

Abstract
GSK3389404 is a liver‐targeted antisense oligonucleotide that inhibits synthesis of hepatitis B surface antigen and all other hepatitis B virus proteins. This first‐in‐human, randomized, double‐blind, phase 1 study assessed the safety and pharmacokinetics of GSK3389404 administered subcutaneously (SC) in healthy subjects. Four single ascending‐dose cohorts (10 mg, 30 mg, 60 mg, and 120 mg) and 3 multiple ascending‐dose cohorts (30 mg, 60 mg, and 120 mg once weekly for 4 weeks) each comprised 6 subjects randomized to GSK3389404 and 2 subjects randomized to placebo. There were no serious adverse events (AEs) or withdrawals due to AEs. The safety profile did not worsen with repeated dosing. The most frequent treatment‐related AEs were injection site reactions (19.0% [n = 8/42], frequency unrelated to dose levels); all were mild (Grade 1) and resolved without dose modification or discontinuation. GSK3389404 administered subcutaneously was readily absorbed with a time to maximum plasma concentration (Tmax) of 1–4 hours and an elimination half‐life of 3–6 hours in plasma. Plasma area under the concentration‐time curve (AUC) and maximum observed concentration (Cmax) were dose‐proportional. Dose‐normalized plasma AUC from time 0 to infinity averaged 69.9 ng·h/(mL·mg dose) across cohorts, and Cmax 9.5 ng/(mL·mg dose). Pharmacokinetic profiles and parameters were comparable between single and multiple dosing. No accumulation was observed with once‐weekly dosing. The metabolite was undetectable in urine and plasma. In the pooled urine, GSK3389404 was estimated to account for <0.1% of the total dose. In summary, GSK3389404 dosing has been tested up to 120 mg for 4 weeks with an acceptable safety and pharmacokinetic profile, supporting further clinical investigation in patients with chronic hepatitis B.
Keywords: GSK3389404, chronic hepatitis B, hepatitis B virus, pharmacokinetics, first‐time‐in‐human
Worldwide, approximately 257 million people live with chronic hepatitis B virus (HBV) infection, defined as the persistence of detectable hepatitis B surface antigen (HBsAg) for more than 6 months.1, 2, 3, 4 Chronic HBV infection may be asymptomatic for many years; however, chronic infection may progress over time to trigger debilitating conditions such as cirrhosis, hepatic decompensation, hepatocellular carcinoma, and ultimately death.1 Viral hepatitis is the seventh leading cause of mortality globally5 with an estimated 887,000 deaths due to HBV in 2015.6
The primary goals of treatment for patients with chronic HBV infection are to improve survival rates and quality of life by preventing disease progression.1, 7 Sustained suppression of HBV replication is associated with reversal of cirrhosis and a reduction in rates of hepatic decompensation.8, 9 HBV cure, by complete eradication of the virus from the host, is not considered an attainable goal at present because of the persistence of HBV covalently closed circular DNA in hepatocyte nuclei.8 For the treatment of chronic HBV infections, “functional cure,” defined as persistent, undetectable HBsAg and HBV DNA in serum, is the preferred end point.1, 8, 9 Loss of HBsAg is the best predictor of sustained remission,8 possibly related to HBsAg interference with immune clearance of HBV.10 The main advantage of HBsAg loss is that it permits discontinuation of antiviral treatment; it is also associated with lower rates of hepatocarcinoma.11 However, HBsAg loss is rarely realized with currently approved therapies, and there is an unmet need for new HBV therapies that can provide sustained HBsAg suppression. Currently approved therapies for treatment of chronic HBV include nucleos(t)ide analogues and interferon. Nucleos(t)ide analogues suppress viral replication but require indefinite or long duration of treatment to maintain the viral suppression. Although interferon has a finite duration of treatment, it is associated with interferon‐related side effects.1
GSK3389404, currently in development for the treatment of chronic HBV, is a 2′‐O‐methoxyethyl antisense oligonucleotide (ASO)12 designed to inhibit the synthesis of HBsAg and all other HBV proteins.13 ASOs are small, single‐stranded nucleic acid sequences that bind with high selectivity to their target RNAs, triggering degradation via an RNase H–dependent pathway (Figure 1). They have shown potent suppression of HBV replication.14 GSK3389404 consists of an ASO targeting the HBV mRNA conjugated via a linker to a trimer of N‐acetylgalactosamine (GalNAc) moieties that targets delivery to hepatocytes through interaction with the predominantly hepatocyte‐expressed asialoglycoprotein receptor.15 GSK3389404 can be considered a prodrug because its metabolite GSK3228836 (the free ASO without the link and GalNAc) is the active drug (Figure 1). In a transgenic HBV mouse model,16 GSK3389404 dose‐dependently reduced serum levels of HBV DNA, HBsAg, and hepatitis B envelope antigen and hepatic levels of HBV RNA and HBV DNA when administered once weekly for 4 weeks.17
Figure 1.
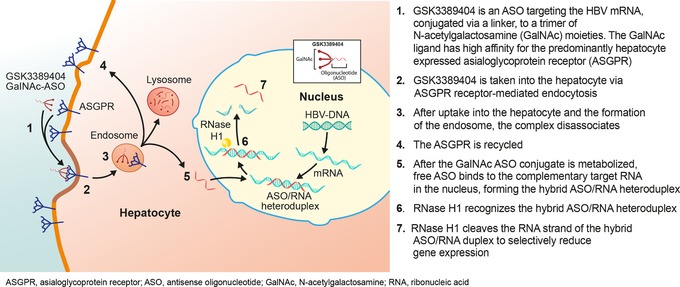
GSK3389404 mode of action. HBV indicates hepatitis B virus.
When delivered by the intravenous or subcutaneous route, phosphorothioate oligonucleotides demonstrate rapid distribution from the plasma compartment into the tissues (notably the liver and kidney), followed by a slow elimination via metabolism and excretion of truncated products.18 GSK3389404 is extensively distributed into the liver by target‐mediated endocytosis and into the kidney by micropinocytosis. Distribution of GSK3389404 and its metabolite GSK3228836 has been studied in the livers of HBV transgenic mice and the liver and kidneys in mice (13‐ and 26‐week toxicology studies) and monkeys (13‐ and 39‐week toxicology studies). GSK3389404 was undetectable in all tissues (<0.25 mg/g), and its metabolite GSK3228836 was quantifiable in both liver and kidney tissue samples at all dose levels during the dosing period. Mean GSK3228836 liver and kidney concentrations generally increased in a dose‐proportional manner, and there were no gender differences. Preclinical studies demonstrated that dosing with GSK3389404 results in a 1.5‐ to 2‐fold higher liver concentration of GSK3228836 when compared with an equivalent dose of GSK3228836, suggesting the advantage of targeted delivery by GalNAc.
The route of metabolism in mouse liver has been described for a similar GalNAc‐conjugated ASO.19 Within 1 hour of administration, the majority of the GalNAc residues had been cleaved to the acyl chain, and by 4 hours postdose, no prodrug (ASO with GalNAc) remained. Between 8 hours and 72 hours postdose, the conjugate was further metabolized to release the free ASO in the liver. In monkeys, after repeat dosing, GSK3389404 is the predominant circulating molecule with its metabolite (GSK3228836) levels at a maximum area under the concentration‐time curve (AUC) of 3.5% of GSK3389404 in the plasma. In tissue, the ratios are reversed with GSK3228836 as the predominant molecule and GSK3389404 being undetectable (<0.25 mg/g), indicating complete metabolism of GSK3389404 to GSK3228836 48 hours after the last dose. Elimination is expected to be via metabolism in the tissue and renal elimination of shortened chain fragments, based on studies with other ASOs.18 The primary route of elimination of GSK3389404 is expected to be via metabolism by endogenous endonucleases. In a recent study a model compound of 2′‐O‐(2‐methoxyethyl)‐modified ASO was shown to be neither a substrate nor an inhibitor of OAT1, OAT3, OCT1, and OCT2.20
Information on GSK's data‐sharing commitments and procedures to request access can be found at https://www.clinicalstudydatarequest.com/.
Rationale/Scientific Objectives
The aims of this phase 1, randomized, double‐blind (sponsor unblinded), multicenter, placebo‐controlled, dose‐escalation study (GSK Study Number 202007, NCT02647281) were to assess the safety and tolerability and the pharmacokinetic (PK) profile of single (part 1) and multiple (part 2) subcutaneous injections of GSK3389404 in healthy subjects.
Methods
Study Design and Procedures
The protocol was reviewed and approved by Bristol Research Ethics Committee Centre, Harrow, London, UK. The study was conducted in accordance with the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use Good Clinical Practice guidelines. Written informed consent was obtained from all subjects before participation. This was a double‐blind (sponsor unblinded) study: some sponsor representatives were unblinded for safety assessment and dose escalation decision making. The study was conducted at 2 centers due to closure of the Quintiles Drug Research Unit, Guy's Hospital after part 1 had been completed. The change in center is not considered significant.
Part 1: Single Ascending Dose
Part 1 was conducted at Quintiles Drug Research Unit, Guy's Hospital, London, UK. Subjects were enrolled in 4 sequential 8‐subject cohorts (Figure 2): A, 10 mg; B, 30 mg; C, 60 mg; and D, 120 mg. In each cohort, subjects were randomized 6:2 to GSK3389404 or placebo. At the beginning of each cohort, 2 subjects received a 1:1 sentinel dose of GSK3389404 and placebo. The remaining subjects were dosed 24 hours later to allow assessment of safety results. Subjects were admitted to the study center on day −1 (the day before the dose, considered part of the 30‐day screening window), and study drug administration occurred on day 1. Subjects remained in the study center until completion of the protocol‐specified procedures on day 4 and then returned to the study center for outpatient visits on days 8, 30, and 60. Doses were escalated in a sequential fashion contingent on the safety and PK profiles of at least 4 subjects who had received GSK3389404 at the previous dose levels up to and including day 4 postdose. Once all subjects in cohort D had reached day 4 with an acceptable safety profile, part 2 of the study was initiated.
Figure 2.
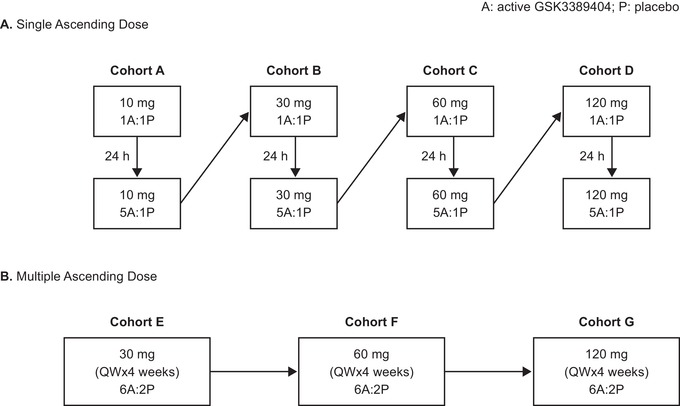
Study design and treatment schedule. A indicates active drug; P, placebo; QW, once weekly.
Part 2: Multiple Ascending Dose
Part 2 was conducted at Hammersmith Medicines Research Centre, London, UK. Subjects were enrolled into 3 sequential cohorts (Figure 2): E, 30 mg; F, 60 mg; and G, 120 mg. Subjects were randomized in a 6:2 ratio to receive GSK3389404 or matching placebo once weekly for 4 weeks. Subjects were admitted to the study center on day −1; study drug administration occurred on day 1, and subjects remained in the study center until completion of the protocol‐specified procedures on day 4. Subjects returned to the study center for outpatient visits on days 8 and 15 for repeat administrations of the study treatment. Subjects were readmitted to the study center on day 21; the final study drug administration occurred on day 22, and subjects were discharged from the study center on day 25, on completion of all the protocol‐specified procedures. Further outpatient visits were scheduled at the study center on days 29, 36, 50, 71, and 113. A phone call only was scheduled on day 92.
In both parts 1 and 2, doses of GSK3389404 were escalated such that the predicted mean plasma AUC(0‐∞) and maximum observed concentration (Cmax) at the next dose did not exceed the threshold values, and the Bayesian predictive probabilities of AUC(0‐∞) and Cmax crossing the threshold value were <50% at the next dose. The threshold value was defined as the gender‐averaged plasma AUC(0‐∞) (492.7 µg·h/mL) and Cmax (52.7 µg/mL) observed at steady state at the no‐observed‐adverse‐effect level dose in the 13‐week monkey toxicity study.
Study Population
Full details of the inclusion and exclusion criteria for eligibility are provided in Supplementary Table S1. Briefly, healthy adult subjects aged 18–55 years with a body weight >50 kg for men and >45 kg for women and a body mass index 18–30 kg/m2 were enrolled in the study. Subjects were healthy as determined by a responsible and experienced physician based on a medical evaluation, including medical history, physical examination, laboratory tests, and electrocardiogram (ECG) with no evidence of cardiac, pulmonary, hepatic, biliary, gastrointestinal, or renal disorders, or cancer within the past 5 years (except localized or in situ cancer of the skin). Subjects were excluded if they had a history of vascular, liver, or pulmonary diseases; a history of regular alcohol consumption within 6 months of the study start; smoking history within 3 months of screening; use of prescription or nonprescription drugs within 7 days (or 5 half‐lives) of the first dose of the study medication; abnormal liver function tests; or positivity for hepatitis C virus.
End Points and Assessments
The primary objectives of this study were to assess the safety, tolerability, and PK of single and multiple doses of GSK3389404. Secondary objectives included estimates of dose proportionality, accumulation and time invariance, and metabolite PK of GSK3389404 following administration of single and multiple doses.
Serial blood samples for PK analysis of GSK3389404 and metabolite (GSK3228836) were collected predose and 0.5, 1, 1.5, 2, 3, 4, 6, 8, 12, 24, 48, and 72 hours postdose (days 1–4) in parts 1 and 2. PK sampling was also conducted on days 8 and 30 in part 1 and days 8 and 15 in part 2 (predose samples). Following the final study drug administration on day 22 (part 2), serial blood samples were collected predose and 0.5, 1, 1.5, 2, 3, 4, 6, 8, 12, 24, 48, and 72 hours postdose (days 22–25), and on days 29, 36, 50, 71, and 113. Urine samples for assessment of the albumin/creatinine ratio were collected at screening, on day −1, predose, and 24 and 72 hours postdose, and on days 8 and 30 in part 1. In part 2, urine samples for albumin/creatinine ratio assessment were collected on day −1, predose, and 24 and 72 hours postdose, and on days 8 and 15 (first dose) and days 29, 36, 50, 71, and 113 (following second dose). Metabolite profiling was also conducted on collected urine samples.
Adverse events (AEs) were recorded from the start of study treatment until the follow‐up visit (day 60 in part 1 and day 113 in part 2). AEs were classified according to maximum severity as follows: Grade 1, mild; Grade 2, moderate; Grade 3, severe; Grade 4, life‐threatening or disabling; Grade 5, death. Serious AEs (SAEs) were predefined as death, disability, or hospitalization. Any SAEs assessed as related to either study participation (eg, protocol‐mandated procedures, invasive tests) or to GSK3389404 were to be recorded from the time of consent up to and including any follow‐up visit. Clinical laboratory safety assessments included chemistry, hematology, urinalysis, physical exam, and ECG.
Analytical Methods
Plasma samples were analyzed for GSK3389404 and GSK3228836 using a validated analytical method based on liquid‐liquid extraction followed by high‐performance liquid chromatography/mass spectrometry/mass spectrometry analysis. The lower limit of quantification for both analytes was 10 ng/mL using a 25‐µL aliquot of human plasma with higher limits of quantification of 10,000 ng/mL. An analogue internal standard ISIS‐440762 was used for both analytes. The separations were performed on a Clarity 3 mm Oligo‐RP 50 × 2 mm analytical column (Phenomenex, Torrance, California) using a Shimadzu, Nexera, 30 Series LC system (Kyoto, Japan). The column was maintained at 70°C with a gradient over 7.8 minutes at 0.2 mL/min. The mobile phases were (A) [EDTA 1 mmol/L (aq):DIPEA (100:0.5)]:DIPEA:HFIP:water (1:0.1:1:98); (B1) [EDTA 1 mmol/L (aq):DIPEA (100:0.5)]: DIPEA:HFIP:Water (1:0.1:1:98):acetonitrile (60:40); and (B2) acetonitrile:water:TEA (65:35:0.5). Mass spectrometry was performed on Sciex API 5000 (Framingham, Massachusetts) operated in negative electrospray ionization mode. Multiple reactions monitoring transitions (m/z) were 885.4 to 94.8 for GSK3389404, 815.4 to 94.8 for GSK3228836, and 773.5 to 94.8 for internal standard ISIS‐440762, respectively.
The within‐run precision was 1.0% to 12.4% relative standard deviation (RSD) for GSK3389404 and 1.7% to 15.5% RSD for GSK3228836. The within‐run accuracy was –2.7% to 9.0% bias for GSK3389404 and –3.5% to 4.4% bias for GSK3228836. The between‐run precision was 4.5% to 9.2% RSD for GSK3389404 and 5.2% to 9.4% RSD for GSK3228836.
Statistical Methods
Two study populations were analyzed in this study: the safety population, which comprised all subjects who received ≥1 dose of GSK3389404 or placebo, and the PK concentration population, which included all subjects who underwent plasma PK sampling and had evaluable postdose PK assay results.
AEs were coded according to the Medical Dictionary for Regulatory Activities. Safety data were analyzed using descriptive statistics.
For single dosing, day 1 data from parts 1 and 2 were combined as appropriate. Descriptive statistics were calculated for all PK parameters and summarized by treatment type. The dose proportionality of GSK3389404 PK parameters (AUC0–24, AUC0–∞, Cmax) from the single‐dose study or day 1 in the multiple‐dose study and from day 22 (AUC0‐τ and Cmax) in the multiple‐dose study was assessed using the power model y = α × doseβ, where y denotes the PK parameter being analyzed, α denotes the intercept, and β denotes the slope. A mixed‐effects model was used in the assessment of steady state, accumulation, and time invariance. Subjects were fit as a fixed effect for the assessment of accumulation and time invariance if the above mixed model failed to converge.
Results
Study Population and Subject Disposition
This study was conducted from December 17, 2015 to January 3, 2017. In part 1 of the study, 141 subjects were screened, and 32 were subsequently randomized to receive GSK3389404 or placebo (Supplementary Figure S1). All 32 subjects completed part 1 and were included in the safety analysis population. In part 2, 44 subjects were screened, and 24 subjects were subsequently randomized to receive GSK3389404 or placebo. One subject (4.2%) receiving GSK3389404 120 mg (cohort G) withdrew consent for personal reasons after receiving all scheduled treatment doses but was included in the analysis. There were no premature discontinuations of the study treatment during part 2, resulting in all 24 subjects being included in the safety analysis population.
Patient demographics and baseline results are summarized in Table 1. In part 1, all 32 subjects (100%) were male with a mean (SD) age of 32.5 (10.0) years and mean (SD) body mass index of 24.7 (2.8) kg/m2. In part 2, all but 2 subjects (92%) were male, mean (SD) age was 37.3 (10.5) years, and mean (SD) body mass index was 23.9 (2.7) kg/m2. Mean body mass index profiles across all cohorts in parts 1 and 2 were comparable.
Table 1.
Demographic Characteristics
| A. Single Ascending Dose (Part 1) | ||||||
|---|---|---|---|---|---|---|
| Demographics | Placebo (n = 8) | Cohort A GSK3389404 10 mg (n = 6) | Cohort B GSK3389404 30 mg (n = 6) | Cohort C GSK3389404 60 mg (n = 6) | Cohort D GSK3389404 120 mg (n = 6) | Total (n = 32) |
| Age, y | ||||||
| Mean (SD) | 28.1 (8.2) | 27.5 (3.8) | 37.5 (12.4) | 32.8 (7.4) | 37.8 (13.4) | 32.5 (10.0) |
| Range (min, max) | 22, 46 | 22, 32 | 22, 51 | 24, 43 | 24, 51 | 22, 51 |
| Sex, male [n (%)] | 8 (100) | 6 (100) | 6 (100) | 6 (100) | 6 (100) | 32 (100) |
| BMI (kg/m2) [mean (SD)] | 23.3 (2.0) | 25.3 (2.8) | 24.9 (4.1) | 24.1 (1.5) | 26.5 (2.7) | 24.7 (2.8) |
| Range (kg/m2) [min, max] | 19.6, 25.8 | 22.7, 29.9 | 20.9, 29.7 | 21.3, 25.8 | 21.8, 29.1 | 19.6, 29.9 |
| Weight (kg) [mean (SD)] | 75.9 (5.5) | 84.8 (11.0) | 75.5 (13.6) | 79.4 (9.8) | 83.2 (7.5) | 79.5 (9.7) |
| Range (kg) [min, max] | 67.6, 81.8 | 67.9, 96.7 | 60.4, 91.3 | 69.6, 93.0 | 74.7, 95.5 | 60.4, 96.7 |
| Race [n (%)] | ||||||
| Whitea | 8 (100) | 3 (50) | 4 (67) | 6 (100) | 6 (100) | 27 (84) |
| Blackb | 0 | 3 (50) | 1 (17) | 0 | 0 | 4 (13) |
| Asianc | 0 | 0 | 1 (17) | 0 | 0 | 1 (3) |
| B. Multiple Ascending Dose (Part 2) | |||||
|---|---|---|---|---|---|
| Demographics | Placebo (QW × 4 weeks) (n = 6) | Cohort E GSK3389404 30 mg (QW × 4 weeks) (n = 6) | Cohort F GSK3389404 60 mg (QW × 4 weeks) (n = 6) | Cohort G GSK3389404 120 mg (QW × 4 weeks) (n = 6) | Total (n = 24) |
| Age, y | |||||
| Mean (SD) | 38.0 (11.8) | 32.2 (6.9) | 40.5 (11.1) | 38.5 (12.2) | 37.3 (10.5) |
| Range (min, max) | 18, 51 | 23, 39 | 24, 50 | 27, 55 | 18, 55 |
| Sex [n (%)] | |||||
| Femaled | 0 | 0 | 2 (33) | 0 | 2 (8) |
| Male | 6 (100) | 6 (100) | 4 (67) | 6 (100) | 22 (92) |
| BMI (kg/m2) | |||||
| [Mean (SD)] | 23.8 (4.0) | 23.7 (1.5) | 24.1 (3.0) | 24.1 (2.2) | 23.9 (2.7) |
| Range (min, max) | 18.9, 29.6 | 21.6, 25.6 | 19.7, 28.0 | 21.5, 27.9 | 18.9, 29.6 |
| Weight (kg) | |||||
| [Mean (SD)] | 73.6 (15.2) | 75.9 (6.4) | 68.0 (10.2) | 74.5 (6.0) | 73.0 (9.9) |
| Range (min, max) | 57.4, 95.8 | 67.0, 83.6 | 56.8, 81.8 | 68.8, 84.4 | 56.8, 95.8 |
| Race [n (%)] | |||||
| Whitea | 4 (67) | 5 (83) | 3 (50) | 6 (100) | 18 (75) |
| Asiane | 0 | 1 (17) | 2 (33) | 0 | 3 (13) |
| Asianc | 1 (17) | 0 | 0 | 0 | 1 (4) |
| Caribbean | 1 (17) | 0 | 0 | 0 | 1 (4) |
| Caribbean British | 0 | 0 | 1 (17) | 0 | 1 (4) |
Safety population: defined as all subjects who received ≥1 dose of GSK3389404 or placebo. BMI indicates body mass index; max, maximum; min, minimum; N, total number of subjects; n, number of subjects with observation; QW, once weekly.
White/European heritage.
African heritage.
South‐East Asian heritage.
Women were all postmenopausal.
Central/South Asian heritage.
PK Profile of Single and Multiple Doses of GSK3389404
Twenty‐three (95.8%) of the 24 subjects who received GSK3389404 in part 1 were included in the PK analysis population. The entire PK profile of 1 subject receiving GSK3389404 30 mg (cohort B) was identified as atypical (the entire profile was flat with a sudden peak in GSK3389404 plasma concentration at 8 hours postdose), and all PK parameters were excluded from the analysis. In part 2, 1 subject receiving GSK3389404 60 mg (cohort F) had an atypical terminal elimination phase characterized by no obvious decline in GSK3389404 plasma concentrations between 6 and 12 hours postdose followed by nonquantifiable levels at 24 hours. All PK parameters of this subject were excluded from the analysis with the exception of values of GSK3389404 Cmax and Tmax.
PK profiles were similar across all cohorts with single (part 1) and multiple (part 2) ascending dosing (Figure 3). GSK3389404 was readily absorbed following subcutaneous administration with mean peak GSK3389404 plasma concentrations (Tmax) occurring 1 to 4 hours postdose, followed by a multiexponential decline in concentration in all cohorts (Table 2). GSK3389404 plasma AUC and Cmax increased proportionally with increasing dose and did not vary markedly with multiple dosing (part 2: first versus fourth dose; Table 2). Mean decline half‐life ranged from approximately 3 to 6 hours across all cohorts. Both decline half‐life and Tmax were independent of dose levels or intervals (day 1 versus day 22) and did not vary markedly with multiple dosing (part 2: first versus fourth dose; Table 2).
Figure 3.
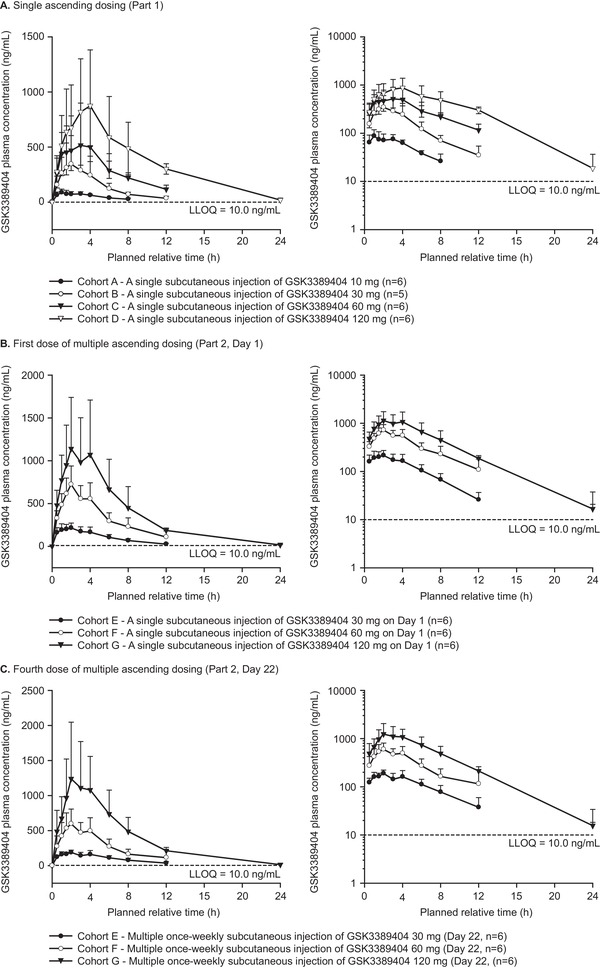
Plasma concentration of GSK3389404. Error bars indicate SDs; values below the quantification limit were entered as 0 and included as such in the calculation of means and SDs.
Table 2.
Pharmacokinetic Parameters for GSK3389404
| Single Ascending Dose (Part 1) | Multiple Ascending Dose (Part 2) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Cohort | A | B | C | D | E | F | G | |||
| Dose (mg) | 10 | 30 | 60 | 120 | 30 | 60 | 120 | |||
| N | 6 | 5 | 6 | 6 | 6 | 6 | 6d | 6 | 6 | 6 |
| Dosing interval for PK evaluation | After first dose | After first dose | After first dose | After first dose | After first dose | After fourth dose | After first dose | After fourth dose | After first dose | After fourth dose |
| Half‐lifea (h) | 4.55 (3.39) | 4.67 (3.55) | 6.07 (3.95) | 4.27 (1.23) | 3.14 (1.06) | 4.77 (3.21) | 3.47 (1.89) | 3.45 (1.60) | 4.04 (1.79) | 3.59 (1.23) |
| Tmax b (h) | 1.04 (1.00–4.02) | 2.00 (1.00–2.12) | 3.00 (1.00–4.00) | 4.00 (3.00–4.00) | 2.00 (1.00–4.00) | 2.00 (1.02–4.00) | 2.00 (2.00–2.05) | 2.00 (1.50–4.00) | 2.00 (1.50–4.00) | 2.00 (2.00–4.00) |
| Cmax a (ng/mL) | 92.5 (23.6) | 350 (257) | 584 (367) | 889 (497) | 236 (64.6) | 198 (39.7) | 723 (216) | 615 (211) | 1280 (634) | 1260 (800) |
| AUC0–∞ a, c (ng·h/mL) | 632 (172) | 2080 (802) | 4750 (1451) | 8150 (3205) | 1420 (292) | 1490 (365) | 5080 (1330) | 3970 (907) | 8480 (3250) | 8920 (3450) |
| CL/Fa (L/h) | 16.8 (4.28) | 15.8 (4.67) | 13.6 (3.69) | 16.2 (4.71) | 22.1 (5.98) | 20.5 (6.39) | 12.5 (3.42) | 15.5 (3.70) | 15.6 (4.79) | 14.4 (3.70) |
Data are presented as arithmetic mean (SD).
Data are presented as median (range).
AUC0–24 for day 22 in cohorts E, F, and G.
N = 5 for half‐life and AUC calculation.
Dose Proportionality, Accumulation, and Time Invariance
Dose proportionality analysis suggested that GSK3389404 AUC and Cmax increased in a dose‐proportional manner following a single subcutaneous injection within the studied dose range of 10 to 120 mg. Slope estimates for AUC0‐24, AUC0‐∞, and Cmax across the cohorts in part 1 and day 1 of part 2 were close to 1.00 (range 0.975–1.08), also indicating dose proportionality (Table 3). Slope estimates for AUC0‐τ and Cmax on day 22 of part 2 were 1.25‐fold greater than dose proportional, although the 90% CIs of Cmax included 1.00 (Table 3).
Table 3.
GSK3389404 Dose Proportionality Analysis Outcomes
| Day (Dose Range) | N | PK Parameter | Slope Estimate | 90%CI | Rsq |
|---|---|---|---|---|---|
| 1 (10–120 mg) | 40 | AUC0‐24 (ng·h/mL) | 1.08 | 0.98–1.18 | 0.8983 |
| 40 | AUC0‐∞ (ng·h/mL) | 1.07 | 0.97–1.18 | 0.8903 | |
| 41 | Cmax (ng/mL) | 0.975 | 0.83–1.12 | 0.7771 | |
| 22 (30–120 mg) | 18 | AUC0–τ (ng·h/mL) | 1.25 | 1.04–1.46 | 0.8727 |
| 18 | Cmax (ng/mL) | 1.25 | 0.96–1.55 | 0.7789 |
CI, confidence interval; N, total number of subjects; PK, pharmacokinetic; Rsq, coefficient of determination.
Mean plasma AUC and Cmax of GSK3389404 after the first and fourth doses in part 2 (day 1 versus day 22) were comparable and did not indicate marked accumulation. Point estimates of accumulation ratio, calculated for AUC0‐τ as AUC0‐τ, day 22/AUC0–168, day 1 (RAUC) and for Cmax as Cmax, day 22/Cmax, day 1, ranged from 85.5% to 109% at the 30‐mg and 120‐mg dose levels, and all 90%CIs included 100% (Table 4). GSK3389404 mean AUC and Cmax values for the 60‐mg dose were approximately 15% lower on day 22 compared with day 1 (Table 4). Similarly, GSK3389404 AUC and Cmax were time invariant between day 22 and day 1 for the 30‐mg and 120‐mg doses (range 108% to 109%) but were 15% lower on day 22 compared with day 1 for the 60‐mg dose.
Table 4.
GSK3389404 Accumulation and Time Invariance Analyses
| Parameter | GSK3389404 Dose | Geometric LS Mean (Day 1) | Geometric LS Mean (Day 22) | Day 22/Day 1 Ratio (%) | 90%CI |
|---|---|---|---|---|---|
| RAUC | 30 mg | 1390 | 1530 | 109 | 93.01–128.52 |
| 60 mg | 4670 | 3970 | 84.9 | 75.60–95.30 | |
| 120 mg | 8040 | 8640 | 108 | 90.73–127.42 | |
| RCmax | 30 mg | 228 | 194 | 85.5 | 72.57–100.61 |
| 60 mg | 692 | 577 | 83.4 | 69.27–100.40 | |
| 120 mg | 1170 | 1110 | 94.9 | 72.39–124.34 | |
| LI | 30 mg | 1390 | 1530 | 109 | 93.01–128.52 |
| 60 mg | 4670 | 3970 | 84.9 | 75.67–95.28 | |
| 120 mg | 8040 | 8640 | 108 | 90.73–127.42 |
CI, confidence interval; LI, time invariance, calculated as [AUC(0‐τ), Day 22/AUC(0‐∞), Day 1]; LS, least squares; RAUC, accumulation ratio, calculated for AUC(0‐τ) as [AUC(0‐τ), Day 22/AUC(0‐168), Day 1]; RCmax, accumulation ratio, calculated for Cmax as [Cmax, Day 22/Cmax, Day 1].
Metabolite PK
The active metabolite of GSK3389404, GSK3228836, was not detected in either urine or plasma extracts. GSK3389404 was the only drug‐related component detected in the pooled human urine from part 1 of the study (0‐ to 24‐hour pool) and was estimated to account for less than 0.1% of the total dose. No drug‐related material was observed in the 48‐hour or 72‐hour single‐dose pooled plasma extracts. GSK3389404 was not detected in the pooled urine from part 2, and no additional drug‐related components were detected in the plasma or urine from part 1 or part 2.
Safety and Tolerability of Single and Multiple Doses of GSK3389404
There were no SAEs and no AEs leading to study withdrawal in either study part. There was no indication of a relationship between dose and AE severity. AEs at the highest GSK3389404 dose of 120 mg were reported in 83% (5/6) of subjects in part 1 and 67% (4/6) of subjects in part 2 (Table 5). AEs were of Grade 1 (mild) or Grade 2 (moderate) severity.
Table 5.
Summary of AEs by Maximum Grade Reported
| A. Single Ascending Dose | |||||
|---|---|---|---|---|---|
| Placebo (N = 8) n (%) | Cohort A GSK3389404 10 mg (N = 6) n (%) | Cohort B GSK3389404 30 mg (N = 6) n (%) | Cohort C GSK3389404 60 mg (N = 6) n (%) | Cohort D GSK3389404 120 mg (N = 6) n (%) | |
| AE | 4 (50) | 3 (50) | 2 (33) | 3 (50) | 5 (83) |
| Grade 1 | 3 (38) | 0 | 1 (17) | 1 (17) | 4 (67) |
| Grade 2 | 1 (13) | 3 (50) | 1 (17) | 2 (33) | 1 (17) |
| Grades 3–5 | 0 | 0 | 0 | 0 | 0 |
| AE leading to study withdrawal | 0 | 0 | 0 | 0 | 0 |
| Serious AE | 0 | 0 | 0 | 0 | 0 |
| Grades 3–4 laboratory abnormality | 0 | 0 | 0 | 1 (17) | 0 |
| B. Multiple ascending dose | ||||
|---|---|---|---|---|
| Placebo (QW × 4 weeks) (N = 6) n (%) | Cohort E GSK3389404 30 mg (QW × 4 weeks) (N = 6) n (%) | Cohort F GSK3389404 60 mg (QW × 4 weeks) (N = 6) n (%) | Cohort G GSK3389404 120 mg (QW × 4 weeks) (N = 6) n (%) | |
| AE | 3 (50) | 5 (83) | 2 (33) | 4 (67) |
| Grade 1 | 2 (33) | 2 (33) | 0 | 2 (33) |
| Grade 2 | 1 (17) | 3 (50) | 2 (33) | 2 (33) |
| Grade 3–5 | 0 | 0 | 0 | 0 |
| AE leading to study withdrawal | 0 | 0 | 0 | 0 |
| Serious AE | 0 | 0 | 0 | 0 |
| Grades 3–4 laboratory abnormality | 0 | 0 | 0 | 0 |
Safety population: defined as all subjects who received ≥1 dose of GSK3389404 or placebo. Data are for the number of subjects reporting ≥1 AE. AE indicates adverse event; QW, once weekly. Grades 3–4 laboratory abnormality is from ad hoc analysis.
Individual AEs are shown in the Appendix (Supplementary Table S2). The most common AEs in subjects receiving GSK3389404 in part 1 were upper respiratory tract infections (20.8% [5/24]), headache (16.7% [4/24]), and injection site reactions (ISR, 16.7% [4/24]). All AEs related to upper respiratory tract infections were reported during the follow‐up period and were not considered related to the study treatment. Two subjects had transient Grade 1 increases in hepatic enzymes (1.28 and 1.72 times the upper limit of normal, respectively; both resolved by the day‐60 follow‐up). The most common AEs in subjects receiving GSK3389404 in part 2 were headache (27.8% [5/18]), ISR (27.8% [5/18]), and nasopharyngitis (22.2% [4/18]). All nasopharyngitis AEs were reported during the follow‐up period and were not considered related to the study treatment.
The most commonly reported treatment‐related AE in subjects receiving GSK3389404 in both parts of the study was ISR (19.0% [8/42]; Table 6). All ISRs were of Grade 1 severity and resolved during the study without the need for treatment, dose modification, or discontinuation. The frequency or severity of ISRs did not increase with multiple dosing. The frequency and severity of ISRs were not correlated with dose levels.
Table 6.
Treatment‐Related Adverse Events
| A. Single Ascending Doses | |||||
|---|---|---|---|---|---|
| System Organ Class/Preferred Term | Placebo (N = 8) n (%) | Cohort A GSK3389404 10 mg (N = 6) n (%) | Cohort B GSK3389404 30 mg (N = 6) n (%) | Cohort C GSK3389404 60 mg (N = 6) n (%) | Cohort D GSK3389404 120 mg (N = 6) n (%) |
| Any related event | 1 (13) | 0 | 2 (33) | 0 | 5 (83) |
| General disorders and administration site conditions | |||||
| Any event | 1 (13) | 0 | 0 | 0 | 3 (50) |
| Injection site bruising | 0 | 0 | 0 | 0 | 1 (17) |
| Injection site erythema | 0 | 0 | 0 | 0 | 2 (33) |
| Injection site pain | 1 (13) | 0 | 0 | 0 | 0 |
| Investigations | |||||
| Any event | 0 | 0 | 0 | 0 | 1 (17) |
| ALT increased | 0 | 0 | 0 | 0 | 1 (17) |
| AST increased | 0 | 0 | 0 | 0 | 1 (17) |
| Nervous system disorders | |||||
| Any event | 0 | 0 | 2 (33) | 0 | 1 (17) |
| Headache | 0 | 0 | 2 (33) | 0 | 1 (17) |
| B. Multiple Ascending Doses | ||||
|---|---|---|---|---|
| System Organ Class/Preferred Term | Placebo (QW × 4 weeks) (N = 6) n (%) | Cohort E GSK3389404 30 mg (QW × 4 weeks) (N = 6) n (%) | Cohort F GSK3389404 60 mg (QW × 4 weeks) (N = 6) n (%) | Cohort G GSK3389404 120 mg (QW × 4 weeks) (N = 6) n (%) |
| Any related event | 1 (17) | 4 (67) | 1 (17) | 2 (33) |
| General disorders and administration site conditions | ||||
| Any event | 1 (17) | 3 (50) | 0 | 2 (33) |
| Injection site erythema | 1 (17) | 2 (33) | 0 | 2 (33) |
| Injection site discomfort | 0 | 1 (17) | 0 | 0 |
| Injection site pain | 0 | 0 | 0 | 1 (17) |
| Injection site pruritus | 0 | 1 (17) | 0 | 0 |
| Injection site swelling | 0 | 1 (17) | 0 | 0 |
| Metabolism and nutrition disorders | ||||
| Any event | 0 | 1 (17) | 0 | 0 |
| Decreased appetite | 0 | 1 (17) | 0 | 0 |
| Nervous system disorders | ||||
| Any event | 1 (17) | 2 (33) | 1 (17) | 0 |
| Dizziness | 0 | 1 (17) | 0 | 0 |
| Headache | 0 | 1 (17) | 1 (17) | 0 |
| Somnolence | 1 (17) | 0 | 0 | 0 |
Safety population: defined as all subjects who received ≥1 dose of GSK3389404 or placebo. ALT indicates alanine aminotransferase; AST, alanine aminotransferase; N, total number of subjects; n, number of subjects with observation; QW, once weekly.
AEs of special interest included potential ASO class effects of thrombocytopenia, activated partial thromboplastin time prolongation, and complement system activation. No hematology AEs, including thrombocytopenia, were detected, and no prolongation of the activated partial thromboplastin time was observed. Analysis of complement pathway markers Bb, C5a, C3, and C4 did not indicate complement activation.
Discussion
This randomized, double‐blind, placebo‐controlled, dose‐escalation trial is the first‐in‐human study of GSK3389404. Results of the PK analysis revealed that GSK3389404 is readily absorbed following subcutaneous injection in healthy subjects with Tmax between 1 and 4 hours postdose. Plasma AUC and Cmax increased in a dose‐proportional manner over the 10‐ to 120‐mg dose range evaluated. GSK3389404 plasma AUC and Cmax were comparable following single and multiple dosing. No accumulation was observed with once‐weekly dosing, likely reflecting the relatively short half‐life (3–6 hours) of the drug.
The dosing interval was selected to be 1 week based on the long half‐life of the active metabolite (GSK3228836) in the liver. GSK3389404 is rapidly and completely converted to GSK3228836 in the liver, with an estimated half‐life of approximately 28 days in monkeys. Liver concentration was fit to a maximum effect model and then extrapolated to human. Therefore, a weekly or less frequent dosing was considered appropriate for the toxicology and clinical studies.
No safety concerns were identified for GSK3389404 over the 10‐ to 120‐mg dose range evaluated. There were no SAEs or withdrawals due to an AE, and the safety profile of GSK3389404 did not worsen with multiple versus single dosing. The most frequent treatment‐related AEs were ISRs, which were all mild in severity and resolved during the study without the need for dose modification or discontinuation. The frequency of ISRs did not increase with multiple dosing, and no dose‐response relationship was observed. Elevations in liver enzymes were observed in 2 patients but were transient, mild in severity, and not associated with clinical signs or symptoms.
ASOs as a class are based on short nucleotide sequences with similar structures and physical and chemical properties that may result in comparable clinical profiles.21, 22, 23 An integrated safety assessment of 2′‐O‐methoxyethyl ASOs across studies of nonhuman primates (NHPs; n = 710) and humans (n = 750) evaluated potential class safety effects.23 The assessment showed no evidence of drug‐associated effects on renal or hepatic function in NHPs or humans. NHPs did show evidence of potential class effects for acute thrombocytopenia and complement activation but are generally considered to be more sensitive to these effects compared with humans.24 In line with these findings, we found no evidence in this study of marked abnormalities in hepatic or renal function, hematology, or complement activation with GSK3389404 in healthy subjects.
The favorable safety profile of GSK3389404 in this study is encouraging and in keeping with the specific targeting of viral mRNA. ASOs as a class are highly target specific, with single nucleotide substitutions affecting their activity.25 The specificity may reduce the possibility of off‐target effects. The targeted delivery of GSK3389404 to hepatocytes is further aided by its GalNAc moiety and therefore may lead to lower doses with similar efficacy versus administration of the ASO without GalNAc.
Limitations of this study include the absence of pharmacodynamic assessments, given that this study was conducted in healthy subjects. The absence of pharmacodynamic data precluded determination of the appropriate GSK3389404 dose from this study. PK parameters could not be correlated with important intrinsic and extrinsic factors such as liver and renal functions given their narrow range in healthy subjects. Finally, characterization of GSK3389404 metabolites was limited by a lower limit of quantification of 10 ng/mL in this study, which resulted in an inability to detect levels of the active metabolite GSK3228836 in the plasma. Future studies will assess metabolite PK with a lower limit of quantification of 1 ng/mL.
Conclusion
In summary, GSK3389404 SC has been tested in doses of up to 120 mg and was found to have dose‐proportional PK plasma exposures (AUC and Cmax) with a half‐life of 3 to 6 hours and no indication of accumulation with weekly administration over 4 weeks in healthy subjects. No safety concerns were identified with GSK3389404 over the 10‐ to 120‐mg dose range, and the data support further clinical investigation in patients with chronic HBV.
Funding
This study (GSK 202007, NCT02647281) was funded by GSK.
Supporting information
Supplemental Figure S1.
Supplemental Table S1.
Supplemental Table S2.
Acknowledgments
Medical writing support was provided by Hannah Greenwood of Fishawack Indicia Ltd, UK, and funded by GlaxoSmithKline (GSK). Stephen Gardner, Joseph Kim, and Soumi Lahiri are acknowledged for their involvement in the study concept and design.
Disclosures
D.G., D.T., J.L., K.H., M.D., M.L., M.P., R.E., R.H., S.B.‐B., S.C., and S.H. are employees of GSK with stock or stock options; J.C. and J.M.R. are employees of GSK; S.O. is an employee of IQVIA; J.S. was an employee of GSK at the time of this study with stock or stock options; J.M.R. was an employee of Quintiles at the time of this study; and F.v.d.B. is an employee of Hammersmith Medicines Research.
References
- 1. European Association for the Study of the Liver . EASL clinical practice guidelines: management of chronic hepatitis B virus infection. J Hepatol. 2012;57(1):167‐185. [DOI] [PubMed] [Google Scholar]
- 2. Ott JJ, Stevens GA, Groeger J, Wiersma ST. Global epidemiology of hepatitis B virus infection: new estimates of age‐specific HBsAg seroprevalence and endemicity. Vaccine. 2012;30(12):2212‐2219. [DOI] [PubMed] [Google Scholar]
- 3. World Health Organization . Global Hepatitis Report. 2017. http://apps.who.int/iris/bitstream/handle/10665/255016/9789241565455-eng.pdf?sequence=1. Accessed May 29, 2018.
- 4. World Health Organization . Guidelines on hepatitis B and C testing. http://apps.who.int/iris/bitstream/handle/10665/254621/9789241549981-eng.pdf;jsessionid=6540EC50346ADDDB67F0A8676708E7DD?sequence=1. Published February 2017. Accessed February 28, 2018.
- 5. Stanaway JD, Flaxman AD, Naghavi M, et al. The global burden of viral hepatitis from 1990 to 2013: findings from the Global Burden of Disease Study 2013. Lancet. 2016;388(10049):1081‐1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. World Health Organization . Hepatitis B fact sheet. http://www.who.int/mediacentre/factsheets/fs204/en/. Updated July 5, 2017. Accessed February 28, 2018.
- 7. European Association for the Study of the Liver . Electronic address, European Association for the Study of the Liver. EASL 2017 Clinical Practice Guidelines on the management of hepatitis B virus infection. J Hepatol. 2017;67(2):370‐398. [DOI] [PubMed] [Google Scholar]
- 8. Terrault N, Bzowej N, Chang K, Hwang J, Jonas M, Murad M. AASLD guidelines for treatment of chronic hepatitis B. Hepatology. 2016;63(1):261‐283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lok AS, Zoulim F, Dusheiko G, Ghany MG. Hepatitis B cure: from discovery to regulatory approval. J Hepatol. 2017;67(4):847‐861. [DOI] [PubMed] [Google Scholar]
- 10. Vanlandschoot P, Leroux‐Roels G. Viral apoptotic mimicry: an immune evasion strategy developed by the hepatitis B virus? Trends Immunol. 2003;24(3):144‐147. [DOI] [PubMed] [Google Scholar]
- 11. Kim GA, Lim YS, An J, et al. HBsAg seroclearance after nucleoside analogue therapy in patients with chronic hepatitis B: clinical outcomes and durability. Gut. 2014;63(8):1325‐1332. [DOI] [PubMed] [Google Scholar]
- 12. WO2014/179627 (PCT/US2014/03643). Compositions and Methods for Modulating HBV and TTR Expression, filed May 1, 2014 in the name of Isis Pharmaceuticals, Inc. https://worldwide.espacenet.com/publicationDetails/originalDocument?CC=WO&NR=2014179627A2&KC=A2&FT=D&ND=5&date=20141106&DB=EPODOC&locale=en_EP#. Accessed February 28, 2019.
- 13. Han K, Kim J, Elston R, et al. Safety, tolerability and pharmacokinetics of GSK3389404, an antisense oligonucleotide for the treatment of chronic hepatitis B (CHB) infection: a randomized, double‐blind, placebo‐controlled, dose‐escalation, first time in human study. American Association for the Study of Liver Disease (AASLD): The Liver Meeting; October 21, 2017; Washington, DC. [Google Scholar]
- 14. Billioud G, Kruse RL, Carrillo M, et al. In vivo reduction of hepatitis B virus antigenemia and viremia by antisense oligonucleotides. J Hepatol. 2016;64(4):781‐789. [DOI] [PubMed] [Google Scholar]
- 15. Huang Y. Preclinical and clinical advances of GalNAc‐decorated nucleic acid therapeutics. Mol Ther Nucleic Acids. 2017;6:116‐132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Guidotti LG, Matzke B, Schaller H, Chisari FV. High‐level hepatitis B virus replication in transgenic mice. J Virol. 1995;69(10):6158‐6169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shihyun You RC, Billioud G, Whitten‐Bauer C, et al. Preclinical evaluation of 5’‐N‐acetylgalactosamine (GalNAc) conjugated anti‐sense oligonucleotides (ASO) against HBV in primary human hepatocytes and HBV transgenic mice. In: The International HBV Meeting of the Molecular Biology of Hepatitis B Viruses; Seoul, South Korea; September 21‐24, 2016. [Google Scholar]
- 18. Geary RS. Antisense oligonucleotide pharmacokinetics and metabolism. Expert Opin Drug Metab Toxicol. 2009;5(4):381‐391. [DOI] [PubMed] [Google Scholar]
- 19. Prakash TP, Graham MJ, Yu J, et al. Targeted delivery of antisense oligonucleotides to hepatocytes using triantennary N‐acetyl galactosamine improves potency 10‐fold in mice. Nucleic Acids Res. 2014;42(13):8796‐8807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yu RZ, Warren MS, Watanabe T, et al. Lack of interactions between an antisense oligonucleotide with 2'‐O‐(2‐methoxyethyl) modifications and major drug transporters. Nucleic Acid Ther. 2016;26(2):111‐117. [DOI] [PubMed] [Google Scholar]
- 21. Chi X, Gatti P, Papoian T. Safety of antisense oligonucleotide and siRNA‐based therapeutics. Drug Discov Today. 2017;22(5):823‐833. [DOI] [PubMed] [Google Scholar]
- 22. Kwoh TJ. An overview of the clinical safety experience of first‐ and second‐generation antisense oligonucleotides In: Crooke ST, ed. Antisense Drug Technology: Principles, Strategies and Applications. 2nd ed. Boca Raton, FL: Taylor & Francis Group; 2008:365‐399. [Google Scholar]
- 23. Crooke ST, Baker BF, Kwoh TJ, et al. Integrated safety assessment of 2'‐O‐methoxyethyl chimeric antisense oligonucleotides in nonhuman primates and healthy human volunteers. Mol Ther. 2016;24(10):1771‐1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shen L, Frazer‐Abel A, Reynolds PR, et al. Mechanistic understanding for the greater sensitivity of monkeys to antisense oligonucleotide‐mediated complement activation compared with humans. J Pharmacol Exp Ther. 2014;351(3):709‐717. [DOI] [PubMed] [Google Scholar]
- 25. Vickers TA, Koo S, Bennett CF, Crooke ST, Dean NM, Baker BF. Efficient reduction of target RNAs by small interfering RNA and RNase H‐dependent antisense agents. A comparative analysis. J Biol Chem. 2003;278(9):7108‐7118. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure S1.
Supplemental Table S1.
Supplemental Table S2.