that stimulates rate‐limiting steps in transcription initiation. In contrast, two unique conserved protein motifs are present in the C terminus of PGC‐1α, including a short serine/arginine‐rich region (RS) and an RNA recognition motif (RRM). The central and C‐terminal domains mediate the interaction with other transcription factors, such as peroxisome proliferator‐activated receptor‐γ(PPARγ), nuclear respiratory factors‐1(NRF1), myocyte enhancer factor 2C (MEF2C) and forkhead box class‐O (FOXO1).5, 12 Different functional PGC‐1α splice variants have been reported.11, 13 NT‐PGC‐1α (amino acids 1–270) is a truncated alternative spliced isoform of PGC‐1α that retains the N‐terminal AD and interacts with nuclear hormone receptors as well (Fig. 1). In contrast to PGC‐1α, NT‐PGC‐1α is relatively stable and is predominantly retained in the cytoplasm because of its lack of a C‐terminal domain.11, 14
Posttranslational Modification of PGC‐1α
Numerous posttranslational modifications impact PGC‐1α.9, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25 Among them, phosphorylation, acetylation, methylation and ubiquitination are predominantly responsible for the transcriptional activity and stability of PGC‐1α (Fig. 2).
Figure 2.

Posttranslational modifications of PGC‐1α. The mainly identified sites for posttranslational modifications, including phosphorylation, acetylation, methylation and ubiquitination of PGC‐1α or NT‐PGC‐1α, are mapped in the protein structure. [Color figure can be viewed at wileyonlinelibrary.com]
Phosphorylation of PGC‐1α
The adenosine monophosphate (AMP)‐activated protein kinase (AMPK), p38 mitogen‐activated protein kinase (MAPK), Akt, S6 kinase and glycogen synthase kinase 3β (GSK3β) are the best‐characterized protein kinases targeting PGC‐1α for phosphorylation.16, 26
Phosphorylation can promote the activation of PGC‐1α. For example, AMPK binds to and activates PGC‐1α in skeletal muscle by direct phosphorylation on Thr177 and Ser538. This increases the transcriptional activity of PGC‐1α and is required for AMPK‐induced gene expression of glucose transporter 4 (GLUT4), mitochondrial genes, as well as PGC‐1α itself.20 The p38 MAPK phosphorylates PGC‐1α on Thr 262, Ser265 and Thr298 resulting in increased protein stability.16
On the other hand, like a two‐edged sword, phosphorylation can also decrease PGC‐1α activity as well. The C‐terminus of PGC‐1α contains multiple potential sites of Akt phosphorylation (RXRXXS/T, where X is any amino acid). Akt suppresses both gluconeogenesis and fatty acid oxidation (FAO) through its phosphorylation and inhibition of PGC‐1α.19 Similarly, when induced by nutrient signals, the gluconeogenic genes program is also inhibited through the phosphorylation of PGC‐1α at Ser568 and Ser572 by serine/threonine kinase S6K1. However, these phosphorylations do not affect the stability of PGC‐1α or other gene programs mediated by PGC‐1α, such as mitochondrial electron transport and FAO.15 Because all the sites phosphorylated by Akt or S6K1 are located within the RS region (amino acids 551–635) speculating that phosphorylation of the RS domain might attenuate the transcriptional activity of PGC‐1α is reasonable.
Unlike the full‐length‐PGC‐1α, NT‐PGC‐1α is likely to be exported to the cytoplasm by interacting with the nuclear protein, chromosomal maintenance 1 (also known as exportin 1, CRM1), through its LxxLL domain (nuclear export sequence). This process can be blocked by protein kinase A‐dependent phosphorylation of NT‐PGC‐1α on Ser194, Ser241 and Thr256. The phosphorylation of NT‐PGC‐1α by PKA not only inhibits CRM1‐mediated export from the nucleus but also affects its transcriptional potential.11
Acetylation modification of PGC‐1α
PGC‐1α is controlled by acetylation to modulate its ability to recruit chromatin‐remodeling complex and initiate gene transcription. PGC‐1α is dynamically acetylated and inhibited by GCN5 (general control nonrepressed protein 5) when deacetylated and activated by SIRT1 (silent information regulator 2 homolog 1).
Although PGC‐1α can interact with several acetyltransferases including GCN5, p300, SRC‐1 and SRC‐3, only GCN5 is characterized in its acetylation and inhibition of PGC‐1α.17 GCN5 is the first discovered histone lysine acetyltransferase, linking histone acetylation to transcriptional activation.27 Besides histone proteins, GCN5 can also acetylate nonhistone proteins, such as PGC‐1α, which harbors multiple acetylation sites spanning the whole sequence of the protein.16 The acetylation status of PGC‐1α could be modulated by GCN5 depending on the amounts of nuclear acetyl‐CoA. In mammals, acetyl‐CoA can be generated from TCA (tricarboxylic acid cycle)‐derived citrate by ACL (ATP citrate lyase). Considering that GCN5, like all other acetyltransferases, requires acetyl‐CoA as a substrate for the acetylation reaction, ACL might connect energy balance to GCN5 and affect the acetylation status of PGC‐1α by controlling the nuclear production of acetyl‐CoA.28
SRC‐3 facilitates PGC‐1α acetylation in muscle and BAT by enhancing the expression of GCN5.18 Under conditions of caloric restriction, decreased expression of SRC‐3 and GCN5 leads to reduced acetylation levels, but enhanced PGC‐1α activity, which promotes the use of fatty acids stored in adipose tissues as a source of energy.18 Although SRC‐1 can also interact with PGC‐1α, whether it affects GCN5 activity in a manner similar to SRC‐3 remains to be further investigated.
To date, SIRT1 and histone deacetylase 3 (HDAC3) are the only identified deacetylases for PGC‐1α. SIRT1 is a member of the Sir2 family of nicotinamide adenine dinucleotide (NAD+)‐dependent histone deacetylases that targets histone H4 at Lys16 and histone H3 at Lys9. As the principal NAD+‐dependent deacetylase in mammalian cells, SIRT1 also deacetylates a wide set of nonhistone proteins, such as p53, p300, FOXO‐1, 3a,4, Ku70 and PGC‐1α.29 Because SIRT1 requires the coenzyme NAD+ as a substrate for its function, when in high energy need, PGC‐1α is activated by SIRT1‐mediated deacetylation presumably in response to changes in the amounts of NAD+ or NADH or the NAD+/NADH ratio in cells. In turn, PGC‐1α promotes the induction of fatty acid utilization and mitochondrial metabolism.23 In addition, AMPK can perceive the energy stress and modulate SIRT1 activity by altering the intracellular level of NAD+. 30 Therefore, AMPK increases PGC‐1α activity by direct phosphorylation and also by inducing SIRT1‐mediated PGC‐1α deacetylation.
Most recently, Emmett et al. reported that HDAC3 can also deacetylate PGC‐1α and reverse GCN5‐mediated repression of PGC‐1α coactivator activity. In BAT, HDAC3 acts as a coactivator of ERRα. Deacetylation of PGC‐1α mediates the coactivation of ERRα induced by HDAC3 and is required for the transcription of oxidative phosphorylation (OXPHOS) genes, which prepares BAT for acute thermogenic challenge.21
All these findings suggest that reversible acetylation of PGC‐1α connects cell energy status to transcriptional regulatory circuits mediating mitochondrial function and guarantees metabolic flexibility.
Methylation of PGC‐1α
Another posttranslational modification contributing to metabolic flexibility is reversible methylation of PGC‐1α. Arginine methylation has been implicated in the regulation of multiple cellular processes, including subcellular localization of proteins, protein–protein interactions and transcriptional activation. PRMT1 is the major protein arginine methyltransferase (PRMT) family member in mammalian cells. It serves as a coactivator for NRs and other DNA‐binding transcription factors, such as p53 and initiator element binding factor (YY1) and mediates chromatin remodeling and initiation of transcription.31, 32 PGC‐1α has been confirmed to be methylated by PRMT1 at arginines 665, 667 and 669. These sites reside in a RERQR sequence within the C‐terminal Glu‐rich E region of PGC‐1α. The methylation of PGC‐1α by PRMT1 strongly enhances the transcription of target genes important for the biogenesis and function of mitochondria.24 Theoretically, the E region of PGC‐1α contains a nuclear localization signal and arginine methylation of PGC‐1α in this region might regulate its nuclear export function.
SET7/9 is a mono‐methyltransferase that methylates histone 3 Lys4 (H3K4), which is predominately found in active chromatin. Although the major role of SET7/9 is believed to regulate gene activation through histone methylation and alteration of chromatin structure, many nonhistone protein substrates of SET7/9 have been discovered, including PGC‐1α, p53, nuclear hormone estrogen receptor alpha (ERα) and DNMT1.9, 33, 34 Lysine‐specific demethylase 1 (LSD1) is the first enzyme identified to be capable of removing the methyl group from methylated lysines in histone proteins. LSD1 reportedly demethylates histone H3 on Lys4 (H3K4) and on Lys9 (H3K9)35, 36 and PGC‐1α is methylated by SET7/9 and demethylated by LSD1 at Lys779.
Most recently, a new mechanism reinforcing the coactivator function of PGC‐1α by methylation was revealed.9 SET7/9 mediates the methylated status of PGC‐1α. In particular, in response to metabolic stress of fasting in liver, the interaction of methylated PGC‐1α (K779Me) with RNA methytransferase 7 (NSUN7) could increase the enrichment of m5C‐modified eRNAs (enhancer RNAs) at enhancers of PGC‐1α‐targeted genes, heightening the transcriptional program of these genes. Accordingly, ablation of SET7/9 and NSUN7 resulted in depletion of the PGC‐1α‐targeted genes.9 Collectively, methylation may link epigenetic circuit with transcriptional initiation of PGC‐1α‐targeted gene programs and promote mitochondrial proliferation under conditions of energy crisis.
Ubiquitination of PGC‐1α
PGC‐1α is a short‐lived protein and its cellular levels are tightly controlled by a dynamic balance of synthesis and degradation. Ubiquitination and subsequent proteasome‐mediated degradation greatly affect the expression level of PGC‐1α. The N‐terminal AD (amino acids 1–185) of PGC‐1α has previously been considered to have no effect on protein stability and subcellular distribution. Ubiquitination of PGC‐1α relies on the integrity of the C‐terminal region, which might interact with PGC‐1α‐specific E3 ligases. Deletion of the C‐terminal fragment of PGC‐1α (amino acids 565–798) was demonstrated to completely block ubiquitination and stabilized the protein. Interestingly, another study revealed that instead of entering into the proteasome for degradation, the polyubiquitinated PGC‐1α was prone to form intranuclear aggregation and the N‐terminal of PGC‐1α was required for targeting the polyubiquitinated PGC‐1α to the proteasome.22 Thereby, intramolecular interactions between the N‐ and C‐terminal regions might cooperatively tune PGC‐1α coactivator function by regulating protein stability and sublocalization.
In fact, multiple posttranslational modifications always act in concert to finely control the activity and stability of PGC‐1α, which is exemplified by the regulation of ubiquitination of PGC‐1α in a phosphorylation‐dependent manner. SCFCdc4 is identified as an E3 ubiquitin ligase that regulates PGC‐1α stability through ubiquitin‐mediated proteolysis. PGC‐1α harbors two Cdc4 phosphodegrons that bind Cdc4 when phosphorylated by GSK3β on T295 and by p38 MAPK on Thr262, Ser265, and Thr298, resulting in SCFCdc4‐dependent ubiquitination and degradation of PGC‐1α in the nucleus.37
Overall, various posttranslational modifications provide an effective and flexible system for the regulation of activity and organelle localization of PGC‐1α, which contributes substantially to its nodal roles in mitochondrial energy metabolism.
The Role of PGC‐1α in Cancer
As a central regulator of energy metabolism, PGC‐1α plays an important role in carcinogenesis and progression (Figure 3). Indeed, accumulating evidence linking both pro‐ and antineoplastic aspects of PGC‐1α in cancers has been reported.38, 39, 40, 41, 42, 43, 44, 45, 46
Figure 3.
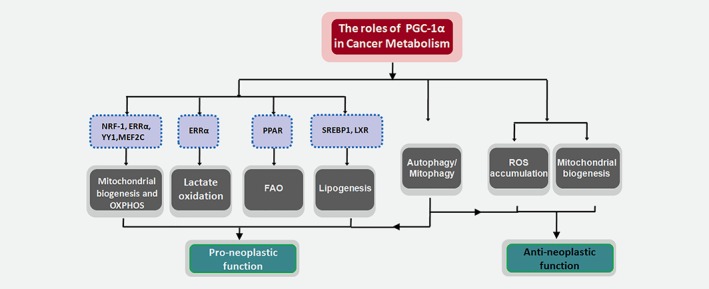
Role of PGC‐1α in the regulation of cancer metabolism. The main altered metabolic pathways regulated by PGC‐1α and accounted for its pro‐ and anti‐neoplastic aspects in cancer cells. [Color figure can be viewed at wileyonlinelibrary.com]
Pro‐neoplastic function of PGC‐1α
In multiple cancers, such as in nonsmall cell lung carcinoma (NSCLC), prostate, cervical, breast, colon cancers and melanoma, increased expression and activity of PGC‐1α is closely associated with the metabolic phenotype and facilitates tumor cell growth, invasion, distal dissemination and chemoresistance.10, 39, 42, 43, 44, 47, 48, 49 In melanoma, expression levels of PGC‐1α are tightly associated with the metabolism, biology and drug sensitivity. PGC‐1α‐positive melanoma cells display increased mitochondrial capacity and survival under oxidative stress conditions, whereas PGC‐1α‐negative cells are more glycolytic and vulnerable to ROS‐inducing drugs.43 In colorectal tumors, chemotherapy induces a SIRT1/PGC‐1α‐dependent increase in OXPHOS and mitochondrial biogenesis that favor tumor survival during treatment. In this case, SIRT1‐mediated de‐acetylation of PGC‐1α activated its transcriptional coactivator activity and promoted the expression of genes involved in OXPHOS.10 Inhibition of SIRT1/PGC‐1α axis sensitized tumor xenografts to chemotherapy.10
Moreover, in human invasive breast cancers, clinical analysis revealed a strong positive correlation between PGC‐1α expression and the formation of distant metastases. Invading cancer cells use PGC‐1α to enhance mitochondrial biogenesis and OXPHOS to ensure the increased production of ATP required for trafficking to distal tissues.42 Consistent with this finding, using large‐scale proteomic analysis, increased expression of PGC‐1α was observed in brain metastatic derived human breast cancer cells.50
To exert its pro‐neoplastic function, PGC‐1α partners with diverse regulatory factors to form complexes and targets gene programs for mitochondrial biogenesis and respiratory capacity, as well as other metabolic pathways such as lipogenesis and FAO that are involved in the metabolic reprogramming of cancer cells.
PGC‐1α and mitochondrial OXPHOS
PGC‐1α interacts with a number of transcription factors and nuclear receptors, including NRF‐1, ERRα(estrogen‐related receptor), YY1 and MEF2C, to increase mitochondrial OXPHOS function under high‐energy need condition.51
ERRα is one of the most well‐characterized transcription factors interacting with PGC‐1α and probably acts as a surrogate ligand for ERRα.52, 53 In response to oncogenic signals, PGC‐1α/ERRα can be recruited to the promoter of genes involved in the TCA cycle and OXPHOS to initiate transcriptional programs, and this could favor pro‐neoplastic outcomes.47, 52, 53, 54 In mouse embryo fibroblasts (MEFs) expressing H‐RasV,12 ectopic PGC‐1αwas reported to rescue ERRαexpression, increase OXPHOS capacity, and promote Ras‐dependent anchorage‐independent growth. Moreover, the interaction with ERRα was required for PGC‐1α to stimulate anchorage‐independent growth.47 Meanwhile, the PGC‐1α/ERRα axis greatly contributes to antioxidant defense mechanisms by regulating the expression of detoxification enzymes like superoxide dismutase 2 (SOD2), glutathione S‐transferase M1 (GSDM1) and enhancing NADPH production through glycolysis and glutamine metabolism. These could protect proliferating cancer cells from oxidative stress damage and confer a survival advantage upon them.50, 52 PGC‐1α/ERRα has also been demonstrated to be a potent inducer of vascular endothelial growth factor (VEGF) to enhance angiogenesis in muscles.55 Due to its capacity for pro‐angiogenesis and promotion of mitochondrial biogenesis and OXPHOS, the PGC‐1α/ERRα axis is strongly implicated in tumor angiogenesis, invasion and metastasis.42, 55, 56
It should also be noted that the PGC‐1α/ERRα axis can mediate lactate oxidation.54 In breast cancer cells, the PGC‐1α/ERRα axis has been demonstrated to upregulate most of the genes involved in lactate metabolism, including lactate dehydrogenase A (LDHA), LDHB and monocarboxylate transporter 1 (MCT1). The anticancer efficacy of PI3K/mTOR inhibitors are ascribed in part to their inhibition of glucose uptake and metabolism. Whereas the activation of PGC‐1α/ERRα axis may increase and utilize lactate as an alternative carbon source to favor the survival of cancer cells and this could confer resistance of tumors to treatment with PI3K/mTOR inhibitor. Thereby, inhibition of the PGC‐1α/ERRα axis in tumors may lead to increased sensitivity to these targeted therapies.54
PGC‐1α and lipid metabolism
Activated fatty acids (FA), FA‐CoAs, are converted to FA carnitines by carnitine palmitoyl transferase 1 (CPT1) and transported into mitochondria. FAs are then repeatedly cleaved to produce NADH, FADH and acetyl‐CoAs, which are coupled to the TCA cycle and electron transport chain (ETC) to produce ATP.4 This process is defined as FAO, which is also named mitochondrial β‐oxidation. Relative to each molecule, fatty acids (such as palmitate) provide three times as much ATP as does glucose. In addition to promoting mitochondrial biogenesis and OXPHOS, PGC‐1α has been shown to drive FAO to maintain energy balance during increased energy demand and help protect mitochondria from the toxic lipid overload.7, 8, 57, 58, 59 These factors could be beneficial for PGC‐1α’s pro‐neoplastic effects.
The promyelocytic leukemia protein (PML) exerts an important role in solid tumors and leukemia pathogenesis. In breast cancer cells, PML acts as a negative regulator of PGC‐1α acetylation, thus rendering PGC‐1α active. When activated, as part of the PPAR transcriptional complex, PGC‐1α promotes FAO to increase ATP levels and provides a selective survival advantage for cancer cells.60 Similarly, the PML/PGC‐1α/PPAR/FAO regulatory machinery was also observed to participate in the maintenance of HSCs (hematopoietic stem cells).61
Overaccumulation of fatty acids in mitochondria leads to incomplete FAO, mitochondrial deficiency and insulin resistance. During catecholamine‐stimulated lipolysis in myoblasts, PGC‐1α was illustrated to interact with SIRT1 and perilipin 5, a lipid droplet coat protein, to form transcriptional complexes. In turn, this complex increased the PGC‐1α‐target gene programs to promote efficient fatty acid catabolism and prevent mitochondrial dysfunction. In this case, perilipin 5 activated the deacetylase activity of SIRT1 by displacing DBC1 (deleted in breast cancer 1), an inhibitor of SIRT1, consequently increasing the transcriptional coactivator activity of PGC‐1α by deacetylation.8
Beyond its ability to promote lipid catabolic metabolism, PGC‐1α can also mediate lipogenesis. PGC‐1α is capable of recruiting transcription factors to initiate fatty acid synthesis programs, such as SREBP1 and liver X receptor (LXR).
Particularly in cancer cells, acetyl‐CoAs derived from glucose and glutamine provide key substrates for fatty acid synthesis. Using the PGC‐1α−/− and PGC‐1α+/+ mouse models, loss of PGC‐1α was shown to reduce both azoxymethane‐induced colon carcinogenesis and diethylnitrosamine‐induced liver carcinogenesis. Based on this model, PGC‐1α was demonstrated to promote tumor growth by enhancing lipogenesis from glucose. Gene expression programs supporting de novo fatty acid synthesis (ACC and FASN) as well as the conversion of glucose into acetyl‐CoA (SLC25A1 and ACLY) have been induced by gain of PGC‐1α. Additionally, the ability of PGC‐1α to promote the TCA cycle and mitochondrial function enables increased substrates available for fatty acid synthesis reactions.62
Antineoplastic function of PGC‐1α
Although the main body of documents supports pro‐tumorigenic activity of PGC‐1α, paradoxical antineoplastic effects also exist for some tumor types.38, 40, 63, 64, 65 In VHL‐deficient clear cell renal cell carcinoma (ccRCC), elevated mitochondrial activity induced by PGC‐1α is tightly associated with increased ROS production, leading to augmented oxidative stress. Thus, ectopic expression of PGC‐1α results in impaired tumor growth and enhanced sensitivity to cytotoxic therapies.40 Similarly, in human cells and mouse models of intestinal cancer, PGC‐1α regulates enterocyte cell fate and protects against tumorigenesis by promoting mitochondrial‐mediated apoptosis through reactive oxygen species (ROS) accumulation.38 Moreover, under glycolytic conditions, increased mitochondrial biogenesis induced by PGC‐1α inhibits cancer cell proliferation.63 Hence, all these findings indicate that the role of PGC‐1α in cancer cell fate determination is not univocal. The elegant function of PGC‐1α depends on the specific tumor context and tumor subpopulation with a particular metabolic phenotype.
PGC‐1α and autophagy/mitophagy
Autophagy is the main process for lysosomal degradation, recycling of proteins and damaged organelles and providing nutrients during stress conditions.66 Recent data have mechanistically linked the expression of PGC‐1α to the autophagic machinery with potential implications in cancer cell metabolic rewiring.67, 68, 69, 70
Overexpressing PGC‐1α promotes the abundance of OXPHOS protein complexes, confers autophagy resistance and stimulates tumor growth in breast carcinoma cells. Under conditions of starvation, overexpression of PGC‐1α failed to upregulate autophagic markers, such as Beclin‐1 and Cathepsin B.68 V600BRAF mutation in melanoma cells switches on a metabolic reprogramming by downregulating PGC‐1α/β, leading to decreased OXPHOS activity and increased glycolytic lactate and ATP levels. The exported lactate creates an acidic tumor environment and the melanoma cells triggers autophagy as a protective and adaptive response to acidic stress.67, 69
As a macroautophagy process, mitophagy is the major mechanism by which damaged mitochondrial components are selective to lysosomal‐mediated degradation and plays a crucial role in mitochondrial quality control.71, 72 PGC‐1α also mediates mitochondrial degradation through transcriptional mechanisms.73 It can interact and stabilize mRNAs of mitostatin, a mitochondrial protein with oncostatic activity. This evokes mitostatin‐dependent mitophagy, resulting in a negative feedback regulation on vascular endothelial growth factor A (VEGFA) production, consequently tumor angiogenic attenuation.70 Thus, although in general, the expression of PGC‐1α counteracts the process of autophagy, it can foster mitophagy to sustain mitochondrial homeostasis in the specific scenario, which further illustrates the complexity of PGC‐1α’s roles in the regulation of autophagic pathways.
Conclusions and Perspectives
Reprogramming energy metabolism represents one of the hallmarks of cancer cells.1, 4, 74, 75, 76 As a core regulator of mitochondrial biogenesis and metabolism, the emerging roles of PGC‐1α implicated in tumorgenesis and progression have been gradually unraveled. The activity and stability of PGC‐1α are subject to dynamic and versatile posttranslational modification in response to metabolic stress and other environmental signals. Diverse posttranslational modifications could very well account for the pleiotropic effects of this coactivator. The expanding discovery of multiple posttranslational modifications would contribute to a more profound understanding of the complex regulatory networks that modulate PGC‐1α signaling.
Indeed, the pro‐neoplastic effect of PGC‐1α mainly depends upon its ability to promote mitochondrial content and function, including mitochondrial biogenesis, mitochondrial respiration and fatty acid utilization. However, in some specific tumor types, PGC‐1α is poorly expressed. Malignant cells often represent an elevated steady state of ROS levels causing them to be sensitive to further increased ROS production. Expression of PGC‐1α restores mitochondrial respiration accompanied by ROS accumulation, which ultimately leads to tumor cell death, especially when intracellular ROS scavenging systems are scarce. In cancer cells, the exquisite function of PGC‐1α depends on the interacting partners of the composition of its complexes, which include sets of enzymes responsible for PGC‐1α posttranslational regulation, transcription factors, and other coactivators or cosuppressors. These complexes will integrate the oncogenic signals to dictate the pro‐ or antineoplastic outcomes of PGC‐1α by enhancing specific transcriptional activation. In addition, the function of PGC‐1α in the regulation of autophagic pathway may also in part account for the fate determination of tumor cells. Therefore, to decipher the exquisite role of PGC‐1α in determining the metabolic response in cancer cells is crucial. This would help better assess the type of cancers that should best benefit from pharmacological targeting of PGC‐1α signaling.
Currently, no commercial PGC‐1α antagonist is available. As indicated earlier, the expression and/or activity of PGC‐1α is amenable to transcriptional and posttranslational regulations by a set of enzymes and transcription factors. Among these regulators, the most well‐validated factors to activate PGC‐1α are ERRα and SIRT1.77 An alternative strategy could be to utilize inhibitors targeting ERRα or SIRT1 to disrupt the interaction between them and PGC‐1α, thus inhibiting the transcription and activation of PGC‐1α. On the other hand, some proteins are responsible for negative regulation of PGC‐1α activity, such as the histone acetyltransferase, GCN5. GCN5 suppresses PGC‐1α activity by acetylation and, theoretically, its agonists should inhibit PGC‐1α signaling. Thus, all these regulators of PGC‐1α might be developed as potential candidates for pharmaceutical intervention targeting PGC‐1α signaling.
In situations in which activation of PGC‐1α is preferred, among others, ERRα agonists or SIRT1 activators that cooperate with PGC‐1α to enhance mitochondrial function should be good candidates. Using gene expression‐based screening, microtubule inhibitor and protein synthesis inhibitors have also been identified as inducers of PGC‐1α and OXPHOS.78 In addition, exercise training can increase PGC‐1α levels in muscle and as a nonpharmacological approach, could prevent skeletal muscle wasting and improve muscle function in cancer cachexia, at least partially through activation of PGC‐1α.13, 79, 80, 81
Considering the master role of PGC‐1α in energy homeostasis, studies on its transcriptional regulatory functions in mitochondrial proliferation and metabolism might provide important insights into our understanding and targeting of rewiring energy metabolism in cancer.
Conflict of interest: No potential conflicts of interest were disclosed.
References
- 1. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011;144:646–74. [DOI] [PubMed] [Google Scholar]
- 2. Jones AW, Yao Z, Vicencio JM, et al. PGC‐1 family coactivators and cell fate: roles in cancer, neurodegeneration, cardiovascular disease and retrograde mitochondria‐nucleus signalling. Mitochondrion 2012;12:86–99. [DOI] [PubMed] [Google Scholar]
- 3. Luo X, Hong L, Cheng C, et al. DNMT1 mediates metabolic reprogramming induced by Epstein‐Barr virus latent membrane protein 1 and reversed by grifolin in nasopharyngeal carcinoma. Cell Death Dis 2018;9:619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Luo X, Cheng C, Tan Z, et al. Emerging roles of lipid metabolism in cancer metastasis. Mol Cancer 2017;16:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Villena JA. New insights into PGC‐1 coactivators: redefining their role in the regulation of mitochondrial function and beyond. FEBS J 2015;282:647–72. [DOI] [PubMed] [Google Scholar]
- 6. Lin J, Handschin C, Spiegelman BM. Metabolic control through the PGC‐1 family of transcription coactivators. Cell Metab 2005;1:361–70. [DOI] [PubMed] [Google Scholar]
- 7. Tan Z, Luo X, Xiao L, et al. The role of PGC1alpha in cancer metabolism and its therapeutic implications. Mol Cancer Ther 2016;15:774–82. [DOI] [PubMed] [Google Scholar]
- 8. Gallardo‐Montejano VI, Saxena G, Kusminski CM, et al. Nuclear Perilipin 5 integrates lipid droplet lipolysis with PGC‐1alpha/SIRT1‐dependent transcriptional regulation of mitochondrial function. Nat Commun 2016;7:12723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Aguilo F, Li S, Balasubramaniyan N, et al. Deposition of 5‐methylcytosine on enhancer RNAs enables the coactivator function of PGC‐1alpha. Cell Rep 2016;14:479–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vellinga TT, Borovski T, de Boer VC, et al. SIRT1/PGC1alpha‐dependent increase in oxidative phosphorylation supports chemotherapy resistance of colon cancer. Clin Cancer Res 2015;21:2870–9. [DOI] [PubMed] [Google Scholar]
- 11. Chang JS, Huypens P, Zhang Y, et al. Regulation of NT‐PGC‐1alpha subcellular localization and function by protein kinase A‐dependent modulation of nuclear export by CRM1. J Biol Chem 2010;285:18039–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Girnun GD. The diverse role of the PPARgamma coactivator 1 family of transcriptional coactivators in cancer. Semin Cell Dev Biol 2012;23:381–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ruas JL, White JP, Rao RR, et al. A PGC‐1alpha isoform induced by resistance training regulates skeletal muscle hypertrophy. Cell 2012;151:1319–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang Y, Huypens P, Adamson AW, et al. Alternative mRNA splicing produces a novel biologically active short isoform of PGC‐1alpha. J Biol Chem 2009;284:32813–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lustig Y, Ruas JL, Estall JL, et al. Separation of the gluconeogenic and mitochondrial functions of PGC‐1{alpha} through S6 kinase. Genes Dev 2011;25:1232–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fernandez‐Marcos PJ, Auwerx J. Regulation of PGC‐1alpha, a nodal regulator of mitochondrial biogenesis. Am J Clin Nutr 2011;93:884S–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jeninga EH, Schoonjans K, Auwerx J. Reversible acetylation of PGC‐1: connecting energy sensors and effectors to guarantee metabolic flexibility. Oncogene 2010;29:4617–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Coste A, Louet JF, Lagouge M, et al. The genetic ablation of SRC‐3 protects against obesity and improves insulin sensitivity by reducing the acetylation of PGC‐1{alpha}. Proc Natl Acad Sci USA 2008;105:17187–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Li X, Monks B, Ge Q, et al. Akt/PKB regulates hepatic metabolism by directly inhibiting PGC‐1alpha transcription coactivator. Nature 2007;447:1012–6. [DOI] [PubMed] [Google Scholar]
- 20. Jager S, Handschin C, St‐Pierre J, et al. AMP‐activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC‐1alpha. Proc Natl Acad Sci USA 2007;104:12017–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Emmett MJ, Lim HW, Jager J, et al. Histone deacetylase 3 prepares brown adipose tissue for acute thermogenic challenge. Nature 2017;546:544–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sano M, Tokudome S, Shimizu N, et al. Intramolecular control of protein stability, subnuclear compartmentalization, and coactivator function of peroxisome proliferator‐activated receptor gamma coactivator 1alpha. J Biol Chem 2007;282:25970–80. [DOI] [PubMed] [Google Scholar]
- 23. Gerhart‐Hines Z, Rodgers JT, Bare O, et al. Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC‐1alpha. EMBO J 2007;26:1913–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Teyssier C, Ma H, Emter R, et al. Activation of nuclear receptor coactivator PGC‐1alpha by arginine methylation. Genes Dev 2005;19:1466–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Housley MP, Udeshi ND, Rodgers JT, et al. A PGC‐1alpha‐O‐GlcNAc transferase complex regulates FoxO transcription factor activity in response to glucose. J Biol Chem 2009;284:5148–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chambers KT, Leone TC, Sambandam N, et al. Chronic inhibition of pyruvate dehydrogenase in heart triggers an adaptive metabolic response. J Biol Chem 2011;286:11155–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Robert T, Vanoli F, Chiolo I, et al. HDACs link the DNA damage response, processing of double‐strand breaks and autophagy. Nature 2011;471:74–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wellen KE, Hatzivassiliou G, Sachdeva UM, et al. ATP‐citrate lyase links cellular metabolism to histone acetylation. Science 2009;324:1076–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dai H, Sinclair DA, Ellis JL, et al. Sirtuin activators and inhibitors: promises, achievements, and challenges. Pharmacol Ther 2018;188:140–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Canto C, Jiang LQ, Deshmukh AS, et al. Interdependence of AMPK and SIRT1 for metabolic adaptation to fasting and exercise in skeletal muscle. Cell Metab 2010;11:213–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Stouth DW, vanLieshout TL, Shen NY, et al. Regulation of skeletal muscle plasticity by protein arginine Methyltransferases and their potential roles in neuromuscular disorders. Front Physiol 2017;8:870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Han HS, Choi D, Choi S, et al. Roles of protein arginine methyltransferases in the control of glucose metabolism. Endocrinol Metab (Seoul) 2014;29:435–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shen C, Wang D, Liu X, et al. SET7/9 regulates cancer cell proliferation by influencing beta‐catenin stability. FASEB J 2015;29:4313–23. [DOI] [PubMed] [Google Scholar]
- 34. Del Rizzo PA, Trievel RC. Substrate and product specificities of SET domain methyltransferases. Epigenetics 2011;6:1059–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ambrosio S, Ballabio A, Majello B. Histone methyl‐transferases and demethylases in the autophagy regulatory network: the emerging role of KDM1A/LSD1 demethylase. Autophagy 2019;15:187–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hosseini A, Minucci S. A comprehensive review of lysine‐specific demethylase 1 and its roles in cancer. Epigenomics 2017;9:1123–42. [DOI] [PubMed] [Google Scholar]
- 37. Anderson RM, Barger JL, Edwards MG, et al. Dynamic regulation of PGC‐1alpha localization and turnover implicates mitochondrial adaptation in calorie restriction and the stress response. Aging Cell 2008;7:101–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. D'Errico I, Salvatore L, Murzilli S, et al. Peroxisome proliferator‐activated receptor‐coactivator 1‐ (PGC1 ) is a metabolic regulator of intestinal epithelial cell fate. Proc Natl Acad Sci USA 2011;108:6603–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Haq R, Shoag J, Andreu‐Perez P, et al. Oncogenic BRAF regulates oxidative metabolism via PGC1α and MITF. Cancer Cell 2013;23:302–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. LaGory EL, Wu C, Taniguchi CM, et al. Suppression of PGC‐1alpha is critical for reprogramming oxidative metabolism in renal cell carcinoma. Cell Rep 2015;12:116–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lim JH, Luo C, Vazquez F, et al. Targeting mitochondrial oxidative metabolism in melanoma causes metabolic compensation through glucose and glutamine utilization. Cancer Res 2014;74:3535–45. [DOI] [PubMed] [Google Scholar]
- 42. LeBleu VS, O'Connell JT, Gonzalez Herrera KN, et al. PGC‐1alpha mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat Cell Biol 2014;16:992–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Vazquez F, Lim JH, Chim H, et al. PGC1alpha expression defines a subset of human melanoma tumors with increased mitochondrial capacity and resistance to oxidative stress. Cancer Cell 2013;23:287–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Shiota M, Yokomizo A, Tada Y, et al. Peroxisome proliferator‐activated receptor gamma coactivator‐1alpha interacts with the androgen receptor (AR) and promotes prostate cancer cell growth by activating the AR. Mol Endocrinol 2010;24:114–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ronai Z. The masters talk: the PGC‐1alpha‐MITF axis as a melanoma energizer. Pigment Cell Melanoma Res 2013;26:294–5. [DOI] [PubMed] [Google Scholar]
- 46. Sancho P, Burgos‐Ramos E, Tavera A, et al. MYC/PGC‐1alpha balance determines the metabolic phenotype and plasticity of pancreatic cancer stem cells. Cell Metab 2015;22:590–605. [DOI] [PubMed] [Google Scholar]
- 47. Fisher KW, Das B, Kortum RL, et al. Kinase suppressor of ras 1 (KSR1) regulates PGC1alpha and estrogen‐related receptor alpha to promote oncogenic Ras‐dependent anchorage‐independent growth. Mol Cell Biol 2011;31:2453–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tennakoon JB, Shi Y, Han JJ, et al. Androgens regulate prostate cancer cell growth via an AMPK‐PGC‐1alpha‐mediated metabolic switch. Oncogene 2014;33:5251–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gopal YN, Rizos H, Chen G, et al. Inhibition of mTORC1/2 overcomes resistance to MAPK pathway inhibitors mediated by PGC1alpha and oxidative phosphorylation in melanoma. Cancer Res 2014;74:7037–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chen EI, Hewel J, Krueger JS, et al. Adaptation of energy metabolism in breast cancer brain metastases. Cancer Res 2007;67:1472–86. [DOI] [PubMed] [Google Scholar]
- 51. Scarpulla RC. Metabolic control of mitochondrial biogenesis through the PGC‐1 family regulatory network. Biochim Biophys Acta 2011;1813:1269–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Deblois G, St‐Pierre J, Giguere V. The PGC‐1/ERR signaling axis in cancer. Oncogene 2013;32:3483–90. [DOI] [PubMed] [Google Scholar]
- 53. Deblois G, Giguere V. Functional and physiological genomics of estrogen‐related receptors (ERRs) in health and disease. Biochim Biophys Acta 2011;1812:1032–40. [DOI] [PubMed] [Google Scholar]
- 54. Park S, Chang CY, Safi R, et al. ERRalpha‐regulated lactate metabolism contributes to resistance to targeted therapies in breast cancer. Cell Rep 2016;15:323–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Arany Z, Foo SY, Ma Y, et al. HIF‐independent regulation of VEGF and angiogenesis by the transcriptional coactivator PGC‐1alpha. Nature 2008;451:1008–12. [DOI] [PubMed] [Google Scholar]
- 56. Dwyer MA, Joseph JD, Wade HE, et al. WNT11 expression is induced by estrogen‐related receptor alpha and beta‐catenin and acts in an autocrine manner to increase cancer cell migration. Cancer Res 2010;70:9298–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. He F, Jin JQ, Qin QQ, et al. Resistin regulates fatty acid Beta oxidation by suppressing expression of peroxisome proliferator activator receptor gamma‐Coactivator 1alpha (PGC‐1alpha). Cell Physiol Biochem 2018;46:2165–72. [DOI] [PubMed] [Google Scholar]
- 58. Pambianco S, Giovarelli M, Perrotta C, et al. Reversal of defective mitochondrial biogenesis in limb‐girdle muscular dystrophy 2D by independent modulation of histone and PGC‐1alpha acetylation. Cell Rep 2016;17:3010–23. [DOI] [PubMed] [Google Scholar]
- 59. Jun HJ, Joshi Y, Patil Y, et al. NT‐PGC‐1alpha activation attenuates high‐fat diet‐induced obesity by enhancing brown fat thermogenesis and adipose tissue oxidative metabolism. Diabetes 2014;63:3615–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Carracedo A, Weiss D, Leliaert AK, et al. A metabolic prosurvival role for PML in breast cancer. J Clin Invest 2012;122:3088–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ito K, Carracedo A, Weiss D, et al. A PML‐PPAR‐delta pathway for fatty acid oxidation regulates hematopoietic stem cell maintenance. Nat Med 2012;18:1350–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Bhalla K, Hwang BJ, Dewi RE, et al. PGC1alpha promotes tumor growth by inducing gene expression programs supporting lipogenesis. Cancer Res 2011;71:6888–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wang X, Moraes CT. Increases in mitochondrial biogenesis impair carcinogenesis at multiple levels. Mol Oncol 2011;5:399–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Burchard J, Zhang C, Liu AM, et al. microRNA‐122 as a regulator of mitochondrial metabolic gene network in hepatocellular carcinoma. Mol Syst Biol 2010;6:402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zhang Y, Ba Y, Liu C, et al. PGC‐1alpha induces apoptosis in human epithelial ovarian cancer cells through a PPARgamma‐dependent pathway. Cell Res 2007;17:363–73. [DOI] [PubMed] [Google Scholar]
- 66. Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell 2011;147:728–41. [DOI] [PubMed] [Google Scholar]
- 67. Ferretta A, Maida I, Guida S, et al. New insight into the role of metabolic reprogramming in melanoma cells harboring BRAF mutations. Biochim Biophys Acta 2016;1863:2710–8. [DOI] [PubMed] [Google Scholar]
- 68. Salem AF, Whitaker‐Menezes D, Howell A, et al. Mitochondrial biogenesis in epithelial cancer cells promotes breast cancer tumor growth and confers autophagy resistance. Cell Cycle 2012;11:4174–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Marino ML, Pellegrini P, Di Lernia G, et al. Autophagy is a protective mechanism for human melanoma cells under acidic stress. J Biol Chem 2012;287:30664–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Neill T, Torres A, Buraschi S, et al. Decorin induces mitophagy in breast carcinoma cells via peroxisome proliferator‐activated receptor gamma coactivator‐1alpha (PGC‐1alpha) and mitostatin. J Biol Chem 2014;289:4952–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Wang K, Klionsky DJ. Mitochondria removal by autophagy. Autophagy 2011;7:297–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kubli DA, Gustafsson AB. Mitochondria and mitophagy: the yin and yang of cell death control. Circ Res 2012;111:1208–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Zhu J, Wang KZ, Chu CT. After the banquet: mitochondrial biogenesis, mitophagy, and cell survival. Autophagy 2013;9:1663–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Tan Z, Xiao L, Tang M, et al. Targeting CPT1A‐mediated fatty acid oxidation sensitizes nasopharyngeal carcinoma to radiation therapy. Theranostics 2018;8:2329–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Luo X, Zhao X, Cheng C, et al. The implications of signaling lipids in cancer metastasis. Exp Mol Med 2018;50:127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Xiao L, Hu Zy, Dong X, et al. Targeting Epstein–Barr virus oncoprotein LMP1‐mediated glycolysis sensitizes nasopharyngeal carcinoma to radiation therapy. Oncogene 2014;33:4568–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Wu Z, Boss O. Targeting PGC‐1 alpha to control energy homeostasis. Expert Opin Ther Targets 2007;11:1329–38. [DOI] [PubMed] [Google Scholar]
- 78. Arany Z, Wagner BK, Ma Y, et al. Gene expression‐based screening identifies microtubule inhibitors as inducers of PGC‐1alpha and oxidative phosphorylation. Proc Natl Acad Sci USA 2008;105:4721–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Padrao AI, Figueira AC, Faustino‐Rocha AI, et al. Long‐term exercise training prevents mammary tumorigenesis‐induced muscle wasting in rats through the regulation of TWEAK signalling. Acta Physiol (Oxf) 2017;219:803–13. [DOI] [PubMed] [Google Scholar]
- 80. Pin F, Busquets S, Toledo M, et al. Combination of exercise training and erythropoietin prevents cancer‐induced muscle alterations. Oncotarget 2015;6:43202–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. White JP, Puppa MJ, Sato S, et al. IL‐6 regulation on skeletal muscle mitochondrial remodeling during cancer cachexia in the ApcMin/+ mouse. Skelet Muscle 2012;2:14. [DOI] [PMC free article] [PubMed] [Google Scholar]