

Summary
The species of the genus Saccharomyces are commonly inhabiting tree bark and the surrounding soil, but their abundance have likely been underestimated due to biases in culturing methods. Metagenomic studies have so far been unable to detect Saccharomyces species in wild environments. Here, we sequenced the mycobiome of soils surrounding different trees at various altitudes in the Italian Alps. To survey for yeasts species belonging to Saccharomyces genus rather than other fungal species, we performed a selectivity step involving the isolation of the internal transcribed spacer (ITS) region that is specific to this yeast group. Reads mapping to Saccharomyces species were detected in all soil samples, including reads for S. mikatae and for S. eubayanus. ITS1 alignment of the S. cerevisiae, S. paradoxus and S. kudriavzevii sequences showed up to three base pair polymorphisms with other known strains, indicating possible new lineages. Basidiomycetous fungi were still the dominant species, compared to the Ascomycota, but the selectivity step allowed for the first time the detection and study of the biodiversity of the Saccharomyces species in their natural environment.
Introduction
The Saccharomyces species are present in a variety of natural environments, including tree bark, soil, fruits and insects guts (Naumov et al., 1998; Sniegowski et al., 2002; Stefanini et al., 2012; Hyma and Fay, 2013; Charron et al., 2014). Although, humans have exploited these species for fermentation and biotechnological purposes for hundreds of years, their ecology, biodiversity and geographic distribution have only recently received attention from the science community. In particular, three new species (S. arboricola, S. eubayanus and S. jurei) have been isolated in the last 10 years, bringing the number of species in the Saccharomyces group to eight (Wang and Bai, 2008; Libkind et al., 2011; Naseeb et al., 2017). Despite the strong association of S. cerevisiae with domestication, wild species have been detected at a low frequency in substrates that are isolated from human activity; these wild species have formed geographically structured lineages that are distinct from the domesticated species (Peter et al., 2018). The relatively broad ecology of these species occurring in low numbers suggests that they may be generalist nomads (Goddard and Greig, 2015). S. paradoxus is a wild species that is frequently associated with oak trees (Quercus spp), commonly residing in sympatry with S. cerevisiae. The lineages of S. paradoxus are differentiated by geographic distance, making this species a model for the study of the natural habitat and evolution of the wild Saccharomyces species (Naumov et al., 1998; Sniegowski et al., 2002; Liti et al., 2009).
Far East Asia harbours the broadest diversity of Saccharomyces species. S. kudriavzevii, S. mikatae and S. arboricola were initially isolated in this region (along with genetically diverged populations of S. cerevisiae and S. eubayanus) from soil, decayed leaves and oak bark. (Naumov et al., 2000; Wang and Bai, 2008; Bing et al., 2014; Peris et al., 2014). The distribution of S. kudriavzevii and S. arboricola has extended out of Far East Asia. S. kudriavzevii strains have been detected in Europe (Portugal and Spain), and S. arboricola have been reported in Australasia (New Zealand) (Sampaio and Goncalves, 2008; Lopes et al., 2010; Gayevskiy and Goddard, 2016). Meanwhile, S. mikatae presence is restricted to Far East Asia, and it has not been isolated in any other region. S. eubayanus is well‐established in South America. Initially isolated from Southern beech trees (Nothofagus spp) in Patagonia, Argentina, S. eubayanus has been described as the cryotolerant parent of the lager hybrid S. pastorianus (S. cerevisiae × S. eubayanus) (Libkind et al., 2011). The lager yeast was thought to be of Patagonian or Chinese origin, as S. eubayanus strains isolated from both regions were highly similar to the non‐S. cerevisiae subgenome of S. pastorianus (Libkind et al., 2011; Bing et al., 2014). Population genomic studies of S. eubayanus and lager strains have shown that the cryotolerant parent is derived from a lineage of Holarctic distribution; however, none of the analysed wild S .eubayanus isolates have been identified as the sole closest relative of the lager yeast (Peris et al., 2016). Most wild S. uvarum have been isolated from South America coexisting with S. eubayanus (Libkind et al., 2011; Rodriguez et al., 2014). The S. uvarum and S. eubayanus South American populations are of high genetic diversity, suggesting that these species are native to the region (Almeida et al., 2014; Peris et al., 2016). S. jurei is the latest addition to the Saccharomyces species, isolated from oak bark and soil in France (Naseeb et al., 2017). Phylogenetic analysis based on 101 concatenated genes has grouped the S. jurei strains in the same clade as S. mikatae. The two species share a reciprocal chromosomal translocation, suggesting a common evolutionary history (Naseeb et al., 2018).
Early studies that sought to isolate Saccharomyces species from oak tree bark, surrounding soil and exudates frequently found S. paradoxus and S. cerevisiae, suggesting that oaks may be the natural habitat of the Saccharomyces species (Naumov et al., 1998; Sniegowski et al., 2002). However, Saccharomyces species were also isolated from other tree species, encouraging researchers to explore different habitats (Sampaio and Goncalves, 2008; Bing et al., 2014; Charron et al., 2014; Sylvester et al., 2015). The current understanding of the natural habitats of the Saccharomyces species relies on specific enrichment culturing methods. Due to differences in the temperature growth profiles of the species, incubation of samples at varying temperatures have allowed researchers to retrieve a wider range of species. For example, the cryotolerant S. kudriavzevii and S. uvarum were isolated at 10 °C (Sampaio and Goncalves, 2008). However, substrate enrichment methods inevitably underestimate the variety of wild species, as competition in the enrichment culture reduces the propagation of some species.
To avoid culturing biases, high‐throughput sequencing platforms have been used to study fungal communities existing in natural substrates (Buee et al., 2009; Pinto et al., 2014; Taylor et al., 2014; Masinova et al., 2017). Kowallik et al. (2015) did not obtain sequences of Saccharomyces species from oak bark using high‐throughput sequencing of environmental DNA (eDNA), even though they isolated S. paradoxus colonies from the same trees by selective plating (Kowallik et al., 2015). A more recent metagenomic study that analysed the yeast community changes in soil and litter affected by biotic and abiotic factors found differences in the basidiomycetes and ascomycetes yeasts among beech (Fagus spp), oak and spruce (Picea spp) trees, but again, species belonging to the Saccharomyces genus were not detected (Masinova et al., 2017). Pyrosequencing of eDNA from grapes in New Zealand vineyards has detected Saccharomyces species, but in very low numbers (~1:20 000 of sequences) (Taylor et al., 2014). A higher number of S. cerevisiae and S. paradoxus sequences were obtained from DNA extracted from grape must, but fewer sequences for these species were found in the eDNA from oak bark and soil (Dashko et al., 2016).
To date, metagenomic studies have eitheir not detected Saccharomyces species in natural environments (i.e., contexts not associated with human activities), or they have detected the sequences of a few species in low numbers in substrates related to fermentation activities. However, Saccharomyces sequences were recently detected in human‐related samples in relatively high abundance (Boix‐Amoros et al., 2017; Nash et al., 2017). These results indicate that wild Saccharomyces species are in low abundance relative to other fungi and yeasts, hampering the understanding of the species' biodiversity.
The sequencing of eDNA has not previously been used to specifically target Saccharomyces species in soil. Here, to capture the diversity of the Saccharomyces species associated with the soil surrounding trees at various altitudes, we employed a selection method based on the specific length of the ITS sequence (~850 bp) of the Saccharomyces species. Using this technique, we were able to enrich eDNA samples for this yeast group prior to sequencing (using Illumina MiSeq). We detected the molecular signals for S. mikatae and S. eubayanus, two species that have not previously been isolated in Europe. This is the first study to use an enrichment step specific for the Saccharomyces genus, prior to deep eDNA sequencing, to assess the biodiversity of these wild species in areas that are remote from domestication.
Results and discussion
Sequences analysis and OTU clustering
To have an overview of Saccharomyces species in their natural habitats, we have exploited the specific ITS region size of these species (Supporting Information Fig. S2) to limit the interference of other fungi, followed by high‐throughput sequencing of the ITS1 using Illumina MiSeq platform. A total of 27 samples comprising three soil replicates from each of nine trees in three patches at different altitudes (i.e., 600 m, 1400 m and 1800–1900 m) were sequenced (Supporting Information Table S1). After the removal of low quality, chimeric and singleton sequences, 13 088 931 sequences of high quality were obtained from the dataset and clustered into a total of 5578 OTUs. The sequences of the three biological replicates were then grouped to represent the fungal population in the soil of different trees (Supporting Information Table S2). Rarefaction analysis of OTUs in each soil sample (Supporting Information Fig. S3) showed an increase in the number of OTUs with an increase in a number of sequences, with all curves approaching saturation, suggesting that OTU richness was captured. Out of these 5578 OTUs, 1720 OTUs belong to Ascomycetes, 1200 OTUs to Basidiomycetes and the remaining 2658 OTUs were assigned to unclassified read (unidentified), reads with no identity in the databases, or to different phyla (Supporting Information Table S3). Although, we targeted a specific section of the soil mycobiome to select for hemiascomycetes (i.e., ITS corresponding to a region around 850 bp), which constituted 17% of total reads of all samples, a high proportion of OTUs, accounting for 31% of all reads, were assigned to the phylum basidiomycota (Supporting Information Table S3). The average ITS region size of Basidiomycota species is ∼600 bp (Porter and Golding, 2011), it is likely that lower size ITS DNA was still present in the gel extracted area corresponding to the Saccharomyces species. Another possibility for the abundance of basidiomycetes is PCR bias towards amplification of shorter length ITS and ITS1 regions (∼600 bp and 214 bp respectively) of basidiomycetes (Porter and Golding, 2011), in contrast to relatively longer regions in Saccharomyces species (∼850 bp and 360 bp respectively) (Naumov et al., 2000). Moreover, PCR primers were reported to be biased towards the amplification of basidiomycetes (primers ITS1, ITS1‐F and ITS5) or ascomycetes (primers ITS2, ITS3 and ITS4) (Bellemain et al., 2010), however, we have reduced this biases by a different combination of primers sequencing in both directions. The amplification bias to a specific Saccharomyces species is unlikely, due to the similarity of the species in the size of ITS and ITS1 (Naumov et al., 2000). A high number of saprotrophic and mycorrhizal fungi fruiting bodies were visually observed in the sampling locations, especially at areas of 1600 m–1800 m altitude, therefore, accounting for the high proportion of basidiomycetes reads in the eDNA. Surveys of fungal diversity in the soil of forests populated with a variety of trees using pyrosequencing revealed the similar dominance of Basidiomycota fungi (Buee et al., 2009).
Our data included samples from both beech and spruce trees at 600 m and 1400 m and no other tree species were sampled at more than one location. Focusing on beech and spruce samples, we found that fungal communities (represented as OTUs) detected in the same location from different tree species were more similar than those collected at different locations from the same tree species (Table 1). This suggests that, even at a moderate spatial scale, location plays a more important role in determining the fungal community than tree species.
Table 1.
Jaccard similarity coefficients for fungal communities collected from different tree species at the same location (rows 1 and 2), and for the same tree species at different locations (rows 3 and 4).
| Trees in comparison | Jaccard similarity coefficient |
|---|---|
| Spruce, 600 m versus beech, 600 m | 0.291 |
| Spruce, 1400 m versus beech, 1400 m | 0.288 |
| Spruce, 600 m versus spruce 1400 m | 0.213 |
| Beech, 600 m versus beech 1400 m | 0.190 |
Higher similarity coefficients indicate more similar communities.
The diversity of Saccharomyces species in soil surrounding trees at varying altitudes
Sequencing of the gel‐extracted ITS1 succeeded in detecting S. kudriavzevii, S. mikatae, S. cerevisiae, S. paradoxus, S. eubayanus, S. jurei and S. uvarum in our samples, representing about 0.1% of all Ascomycota sequences, therefore, these species are rare in wild environments relative to other fungal species (Fig. 1). The identity of Saccharomyces species obtained using QIIME 12_11 ITS database was manually confirmed by comparison to NCBI database using Blast. Most Saccharomyces species reads showed 99% similarity to the corresponding NCBI GenBank reference, with 100% sequence coverage (Table 2). The taxonomy of three Saccharomyces species reads assigned by QIIME 12_11 database were not the same as the GenBank reference. OTU 1152 was assigned to S. bayanus; however, the sequences corresponded to ‘uncultured fungus clone’. Alignment of reads assigned as S. spencerorum (OTU 1159, Supporting Information Table S2), a species that have been renamed to the genus Kazachstania as K. spencerorum (Kurtzman and Robnett, 2003), to GenBank references resulted in 99% similarity to S. jurei NCYC 3947T. OTU 1159 aligned to the ITS1 region of S. jurei NCYC 3947T, however, did not align to the ITS1 of K. spencerorum (Supporting Information Fig. S4). Reads of OTU 1158 were assigned as S. uvarum instead of S. pastorianus as the sequences were identified as S. uvarum against the GenBank database. Moreover, the sequence of OTU 1158 showed similarity to ITS1 of S. uvarum NRRL Y‐17034T in the two regions that differentiate S. uvarum from S. pastorianus NRRL Y‐2717NT and S. bayanus NRRL Y‐12624T (Supporting Information Fig. S5). QIIME 12_11 can misidentify some Saccharomyces species reads due to errors in taxonomy assignment in the database and lack of recent updates of ITS sequences databases (Vilgalys, 2003). We were able to overcome the poor taxonomic annotation of the ITS1 sequences deposited in the QIIME 12_11 database by the alignment of the individual Saccharomyces sequences to the reference sequences of the NCBI database. Previous attempts to study the diversity of Saccharomyces species in oak trees bark in nature through pyrosequencing did not obtain any reads corresponding to Saccharomyces (Kowallik et al., 2015). This indicates that the selectivity step used in this study is effective in capturing sequences of most Saccharomyces species present in nature.
Figure 1.
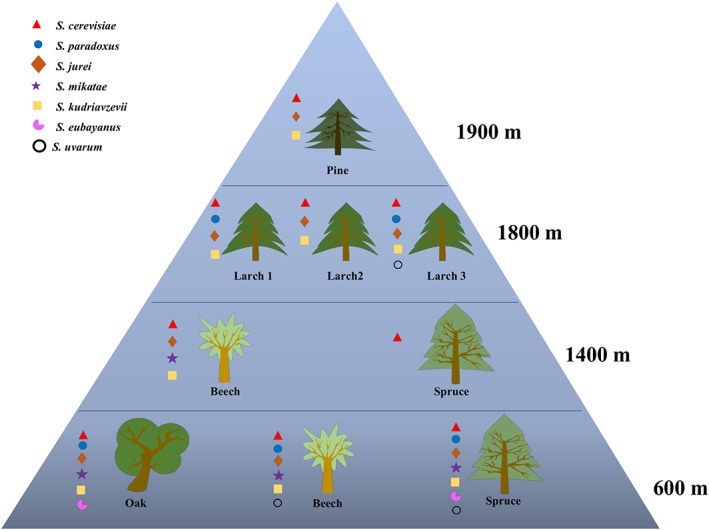
Distribution of Saccharomyces species in the mycobiome of soil surrounding different tree species at elevated altitudes.
The presence of species is expressed as Operational taxonomic units (OTUs) which consist of at least two identical reads. Three individual Larch trees were sampled in the area of 1800 m altitude.
Table 2.
Operational taxonomic units of Saccharomyces species in the dataset aligned to reference sequences of NCBI database.
| OTU | Species (Accession number) | Similarity to reference species (%) | Coverage (%) |
|---|---|---|---|
| OTU 1153 | S. cerevisiae (MG241531) | 99 | 100 |
| OTU 1157 | S. paradoxus (MH032820) | 99 | 99 |
| OTU 1156 | S. mikatae (KY105203) | 99 | 100 |
| OTU 1159 | S. jurei (HG764814) | 99 | 100 |
| OTU 1155 | S. kudriavzevii (CP030973) | 99 | 100 |
| OTU 1154 | S. eubayanus (CP030956) | 100 | 100 |
| OTU 1158 | S. uvarum (MH459413) | 99 | 100 |
Sequences of S. eubayanus were found in the soil of oak and spruce at 600 m (Fig. 1). This study presents the first evidence of S. eubayanus presence in Europe based on sequencing of the mycobiome. Extensive sampling of different tree species in South America and China resulted in the isolation of a population of S. uvarum existing in sympatry with S. eubayanus (Libkind et al., 2011; Bing et al., 2014). In our data, S. uvarum (detected as S. pastorianus QIIME 12_11 ITS database and manually corrected) co‐occurred with S. eubayanus in soil from spruce at 600 m. S. uvarum was also detected in soil from a beech tree at 600 m and larch 3 at 1800 m (Fig. 1).
S. kudriavzevii and S. jurei were each present in eight out of nine trees sampled (S. jurei and S. kudriavzevii were absent from spruce soil at 1400 m), while S. cerevisiae was in the soil of all trees. Interestingly, S. paradoxus, which is the Saccharomyces species most commonly isolated from natural environments (Naumov et al., 1998; Sniegowski et al., 2002; Sampaio and Goncalves, 2008), was detected in only five out of nine trees.
In agreement with previous results on the coexistence of S. cerevisiae and S. paradoxus, both species were recorded in the soil of trees at 600 m and two trees at 1800 m (Fig. 1) (Naumov et al., 1998; Sniegowski et al., 2002; Sweeney et al., 2004). Although, S. cerevisiae has been thoroughly domesticated, wild strains forming distinct populations have been isolated from natural substrates such as soil, bark and tree exudates (Naumov et al., 1998; Sniegowski et al., 2002; Fay and Benavides, 2005; Wang et al., 2012; Almeida et al., 2015). Phylogenetic analysis based on single nucleotide polymorphisms (SNPs) of a large number of S. cerevisiae from different habitats and geographical regions revealed domesticated population and wild population as two separated lineages separated by mosaic strains of admixture lineages that are human‐related (Peter et al., 2018). However, previous metagenomics studies on fungal diversity in soil and bark of different tree species have failed to detect S. cerevisiae (Buee et al., 2009; Cordier et al., 2012; Kowallik et al., 2015; Masinova et al., 2017), probably due to the higher number of reads corresponding to the more abundant fungi in these areas. The latest addition to the Saccharomyces species, S. jurei, had been isolated in the St Auban region of France (Naseeb et al., 2017). Our results show that S. jurei is also present in other pre‐alpine and alpine environments.
S. mikatae reads were detected in soil samples of all trees at 600 m and in the soil of beech at 1400 m. Sequences of this species were detected recently in grape must from European vineyards in Slovenia (Dashko et al., 2016). Our study is the first to demonstrate the presence of wild S. mikatae in areas without domestication activities.
The soil of oak tree harboured over twofold the number of Saccharomyces species reads in comparison to samples of different trees at the same altitude (Table 3), which may indicate the oak tree as the preferred habitat of these species. Despite the isolation of Saccharomyces species from different tree species, (Libkind et al., 2011; Bing et al., 2014; Rodriguez et al., 2014; Gayevskiy and Goddard, 2016), most Saccharomyces species show a relatively strong association with oak tree, especially in the Northern Hemisphere (Sniegowski et al., 2002; Sampaio and Goncalves, 2008; Charron et al., 2014; Sylvester et al., 2015).
Table 3.
Number of Saccharomyces reads in soil samples surrounding trees at varying altitudes.
| Tree, altitude | S. cerevisiae | S. paradoxus | S. jurei | S. mikatae | S. kudriavzevii | S. eubayanus | S. uvarum |
|---|---|---|---|---|---|---|---|
| Oak 600 m | 24 | 18 | 24 | 132 | 316 | 1 | 0 |
| Beech 600 m | 85 | 7 | 23 | 41 | 50 | 0 | 9 |
| Spruce 600 m | 21 | 6 | 13 | 29 | 42 | 2 | 2 |
| Beech 1400 m | 13 | 0 | 3 | 27 | 183 | 0 | 0 |
| Spruce 1400 m | 2 | 0 | 0 | 0 | 0 | 0 | 0 |
| Larch 1 1800 m | 2 | 2 | 2 | 0 | 7 | 0 | 0 |
| Larch 2 1800 m | 3 | 0 | 65 | 0 | 158 | 0 | 0 |
| Larch 3 1800 m | 6 | 10 | 2 | 0 | 44 | 0 | 3 |
| Pine 1900 m | 1 | 0 | 1 | 0 | 8 | 0 | 0 |
The best model for our read count data included effects of species, patch and the interaction between species and patch (likelihood ratio of the second best to the best model, < 10−10). Saccharomyces species in our samples had different abundances (expressed as number of reads), and the relative abundances of the species differed among patches (Table 3). Across most samples in most patches, S. kudriavzevii was more abundant than any other species (Fig. 2, Table 3). The presence of S. kudriavzevii in most soil samples coupled with its abundance relative to other Saccharomyces species suggests that it is well established in the sampling area (Table 3).
Figure 2.
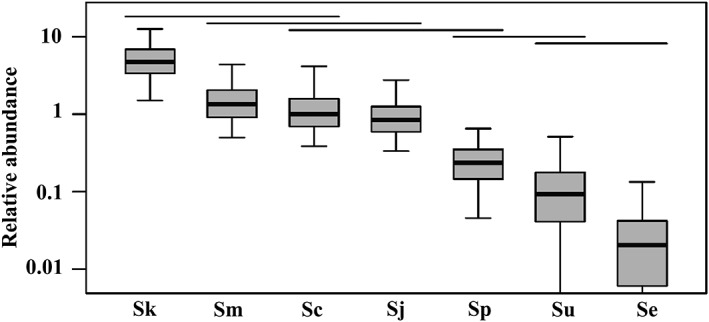
Relative abundance of seven Saccharomyces species across all samples in our study.
Saccharomyces species are abbreviated as: Sk (S. kudriavzevii), Sm (S. mikatae), Sj (S. jurei), Sc (S. cerevisiae), Sp (S. paradoxus), Su (S.uvarum) and Se (S. eubayanus). Abundance is reported relative to the abundance of S. cerevisiae in the observed data. Box plots show 50% (boxes) and 95% (whiskers) confidence intervals around the observed relative abundance. Our bootstrap analysis could not identify a lower bound for the 95% confidence interval around the abundance of S. uvarum or S. eubayanus. The abundance of species under the same horizontal line are not significantly different (study‐wide type I error rate α = 0.05).
S. paradoxus is geographically widely distributed and can be readily isolated in high abundance from tree bark and surrounding soil using enrichment culturing protocols (Naumov et al., 1998; Sniegowski et al., 2002; Liti et al., 2009; Charron et al., 2014). However, in our samples, S. paradoxus was less abundant than S. kudriavzevii or S. mikatae. Thus, either S. paradoxus was less common in our samples than in previous studies, or the use of enrichment cultures to evaluate the diversity of wild Saccharomyces species is biased towards detecting S. paradoxus. The latter might be true if S. paradoxus is able to outcompete other Saccharomyces species in laboratory enrichment cultures. In fact, Kowallik et al. (2015) reported the inhibition of S. paradoxus growth in its natural habitat caused by the surrounding microbial community. S. eubayanus was less abundant in our samples than any other species except possibly S. uvarum (Fig. 2). The low amount of reads found for S. eubayanus may explain why it has not previously been isolated in Europe.
Saccharomyces species were more abundant in the patch at 600 m than in the patches 1400 m or 1800–1900 m (Fig. 3), indicating a possible consequence of temperature decreases with altitude, which can shape ecological communities (Korner, 2007). We obtained no S. mikatae reads, and few S. cerevisiae reads from soils collected above 1400 m (Table 3). The optimum growth temperatures of S. mikatae and S. cerevisiae are approximately 29 °C and 32 °C respectively (Salvado et al., 2011). We hypothesise that the thermosensitive nature of these species may have contributed to their reduced abundance in the high‐altitude patch. S. kudriavzevii and S. jurei appear to be relatively more abundant than other Saccharomyces species in soil collected at altitude 1800 m (Table 3). The abundance of the species at high altitudes is correlated with their temperature growth profile, S. kudriavzevii is characterized as a cryotolerant species grows optimally at approximately 23 °C and S. jurei is able to grow at high and low temperatures with optimum growth temperature higher than S. kudriavzevii (∼25°C–30°C) (Salvado et al., 2011; Naseeb et al., 2018).
Figure 3.
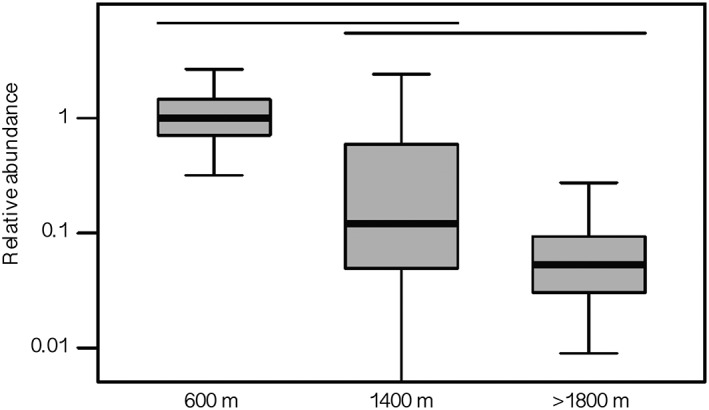
Relative abundance of Saccharomyces in patches at three different altitudes.
Abundance is reported relative to the abundance of Saccharomyces in the patch at 600 m. Box plots show 50% (boxes) and 95% (whiskers) confidence intervals around the observed relative abundance. Our bootstrap analysis could not identify a lower bound for the 95% confidence interval around the abundance of Saccharomyces in the patch at 1400 m. Abundance in patches under the same horizontal line are not significantly different (study‐wide type I error rate α = 0.05).
Saccharomyces reads alignment with genetically distinct populations
We aligned the ITS1 sequences in our samples with those of known Saccharomyces species and found unique base pair differences in S. cerevisiae, S. paradoxus and S. kudriavzevii. This suggests the presence of new distinct Italian lineage of these species.
Diverged populations within wild Saccharomyces species have been related to different geographical origins (Liti et al., 2009; Wang et al., 2012; Almeida et al., 2014; Peris et al., 2014; Peris et al., 2016). Recently, whole‐genome sequencing of over 1000 S. cerevisiae strains revealed strains clustering into lineages that are associated with ecological niche, geographical origin and domestication (Peter et al., 2018). Liti et al. identified five genetically diverged wild and domesticated populations of S. cerevisiae (West African, Malaysian, Sake, North American and Wine/European) (Liti et al., 2009). Later, eight distinct lineages of wild S. cerevisiae were identified from different regions of China (Wang et al., 2012). Here, we report a S. cerevisiae population in which the OTU 1153 sequence possess three unique base pair difference from previously reported S. cerevisiae populations and is closely related to the European strain indicated by an identical nucleotide in the ITS1 sequence that is specific to the strains (Supporting Information Fig. S6).
S. paradoxus strains are clustered into four genetically distinct populations correlated with the geographical origin (Liti et al., 2009). Alignment of our S. paradoxus ITS1 sequence with those from the European, American, Far Eastern and Hawaiian populations shows the segregation of S. paradoxus OTU 1157 by three base pairs (Supporting Information Fig. S7), indicating that this is a potentially distinct lineage. However, the ITS1 sequence of OTU 1157 shares the same base pair with the European (CBS 432) and Far Eastern (N 45) populations that separate these species from the American (YPS138) and Hawaiian strains (UWOPS91.917.1).
Natural isolates of S. kudriavzevii are distributed into three genetically diverged populations. The European population includes Spanish and Portuguese isolates, a Japanese population consisting of the type strain IFO 1802T, and a second Japanese population that includes IFO 1803 that is diverged from IFO 1802T by 4% (Hittinger et al., 2010; Lopes et al., 2010). The different populations of S. kudriavzevii differ in the ITS1 sequence by two nucleotides (Supporting Information Fig. S8) The ITS1 sequence of S. kudriavzevii OTU 1155 is more similar to the ITS1 of the Spanish strain CA111 than to the Portuguese strain (ZP591), as indicated by a single common base pair between the strains (Supporting Information Fig. S8). However, the ITS1 sequence of the Italian population differed from all known sequences (i.e., European and Asian strains) by two unique base pair. The presence of unique base pair substitutions in the Italian S. kudriavzevii ITS1 sequence again points to a new potential European lineage of S. kudriavzevii.
S. eubayanus (OTU 1154) ITS1 sequence was not aligned with ITS1 of previously identified populations of S. eubayanus as the sequence of this loci is identical in the known populations (Peris et al., 2014). The ITS1 sequence of S. uvarum differs from S. eubayanus by one base pair (Peris et al., 2014). This base pair difference was also detected between S. eubayanus OTU 1154 and S. uvarum NRRL Y‐17034T in our samples (Supporting Information Fig. S9).
Population structure studies are not possible yet for S. mikatae and S. jurei since S. mikatae strains (IFO1815T and IFO 1816) were isolated only in Asia, while S. jurei strains (NCYC3947T and NCYC 3962) were only isolated in France (Naumov et al., 2000; Naseeb et al., 2017). Here, S. mikatae OTU 1156 and S. jurei OTU 1159 showed one and two base pair substitution, respectively, with their type strains (Table 2).
Conclusion
We have demonstrated that high‐throughput sequencing of the ITS1 regions amplified from size‐specific ITS is effective in capturing the diversity of wild Saccharomyces species in their habitat. This method allowed for the first time the detection of most natural Saccharomyces species including S. eubayanus and S. mikatae, which had not previously been isolated from Europe, and S. jurei which is a newly discovered species originally isolated in France. Potential signs of S. eubayanus in the Italian Alps will encourage focused field work to isolate this species in Europe where brewing initiated in the 1400s. A general high number of fungal OTUs shared between the same patch rather than the tree type suggests that the fungal soil community depends more strongly on the soil location than the tree type. However, Saccharomyces species reads were noticeably more abundant in soil of oak tree than in soil of other trees at the same sampling location, which refers to a possible specificity of Saccharomyces species to oak tree. S. kudriavzevii, S. cerevisiae and S. paradoxus sequences of ITS1 show up to three base pair differences from the ITS1 sequences of specific strains belonging to previously described populations, suggesting that they may represent novel Italian lineages.
Our results suggest that culturing methods may have introduced bias into the isolation of some Saccharomyces species, since S. paradoxus, which has been widely isolated by enrichment culturing, had lower abundance than S. kudriavzevii and S. mikatae in our data. However, we cannot exclude the possibility that high abundance of S. kudriavzevii and S. mikatae may be specific to the sampling regions.
Overall, by using a targeted metagenomic approach, we were able for the first time to detect the species belonging to Saccharomyces genus in their natural habitat. Moreover, our results suggest the presence of new Saccharomyces populations and our method can be applied to systematic sampling to gain a better understanding of the ecology and evolution of Saccharomyces species.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
Appendix S1: Supporting information.
Table S1. Location of soil samples collected surrounding different tree species at varying altitudes.
Table S2. OTUs count of fungal species across all samples of soil.
Table S3. Abundance and diversity of fungal phyla.
Acknowledgements
The authors would like to thank the Genomic Technology Core Facility at the University of Manchester for carrying out sequencing of the samples using Illumina MiSeq. We thank Kobchai Dungrattanalert for his help in the data analysis. HA is supported by a scholarship funded by the Kuwait government through Kuwait University. SN is supported through BBSRC funding (BB/L021471/1). RTG received funding from the European Union's Horizon 2020 research and innovation programme under grant agreement No 767015.
The copyright line for this article was changed on 11 September 2019 after original online publication.
References
- Almeida, P. , Goncalves, C. , Teixeira, S. , Libkind, D. , Bontrager, M. , Masneuf‐Pomarede, I. , et al (2014) A Gondwanan imprint on global diversity and domestication of wine and cider yeast Saccharomyces uvarum . Nat Commun 5: 4044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida, P. , Barbosa, R. , Zalar, P. , Imanishi, Y. , Shimizu, K. , Turchetti, B. , et al (2015) A population genomics insight into the Mediterranean origins of wine yeast domestication. Mol Ecol 24: 5412–5427. [DOI] [PubMed] [Google Scholar]
- Bellemain, E. , Carlsen, T. , Brochmann, C. , Coissac, E. , Taberlet, P. , and Kauserud, H. (2010) ITS as an environmental DNA barcode for fungi: an in silico approach reveals potential PCR biases. BMC Microbiol 10: 189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bing, J. , Han, P.J. , Liu, W.Q. , Wang, Q.M. , and Bai, F.Y. (2014) Evidence for a Far East Asian origin of lager beer yeast. Curr Biol 24: R380–R381. [DOI] [PubMed] [Google Scholar]
- Boix‐Amoros, A. , Martinez‐Costa, C. , Querol, A. , Collado, M.C. , and Mira, A. (2017) Multiple approaches detect the presence of fungi in human Breastmilk samples from healthy mothers. Sci Rep 7: 13016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buee, M. , Reich, M. , Murat, C. , Morin, E. , Nilsson, R.H. , Uroz, S. , and Martin, F. (2009) 454 pyrosequencing analyses of forest soils reveal an unexpectedly high fungal diversity. New Phytol 184: 449–456. [DOI] [PubMed] [Google Scholar]
- Charron, G. , Leducq, J.B. , Bertin, C. , Dube, A.K. , and Landry, C.R. (2014) Exploring the northern limit of the distribution of Saccharomyces cerevisiae and Saccharomyces paradoxus in North America. FEMS Yeast Res 14: 281–288. [DOI] [PubMed] [Google Scholar]
- Cordier, T. , Robin, C. , Capdevielle, X. , Fabreguettes, O. , Desprez‐Loustau, M.L. , and Vacher, C. (2012) The composition of phyllosphere fungal assemblages of European beech (Fagus sylvatica) varies significantly along an elevation gradient. New Phytol 196: 510–519. [DOI] [PubMed] [Google Scholar]
- Dashko, S. , Liu, P. , Volk, H. , Butinar, L. , Piskur, J. , and Fay, J.C. (2016) Changes in the relative abundance of two Saccharomyces species from oak forests to wine fermentations. Front Microbiol 7: 215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fay, J.C. , and Benavides, J.A. (2005) Evidence for domesticated and wild populations of Saccharomyces cerevisiae . PLoS Genet 1: 66–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gayevskiy, V. , and Goddard, M.R. (2016) Saccharomyces eubayanus and Saccharomyces arboricola reside in North Island native New Zealand forests. Environ Microbiol 18: 1137–1147. [DOI] [PubMed] [Google Scholar]
- Goddard, M.R. , and Greig, D. (2015) Saccharomyces cerevisiae: a nomadic yeast with no niche? FEMS Yeast Res 15: 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hittinger, C.T. , Goncalves, P. , Sampaio, J.P. , Dover, J. , Johnston, M. , and Rokas, A. (2010) Remarkably ancient balanced polymorphisms in a multi‐locus gene network. Nature 464: 54–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyma, K.E. , and Fay, J.C. (2013) Mixing of vineyard and oak‐tree ecotypes of Saccharomyces cerevisiae in North American vineyards. Mol Ecol 22: 2917–2930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korner, C. (2007) The use of ‘altitude’ in ecological research. Trends Ecol Evol 22: 569–574. [DOI] [PubMed] [Google Scholar]
- Kowallik, V. , Miller, E. , and Greig, D. (2015) The interaction of Saccharomyces paradoxus with its natural competitors on oak bark. Mol Ecol 24: 1596–1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtzman, C.P. , and Robnett, C.J. (2003) Phylogenetic relationships among yeasts of the ‘Saccharomyces complex’ determined from multigene sequence analyses. FEMS Yeast Res 3: 417–432. [DOI] [PubMed] [Google Scholar]
- Libkind, D. , Hittinger, C.T. , Valerio, E. , Goncalves, C. , Dover, J. , Johnston, M. , et al (2011) Microbe domestication and the identification of the wild genetic stock of lager‐brewing yeast. Proc Natl Acad Sci USA 108: 14539–14544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liti, G. , Carter, D.M. , Moses, A.M. , Warringer, J. , Parts, L. , James, S.A. , et al (2009) Population genomics of domestic and wild yeasts. Nature 458: 337–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes, C.A. , Barrio, E. , and Querol, A. (2010) Natural hybrids of S. cerevisiae x S. kudriavzevii share alleles with European wild populations of Saccharomyces kudriavzevii . FEMS Yeast Res 10: 412–421. [DOI] [PubMed] [Google Scholar]
- Masinova, T. , Bahnmann, B.D. , Vetrovsky, T. , Tomsovsky, M. , Merunkova, K. , and Baldrian, P. (2017) Drivers of yeast community composition in the litter and soil of a temperate forest. FEMS Microbiol Ecol 93: 2. [DOI] [PubMed] [Google Scholar]
- Naseeb, S. , Alsammar, H. , Burgis, T. , Donaldson, I. , Knyazev, N. , Knight, C. , and Delneri, D. (2018) Whole genome sequencing, de novo assembly and phenotypic profiling for the new budding yeast species Saccharomyces jurei . G3: Genes|Genomes|Genetics 8: 2967–2977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naseeb, S. , James, S.A. , Alsammar, H. , Michaels, C.J. , Gini, B. , Nueno‐Palop, C. , et al (2017) Saccharomyces jurei sp. nov., isolation and genetic identification of a novel yeast species from Quercus robur . Int J Syst Evol Microbiol 67: 2046–2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nash, A.K. , Auchtung, T.A. , Wong, M.C. , Smith, D.P. , Gesell, J.R. , Ross, M.C. , et al (2017) The gut mycobiome of the human microbiome project healthy cohort. Microbiome 5: 153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naumov, G.I. , Naumova, E.S. , and Sniegowski, P.D. (1998) Saccharomyces paradoxus and Saccharomyces cerevisiae are associated with exudates of North American oaks. Can J Microbiol 44: 1045–1050. [PubMed] [Google Scholar]
- Naumov, G.I. , James, S.A. , Naumova, E.S. , Louis, E.J. , and Roberts, I.N. (2000) Three new species in the Saccharomyces sensu stricto complex: Saccharomyces cariocanus, Saccharomyces kudriavzevii and Saccharomyces mikatae . Int J Syst Evol Microbiol 50: 1931–1942. [DOI] [PubMed] [Google Scholar]
- Peris, D. , Sylvester, K. , Libkind, D. , Goncalves, P. , Sampaio, J.P. , Alexander, W.G. , and Hittinger, C.T. (2014) Population structure and reticulate evolution of Saccharomyces eubayanus and its lager‐brewing hybrids. Mol Ecol 23: 2031–2045. [DOI] [PubMed] [Google Scholar]
- Peris, D. , Langdon, Q.K. , Moriarty, R.V. , Sylvester, K. , Bontrager, M. , Charron, G. , et al (2016) Complex ancestries of lager‐brewing hybrids were shaped by standing variation in the wild yeast Saccharomyces eubayanus . PLoS Genet 12: e1006155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter, J. , De Chiara, M. , Friedrich, A. , Yue, J.X. , Pflieger, D. , Bergstrom, A. , et al (2018) Genome evolution across 1,011 Saccharomyces cerevisiae isolates. Nature 556: 339–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto, C. , Pinho, D. , Sousa, S. , Pinheiro, M. , Egas, C. , and Gomes, A.C. (2014) Unravelling the diversity of grapevine microbiome. PLoS One 9: e85622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter, T.M. , and Golding, G.B. (2011) Are similarity‐ or phylogeny‐based methods more appropriate for classifying internal transcribed spacer (ITS) metagenomic amplicons? New Phytol 192: 775–782. [DOI] [PubMed] [Google Scholar]
- Rodriguez, M.E. , Perez‐Traves, L. , Sangorrin, M.P. , Barrio, E. , and Lopes, C.A. (2014) Saccharomyces eubayanus and Saccharomyces uvarum associated with the fermentation of Araucaria araucana seeds in Patagonia. FEMS Yeast Res 14: 948–965. [DOI] [PubMed] [Google Scholar]
- Salvado, Z. , Arroyo‐Lopez, F.N. , Guillamon, J.M. , Salazar, G. , Querol, A. , and Barrio, E. (2011) Temperature adaptation markedly determines evolution within the genus Saccharomyces . Appl Environ Microbiol 77: 2292–2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampaio, J.P. , and Goncalves, P. (2008) Natural populations of Saccharomyces kudriavzevii in Portugal are associated with oak bark and are sympatric with S. cerevisiae and S. paradoxus . Appl Environ Microbiol 74: 2144–2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sniegowski, P.D. , Dombrowski, P.G. , and Fingerman, E. (2002) Saccharomyces cerevisiae and Saccharomyces paradoxus coexist in a natural woodland site in North America and display different levels of reproductive isolation from European conspecifics. FEMS Yeast Res 1: 299–306. [DOI] [PubMed] [Google Scholar]
- Stefanini, I. , Dapporto, L. , Legras, J.L. , Calabretta, A. , Di Paola, M. , De Filippo, C. , et al (2012) Role of social wasps in Saccharomyces cerevisiae ecology and evolution. Proc Natl Acad Sci USA 109: 13398–13403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweeney, J.Y. , Kuehne, H.A. , and Sniegowski, P.D. (2004) Sympatric natural Saccharomyces cerevisiae and S. paradoxus populations have different thermal growth profiles. FEMS Yeast Res 4: 521–525. [DOI] [PubMed] [Google Scholar]
- Sylvester, K. , Wang, Q.M. , James, B. , Mendez, R. , Hulfachor, A.B. , and Hittinger, C.T. (2015) Temperature and host preferences drive the diversification of Saccharomyces and other yeasts: a survey and the discovery of eight new yeast species. FEMS Yeast Res 15: 1–16. [DOI] [PubMed] [Google Scholar]
- Taylor, M.W. , Tsai, P. , Anfang, N. , Ross, H.A. , and Goddard, M.R. (2014) Pyrosequencing reveals regional differences in fruit‐associated fungal communities. Environ Microbiol 16: 2848–2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilgalys, R. (2003) Taxonomic misidentification in public DNA databases. New Phytologist 160: 4–5. [DOI] [PubMed] [Google Scholar]
- Wang, Q.M. , Liu, W.Q. , Liti, G. , Wang, S.A. , and Bai, F.Y. (2012) Surprisingly diverged populations of Saccharomyces cerevisiae in natural environments remote from human activity. Mol Ecol 21: 5404–5417. [DOI] [PubMed] [Google Scholar]
- Wang, S.A. , and Bai, F.Y. (2008) Saccharomyces arboricolus sp. nov., a yeast species from tree bark. Int J Syst Evol Microbiol 58: 510–514. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supporting information.
Table S1. Location of soil samples collected surrounding different tree species at varying altitudes.
Table S2. OTUs count of fungal species across all samples of soil.
Table S3. Abundance and diversity of fungal phyla.