

Summary at a Glance
The study revealed that 69% of families with hereditary nephritis that was difficult to diagnose clinicopathologically had heterozygous mutations of COL4A3/A4 (TBMN/ADAS). The finding suggests the importance of genetic testing in appropriate patients.
Keywords: autosomal dominant Alport syndrome, COL4A3, COL4A4, thin basement membrane nephropathy, type IV collagen α5 chain
ABSTRACT
Aim
Type IV collagen nephropathies include Alport Syndrome and thin basement membrane nephropathy (TBMN), which are caused by mutations in COL4A3/A4/A5 genes. Recently, reports of patients with heterozygous mutations in COL4A3/A4 have been increasing. The clinical course of these patients has a wide variety, and they are diagnosed as TBMN, autosomal dominant Alport syndrome (ADAS), or familial focal segmental glomerular sclerosis. However, diagnosis, frequency and clinicopathological manifestation of them remains unclear. We tested COL4A3/A4/A5 genes in patients with hereditary nephritis that was difficult to diagnose clinicopathologically, and investigated who should undergo such testing.
Methods
We performed immunostaining for α5 chain of type IV collagen [α5 (IV)] in 27 patients from 21 families who fitted the following criteria: (i) haematuria and proteinuria (± renal dysfunction); (ii) family history of haematuria, proteinuria, and/or renal dysfunction (autosomal dominant inheritance); (iii) no specific glomerulonephritis; and (iv) thinning, splitting, or lamellation of the glomerular basement membrane (GBM) on electron microscopy. Then we performed genetic testing in 19 patients from 16 families who showed normal α5 (IV) patterns. We conducted a retrospective analysis of their clinicopathological findings.
Results
Among 16 families, 69% were detected heterozygous mutations in COL4A3/A4, suggesting the diagnosis of TBMN/ADAS. Twenty‐one percent of patients developed end stage renal disease. All patients showed thinning of GBM, which was accompanied by splitting or lamellation in seven patients.
Conclusion
A considerable fraction of patients with hereditary nephritis that is difficult to diagnose clinicopathologically have TBMN/ADAS. It is important to recognize TBMN/ADAS and perform genetic testing in appropriate patients.
Familial glomerular haematuria is a heterogeneous condition which include Alport Syndrome (AS) and thin basement membrane nephropathy (TBMN). Since AS and TBMN are mainly caused by the mutation of COL4A3, COL4A4, or COL4A5 genes which encode α3, α4, and α5 chains of type IV collagen respectively, they are also called ‘type IV collagen nephropathies’.1 AS is a hereditary nephropathy characterized by progressive renal failure with ultrastructural changes of the glomerular basement membrane (GBM), sensorineural deafness, and variable ocular abnormalities.2 AS is genetically heterogeneous and there are three modes of inheritance, with this syndrome being X‐linked in 80% of patients, autosomal recessive in 15%, and autosomal dominant in less than 5%.3 X‐linked Alport syndrome (XLAS) is the most common form and is due to mutations of COL4A5 located at Xq22.3.4 Typically, affected males develop end‐stage renal disease (ESRD) before 30 years of age and they also frequently have hearing loss.5 Immunohistochemical evaluation of renal biopsy specimens shows lack of the α5 chain of type IV collagen [α5 (IV)]. On the other hand, Autosomal dominant Alport syndrome (ADAS) is caused by heterozygous mutations of either COL4A3 or COL4A4. In patients with ADAS, progression of renal dysfunction is slower and the frequency of deafness and ocular abnormalities is also lower than in typical XLAS.6 Immunohistochemistry of renal biopsy specimens shows normal α5 (IV) staining, so it is difficult to make an accurate diagnosis of ADAS from the clinicopathological findings and genetic testing is necessary. Although ADAS has been thought to be an extremely rare condition, reports have been increasing in recent years, suggesting that there may be undiagnosed patients.7, 8, 9 On the other hand, heterozygous COL4A3/A4 mutations are also found in about 40% of patients with TBMN.10 Although their clinical course has classically been believed to be benign, some authors reported that a part of patients with TBMN develop proteinuria and renal impairment.11, 12 Furthermore, recent studies revealed that some patients with heterozygous COL4A3/A4 mutations develop focal segmental glomerular sclerosis (FSGS) in their later life on the basis of TBMN, which expand the spectrum of type IV collagen nephropthy.13, 14, 15, 16, 17 Thus, diagnosis, frequency and clinicopathological manifestation of patients with heterozygous mutations in the COL4A3/A4 genes remains largely unknown.
In this study, we performed genetic testing of COL4A3, COL4A4, and COL4A5 in patients with hereditary nephritis that was difficult to diagnose from clinicopathological findings, and investigated clinicopathological characteristics of patients who should undergo genetic testing.
METHODS
We investigated 26 patients (from 22 families) who underwent renal biopsy at Toranomon Hospital and Toranomon Kajigaya Hospital from June 1985 to September 2015 and fulfilled all of the following four criteria: (i) haematuria and proteinuria with or without renal dysfunction; (ii) a family history of haematuria, proteinuria, and/or renal dysfunction (especially showing autosomal dominant inheritance); (iii) no specific glomerulonephritis identified by light microscopy and immunofluorescent staining of a renal biopsy specimen; and (iv) thinning, splitting, or lamellation of the glomerular basement membrane (GBM) on electron microscopy (Fig. 1). Measurement of the GBM was performed on at least three photomicrographs at a magnification of 3000×. We measured GBM width at 10 points in at least five capillary loops, and GBM thinning was defined by a minimum width of less than 264 nm.
Figure 1.
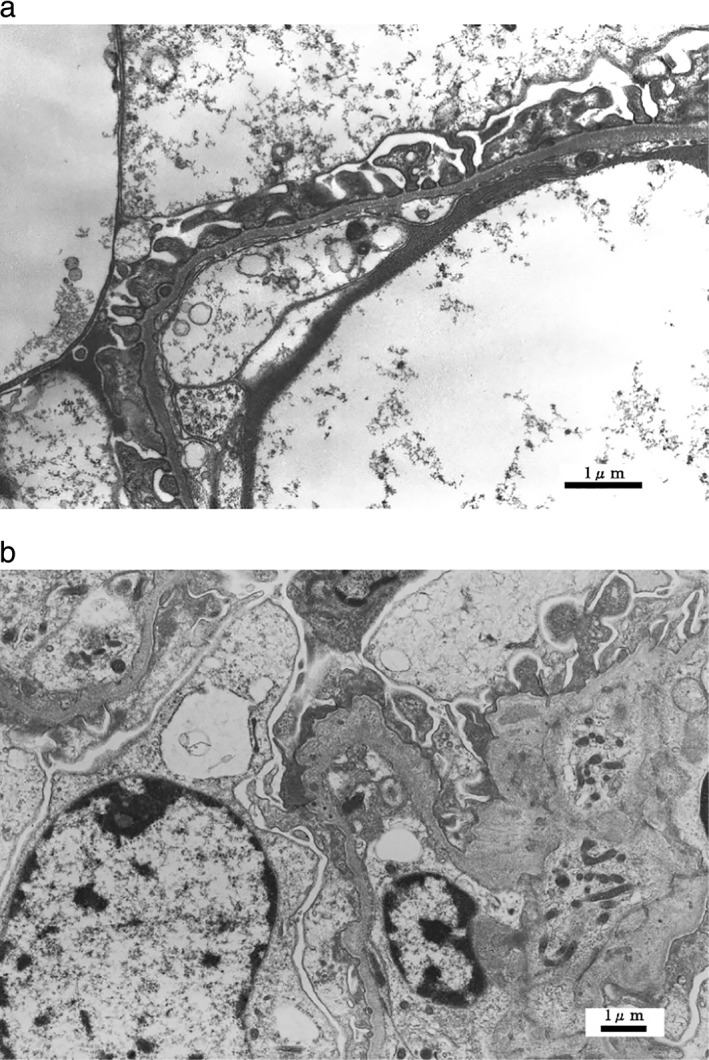
Typical glomerular basement membrane (GBM) changes on electron microscopy. (a) Thinning of the GBM (×6000), (b) Lamellation of the GBM (×6000).
We performed immunohistochemical staining for α5 (IV) using renal biopsy specimens of all patients for whom specimens were available. Twenty‐one patients from 18 families showed a normal pattern of α5 (IV) staining that excluded typical XLAS and they were classified as having hereditary nephritis that was ‘difficult to diagnose clinicopathologically’. Among them, we performed genetic testing of COL4A3, COL4A4, and COL4A5 in 19 patients from 16 families who consented to such testing. For patients in whom we could not carry out α5 (IV) staining of renal biopsy specimens, we confirmed that at least one family member fulfilled the above four criteria and showed normal α5 (IV) staining of a biopsy specimen. Retrospective analysis of clinical and pathological findings was performed using data from the medical records. This study was approved by the Ethics Committee of Toranomon hospital (2015‐8) and all subjects gave written informed consent.
Genetic analysis
Blood samples were collected from patients and genomic DNA was isolated from peripheral blood leukocytes using the Quick Gene Mini 80 system (Wako Pure Chemical Industries, Tokyo, Japan) according to the manufacturer’s instructions. NGS samples were prepared using a HaloPlex target enrichment system kit by following the manufacture’s instruction (Agilent Technologies, Santa Clara, CA, USA). Briefly, digested 225 ng of genomic DNA were hybridized at 54°C for 16 h with custom‐designed NGS probes to capture genes for inherited kidney diseases such as COL4A3, COL4A4, COL4A5, CLCN5, OCRL and other FSGS causative genes. Amplified target libraries were sequenced with 150 bp pair‐end reads on a MiSeq platform (Illumina, San Diego, CA, USA), and followed by variant analysis on a SureCall 3.0 (Agilent). We conducted Sanger’s sequencing for all variants detected by NGS analysis.
RESULTS
Clinical and genetic findings
Among 19 patients from 16 families, 69% (14 patients from 11 families) showed heterozygous mutations in COL4A3 or COL4A4 gene, suggesting the diagnosis of TBMN/ADAS. XLAS was diagnosed in 25% (four patients from four families). One patient did not show mutation of COL4A3, COL4A4, or COL4A5, and further investigation finally led to a diagnosis of Dent disease by targeted sequencing using NGS.
Table 1 shows the results of genetic testing in the 14 patients with TBMN/ADAS. In four of the 11 affected families, ADAS was caused by mutation of the COL4A3 gene, while there was mutation of COL4A4 in seven families. Three mutations were reported to be causative mutation for ARAS in previous studies.18, 19, 20 Six of the mutations were novel mutations.
Table 1.
Results of genetic testing in 14 patients from 11 thin basement membrane nephropathy (TBMN)/Autosomal dominant Alport syndrome (ADAS) families
| Patient No. | Family No. | Gene | Exon/Intron | Nucleotide change | Amino acid change | Previous report of ARAS |
|---|---|---|---|---|---|---|
| 1 | 1 | COL4A4 | Exon 20 | c.1323_1340del | 18 bp deletion | 18 |
| 2 | 1 | COL4A4 | Exon 20 | c.1323_1340del | 18 bp deletion | 18 |
| 3 | 2 | COL4A4 | Exon 48 | c.4847T>G[Link] | p.Leu1616Arg | ‐ |
| 4 | 2 | COL4A4 | Exon 48 | c.4847T>G[Link] | p.Leu1616Arg | ‐ |
| 5 | 3 | COL4A4 | Exon 14 | c.827G>C[Link] | p.Gly276Ala | ‐ |
| 6 | 3 | COL4A4 | Exon 14 | c.827G>C[Link] | p.Gly276Ala | ‐ |
| 7 | 4 | COL4A4 | Exon 44 | c.4129C>T | p.Arg1377X | 19 |
| 8 | 5 | COL4A4 | Exon 18 | c.1057G>C[Link] | p.Gly353Arg | ‐ |
| 9 | 6 | COL4A4 | Exon 14 | c.827G>C[Link] | p.Gly276Ala | ‐ |
| 10 | 7 | COL4A4 | Intron 38 | c.3577+1G>A[Link] | ‐ | |
| 11 | 8 | COL4A3 | Exon 47 | c.4207G>A[Link] | p.Gly1403Arg | ‐ |
| 12 | 9 | COL4A3 | Exon 40 | c.3464G>A | p.Gly1155Asp | 20 |
| 13 | 10 | COL4A3 | Exon 40 | c.3464G>A | p.Gly1155Asp | 20 |
| 14 | 11 | COL4A3 | Exon 33 | c.2863G>A[Link] | p.Gly955Arg | ‐ |
†Novel mutation.
The clinical features of the 14 patients with TBMN/ADAS are detailed in Table 2. There were eight males and six females, and the median age at the time of genetic testing was 54.5 years (range: 29–69 years). The median age at the diagnosis of haematuria was 22 years (range: 5–44 years), and the median age at detection of proteinuria was 21.5 years. An episode of macroscopic haematuria was noted after upper respiratory infection in two patients (14.3%). In 57.2% of the patients, renal dysfunction (stage >3 chronic kidney disease (CKD)) was evident at the time of genetic diagnosis. Three patients (21.5%) had reached end stage renal disease (ESRD) at 42, 58, and 59 years old and all of them were men. Renal biopsy was performed multiple times in 11 patients (78.6%), with the average number of biopsies being 1.9. The median age at the first biopsy was 28.5 years (range: 6–56 years), and there was an interval of 16.1 years (range: 0.2–41.9 years) from the first biopsy to final diagnosis. Two patients had hearing loss (14.2%) and none had ocular changes. Five patients (35.7%) had hypertension at the time of genetic diagnosis, which accounted for 62.5% of patients with stage >3CKD. The detection of proteinuria and kidney dysfunction was prior to the detection of hypertension in all patients. None of our patients had diabetes. Family trees are shown in Figure 2. The clinical manifestation of patients varied from isolated haematuria, proteinuria and mild kidney dysfunction to ESRD in many families.
Table 2.
Clinicopathological manifestations of 14 patients with thin basement membrane nephropathy (TBMN)/Autosomal dominant Alport syndrome (ADAS)
| Patient No (Family No) | Age Sex | sCr (mg/dL) | eGFR (mL/min) | Proteinuria (g/gCre) | Haematuria/Proteinuria | Age | sCr (mg/dL) | eGFR (mL/min) | Proteinuria (g/gCre) | RBC (/HPF) | Light microscopy | Immunofluorescence | Electron microscopy | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (at genetic diagnosis) | (age at detection) | (at last kidney biopsy) | Diagnosis | GS/TG | IgG/A/M | α5 (IV) | (GBM thickness, average: range; nm) | ||||||||
| 1 (1) | 66M | ESRD (59 years) | ESRD | ESRD | 27/34 | 44 | 2.0 | 35.4 | 0.6 | 6–10 | NSc | 8/16 | Negative | ND | TBM (205:151–252) lamellation, splitting |
| 2 (1) | 29M | 0.91 | 81.8 | 1.3 | 6/18 | 29 | 0.9 | 81.8 | 1.3 | Many | MGA | 1/28 | IgM | Positive | TBM (204:189–252) lamellation |
| 3 (2) | 67F | 1.24 | 34.0 | 1.7 | 23/23 | 57 | 0.5 | 86.0 | 0.7 | 11–30 | MGA | 3/27 | Negative | Positive | TBM (262:230–320) |
| 4 (2) | 42M | 0.89 | 75.4 | 0.3 | 15/15 | 17 | 1.0 | 87.5 | 0.3 | 1–5 | MGA | 0/8 | Negative | ND | TBM (305:252–377) |
| 5 (3) | 61M | ESRD (58 years) | ESRD | ESRD | 31/31 | 43 | 1.5 | 40.8 | 0.3 | Many | NSc | 5/10 | IgM | ND | TBM (242:192–282) lamellation |
| 6 (3) | 30F | 0.40 | 148.6 | 0.5 | 12/29 | 21 | 0.5 | 129.5 | 0.1 | 11–30 | MGA | 4/46 | Negative | Positive | TBM (262:256–282) lamellation |
| 7 (4) | 54M | 1.25 | 48.6 | 0.3 | 23/23 | 46 | 1.0 | 65.1 | 0.8 | 11–30 | MGA | 1/12 | Negative | Positive | TBM (216:154–256) |
| 8 (5) | 49F | 0.82 | 58.3 | 2.2 | 9/20 | 49 | 0.8 | 64.7 | 0.8 | 11–30 | FSGS | 7/31 | IgM | Positive | TBM (275:230–358) |
| 9(6) | 64F | 0.66 | 68.8 | 1.4 | 42/42 | 55 | 0.4 | 124.3 | 3.0 | 6–10 | MGA | 0/7 | Negative | Positive | TBM (251:230–282) |
| 10 (7) | 61F | 0.59 | 78.9 | 2.9 | 35/35 | 52 | 0.5 | 99.0 | 1.8 | Many | MGA | 3/37 | Negative | Positive | TBM (256:196–327) lamellation, splitting |
| 11 (8) | 33M | 0.98 | 73.4 | 1.3 | 5/7 | 33 | 1.0 | 73.4 | 1.3 | Many | MGA | 2/6 | Negative | Positive | TBM (238:192–282) |
| 12 (9) | 69F | 1.76 | 22.9 | 3.6 | 44/44 | 62 | 1.1 | 39.7 | 8.0 | 11–30 | NSc | 7/11 | Negative | Positive | TBM (220:200–240) lamellation |
| 13 (10) | 55M | 1.18 | 51.5 | 0.4 | 13/13 | 52 | 1.1 | 57.7 | 0.5 | Many | MGA | 2/17 | Negative | Positive | TBM (149:131–196) |
| 14 (11) | 50M | ESRD (42 years) | ESRD | ESRD | 6/6 | 30 | 1.4 | 70.0 | 5.4 | Many | MGA | 1/13 | Negative | Positive | TBM (256:200–293) lamellation, splitting |
FSGS, focal segmental glomerular sclerosis; GBM, glomerular basement membrane; GS, global sclerosis; MGA, minimal glomerular abnormality; ND, not determined; NSc, nephrosclerosis; TG, total glomeruli.
Two patients had hearing loss (patient 12 and 14) and none had ocular changes.
Figure 2.
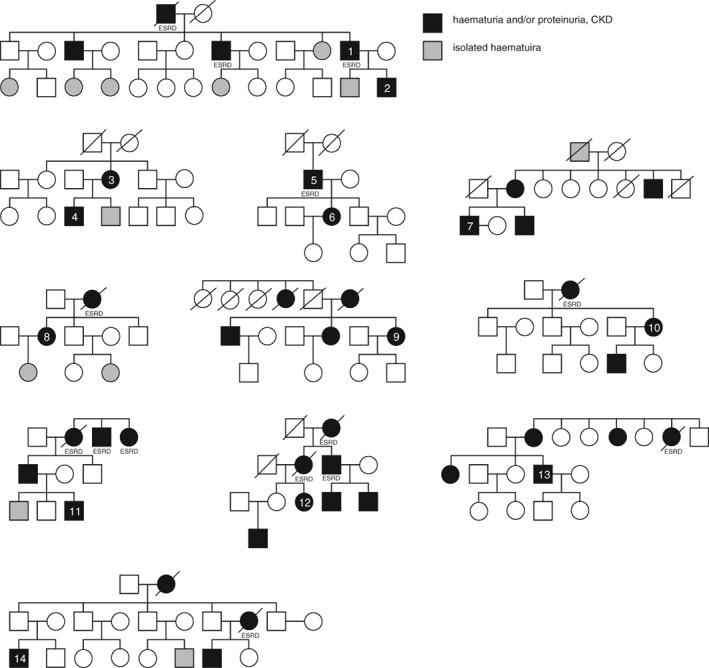
Family trees of the patients with thin basement membrane nephropathy (TBMN)/Autosomal dominant Alport syndrome (ADAS). Black shows patients with hematuria and or proteinuria, chronic kidney disease and gray shows patients with isolated hematuria.
Pathological findings
Pathological findings of the 14 patients with TBMN/ADAS are listed in Table 2. In patients who underwent renal biopsy more than once, the findings of the last biopsy were analyzed. On light microscopy, 10 patients had minor glomerular abnormalities, one patient displayed focal segmental glomerular sclerosis, and three patients showed nephrosclerosis. Immunofluorescent staining revealed nonspecific IgM deposits in three patients. Immunohistochemical staining for α5 (IV) showed a normal pattern in all 11 patients in whom it was performed. While α5 (IV) staining was not performed in three patients, we confirmed that at least one of their family members showed a normal pattern of α5 (IV) staining in a renal biopsy specimen. Thinning of the GBM was identified in all 14 patients by electron microscopy, while thickening and splitting of the GBM was noted in seven patients (50%). Among 11 patients who underwent kidney biopsy multiple times, only two patients were evaluated by electron microscopy in both biopsies (Patient 2 and 12). In patient 2, the manifestation of GBM did not differ in 10 years, and in patient 12, lamellation of GBM was more noticeable after 12 years. Skin biopsy was performed in three patients which all showed a normal pattern of α5 (IV) staining.
DISCUSSION
In this study, we found 14 patients (11 families) with heterozygous mutations in the COL4A3 or COL4A4 genes, suggesting the diagnosis of TBMN/ADAS. They accounted for 69% of patients with hereditary nephritis that was ‘difficult to diagnose’ who fulfilled the following five criteria: (i) haematuria and proteinuria with or without renal dysfunction; (ii) a family history of haematuria, proteinuria, and/or renal dysfunction (especially showing autosomal dominant inheritance); (iii) no specific glomerulonephritis identified by light microscopy and immunofluorescent staining of a renal biopsy specimen; (iv) thinning, splitting, or lamellation of GBM on electron microscopy; and (v) normal immunohistochemical staining of a renal biopsy specimen for α5 (IV). Our result suggests that autosomal dominant forms of type IV collagen nephropathy (TBMN/ADAS) appears more frequent than previously reported. The detection of these patients could be improved by genetic testing based on our criteria (Fig. 3).
Figure 3.
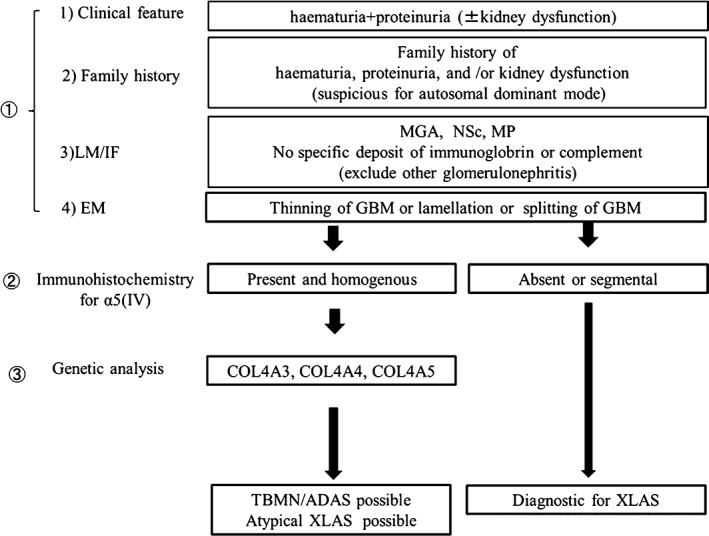
Proposed criteria for thin basement membrane nephropathy (TBMN)/Autosomal dominant Alport syndrome (ADAS) genetic testing. It is important to perform genetic testing of those patients fulfilling this criteria.
The spectrum of type IV collagen nephropathy caused by heterozygous mutation of COL4A3/A4 has been widening and remains debatable. Some authors use the diagnosis of ADAS, others use TBMN, or autosomal dominant later‐onset Alport‐related nephropathy, or even familial FSGS, in the presence of FSGS pathologically.
In 1997, Jefferson et al. reported the first convincing evidence of an ADAS family.21 Although ADAS has been thought to be an extremely rare condition, the reports of patients with ADAS have been increasing during the last decade, mainly from Europe and Japan.7, 8, 22, 23 Recently, Kamiyoshi et al. investigated the detailed characteristics of 16 ADAS families and concluded that ADAS accounted for 5% of AS.23 On the other hand, Fallerini et al. reported a much higher frequency of ADAS (31%: 15/48 families) and suggested the possible existence of undiagnosed ADAS patients.7 However, in fact, many patients with heterozygous COL4A3/A4 mutations, but without ADAS diagnosis, develop severe renal failure and even ESRD. Heterozygous COL4A3/A4 mutations are also found in about 40% of patients with TBMN.10 Some studies have shown that renal function can decline in patients with TBMN during long‐term follow‐up, suggesting that this disease might not be as benign as was previously believed.11, 12 Voskarides et al. reported eight families with heterozygous COL4A3 mutation and dual diagnosis of FSGS and TBMN, showing a wide phenotypic spectrum that ranged from isolated haematuria to ESRD.13 More recently, Malone et al. reported that 10% of their cohort of patients with the diagnosis of familial FSGS had heterozygous mutation in COL4A3/A4 gene.14 Furthermore, Papazachariou et al. reported that 12 out of 24 families (50%) with familial microscopic haematuria had heterozygous mutation in COL4A3/A4 genes, 12% of those developed ESRD.24 They used the diagnosis of autosomal dominant later‐onset Alport‐related nephropathy, and congruent with our study, they mention the difficulty of clinicopathological diagnosis of these patients.15, 24
The median age at diagnosis of haematuria and proteinuria was 22 years and 21.4% developed ESRD at the age of 40–50 years. The age at which the patients with heterozygous mutation in COL4A3/A4 develop ESRD varies between the reports. Some researchers report that 24.3% of ADAS patients develop ESRD by the age of 51 years, which is consistent with our reports.6 On the other hand, others report that 35% of patients with heterozygous COL4A3/A4 mutations develop ESRD by the age of 70 years.25 Although renal dysfunction in patients with heterozygous mutation in COL4A3/A4 gene is milder than in patients with XLAS, it is noteworthy that many patients still developed ESRD. Although 62.5% of patients with stage >3 CKD had hypertension at the time of genetic diagnosis, the detection of proteinuria and kidney dysfunction was prior to the detection of hypertension in all patients, which suggests that hypertension could not be the main cause leading to renal dysfunction. In our series, only 14% of patients had sensorineural hearing loss and there were no ocular abnormalities. These results are also similar to a previous report that hearing loss and ocular abnormalities occurred in 13.3% and 0% of patients with ADAS, respectively.6 The low frequency of typical extra‐renal manifestations of AS is one of the factors that makes diagnosis of patients with heterozygous mutation in COL4A3/A4 gene difficult.
In the present study, half of our patients showed isolated thinning of the GBM. Van der loop et al. reported lamellation or splitting of the GBM in all four ADAS patients they investigated by renal biopsy,22 while Kamiyoshi et al. reported that isolated thinning of the GBM was found in 44% of ADAS patients.23 Although GBM thinning is a pathological feature of TBMN, isolated thinning of the GBM is known to occur in patients with XLAS, especially at an early stage of the disease.26 In our cohort, only one patient showed FSGS (Patient 8). Interestingly, many patients diagnosed as familial FSGS with heterozygous mutation in COL4A3/A4 genes are accompanied by the thinning of GBM, suggesting the causal relationship between TBMN and FSGS.13, 14, 15, 25, 27
Although the spectrum of type IV collagen nephropathy caused by heterozygous mutation in COL4A3/A4 genes is still controversial, it is clear that the patients with these mutations are more frequent than previously thought, and they show a variety of clinical courses with some reaching ESRD, regardless of what name we give to the diagnosis. It is important for nephrologists to recognize this condition and make an early diagnosis, so that adequate treatment can be provided to delay the decline of renal function. The combination of comprehensive clinical evaluation and careful follow‐up, pedigree analysis, histopathology (including electron microscopy, α5 (IV) staining) and molecular studies are required for an accurate diagnosis. However, we also have to emphasize that we should perform genetic analysis before performing kidney biopsy in the future, although it is still common to perform kidney biopsy prior to genetic analysis due to lack of recognition of this condition.
In the present study, XLAS patients accounted for 25% of hereditary nephritis that was ‘difficult to diagnose’. Although our criteria excluded patients with typical XLAS based on the family history, symptoms, or absence of immunostaining for α5 (IV), XLAS and ADAS cannot necessarily be distinguished by obtaining a family history, and some XLAS patients have mild clinical features and a normal pattern of α5 (IV) staining. Female XLAS patients are known to have less severe renal dysfunction than male XLAS patients, and they show a normal or mosaic α5 (IV) staining pattern.3 Moreover, some male XLAS patients, especially those with missense mutation in COL4A5 gene, have a milder clinical phenotype than typical male XLAS patients, and α5 (IV) staining pattern is normal in these patients.28 It is important for nephrologists to be able to recognize these ‘atypical XLAS patients’.
Our research has some limitations. First, the number of subjects is small and they were enrolled from only two hospitals. Second, although we conducted targeted resequencing to search for modifier genes, we could not identify any modifier genes (Table S1). However, since there is a detection limit of modifier genes with targeted sequence, it is possible that modifier genes will be detected by whole exome sequencing. Further investigations are needed to solve this problem. Third, although some authors report that unusual deep intronic mutations in the COL4A5 gene cause XLAS by reverse transcription polymerase chain reaction (RT‐PCR) mRNA analysis, which cannot be detected by NGS,29 we have not performed such analysis in this study. We cannot exclude the possibility that our patients have another pathogenic deep intron mutation in either of COL4A3, COL4A4, and COL4A5 genes.
Conclusion
Undiagnosed autosomal dominant form of type IV collagen nephropathy (TBMN/ADAS) exists among patients with hereditary nephritis that is ‘difficult to diagnose clinicopathologically’. It is important for nephrologists to recognize these conditions and perform genetic testing of appropriate patients to reach an early diagnosis and provide adequate treatment.
DISCLOSURE
All authors declare no financial conflicts of interest.
Supporting information
Table S1. Targeted resequencing to search for modifier genes. Forty‐five podocyte‐related genes in this table were screened by targeted sequencing that are known to be causative of inherited focal segmental glomerulosclerosis or Alport syndrome.
ACKNOWLEDGEMENTS
This study was partially funded by the Okinaka Memorial Institute for Medical Research. Research idea and study design: YU; data acquisition: AI; data analysis/interpretation: AI, KN, NS; Each author contributed important intellectual content during manuscript drafting or revision and accepts accountability for the overall work by ensuring that questions pertaining to the accuracy or integrity of any portion of the work are appropriately investigated and resolved. We thank Masanori Suzuki for performing electronmicroscopic analysis.
The copyright line for this article was changed on 12 September 2019 after original online publication.
REFERENCES
- 1. Deltas C, Pierides A, Voskarides K. Molecular genetics of familial hematuric diseases. Nephrol. Dial. Transplant. 2013; 28: 2946–60. [DOI] [PubMed] [Google Scholar]
- 2. Flinter F. Alport's syndrome. J. Med. Genet. 1997; 34: 326–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kashtan CE. Alport syndrome and the X chromosome: Implications of a diagnosis of Alport syndrome in females. Nephrol. Dial. Transplant. 2007; 22: 1499–505. [DOI] [PubMed] [Google Scholar]
- 4. Hostikka SL, Eddy RL, Byers MG, Hoyhtya M, Shows TB, Tryggvason K. Identification of a distinct type IV collagen alpha chain with restricted kidney distribution and assignment of its gene to the locus of X chromosome‐linked Alport syndrome. Proc. Natl. Acad. Sci. U. S. A. 1990; 87: 1606–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jais JP, Knebelmann B, Giatras I et al X‐linked Alport syndrome: Natural history in 195 families and genotype‐phenotype correlations in males. J. Am. Soc. Nephrol. 2000; 11: 649–57. [DOI] [PubMed] [Google Scholar]
- 6. Marcocci E, Uliana V, Bruttini M et al Autosomal dominant Alport syndrome: Molecular analysis of the COL4A4 gene and clinical outcome. Nephrol. Dial. Transplant. 2009; 24: 1464–71. [DOI] [PubMed] [Google Scholar]
- 7. Fallerini C, Dosa L, Tita R et al Unbiased next generation sequencing analysis confirms the existence of autosomal dominant Alport syndrome in a relevant fraction of cases. Clin. Genet. 2014; 86: 252–7. [DOI] [PubMed] [Google Scholar]
- 8. Rosado C, Bueno E, Felipe C, Valverde S, Gonzalez‐Sarmiento R. Study of the true clinical progression of autosomal dominant Alport syndrome in a European population. Kidney Blood Press. Res. 2015; 40: 435–42. [DOI] [PubMed] [Google Scholar]
- 9. Moriniere V, Dahan K, Hilbert P et al Improving mutation screening in familial hematuric nephropathies through next generation sequencing. J. Am. Soc. Nephrol. 2014; 25: 2740–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Haas M. Alport syndrome and thin glomerular basement membrane nephropathy: A practical approach to diagnosis. Arch. Pathol. Lab. Med. 2009; 133: 224–32. [DOI] [PubMed] [Google Scholar]
- 11. Savige J, Rana K, Tonna S, Buzza M, Dagher H, Wang YY. Thin basement membrane nephropathy. Kidney Int. 2003; 64: 1169–78. [DOI] [PubMed] [Google Scholar]
- 12. Dische FE, Weston MJ, Parsons V. Abnormally thin glomerular basement membranes associated with hematuria, proteinuria or renal failure in adults. Am. J. Nephrol. 1985; 5: 103–9. [DOI] [PubMed] [Google Scholar]
- 13. Voskarides K, Damianou L, Neocleous V et al COL4A3/COL4A4 mutations producing focal segmental glomerulosclerosis and renal failure in thin basement membrane nephropathy. J. Am. Soc. Nephrol. 2007; 18: 3004–16. [DOI] [PubMed] [Google Scholar]
- 14. Malone AF, Phelan PJ, Hall G et al Rare hereditary COL4A3/COL4A4 variants may be mistaken for familial focal segmental glomerulosclerosis. Kidney Int. 2014; 86: 1253–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Papazachariou L, Demosthenous P, Pieri M et al Frequency of COL4A3/COL4A4 mutations amongst families segregating glomerular microscopic hematuria and evidence for activation of the unfolded protein response. Focal and segmental glomerulosclerosis is a frequent development during ageing. PLoS ONE 2014; 9: e115015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gast C, Pengelly RJ, Lyon M et al Collagen (COL4A) mutations are the most frequent mutations underlying adult focal segmental glomerulosclerosis. Nephrol. Dial. Transplant. 2016; 31: 961–70. [DOI] [PubMed] [Google Scholar]
- 17. Pierides A, Voskarides K, Athanasiou Y et al Clinico‐pathological correlations in 127 patients in 11 large pedigrees, segregating one of three heterozygous mutations in the COL4A3/ COL4A4 genes associated with familial haematuria and significant late progression to proteinuria and chronic kidney disease from focal segmental glomerulosclerosis. Nephrol. Dial. Transplant. 2009; 24: 2721–9. [DOI] [PubMed] [Google Scholar]
- 18. Boye E, Mollet G, Forestier L et al Determination of the genomic structure of the COL4A4 gene and of novel mutations causing autosomal recessive Alport syndrome. Am. J. Hum. Genet. 1998; 63: 1329–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Buzza M, Dagher H, Wang YY et al Mutations in the COL4A4 gene in thin basement membrane disease. Kidney Int. 2003; 63: 447–53. [DOI] [PubMed] [Google Scholar]
- 20. Oka M, Nozu K, Kaito H et al Natural history of genetically proven autosomal recessive Alport syndrome. Pediatr. Nephrol. 2014; 29: 1535–44. [DOI] [PubMed] [Google Scholar]
- 21. Jefferson JA, Lemmink HH, Hughes AE et al Autosomal dominant Alport syndrome linked to the type IV collage alpha 3 and alpha 4 genes (COL4A3 and COL4A4). Nephrol. Dial. Transplant. 1997; 12: 1595–9. [DOI] [PubMed] [Google Scholar]
- 22. van der Loop FT, Heidet L, Timmer ED et al Autosomal dominant Alport syndrome caused by a COL4A3 splice site mutation. Kidney Int. 2000; 58: 1870–5. [DOI] [PubMed] [Google Scholar]
- 23. Kamiyoshi N, Nozu K, XJ F et al Genetic, clinical, and pathologic backgrounds of patients with autosomal dominant Alport syndrome. Clin. J. Am. Soc. Nephrol. 2016; 11: 1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Papazachariou L, Papagregoriou G, Hadjipanagi D et al Frequent COL4 mutations in familial microhematuria accompanied by later‐onset Alport nephropathy due to focal segmental glomerulosclerosis. Clin. Genet. 2017. Epub ahead of print [DOI] [PubMed] [Google Scholar]
- 25. Deltas C, Savva I, Voskarides K, Papazachariou L, Pierides A. Carriers of autosomal recessive Alport syndrome with thin basement membrane nephropathy presenting as focal segmental glomerulosclerosis in later life. Nephron 2015; 130: 271–80. [DOI] [PubMed] [Google Scholar]
- 26. Heidet L, Gubler MC. The renal lesions of Alport syndrome. J. Am. Soc. Nephrol. 2009; 20: 1210–5. [DOI] [PubMed] [Google Scholar]
- 27. Voskarides K, Pierides A, Deltas C. COL4A3/COL4A4 mutations link familial hematuria and focal segmental glomerulosclerosis. Glomerular epithelium destruction via basement membrane thinning? Connect. Tissue Res. 2008; 49: 283–8. [DOI] [PubMed] [Google Scholar]
- 28. Hashimura Y, Nozu K, Kaito H et al Milder clinical aspects of X‐linked Alport syndrome in men positive for the collagen IV alpha5 chain. Kidney Int. 2014; 85: 1208–13. [DOI] [PubMed] [Google Scholar]
- 29. King K, Flinter FA, Nihalani V, Green PM. Unusual deep intronic mutations in the COL4A5 gene cause X linked Alport syndrome. Hum. Genet. 2002; 111: 548–54. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Targeted resequencing to search for modifier genes. Forty‐five podocyte‐related genes in this table were screened by targeted sequencing that are known to be causative of inherited focal segmental glomerulosclerosis or Alport syndrome.