

Abstract
Primary sclerosing cholangitis (PSC) represents a major unmet medical need. In a phase II double‐blind, placebo‐controlled study, we tested the safety and efficacy of cilofexor (formerly GS‐9674), a nonsteroidal farnesoid X receptor agonist in patients without cirrhosis with large‐duct PSC. Patients were randomized to receive cilofexor 100 mg (n = 22), 30 mg (n = 20), or placebo (n = 10) orally once daily for 12 weeks. All patients had serum alkaline phosphatase (ALP) > 1.67 × upper limit of normal and total bilirubin ≤ 2 mg/dL at baseline. Safety, tolerability, pharmacodynamic effects of cilofexor (serum C4 [7α‐hydroxy‐4‐cholesten‐3‐one] and bile acids), and changes in liver biochemistry and serum fibrosis markers were evaluated. Overall, 52 patients were randomized (median age 43 years, 58% male, 60% with inflammatory bowel disease, 46% on ursodeoxycholic acid). Baseline median serum ALP and bilirubin were 348 U/L (interquartile range 288‐439) and 0.7 mg/dL (0.5‐1.0), respectively. Dose‐dependent reductions in liver biochemistry were observed. At week 12, cilofexor 100 mg led to significant reductions in serum ALP (median reduction −21%; P = 0.029 versus placebo), gamma‐glutamyl transferase (−30%; P < 0.001), alanine aminotransferase (ALT) (−49%; P = 0.009), and aspartate aminotransferase (−42%; P = 0.019). Cilofexor reduced serum C4 compared with placebo; reductions in bile acids were greatest with 100 mg. Relative reductions in ALP were similar between ursodeoxycholic acid–treated and untreated patients. At week 12, cilofexor‐treated patients with a 25% or more relative reduction in ALP had greater reductions in serum alanine aminotransferase, aspartate aminotransferase, gamma‐glutamyl transferase, tissue inhibitor of metalloproteinase 1, C‐reactive protein, and bile acids than nonresponders. Adverse events were similar between cilofexor and placebo‐treated patients. Rates of grade 2 or 3 pruritus were 14% with 100 mg, 20% with 30 mg, and 40% with placebo. Conclusion: In this 12‐week, randomized, placebo‐controlled study, cilofexor was well tolerated and led to significant improvements in liver biochemistries and markers of cholestasis in patients with PSC.
Abbreviations
- AE
adverse event
- ALP
alkaline phosphatase
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- C4
7α‐hydroxy‐4‐cholesten‐3‐one
- CRP
C‐reactive protein
- CYP7A1
cholesterol 7‐hydroxylase
- ELF
Enhanced Liver Fibrosis
- FGF19
fibroblast growth factor 19
- FXR
farnesoid X receptor
- GCA
glycocholic acid
- GCDCA
glycochenodeoxycholic acid
- GGT
gamma‐glutamyl transferase
- HDL‐C
high‐density lipoprotein cholesterol
- IBD
inflammatory bowel disease
- IQR
interquartile range
- LC/MS‐MS
liquid chromatography–tandem mass spectrometry
- LDL‐C
low‐density lipoprotein cholesterol
- MRCP
magnetic resonance cholangiopancreatography
- PSC
primary sclerosing cholangitis
- qd
once daily
- TIMP‐1
tissue inhibitor of metalloproteinase 1
- UDCA
ursodeoxycholic acid
- ULN
upper limit of normal
Primary sclerosing cholangitis (PSC) is a chronic and progressive cholestatic liver disease whose pathogenesis remains poorly understood.1 PSC is characterized histologically by chronic inflammation and fibro‐obliterative destruction of intrahepatic and/or extrahepatic bile ducts, resulting in progressive biliary fibrosis and ultimately cirrhosis. Although various pharmacologic interventions have been studied, no approved medical therapies exist that have reduced rates of clinical outcomes such as hepatic decompensation, cholangiocarcinoma, transplantation, or mortality. Although ursodeoxycholic acid (UDCA) is associated with improvement in biochemical markers of cholestasis, including a reduction in serum alkaline phosphatase (ALP), in most PSC patients, the clinical benefits of such responses remain unclear.2, 3, 4, 5 Recent analyses have identified lower baseline levels and/or improvements in ALP over time as important markers of improved prognosis.6, 7, 8, 9 However, a study of high‐dose UDCA demonstrated worse clinical outcomes compared with placebo, despite reductions in serum ALP.3 These seemingly contradictory findings illustrate the complexity of identifying surrogate markers of clinical benefit in PSC and also highlight the major unmet need for novel therapies to treat this condition.
A hallmark of PSC is the presence of cholestasis. Thus, one potential therapeutic target may be activation of the farnesoid X receptor (FXR), a ligand‐activated nuclear hormone receptor that is the key regulator of bile acid synthesis, conjugation, and excretion.10, 11 FXR is highly expressed in the liver, gallbladder, intestines, and kidney. Activation of FXR within the intestine by bile acids or FXR agonists leads to release of fibroblast growth factor 19 (FGF19), a key hormonal regulator of postprandial metabolism.12, 13 FGF19 travels to the liver through the portal circulation, binds to the FGFR4/βKlotho receptor complex, and activates a signaling cascade that results in the down‐regulation of cholesterol 7α‐hydroxylase (CYP7A1) expression, thereby inhibiting the rate‐limiting step in bile acid synthesis.14 Direct activation of FXR in the liver can also regulate expression of a host of genes involved in bile acid metabolism. Specifically, FXR activation increases the expression of transporters responsible for canalicular and basolateral bile acid efflux, while inhibiting bile acid synthesis and basolateral bile acid uptake by hepatocytes. These multifaceted effects of FXR agonism, both indirect through FGF19 induction and direct at the level of hepatocytes, suggest that FXR may be a feasible therapeutic target for mitigating the cholestasis characteristic of PSC.
In this 12‐week, phase II, randomized, placebo‐controlled study, we evaluated the safety and efficacy of cilofexor (formerly GS‐9674) in PSC patients without cirrhosis. Cilofexor is an oral, potent (EC50 43 nM), and selective nonsteroidal FXR agonist that has demonstrated anti‐inflammatory and antifibrotic effects and reduced portal pressure in preclinical models of liver fibrosis.15 Here, we report that cilofexor had a favorable safety profile and led to significant reductions in serum ALP, other liver biochemistry tests, and markers of cholestasis in a dose‐dependent fashion. Cilofexor was well tolerated, and importantly, did not exacerbate the pruritus characteristic of PSC. These findings indicate that cilofexor may have potential therapeutic benefit in patients with PSC.
Patients and Methods
Study Population
Adult patients aged 18 to 70 years with a diagnosis of classic, large‐duct PSC based on cholangiogram (magnetic resonance cholangiopancreatography [MRCP], endoscopic retrograde cholangiopancreatography, or percutaneous transhepatic cholangiogram) within the previous 12 months were eligible. All patients had a serum ALP greater than 1.67 × the upper limit of normal (ULN), platelet count of 150,000/mL3 or higher, serum albumin of 3.3 g/dL or more, and serum creatinine of less than or equal to ULN (≤1.14 mg/dL). Patients with a history of inflammatory bowel disease (IBD) were required to have a colonoscopy within 6 months of screening, demonstrating no evidence of active disease and a partial Mayo score of less than 2 with a score on the rectal bleeding domain of less than 1 during screening.
For patients on UDCA treatment, the dose of UDCA must have been stable for at least 12 months before screening and had to remain stable through the end of the treatment. In patients not on UDCA, there could be no UDCA use for at least 12 months before screening through the end of treatment. For patients receiving biologic treatments (e.g., antitumor necrosis factor or anti‐integrin monoclonal antibodies), immunosuppressants, or systemic corticosteroids, the dose had to be stable for at least 3 months before screening and anticipated to remain stable throughout the trial. Exclusion criteria included histologic or imaging evidence of cirrhosis, previous history of decompensated liver disease, or a FibroSure/FibroTest (LabCorp, Burlington, NC) result of at least 0.75 (unless a biopsy within 12 months excluded cirrhosis). Patients with alanine aminotransferase (ALT) greater than 10 × ULN, total bilirubin greater than 2 × ULN, or an international normalized ratio greater than 1.2 were also excluded. Complete eligibility criteria are provided in the Supporting Information.
Study Design
In this multicenter, double‐blind, randomized, placebo‐controlled, phase II trial, eligible patients were randomized in a 2:2:1 ratio to receive treatment with cilofexor 100 mg orally once daily (qd), cilofexor 30 mg qd, or placebo qd for 12 weeks (ClinicalTrials.gov NCT02943460). Randomization was stratified by the presence or absence of UDCA treatment. After screening, study visits occurred at baseline and weeks 1, 2, 4, 8, and 12 of treatment, and a follow‐up visit 4 weeks after discontinuing treatment. Patients who completed the 12‐week blinded study phase without permanently discontinuing the study drug were eligible to participate in an optional, 96‐week open‐label extension phase, during which all patients were administered cilofexor 100 mg daily. In this manuscript, we report findings from the 12‐week blinded phase of the study.
Assessments
Safety was evaluated by assessment of clinical laboratory tests, physical examinations, and by documentation of adverse events (AEs). All safety data were collected from the time of the first dose of study drug to 30 days after the last dose of study drug. MRCP was performed at baseline and read centrally by an expert radiologist.16 Serum markers of liver injury and function including ALP, ALT, aspartate aminotransferase (AST), bilirubin, and gamma‐glutamyl transferase (GGT) were assessed at baseline and weeks 1, 2, 4, 8, and 12. Noninvasive markers of fibrosis, including the Enhanced Liver Fibrosis (ELF) test (Siemens, Tarrytown, NY) and its components (tissue inhibitor of metalloproteinase 1 [TIMP‐1], procollagen III N‐terminal peptide [PIII‐NP], and hyaluronic acid), and FibroSure/FibroTest were measured at baseline and week 12. Blood samples to evaluate the pharmacodynamic effects of cilofexor were collected to assess changes in fasting plasma FGF19 and 7α‐hydroxy‐4‐cholesten‐3‐one (C4), an intermediate in the biochemical synthesis of bile acids from cholesterol by CYP7A1. Total serum bile acids were measured by an enzymatic reaction (Covance, Princeton, NJ) at baseline and week 12. In addition, total, primary, and secondary bile acids were measured by liquid chromatography–tandem mass spectrometry (LC/MS‐MS) (Metabolon, Durham, NC) to assess the concentration of individual bile acid species. The contribution of UDCA and its conjugates to the bile acid pool were removed for these analyses. A fasting metabolic and lipid profile including total cholesterol, low‐density lipoprotein cholesterol (LDL‐C), high‐density lipoprotein cholesterol (HDL‐C), triglycerides, and glucose were collected at baseline and weeks 1, 4, 8, and 12. Liver stiffness by transient elastography (FibroScan; Echosens, Paris, France) was assessed at baseline and week 12.
Statistical Analyses
Given the exploratory nature of this study, a formal sample‐size calculation was not performed. The planned number of patients was based on clinical experience with similar phase II studies. Safety and efficacy were assessed in all patients who were randomized and received at least one dose of study drug. The primary endpoint of the study was the safety of cilofexor in patients with PSC, as evaluated by examining the incidence of treatment‐emergent AEs, including serious adverse events (SAEs), and clinical laboratory tests. Exploratory efficacy endpoints included changes from baseline in serum ALP, GGT, ALT, AST, pharmacodynamic indicators of FXR agonism (FGF19, C4, and total and primary bile acids), and noninvasive measures of fibrosis (ELF, FibroScan). The Wilcoxon rank sum test was used to compare changes from baseline in continuous parameters at week 12 between cilofexor‐treated groups versus placebo. Univariate logistic regression was also used to explore baseline factors associated with ALP response, defined as at least a 25% relative reduction from baseline to week 12 of therapy. An ALP reduction of this magnitude has been associated with a reduced risk of PSC‐related clinical events during follow‐up.6 All statistical analyses were performed using SAS, version 9.4 (SAS Institute Inc., Cary, NC).
Study Oversight
The study was approved by the institutional review board or independent ethics committee at participating sites and conducted in compliance with the Declaration of Helsinki, Good Clinical Practice guidelines, and local regulatory requirements. Written informed consent was obtained from all patients prior to study initiation. The study was designed and conducted by the sponsor (Gilead Sciences) in collaboration with the principal investigator, according to the protocol. The sponsor collected the data and elected to conduct an additional unplanned interim analysis with measures to ensure data integrity after all patients had discontinued or completed 12 weeks of study treatment. The sponsor also monitored study conduct and performed the statistical analyses. An independent data safety monitoring committee reviewed the progress of the study. All authors had access to the data and assumed responsibility for the integrity and completeness of the reported data. All authors reviewed and approved the final manuscript.
Results
Baseline Characteristics
Between November 29, 2016, and November 9, 2017, a total of 105 patients were screened and 52 patients were randomized and treated across 24 sites in North America and Europe (United States, 16; Canada, 3; United Kingdom, 4; and Austria, 1) (Supporting Fig. S1). Reasons for screen failure are provided in the Supporting Information. Overall, demographics and baseline characteristics were similar between the treatment groups (Table 1). Most of the patients were male (58%); the median age was 43 years (interquartile range [IQR] 35, 52); and 60% had concomitant IBD. Sixty percent of patients had intrahepatic and extrahepatic bile duct involvement on MRCP, and 46% were on UDCA. Median (IQR) serum ALP at baseline was 348 U/L (288, 439) or 2.78 ULN (2.29, 3.66), and serum bilirubin was 0.7 mg/dL (0.5, 1.0). The median ELF score and liver stiffness by FibroScan were 9.38 (8.91, 9.88) and 9.4 kPa (6.8, 10.6), respectively, in keeping with moderate to severe fibrosis.
Table 1.
Demographics and Baseline Characteristics
|
Cilofexor 100 mg (n = 22) |
Cilofexor 30 mg (n = 20) |
Placebo (n = 10) |
Total (n = 52) |
||
|---|---|---|---|---|---|
| Demographics | Age (years) | 43 (36, 47) | 46 (35, 57) | 39 (33, 52) | 43 (35, 52) |
| Male, n (%) | 11 (50%) | 14 (70%) | 5 (50%) | 30 (58%) | |
| White, n (%) | 17 (77%) | 15 (75%) | 7 (70%) | 39 (75%) | |
| Diabetes, n (%) | 6 (27%) | 2 (10%) | 1 (10%) | 9 (17%) | |
| Weight (kg) | 73.5 (67.5, 89.1) | 79.8 (68.4, 95.9) | 82.2 (63.0, 83.3) | 77.9 (67.4, 88.6) | |
| BMI (kg/m2) | 25.8 (23.2, 30.3) | 25.9 (22.8, 29.9) | 25.8 (23.9, 29.6) | 25.8 (23.2, 29.9) | |
| IBD, n (%) | 13 (59%) | 11 (55%) | 7 (70%) | 31 (60%) | |
| UDCA, n (%) | 10 (46%) | 9 (45%) | 5 (50%) | 24 (46%) | |
| Liver biochemistry | ALP (U/L) | 350 (312, 387) | 344 (271, 460) | 380 (265, 547) | 348 (288, 439) |
| ALP (× ULN) | 2.87 (2.45, 3.51) | 2.73 (2.15, 3.74) | 3.31 (2.05, 4.33) | 2.78 (2.29, 3.66) | |
| GGT (U/L) | 305 (192, 542) | 564 (255, 910) | 377 (224, 622) | 423 (203, 628) | |
| Total bilirubin (mg/dL) | 0.6 (0.5, 1.1) | 0.8 (0.6, 1.0) | 0.6 (0.5, 0.9) | 0.7 (0.5, 1.0) | |
| ALT (U/L) | 110 (83, 156) | 119 (60, 197) | 77 (59, 123) | 109 (63, 156) | |
| ALT (× ULN) | 2.90 (1.92, 4.46) | 3.01 (1.74, 4.81) | 2.01 (1.60, 2.86) | 2.67 (1.70, 4.12) | |
| AST (U/L) | 67 (52, 98) | 75 (44, 104) | 59 (47, 76) | 64 (47, 99) | |
| AST (× ULN) | 1.92 (1.49, 2.87) | 2.11 (1.29, 2.90) | 1.74 (1.31, 2.10) | 1.83 (1.34, 2.78) | |
| Albumin (g/dL) | 4.4 (4.2, 4.5) | 4.5 (4.2, 4.7) | 4.6 (4.2, 4.7) | 4.4 (4.2, 4.7) | |
| Fibrosis and inflammation | ELF | 9.26 (8.73, 9.66) | 9.77 (9.26, 10.31) | 9.09 (8.87, 9.60) | 9.38 (8.91, 9.88) |
| FibroTest | 0.29 (0.27, 0.44) | 0.47 (0.39, 0.57) | 0.34 (0.23, 0.51) | 0.40 (0.28, 0.51) | |
| CRP (ug/mL) | 0.27 (0.15, 0.51) | 0.26 (0.10, 0.73) | 0.19 (0.12, 0.46) | 0.25 (0.13, 0.57) | |
| Liver stiffness by FibroScan (kPa) | 7.3 (6.2, 10.6) | 10.1 (6.9, 12.5) | 9.8 (7.9, 10.1) | 9.4 (6.8, 10.6) | |
| Bile acid homeostasis | FGF19 (pg/mL) | 102 (66, 171) | 118 (61, 174) | 115 (107, 156) | 112 (66, 168) |
| C4 (ng/mL) | 10.4 (5.1, 23.5) | 18.7 (9.8, 30.0) | 18.9 (9.3, 27.1) | 13.2 (7.3, 27.1) | |
| Total bile acids by enzymatic assay (umol/L)* | 19.6 (10.3, 33.1) | 15.3 (9.4, 32.2) | 13.7 (6.4, 17.0) | 16.9 (9.6, 30.7) | |
| Total bile acids by LC‐MS/MS (ng/ml)* | 5601.6 (2886.8, 12787.3) | 5424.4 (2381.3, 12245.1) | 5042.0 (1580.6, 5622.2) | 5169.6 (2509.7, 10772.8) | |
| Duct involvement on MRCP† | Intrahepatic and extrahepatic ducts | 11 (50%) | 14 (70%) | 6 (60%) | 31 (60%) |
| Intrahepatic ducts only | 8 (36%) | 4 (20%) | 3 (30%) | 15 (29%) | |
| Normal biliary tree | 2 (9%) | 0 | 0 | 2 (4%) | |
| Metabolism | Glucose (mg/dL) | 87 (81, 98) | 87 (82, 94) | 84 (79, 90) | 87 (82, 96) |
| Cholesterol (mg/dL) | 209 (178, 256) | 240 (200, 274) | 219 (188, 258) | 218 (184, 263) | |
| LDL‐C (mg/dL) | 111 (87, 132) | 139 (102, 166) | 123 (94, 153) | 122 (94, 153) | |
| HDL‐C (mg/dL) | 82 (64, 97) | 75 (64, 86) | 75 (58, 87) | 77 (63, 91) | |
All data are median (IQR) or n (%).
Abbreviation: BMI, body mass index.
Serum total bile acids include UDCA and conjugates.
Baseline MRCP images missing in 4 patients (cilofexor 100 mg, n = 1; cilofexor 30 mg, n = 2; placebo, n = 1).
Cilofexor Reduces Serum ALP in a Dose‐Dependent Manner
At the end of the 12‐week double‐blind phase, significant and dose‐dependent reductions in serum ALP concentration were observed in patients who received cilofexor compared with those who received placebo (Table 2 and Fig. 1A). The median (IQR) absolute difference from baseline to week 12 in serum ALP was −73 U/L (−106, −14) in the cilofexor 100 mg group (P = 0.026 versus placebo), −21 U/L (−60, 40) in the cilofexor 30 mg group (P = 0.37 versus placebo), and +8 U/L (−40, 118) in the placebo group (Fig. 1B). Similarly, relative reductions in serum ALP from baseline were greatest in patients who received cilofexor. The median (IQR) relative difference from baseline to week 12 in serum ALP was −20.5% (−30.2, −3.5) in the cilofexor 100 mg group (P = 0.029 versus placebo), −6.1% (−17.6, 16.8) in the cilofexor 30 mg group (P = 0.32 versus placebo), and +3.4% (−7.2, 18.6) in the placebo group (Fig. 1C). Changes in serum ALP relative to the ULN are provided in Fig. 1C.
Table 2.
Median Relative (%) Changes in Liver Biochemistry and Biomarkers from Baseline to Week 12
|
Cilofexor 100 mg (n = 22) |
Cilofexor 30 mg (n = 20) |
Placebo (n = 10) |
P Values | ||
|---|---|---|---|---|---|
| 100 mg Versus Placebo | 30 mg Versus Placebo | ||||
| ALP | −20.5 (−30.2, −3.5) | −6.1 (−17.6, 16.8) | 3.4 (−7.2, 18.6) | 0.029 | 0.32 |
| UDCA use | −18.6 (−35.2, −3.5) | −7.8 (−14.3, −1.6) | 1.3 (−7.2, 18.6) | 0.12 | 0.27 |
| No UDCA use | −20.5 (−27.9, 0.5) | 0.0 (−19.2, 21.0) | 5.6 (−5.8, 14.7) | 0.23 | 1.00 |
| ≥ 25% ALP reduction, % (n/N) | 35% (7/20) | 5% (1/19) | 10% (1/10) | 0.21* | 1.00* |
| Absolute ALP change, U/L | −73 (−106, −14) | −21 (−60, 40) | 8 (−40, 118) | 0.026 | 0.37 |
| GGT | −30.3 (−47.8, −21.8) | −16.3 (−29.7, −7.2) | 1.1 (−6.1, 15.0) | < 0.001 | 0.003 |
| ALT | −49.4 (−60.7, −22.5) | −26.2 (−36.8, 1.7) | −12.9 (−22.9, −12.1) | 0.009 | 0.24 |
| AST | −42.3 (−51.1, −10.9) | −22.5 (−34.7, 20.4) | −10.8 (−24.6, 10.5) | 0.019 | 0.32 |
| Total bilirubin | 0.0 (−22.1, 29.2) | 14.3 (−11.1, 34.5) | −11.0 (−27.3, 25.0) | 0.58 | 0.35 |
| Fasting FGF19 | −16.6 (−39.0, 13.6) | 25.9 (−37.6, 93.7) | −43.5 (−51.0, −20.5) | 0.16 | 0.044 |
| Fasting C4 | −23.2 (−71.2, 25.7) | −30.5 (−50.5, 5.6) | 10.0 (−14.3, 29.7) | 0.21 | 0.024 |
| Total bile acids † | −38.6 (−55.6, 19.5) | 0.0 (−41.5, 57.1) | 5.5 (−12.4, 44.6) | 0.17 | 0.67 |
| Primary bile acids ‡ | −45.1 (−65.8, 18.3) | −5.3 (−53.1, 78.3) | 4.3 (−35.3, 32.4) | 0.15 | 0.92 |
| Secondary bile acids ‡ | −39.6 (−62.2, −1.7) | 7.4 (−22.4, 16.4) | 47.5 (22.6, 92.0) | 0.013 | 0.17 |
| ELF | −0.4 (−2.9, 3.0) | 0.1 (−1.9, 4.7) | 1.2 (−1.9, 2.8) | 1.00 | 0.55 |
| TIMP‐1 | −8.2 (−13.2, 3.5) | 3.8 (−12.7, 17.7) | 0.3 (−5.2, 15.5) | 0.063 | 0.73 |
| Hyaluronic acid | −4.3 (−20.6, 17.7) | 10.0 (−17.7, 56.5) | 9.9 (−2.2, 14.3) | 0.69 | 0.59 |
| PIII‐NP | 2.2 (−6.0, 18.2) | −5.4 (−12.8, 22.2) | −6.2 (−30.8, 15.5) | 0.23 | 0.30 |
| Liver stiffness by FibroScan | −6.7 (−22.2, 13.2) | −9.7 (−15.8, 33.8) | −5.8 (−27.0, 10.1) | 0.76 | 0.52 |
| CRP | −12.2 (−65.7, 40.5) | −28.7 (−43.8, 14.8) | 8.5 (−23.1, 30.5) | 0.56 | 0.085 |
| Total cholesterol | −2.1 (−9.2, 6.1) | −0.4 (−8.7, 3.4) | 5.4 (−1.3, 9.0) | 0.23 | 0.22 |
| LDL‐C | 1.5 (−6.6, 10.3) | −1.9 (−7.9, 14.8) | 0.7 (−7.2, 5.4) | 0.60 | 0.89 |
| HDL‐C | −10.8 (−17.1, 2.8) | 3.4 (−17.3, 7.1) | 15.5 (−7.2, 22.4) | 0.040 | 0.12 |
| Triglycerides | 8.0 (−13.5, 23.9) | −2.0 (‐16.1, 6.5) | −21.2 (−31.6, 17.4) | 0.19 | 0.33 |
| Glucose | 0.0 (−4.3, 9.5) | −4.0 (−7.8, 3.2) | −3.0 (−8.5, 0.0) | 0.23 | 0.89 |
Unless indicated, all data are median relative (%) changes from baseline, and P values are from Wilcoxon rank‐sum test. P values in boldface are <0.05.
P value by Fisher exact test.
Total bile acids by enzymatic assay.
Serum bile acid species quantified by LC/MS‐MS. UDCA and conjugates removed from secondary bile acids.
Figure 1.
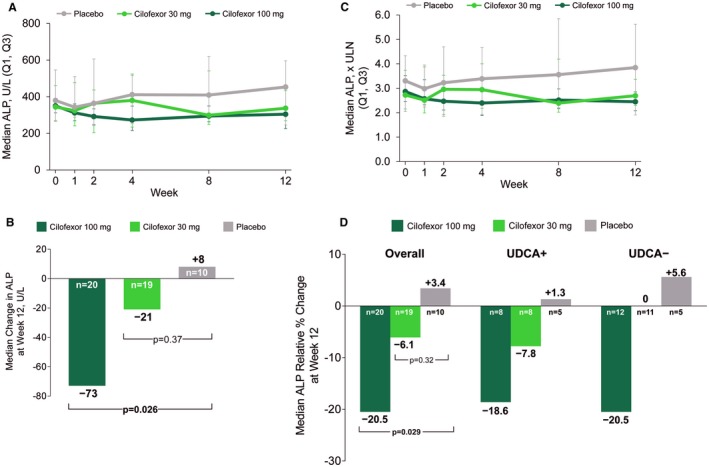
Cilofexor improves serum ALP in patients with PSC. (A) Median (IQR) serum ALP between baseline and week 12 of the double‐blind phase of the study. (B) Median absolute change in serum ALP from baseline to week 12 of therapy. P values versus placebo are according to Wilcoxon rank‐sum test. (C) Median (IQR) change in serum ALP relative to the ULN between baseline and week 12 of therapy. (D) Median relative (percentage) change in serum ALP from baseline to week 12 of therapy (overall and according to UDCA treatment). P values versus placebo are according to Wilcoxon rank‐sum test.
ALP Responses are Independent of UDCA Treatment and Other Baseline Factors
An ALP response, defined as at least a 25% relative reduction in serum ALP from baseline to week 12, was observed more frequently in patients treated with cilofexor 100 mg (35%) than those treated with cilofexor 30 mg (5.3%) or placebo (10%; P = 0.21 versus cilofexor 100 mg). In univariate logistic regression analyses, no baseline factor was associated with the likelihood of ALP response in cilofexor‐treated patients (Supporting Table S1). Specifically, age, sex, history of IBD, intrahepatic duct involvement on MRCP, ALP and other liver biochemistry, fibrosis markers (e.g., ELF, liver stiffness), and markers of bile acid homeostasis (e.g., FGF19, C4, bile acids) were not associated with ALP response. Similarly, UDCA treatment was not associated with ALP response (odds ratio 0.83; 95% confidence interval 0.17, 4.11). The relative reduction in serum ALP between baseline and week 12 in the cilofexor 100 mg group was similar between UDCA‐treated (n = 8) and untreated (n = 12) patients (median, −18.6% versus −20.5%; P = 0.85) (Table 2, Fig. 1D).
Impact of Cilofexor Treatment on Other Liver Biochemistry and Biomarkers
Treatment with cilofexor for 12 weeks also led to significant reductions in serum ALT, AST, and GGT (Table 2, Fig. 2A, and Supporting Fig. S2). From baseline to week 12, the median (IQR) relative reduction in serum ALT was −49.4% (−60.7, −22.5) in the cilofexor 100 mg group (P = 0.009 versus placebo), −26.2% (−36.8, 1.7) in the cilofexor 30 mg group (P = 0.24 versus placebo), and −12.9% (−22.9, −12.1) in the placebo group. Cilofexor 100 mg also significantly reduced serum GGT. At week 12, the median (IQR) relative change in serum GGT was −30.3% (−47.8, −21.8) in the cilofexor 100 mg group (P < 0.001 versus placebo), −16.3% (−29.7, −7.2) in the cilofexor 30 mg group (P = 0.003 versus placebo), and +1.1% (−6.1, 15.0) in the placebo group. Median (IQR) changes from baseline in serum C‐reactive protein (CRP) were greatest in cilofexor‐treated patients (100 mg group: −12.2% [−65.7, 40.5]; 30 mg group: −28.7% [−43.8, 14.8]), although these differences were not statistically significant compared with placebo (+8.5% [−23.1, 30.5]; Table 2).
Figure 2.
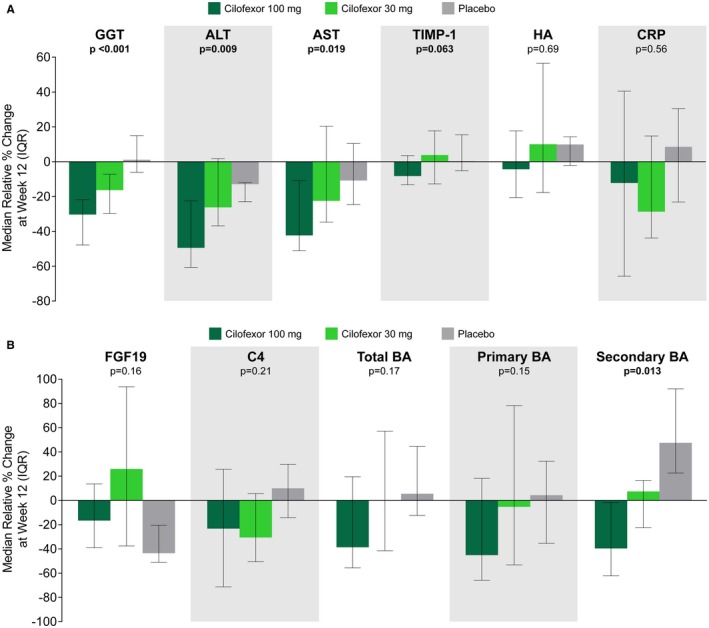
Effect of cilofexor on liver biochemistry and markers of fibrosis and bile acid homeostasis. (A) Cilofexor 100 mg leads to improvement in serum GGT, ALT, AST, and TIMP‐1 compared with placebo. P values for cilofexor 100 mg versus placebo are according to Wilcoxon rank‐sum test. (B) Cilofexor leads to reductions in C4, total bile acids, primary bile acids, and secondary bile acids compared with placebo. Total bile acids by enzymatic assay and bile acid species by LC/MS‐MS. UDCA and its conjugates have been removed from secondary bile acids. P values for cilofexor 100 mg versus placebo are according to Wilcoxon rank‐sum test. Abbreviations: BA, bile acid; HA, hyaluronic acid.
Liver stiffness and noninvasive markers of fibrosis including the ELF score have been associated with prognosis in patients with PSC.17 Between baseline and week 12, changes in overall ELF score, PIII‐NP, hyaluronic acid, and liver stiffness did not differ between treatment groups (Table 2). However, in patients treated with cilofexor 100 mg, a trend toward reduced serum levels of TIMP‐1 was observed (median, −8.2% versus +0.3% in the placebo group; P = 0.063).
Pharmacodynamic Markers of Bile Acid Homeostasis
We also evaluated markers indicative of FXR agonism by cilofexor treatment affecting bile acid homeostasis. Compared with placebo treatment, fasting plasma levels of FGF19 were increased in patients treated with cilofexor 30 mg, but not cilofexor 100 mg (Table 2 and Fig. 2B). Although serum levels of C4, the metabolite produced subsequent to the conversion of cholesterol to 7‐alpha‐hydroxycholesterol by CYP7A1, declined in both cilofexor treatment groups, the total, primary, and secondary bile acids decreased only during therapy with cilofexor 100 mg. Compared with the placebo group, median relative reductions in C4 (+10.0% versus −23.2%; P = 0.21), total bile acids (+5.5% versus −38.6%; P = 0.17), primary bile acids (+4.3% versus −45.1%; P = 0.15), and secondary bile acids (+47.5% versus −39.6%; P = 0.013) were greater in patients treated with cilofexor 100 mg (Table 2 and Fig. 2B).
Considering the observed reductions in bile acids among patients treated with cilofexor 100 mg, we analyzed changes in the concentrations of the various bile acid species to identify the main drivers of these reductions. First, we observed that more than 50% of the bile acid pool at baseline consisted of the conjugated primary bile acids, glycocholic acid (GCA) and glycochenodeoxycholic acid (GCDCA). At week 12, significant reductions in the absolute concentrations of most bile acid species were observed (Fig. 3A); however, no substantial changes in bile acid composition were noted (Fig. 3B). For example, the median (IQR) relative changes of GCA and GCDCA from baseline to week 12 in the cilofexor 100 mg group were −58.1% (−68.1, −20.8) and −41.7% (−69.8, 0.2), respectively (P = 0.03 and P = 0.18 versus placebo). These data suggest that the effects of cilofexor on serum bile acid concentrations are largely due to inhibition of bile acid synthesis.
Figure 3.
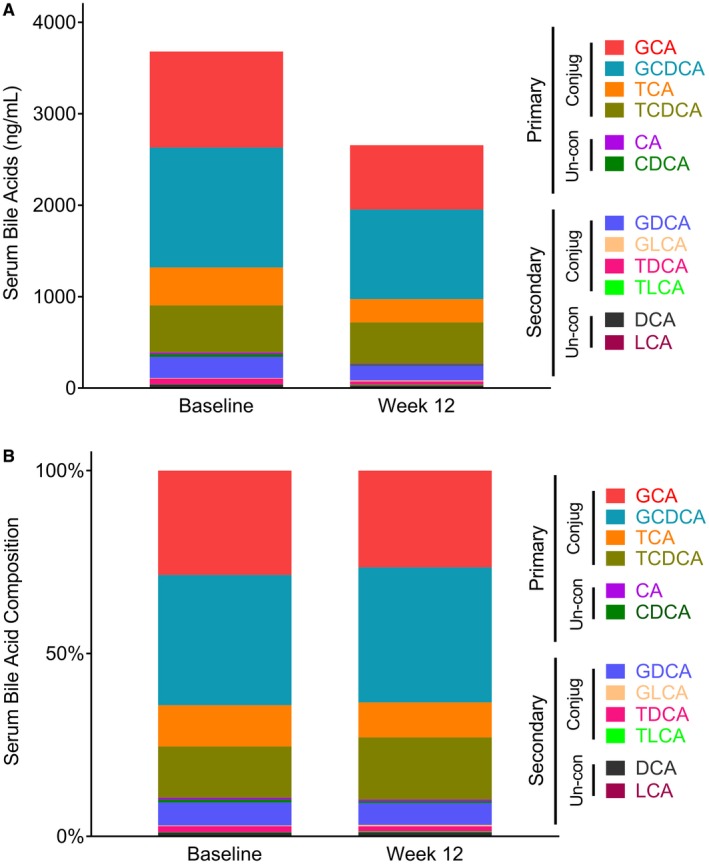
Cilofexor 100 mg lowers serum bile acids, but does not alter bile acid composition. (A) Absolute levels of serum bile acid species before and after 12 weeks of treatment with cilofexor 100 mg daily. UDCA and its conjugates have been removed. (B) Relative contribution of serum bile acid species before and after 12 weeks of treatment with cilofexor 100 mg daily. UDCA and its conjugates have been removed. Abbreviations: CA, cholic acid; CDCA, chenodeoxycholic acid; DCA, deoxycholic acid; GCA, glycocholic acid; GCDCA: glycochenodeoxycholic acid; GDCA, glycolithocholic acid; GLCA, glycolithocholic acid; LCA, lithocholic acid; TCA: taurocholic acid; TCDCA, taurochenodeoxycholic acid; TDCA, taurodeoxycholic acid; TLCA, taurolithocholic acid.
Consistency of Response With Cilofexor
To evaluate the consistency of response with cilofexor treatment, we evaluated the relative changes in liver biochemistry and other biomarkers according to achievement of an ALP response. Compared with nonresponders (n = 31), responders to cilofexor (n = 8) had greater relative reductions in GGT, ALT, and AST; total, primary, and secondary bile acids; TIMP‐1; hyaluronic acid; and CRP (Fig. 4).
Figure 4.
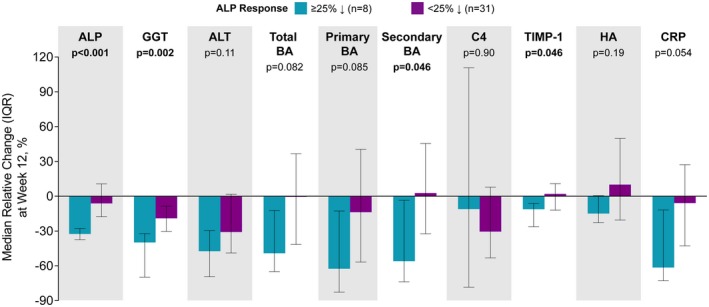
Relative changes in liver biochemistry, markers of fibrosis, and other biomarkers according to achievement of an ALP response, defined as at least a 25% relative reduction in serum ALP from baseline to week 12, in cilofexor‐treated patients. Responder subgroup includes 7 patients treated with cilofexor 100 mg and 1 patient treated with cilofexor 30 mg. P values are according to Wilcoxon rank‐sum test. Abbreviations: BA, bile acid; HA, hyaluronic acid.
Safety and Tolerability of Cilofexor Treatment
Treatment with cilofexor for 12 weeks was generally well tolerated. In total, 5 patients did not complete the study drug treatment: 3 patients (14%) discontinued cilofexor 100 mg due to AEs (n = 1 each of pruritus, acute kidney injury, and elevated ALP); 1 patient (5%) discontinued cilofexor 30 mg due to patient decision; and 1 placebo‐treated patient (10%) discontinued due to elevated liver biochemistry. AEs, which were generally grade 1 or 2 in severity and deemed not to be treatment‐related by the investigator, were similar between treatment groups (Table 3). Treatment‐emergent pruritus occurred in 36% (8 of 22) of patients treated with cilofexor 100 mg, 25% (5 of 20) treated with cilofexor 30 mg, and 60% (6/10) of patients treated with placebo. Treatment‐emergent, grade 2‐3 pruritus was observed in 14% (3 of 22) of patients treated with cilofexor 100 mg, 20% (4 of 20) with 30 mg, and 40% (4 of 10) with those who received placebo. Three SAEs occurred, all in patients treated with cilofexor 100 mg (rib fracture, diarrhea/dehydration, and acute kidney injury), but none were deemed treatment‐related by the investigator. No deaths occurred during the study.
Table 3.
Treatment–Emergent AEs Reported for at Least 2 Patients in Any Treatment Group by Preferred Term
| Preferred Term |
Cilofexor 100 mg (n = 22) |
Cilofexor 30 mg (n = 20) |
Placebo (n = 10) |
|---|---|---|---|
| Number (%) of patients with any AE | 18 (81.8%) | 13 (65.0%) | 10 (100.0%) |
| Pruritus | 8 (36.4%) | 5 (25.0%) | 6 (60.0%) |
| Nasopharyngitis | 5 (22.7%) | 5 (25.0%) | 2 (20.0%) |
| Abdominal pain upper | 3 (13.6%) | 2 (10.0%) | 1 (10.0%) |
| Fatigue | 3 (13.6%) | 2 (10.0%) | 2 (20.0%) |
| Abdominal discomfort | 2 (9.1%) | 0 | 1 (10.0%) |
| Abdominal distension | 2 (9.1%) | 0 | 1 (10.0%) |
| Blood ALP increased | 2 (9.1%) | 0 | 1 (10.0%) |
| Constipation | 2 (9.1%) | 0 | 0 |
| Diarrhea | 2 (9.1%) | 1 (5.0%) | 0 |
| Dizziness | 2 (9.1%) | 1 (5.0%) | 0 |
| Electrocardiogram abnormal | 2 (9.1%) | 0 | 0 |
| Upper respiratory tract infection | 2 (9.1%) | 0 | 1 (10.0%) |
| Viral infection | 2 (9.1%) | 0 | 0 |
| ALT increased | 1 (4.5%) | 0 | 2 (20.0%) |
| Back pain | 1 (4.5%) | 2 (10.0%) | 0 |
| Headache | 1 (4.5%) | 4 (20.0%) | 2 (20.0%) |
| Muscle spasms | 1 (4.5%) | 2 (10.0%) | 0 |
| Pyrexia | 1 (4.5%) | 2 (10.0%) | 0 |
| AST increased | 0 | 0 | 2 (20.0%) |
| Nausea | 0 | 1 (5.0%) | 3 (30.0%) |
Adverse events were coded according to MedDRA Version 20.1.
Most patients had at least one laboratory abnormality, most of whom were grade 1 or 2 in severity (Supporting Table S2). Overall, the most common grade 3 or 4 laboratory abnormalities were elevated liver biochemistry results, specifically, elevated AST (13.5%, 7 of 52), ALT (9.6%, 5 of 52), and ALP (7.7%, 4 of 52). A similar proportion of patients met prespecified criteria for withholding of study medication due to abnormal liver biochemistry: cilofexor 100 mg, 9.1% (2 of 22); cilofexor 30 mg, 5.0% (1 of 20); and placebo, 10.0% (1 of 10). Changes in serum total cholesterol, LDL‐C, and triglycerides did not differ between cilofexor‐treated and placebo‐treated patients (Table 2). However, a median relative reduction in HDL‐C of 10.8% was observed in the cilofexor 100 mg group compared with an increase of 15.5% in the placebo group (P = 0.040).
Discussion
In this double‐blind, placebo‐controlled, randomized study in patients without cirrhosis with large‐duct PSC, the nonsteroidal FXR agonist cilofexor was safe, well tolerated, and improved biochemical markers of cholestasis and inflammation. The main findings of this study included significant dose‐dependent reductions in serum concentrations of ALP, GGT, ALT, and AST within 12 weeks of treatment with cilofexor compared with placebo. The effects on ALP were independent of UDCA use, present in nearly half of the included patients. These data confirm that the mechanism of action of cilofexor is distinct from that of UDCA, which does not have relevant FXR agonistic activity. In this regard, the 100‐mg dose of cilofexor led to reductions in serum C4 and plasma bile acids, but did not alter bile acid composition, which is consistent with inhibition of bile acid biosynthesis through suppression of CYP7A1 activity. However, fasting plasma levels of FGF19 did not increase at this dose, as would have been expected due to activation of intestinal FXR. This finding is due to our assessment of FGF19 in the predose, fasted state. Indeed, data from healthy volunteers has confirmed a rapid, dose‐dependent increase in FGF19 levels when measured serially following cilofexor administration.18 Interestingly, a recent study that included healthy volunteers administered a different nonsteroidal FXR agonist (Px‐102), suggesting that these compounds may reduce bile acid synthesis independent of FGF19 production.19 Finally, although changes in liver stiffness by FibroScan and ELF scores were not observed, patients treated with cilofexor 100 mg had a trend toward reduced serum levels of the pro‐fibrogenic cytokine TIMP‐1, suggesting a potential antifibrotic effect of this treatment. Future trials of longer duration with liver histology will be necessary to confirm this finding.
A significant finding of this study was the dose‐dependent reduction in serum ALP observed in cilofexor‐treated patients. Following 12 weeks of therapy, 35% of patients treated with cilofexor 100 mg had an ALP response, defined as at least a 25% relative reduction from baseline, compared with only 10% of placebo‐treated patients. Consistent with improved cholestasis, responders to cilofexor had greater improvements in other markers of cholestasis (e.g., GGT, total bile acids, primary bile acids), hepatic inflammation (e.g., ALT, AST, CRP), and fibrosis (e.g., TIMP‐1, hyaluronic acid). Although the consistency of these responses supports the efficacy of cilofexor, long‐term studies are necessary to confirm that reduction in the surrogate endpoint of serum ALP is associated with improved clinical outcomes. Indeed, given the chronicity of PSC and the lack of accepted biomarkers associated with disease progression, endpoints that define clinical utility of any pharmacologic intervention in PSC remain undefined.20, 21 Based on recent guidance, a composite endpoint that includes serum ALP response along with assessment of fibrosis (e.g., through elastography or biopsy) may be sufficient for this purpose.21 Although the association between fibrosis and clinical outcomes in PSC is clear, data describing the link between ALP reduction and clinical events are evolving. For example, retrospective cohort studies, including post hoc analyses of randomized trials evaluating UDCA, have shown that serum ALP at baseline and 1 year after diagnosis, as well as improvement over time, are associated with better clinical outcomes.6, 7, 8, 9, 22, 23 For example, in the Scandinavian UDCA trial, ALP normalization and/or a 40% or more relative reduction at 1 year was associated with a reduced risk of death, transplantation, or cholangiocarcinoma, regardless of UDCA use.7 The 40% reduction threshold used in the Scandinavian study was chosen based on data from PBC, whereas other thresholds were not evaluated. However, in a large observational study of 692 patients with PSC in the Netherlands (Epi PSC PBC), a 25% relative reduction (versus no change) in serum ALP 1 year after diagnosis was associated with a greater than 25% relative reduction in the risk of liver transplantation or PSC‐related death over a median follow‐up of 100 months.6 These findings suggest that serum ALP could be an important prognostic marker in PSC, and that interventions that reduce ALP may have meaningful clinical benefit.
Previous studies of FXR agonists in patients with PBC and NASH have reported pruritus and changes in serum lipids as potential drawbacks to this class of agents.22, 24 In the current study, cilofexor was safe and well tolerated. Overall, the incidence of treatment‐emergent AEs was similar between cilofexor‐treated and placebo‐treated patients. The most frequently reported AE across the three study groups—including placebo—was pruritus, confirming the importance of this symptom in patients with PSC. Over 12 weeks of treatment, moderate to severe (grade 2‐3) pruritus was observed in 14%‐20% of cilofexor‐treated patients compared with 40% of those who received placebo. One patient treated with cilofexor 100 mg discontinued study medication due to pruritus, which occurred soon after initiating therapy. Larger and longer‐term studies will be necessary to fully characterize the potential effect of cilofexor on pruritus in PSC, which may differ from steroidal, bile acid–derived FXR ligands such as obeticholic acid. Finally, although no changes were observed in total cholesterol or LDL‐C, patients treated with cilofexor 100 mg experienced an approximately 10% decrease in HDL‐C over 12 weeks of treatment. The relevance of this change with respect to cardiovascular risk warrants further evaluation.
Our study has several limitations. First, patients with cirrhosis were excluded because the pharmacokinetics of cilofexor in the setting of cirrhosis were unknown at the time of study initiation. However, current data suggest that patients with compensated cirrhosis have minimal alterations in the pharmacokinetic and pharmacodynamic profiles of cilofexor compared with healthy controls.18 Given the absence of approved pharmacologic therapy for PSC, targeting those at risk of progressing to cirrhosis remains an important area of unmet need. Additional trials will evaluate the safety and efficacy of cilofexor in patients with cirrhosis due to PSC, as are currently ongoing for nonalcoholic steatohepatitis (NCT03449446). A second limitation is that we included only patients with classic large‐duct PSC. The generalizability of our findings to other populations, including patients with small‐duct PSC, overlap syndrome of PSC with autoimmune hepatitis, immunoglobulin‐4‐related sclerosing cholangitis, and pediatric patients, is unclear. Moreover, our study cohort had a lower prevalence of IBD than the broader PSC population, and all patients with IBD had quiescent disease. Reassuringly, no patient had exacerbation of their IBD or developed new onset IBD during the 12‐week study. Indeed, preclinical studies have suggested that FXR agonism improves intestinal barrier function in animal models of fibrosis and IBD. These observations require clinical evaluation.
In conclusion, this randomized phase II study demonstrates that the nonsteroidal FXR agonist cilofexor was well tolerated and improved markers of cholestasis, liver biochemistry, C4, and serum bile acids in patients with PSC. These effects were independent of UDCA use, thereby pointing to a distinct and potentially complementary mechanism of action. Assessment of whether cilofexor affects clinically relevant endpoints associated with PSC, such as progression to cirrhosis, hepatic decompensation, cholangiocarcinoma, transplantation, and mortality, await longer‐term controlled studies.
Supporting information
Supported by Gilead Sciences.
ClinicalTrials.gov identifier: NCT02943460.
Potential conflict of interest: Dr. Eksteen is on the speakers’ bureau for Pfizer and Intercept. Dr. Landis received grants from Gilead, NGM, Enanta, and High Tide. Dr. Muir consults and received grants from AbbVie, Dova, Gilead, and Merck; and he received grants from NGM and Novartis. Dr. Gulamhusein consults and advises Intercept. Dr. Kowdley consults, advises, is on the speakers’ bureau, and received grants from Gilead; he consults, is on the speakers’ bureau, and received grants from Intercept; he received grants from High Tide; and he received royalties from Up‐to‐Date. Dr. Hameed advises Mallinckrodt and Surrozen; and he received grants from Gilead, Dova, Salix, Intercept, Conatus, and Genfit. Dr. Caldwell received grants from Gilead, Genfit, Vital Therapy, Galmed, Zydus, Bristol‐Myers Squibb, Intercept, Mallinckrodt, and Dova; and he holds intellectual property rights with Halyard. Dr. Agarwal advises, is on the speakers’ bureau, and received grants from Gilead; he is on the speakers’ bureau and received grants from MSD; he consults for Shionogi; he advises Vir and Arbutus. Dr. Trauner consults, is on the speakers’ bureau, and received grants from Gilead and MSD; he consults and received grants from Falk, Albireo, and Intercept; he is on the speakers’ bureau and received grants from Roche; he consults for Phenex, Novartis, Bristol‐Myers Squibb, and Regulus; he is on the speakers’ bureau for Falk Foundation; and he received grants from Takeda. Dr. Bowlus advises and received grants from Gilead, Intercept, Cymabay, and Eli Lilly; he advises Parvus, Pliant, BiomX, and Patara; and he received grants from Takeda, Bristol‐Myers Squibb, GlaxoSmithKline, Genkyotex, and Arena. Dr. Shiffman advises, is on the speakers’ bureau, and received grants from AbbVie, Bristol‐Myers Squibb, Gilead, Intercept, and Merck; he advises and is on the speakers’ bureau for Bayer, Dova, Salix, and Shionogi; he is on the speakers’ bureau and received grants from Enanta; he consults for Optum Rx; he is on the speakers’ bureau for Easai and Daiichi Sankyo; and he received grants from Conatus, CymaBay, Exalenz, Galectin, Genfit, Genkyotex, Immuron, NGM, Novartis, and Shire. Dr. Billin, Dr. Chung, Dr. Subramanian, Dr. Li, Dr. Myers, Dr. Lu, Dr. Xu, and Dr. Chuang are employed by and own stock in Gilead.
Contributor Information
Michael Trauner, Email: michael.trauner@meduniwien.ac.at.
Kris V. Kowdley, Email: kris.kowdley@swedish.org.
References
- 1. Lazaridis KN, LaRusso NF. Primary sclerosing cholangitis. N Engl J Med 2016;375:1161‐1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chapman RW. Primary sclerosing cholangitis: what is the role of ursodeoxycholic acid in therapy for PSC? Nat Rev Gastroenterol Hepatol 2010;7:74‐75. [DOI] [PubMed] [Google Scholar]
- 3. Lindor KD, Kowdley KV, Luketic VA, Harrison ME, McCashland T, Befeler AS, et al. High‐dose ursodeoxycholic acid for the treatment of primary sclerosing cholangitis. Hepatology 2009;50:808‐814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Olsson R, Boberg KM, de Muckadell OS, Lindgren S, Hultcrantz R, Folvik G, et al. High‐dose ursodeoxycholic acid in primary sclerosing cholangitis: a 5‐year multicenter, randomized, controlled study. Gastroenterology 2005;129:1464‐1472. [DOI] [PubMed] [Google Scholar]
- 5. Beuers U, Spengler U, Kruis W, Aydemir U, Wiebecke B, Heldwein W, et al. Ursodeoxycholic acid for treatment of primary sclerosing cholangitis: a placebo‐controlled trial. Hepatology 1992;16:707‐714. [DOI] [PubMed] [Google Scholar]
- 6. de Vries EM, Wang J, Leeflang MM, Boonstra K, Weersma RK, Beuers UH, et al. Alkaline phosphatase at diagnosis of primary sclerosing cholangitis and 1 year later: evaluation of prognostic value. Liver Int 2016;36:1867‐1875. [DOI] [PubMed] [Google Scholar]
- 7. Lindstrom L, Hultcrantz R, Boberg KM, Friis‐Liby I, Bergquist A. Association between reduced levels of alkaline phosphatase and survival times of patients with primary sclerosing cholangitis. Clin Gastroenterol Hepatol 2013;11:841‐846. [DOI] [PubMed] [Google Scholar]
- 8. Rupp C, Rossler A, Halibasic E, Sauer P, Weiss KH, Friedrich K, et al. Reduction in alkaline phosphatase is associated with longer survival in primary sclerosing cholangitis, independent of dominant stenosis. Aliment Pharmacol Ther 2014;40:1292‐1301. [DOI] [PubMed] [Google Scholar]
- 9. Al Mamari S, Djordjevic J, Halliday JS, Chapman RW. Improvement of serum alkaline phosphatase to <1.5 upper limit of normal predicts better outcome and reduced risk of cholangiocarcinoma in primary sclerosing cholangitis. J Hepatol 2013;58:329‐334. [DOI] [PubMed] [Google Scholar]
- 10. Chiang JY. Bile acids: regulation of synthesis. J Lipid Res 2009;50:1955‐1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chiang JY, Kimmel R, Weinberger C, Stroup D. Farnesoid X receptor responds to bile acids and represses cholesterol 7alpha‐hydroxylase gene (CYP7A1) transcription. J Biol Chem 2000;275:10918‐10924. [DOI] [PubMed] [Google Scholar]
- 12. Inagaki T, Choi M, Moschetta A, Peng L, Cummins CL, McDonald JG, et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab 2005;2:217‐225. [DOI] [PubMed] [Google Scholar]
- 13. Kliewer SA, Mangelsdorf DJ. Fibroblast growth factor 21: from pharmacology to physiology. Am J Clin Nutr 2010;91:254S‐257S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Song KH, Li T, Owsley E, Strom S, Chiang JY. Bile acids activate fibroblast growth factor 19 signaling in human hepatocytes to inhibit cholesterol 7alpha‐hydroxylase gene expression. Hepatology 2009;49:297‐305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Schwabl P, Budas G, Hambruch E, Supper P, Burnet M, Lileset J, et al. The FXR agonist GS‐9674 reduces fibrosis and portal hypertension in a rat model of NASH [Abstract]. J Hepatol 2018;68:S471‐S472. [Google Scholar]
- 16. Muir AJ, Taghipour M, Hassanzadeh E, Sahni VA, Sainani N, Shiffman ML, et al. Radiologic progression in primary sclerosing cholangitis by two‐dimensional magnetic resonance cholangiopancreatography: prospective data from a randomized controlled trial [Abstract]. Hepatology 2017;66(Suppl 1):156A. [Google Scholar]
- 17. Vesterhus M, Hov JR, Holm A, Schrumpf E, Nygård S, Godang K, et al. Enhanced liver fibrosis score predicts transplant‐free survival in primary sclerosing cholangitis. Hepatology 2015;62:188‐197. [DOI] [PubMed] [Google Scholar]
- 18. Djedjos CS, Kirby BJ, Billin A, Gosink JJ, Song Q, Srihari R, et al. Pharmacodynamic effects of the oral, nonsteroidal farnesoid X receptor agonist GS‐9674 in healthy volunteers [Abstract]. Hepatology 2016;63(Suppl 1):543A. [Google Scholar]
- 19. Al‐Khaifi A, Rudling M, Angelin B. An FXR agonist reduces bile acid synthesis independently of increases in FGF19 in healthy volunteers. Gastroenterology 2018;155:1012‐1016. [DOI] [PubMed] [Google Scholar]
- 20. Ponsioen CY, Chapman RW, Chazouilleres O, Hirschfield GM, Karlsen TH, Lohse AW, et al. Surrogate endpoints for clinical trials in primary sclerosing cholangitis: review and results from an International PSC Study Group consensus process. Hepatology 2016;63:1357‐1367. [DOI] [PubMed] [Google Scholar]
- 21. Ponsioen CY, Lindor KD, Mehta R, Dimick‐Santos L. Design and endpoints for clinical trials in primary sclerosing cholangitis. Hepatology 2018;68:1174‐1188. [DOI] [PubMed] [Google Scholar]
- 22. Hirschfield GM, Mason A, Luketic V, Lindor K, Gordon SC, Mayo M, et al. Efficacy of obeticholic acid in patients with primary biliary cirrhosis and inadequate response to ursodeoxycholic acid. Gastroenterology 2015;148:751‐761. [DOI] [PubMed] [Google Scholar]
- 23. Stanich PP, Bjornsson E, Gossard AA, Enders F, Jorgensen R, Lindor KD. Alkaline phosphatase normalization is associated with better prognosis in primary sclerosing cholangitis. Dig Liver Dis 2011;43:309‐313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Neuschwander‐Tetri BA, Loomba R, Sanyal AJ, Lavine JE, Van Natta ML, Abdelmalek MF, et al. Farnesoid X nuclear receptor ligand obeticholic acid for non‐cirrhotic, non‐alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo‐controlled trial. Lancet 2015;385:956‐965. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials