

Abstract
In the last decade, cancer immunotherapy has emerged as an effective alternative to traditional therapies such as chemotherapy and radiation. In contrast to the latter, cancer immunotherapy has the potential to distinguish between cancer and healthy cells, and thus to avoid severe and intolerable side‐effects, since the cancer cells are effectively eliminated by stimulated immune cells. The cytosolic nucleotide‐binding oligomerization domains 1 and 2 receptors (NOD1 and NOD2) are important components of the innate immune system and constitute interesting targets in terms of strengthening the immune response against cancer cells. Many NOD ligands have been synthesized, in particular NOD2 agonists that exhibit favorable immunostimulatory and anticancer activity. Among them, mifamurtide has already been approved in Europe by the European Medicine Agency for treating patients with osteosarcoma in combination with chemotherapy after complete surgical removal of the primary tumor. This review is focused on NOD receptors as promising targets in cancer immunotherapy as well as summarizing current knowledge of the various NOD ligands exhibiting antitumor and even antimetastatic activity in vitro and in vivo.
Keywords: adjuvants, cancer immunotherapy, immunotherapeutics, NOD1 agonists, NOD1 antagonists, NOD2 agonists, NOD2 antagonists
Abbreviations
- ActD
actinomycin D
- AP‐1
activator protein 1
- AOM
azoxymethane
- ASC
apoptosis‐associated speck‐like protein containing caspase recruitment domain
- ATG16L1
autophagy‐related 16‐like 1
- Bid
BH3‐interacting domain death agonist
- CARD
caspase recruitment domain
- CCL2
chemokine ligand 2
- CDDP
cisplatin
- cIAP
cellular inhibitor of apoptosis protein
- CRC
colorectal cancer
- CSF
colony‐stimulating factor
- CTX
cyclophosphamide
- DAMP
danger‐associated molecular pattern
- DC
dendritic cell
- DMPG
dimyristoylphosphatidylglycerol
- DOX
doxorubicin
- DSPC
distearoylphosphatidylcholine
- DSS
dextran sulfate sodium
- DTX
docetaxel
- ERK
extracellular signal‐regulated kinase
- FCA
Freund's complete adjuvant
- GC
gastric cancer
- GDP
glycerol dipalmitate
- HNSCC
head and neck squamous cell carcinoma
- IAP
inhibitor of the apoptosis
- ICAM‐1
intercellular adhesion molecule‐1
- ICD
immunogenic cell death
- iE‐DAP
d‐glutamyl‐meso‐diaminopimelic acid
- IFN
interferon
- IFO
ifosfamide
- IKK
IκB kinase
- IκB
protein inhibitor of NF‐κB
- IL
interleukin
- IRF
interferon regulatory factor
- JNK
c‐Jun N‐terminal kinase
- LLC
Lewis lung carcinoma
- LPS
lipopolysaccharide
- LRR
leucine‐rich repeat
- mAb
monoclonal antibody
- MAPK
mitogen‐associated protein kinase
- MAVS
mitochondrial antiviral signaling
- MBSA
maleylated BSA
- MDP
muramyl dipeptide
- MDSC
myeloid‐derived suppressor cell
- MMP
matrix metalloproteinase
- MoDC
monocyte‐derived dendritic cell
- MTX
methotrexate
- NACHT
nucleotide‐binding domain
- NEMO
nuclear factor κB essential modulator
- NLR
nucleotide‐binding oligomerization domain‐like receptor
- NK
natural killer
- NLRP
NACHT‐, LRR‐, and PYD‐domain–containing protein
- NOD
nucleotide‐binding oligomerization domain
- OSCC
oral squamous cell carcinoma
- PAMP
pathogen‐associated molecular pattern
- PBMC
peripheral blood mononuclear cell
- PC
pancreatic cancer
- PGN
peptidoglycan
- PROK2
prokineticin 2
- PRR
pattern‐recognition receptor
- PS
phosphatidylserine
- PTX
paclitaxel
- RIPK2
receptor‐interacting serine/threonine‐protein kinase 2
- PolyG
polyguanylic acid
- SAR
structure‐activity relationship
- STAT1
signal transducer and activator of transcription 1
- S100A8
S100 calcium‐binding protein A8
- TAB
transforming growth factor binding protein
- TAK1
transforming growth factor β‐activated kinase 1
- TBK1
TRAF‐associated nuclear factor‐κB activator–binding kinase 1
- TGF
transforming growth factor
- Th1
type 1T helper
- TIMP1
tissue inhibitor of metalloproteinase 1
- TLR
Toll‐like receptor
- TME
tumor microenvironment
- TNF‐α
tumor necrosis factor α
- TRAF
tumor necrosis factor receptor–associated factor
- Tri‐DAP
l‐alanyl‐γ‐d‐glutamyl‐meso‐DAP
- XIAP
X‐linked inhibitor of apoptosis protein
1. INTRODUCTION
The connection between the immune system and cancer was first proposed by Rudolph Virchow in 1863 when he observed the presence of leukocytes in neoplastic tissues.1 A few years later, in 1891, William Coley laid the groundwork for modern immunotherapy with the discovery that the use of a bacterial vaccine in treating primarily inoperable sarcoma resulted in a cure rate greater than 10%.2 A similar attempt was made in 1976 when Morales et al3 used BCG, a live attenuated strain of bacteria Mycobacterium bovis, for treating bladder cancer. In fact, BCG injection still represents the main intravesical immunotherapy for treating superficial bladder cancer.4 The field of cancer immunotherapy has been intensively developed over the last decade as evidenced by an increasing number of newly approved immunotherapeutics for human use.5 In general, cancer immunotherapeutics are classified as passive or active, based on their ability to activate the host's immune system against cancer cells. Passive immunotherapeutics are designed to act like certain components of the host's immune system whereas active immunotherapeutics stimulate the host's immune system to induce its own response against cancer cells.6, 7 The group of passive immunotherapeutics comprises tumor‐targeting monoclonal antibodies (mAbs), adoptive cell transfer and oncolytic viruses, while active immunotherapeutics comprise dendritic cell‐based immunotherapeutics, cancer vaccines, immunostimulatory cytokines, immunomodulatory mAbs, inhibitors of immunosuppressive metabolism, immunogenic cell death (ICD) inducers, and pattern‐recognition receptor (PRR) ligands (Figure 1).6, 7 The latter group, in particular, has a lot of untapped potential for use as an adjunct to current cancer therapies, as evidenced by recent discoveries.8
Figure 1.
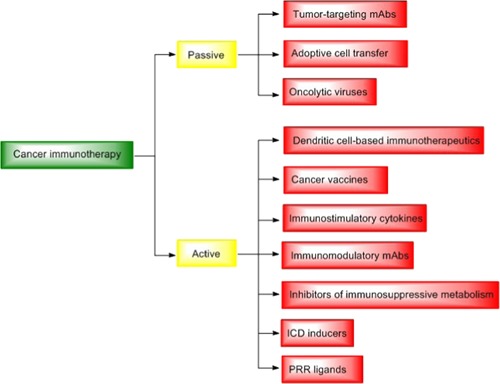
Cancer immunotherapies. Cancer immunotherapies are broadly classified as passive or active, based on their ability to activate host's immune system against cancer cells. The group of passive immunotherapeutics consist of tumor‐targeting mAbs, adoptive cell transfer, and oncolytic viruses, while active immunotherapeutics comprise dendritic cell‐based immunotherapeutics, cancer vaccines, immunostimulatory cytokines, immunomodulatory mAbs, inhibitors of immunosuppressive metabolism, ICD inducers, and PRR ligands. ICD, immunogenic cell death; mAbs, monoclonal antibodies; PRR, pattern‐recognition receptor [Color figure can be viewed at wileyonlinelibrary.com]
PRRs are evolutionarily conserved proteins in the innate immune system that are involved in the recognition of various pathogen‐ or danger‐associated molecular patterns (PAMPs and DAMPs).9 They are subdivided into five families including Toll‐like receptors (TLRs), RIG‐I‐like receptors, NOD‐like receptors (NLRs), AIM2‐like receptors and C‐type lectins.10 NOD1 and NOD2 are the most studied members of the NLR family and sense conserved peptidoglycan (PGN) fragments found in bacterial cell walls. NOD1 senses meso‐diaminopimelic acid (DAP)‐containing fragments of PGN, such as the d‐glutamyl‐meso‐diaminopimelic acid (iE‐DAP) and l‐alanyl‐γ‐d‐glutamyl‐meso‐DAP (Tri‐DAP) found in Gram‐negative bacteria and certain Gram‐positive bacteria. NOD2, however, is activated by N‐(acetylmuramyl)‐l‐alanyl‐d‐isoglutamine (muramyl dipeptide [MDP]), a small PGN fragment found in various Gram‐negative bacteria and Gram‐positive bacteria.11, 12, 13, 14 Due to their ability to detect bacterial PGN and, consequently, to activate an inflammatory response, NODs are considered as key molecules in host defense and inflammation.15 In humans, for example, mutations in genes encoding NOD2 are associated with inflammatory diseases including Blau syndrome and early onset sarcoidosis16, 17 as well as with increased risk of developing Crohn's disease.18 Despite the well‐known role of NODs in pathogen recognition and inflammation, they are also of great importance in the process of cancer development, as evidenced by findings that mutations in the genes encoding NODs are also associated with increased risk of several cancers.15, 19, 20 Moreover, NODs are also targets of interest in terms of strengthening the immune response against cancer cells. Specifically, NOD agonists possess the ability to stimulate anticancer activity of immune cells, in particular monocytes and macrophages. A NOD2 agonist, mifamurtide, has already been approved in immunotherapy for patients with osteosarcoma, in combination with chemotherapy following complete surgical resection of the primary tumor. Other NOD agonists, as well as NOD antagonists, have been under investigation in preclinical and clinical studies.
The purpose of this review is, therefore, to evaluate NOD receptors as new targets in cancer immunotherapy and to highlight the NOD1 and NOD2 agonists as well as antagonists reported to exhibit anticancer activity.
2. NOD1 AND NOD2 PROTEINS—EXPRESSION, STRUCTURE, AND SIGNALING
2.1. Expression
NOD1 and NOD2 are intracellular proteins encoded by the CARD4 gene found on chromosome 7p14‐15, and the CARD15 gene found on chromosome 16q12.21 Both receptors are located in the cell cytosol and, in certain cells, also at the plasma membrane.22 Their recruitment to the cell membrane has been observed in various epithelial cells and recognized as a crucial event for activation of the NF‐κB signaling pathway following bacterial PGN binding.23, 24 Interestingly, although similar in terms of cell localization, NOD1 and NOD2 are very differently expressed in cells and tissues throughout the body. NOD1 is extensively expressed in a variety of cell types, whereas NOD2 has been found mostly in professional immune cells (macrophages,25 dendritic cells,26 and Paneth cells27), osteoblasts,28 keratinocytes,29 intestinal stem cells,30 and various epithelial cells.31, 32, 33
2.2. Structure
NOD1 and NOD2 are multiple domain proteins consisting of a C‐terminal, leucine‐rich repeat domain (LRR) (also widely accepted as the bona fide sensor domain that is responsible for recognition of ligands), a centrally located nucleotide‐binding oligomerization domain (NACHT) that mediates self‐oligomerization and is crucial for NOD activation, and one (NOD1) or two (NOD2) N‐terminal caspase recruitment domains (CARDs) that interact with downstream signaling molecules.34, 35 Normally, NODs are kept in a monomeric autoinhabitable state in the cell cytosol, being activated following ligand binding.19 In addition to being able to recognize ligands in the cytosol, NODs are capable of trafficking dynamically to the cell membrane and of recognizing bacteria at the point of entry.36
2.3. Signaling
2.3.1. The canonical signaling pathway (NF‐κB and MAPK)
On activation by their native ligands, NODs undergo conformational changes and self‐oligomerization through homophilic CARD‐CARD interactions, allowing the recruitment and activation of the CARD‐containing adaptor receptor‐interacting serine/threonine‐protein kinase 2 (RIPK2). The latter is important for downstream signal transduction.37, 38, 39 In an established protein complex, RIPK2 is later polyubiquitinated by several E3 ubiquitin ligases, namely tumor necrosis factor receptor–associated factors (TRAFs), cellular inhibitor of apoptosis protein (cIAP) 1, cIAP2, and X‐linked inhibitor of apoptosis protein (XIAP).40, 41, 42, 43, 44, 45 Polyubiquitin chains attached to RIPK2 then facilitate the formation and activation of a protein complex consisting of tumor growth factor β‐activated kinase 1 (TAK1) and TAK1‐binding proteins (TAB) 1 to 3.46, 47 TAK1 is an upstream activator of the inhibitory κB kinase (IKK) complex.48 This activation leads to the phosphorylation and degradation of a protein inhibitor of NF‐κB (IκB), resulting in translocation of NF‐κB to the nucleus and transcription of NF‐κB target genes.42, 46, 49 On the other hand, TAK1 also activates three mitogen‐activated protein kinases (MAPKs), namely p38, extracellular signal‐regulated kinase (ERK), and c‐Jun N‐terminal kinase (JNK), resulting in the activation of activator protein 1 (AP‐1) transcription factor.50 Moreover, RIPK2 also interacts with the IKKγ/NEMO subunit of IKK complex, resulting in ubiquitination of IKKγ/NEMO and activation of the IKK complex, which is important for downstream NF‐κB activation (Figure 2).15
Figure 2.
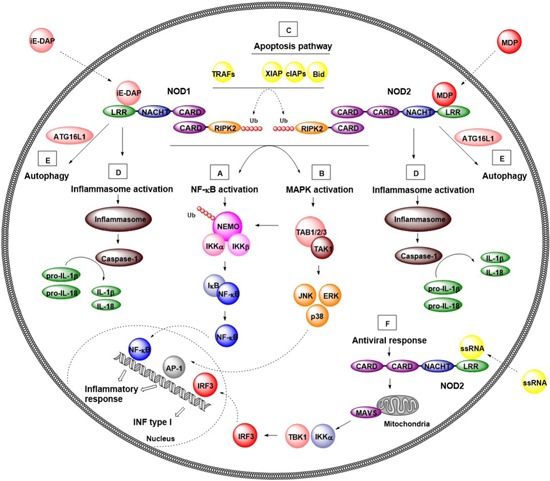
Canonical and noncanonical signaling pathways of NOD1 and NOD2. A, B, NF‐κB and MAPK signaling pathways. NOD1 and NOD2 recognize bacterial PGN fragments, iE‐DAP and MDP, respectively. After ligand recognition, NODs undergo conformational changes and self‐oligomerization through homophilic CARD‐CARD interactions, allowing the recruitment and activation of the adaptor protein RIPK2. In an established protein complex, RIPK2 is polyubquitinated by TRAFs and cIAPs, allowing the recruitment of polyubiquitinated NEMO or TAK1 to the established protein complex. On one hand, NEMO triggers activation of the NF‐κB pathway by phosphorylation of IκB resulting in release NF‐κB transcription factor. The latter translocates to the nucleus where causes induction of pro‐inflammatory genes. On the other hand, TAK1 recruits TAB1/2/3 activating both NF‐κB and MAPK pathway (JNK, ERK, and p38). Activated MAPKs translocate to the nucleus and activate AP‐1 transcription factor resulting transcription of genes involved in inflammatory response. C, Apoptosis pathway. NODs have been reported to interact with the apoptotic pathway indirectly through IAP family of proteins (cIAP1, cIAP2, and XIAP) and proapoptotic protein Bid. D, Inflammasome activation. NODs associate with several other NLRs (NLRP1 and NLRP3) to form inflammasome protein complexes. After detection of their cognate ligand, NLRs interact with ASC protein and procaspase‐1, resulting in formation of inflammasomes. Once formed, inflammasomes activate procaspase‐1, which in turn proteolytically processes pro‐inflammatory cytokines IL‐1β and IL‐18. E, Autophagy. Activated NODs recruit the autophagy protein ATG16L1 to the cell membrane and facilitate the formation of autophagosome around invading bacteria. F, Antiviral response. Viral ssRNA activates NOD2, which translocates to the mitochondria and supposedly binds to MAVS. This promotes formation of a complex with TBK1 and IKKα, which enables activation of interferon regulatory factor (IRF) 3 resulting in production of IFN type I. ASC, apoptosis‐associated speck‐like protein containing caspase recruitment domain; CARD, caspase recruitment domain; cIAP, cellular inhibitor of apoptosis protein; ERK, extracellular signal‐regulated kinase; IAP, inhibitor of the apoptosis; iE‐DAP, d‐glutamyl‐meso‐diaminopimelic acid; IFN, interferon; IKK, IκB kinase; IκB, protein inhibitor of NF‐κB; IL, interleukin; JNK, c‐Jun N‐terminal kinase; NOD, nucleotide‐binding oligomerization domain; NF‐κB, nuclear factor κB; MAPK, mitogen‐associated protein kinase; MAVS, mitochondrial antiviral signaling; MDP, muramyl dipeptide; NEMO, nuclear factor κB essential modulator; NLRs, nucleotide‐binding oligomerization domain‐like receptors; PGN, peptidoglycan; RIPK2, receptor‐interacting serine/threonine‐protein kinase 2; TAB, transforming growth factor binding protein; TAK1, transforming growth factor β‐activated kinase 1; TBK1, TRAF‐associated nuclear factor‐κB activator–binding kinase 1; TRAFs, tumor necrosis factor receptor–associated factors; XIAP, X‐linked inhibitor of apoptosis protein [Color figure can be viewed at wileyonlinelibrary.com]
In addition to NF‐κB and MAPK signaling pathways, NODs are involved in the activation of other innate immunity systems such as autophagy, apoptosis, inflammasome activation, and even antiviral response, as described briefly below.
2.3.2. Noncanonical signaling pathways
Apoptosis pathway
NODs have been reported to interact indirectly with the apoptotic pathway through the inhibitor of the apoptosis (IAP) family of proteins (cIAP1, cIAP2, and XIAP) as with the proapoptotic protein BH3‐interacting domain death agonist (Bid) (Figure 2).40, 43, 51, 52 IAP proteins participate in NOD signaling by polyubiquitinating RIPK2 and consequently stimulating NF‐κB activation and stress kinases activities,40, 43, 51 while Bid has been suggested to bridge the NODs to the IKK complex thereby impacting NF‐κB and ERK activation.52 Furthermore, it has been demonstrated that stimulation of NOD1 activates caspase 8, which has been linked to its underlying antitumor activity.53, 54
Autophagy
Autophagy is a highly conserved degradation process in eukaryotic cells, vitally involved in the normal functioning of the innate immune system.55, 56, 57, 58 It has recently been discovered that NOD‐mediated recognition of bacteria induces autophagy and bacterial clearance.59, 60, 61 NODs have been shown to recruit the autophagy protein ATG16L1 to the cell membrane, to target bacteria at the point of entry, independently of RIPK2 (Figure 2).61 In addition to their role in sensing bacteria, NODs are involved in autophagosome formation. It has been demonstrated that autophagosome formation is induced in epithelial cells, fibroblasts or dendritic cells (DCs) on stimulation by NOD agonists.59, 60, 61 In contrast to the cell membrane targeting function, the induction of autophagy by NOD2 is an RIPK2‐dependent process leading to downstream ERK and p38 activation.60 It should be noted that signaling through RIPK2 deactivates protein phosphatase 2A, which negatively regulates NOD‐dependent autophagy.60
Inflammasome activation
NODs associate with several other NLRs, such as NLRP1 and NLRP3 to form inflammasome protein complexes.62, 63, 64 Inflammasomes are intracellular multiprotein complexes that detect pathogens as well as various sterile stressors including self‐derived DAMPs, alum, asbestos, silica, alloy particles, UV radiation, and skin irritants.10 After detection of PAMPs or DAMPs, NLRs interact with adaptor protein ASC (apoptosis‐associated speck‐like protein containing a CARD) and procaspase‐1, resulting in the formation of multiprotein inflammasome complexes. Once formed, inflammasomes activate procaspase‐1, which in turn proteolytically processes pro‐inflammatory cytokines interleukin (IL)‐1β and IL‐18 (Figure 2).65
Induction of an antiviral response
In addition to the previously described signaling pathways, studies have also provided evidence for PGN‐independent role of NODs.66, 67 Specifically, NOD2 can act as a cytoplasmic viral PRR that activates an antiviral response, resulting in type I interferon (IFN) production. After detection of viral ssRNA, NOD2 translocates to the mitochondria where it supposedly interacts with mitochondrial antiviral signaling (MAVS) protein. This promotes the formation of a complex with serine/threonine‐protein kinases TBK1 and IKKα, enabling activation interferon regulatory factor (IRF) 3 and resulting in the production of IFN‐β (Figure 2).15, 67
3. THE ROLE OF NOD PROTEINS IN CANCER DEVELOPMENT
Although NODs were initially recognized as receptors for pathogen recognition within the scope of the innate immune response, recent findings have further confirmed their involvement in mechanisms underlying cancer development. On the one hand, NOD activation can prevent, inhibit, or block carcinogenesis by controlling epithelial cell regeneration while, on the other hand, it can promote carcinogenesis via the production of pro‐inflammatory cytokines that contribute to chronic inflammation.21, 68 Furthermore, increased cancer risk is also associated with the presence of polymorphisms in genes CARD4 and CARD15.21 These polymorphisms can produce altered NODs with disrupted cytokine‐producing profiles and therefore pose an increased risk, causing inflammation and cancer. Briefly, NOD2 gene polymorphisms have been associated with increased risk of lymphoma, colorectal, gastric, breast, ovarian, lung, and laryngeal cancers while NOD1 gene polymorphisms have been linked to increased risk of lymphoma, gastric, colorectal, ovarian, prostate, and lung cancer, as well as the cancer types whose etiology is related to Crohn's disease and sarcoidosis.21 NODs have been studied to a greater extent in cancers of the gastrointestinal tract, such as colorectal cancer (CRC) and gastric cancer (GC), although studies in breast cancer, oral squamous cell carcinoma (OSCC), head and neck squamous cell carcinoma (HNSCC), and pancreatic cancer (PC) have also been conducted (Table 1). Stimulation of NOD1 and NOD2 was found to be protective in inflammation‐induced CRC,69, 70, 71, 80 whereas there was no straightforward answer as to whether activation of NOD1 in the stomach promotes or prevents the development of GC.72, 73, 74 Moreover, NOD1 was found to be upregulated in PC,79 HNSCC,77, 78 OSCC,76 and GC,73, 74 as opposed to certain studies that reported NOD1 downregulation in the cases of OSCC75 and GC.72 NOD2 was also found to be upregulated in GCs.74
Table 1.
Role of NOD proteins and their expression level in different types of cancer
| Role of NOD proteins and their expression level | ||||
|---|---|---|---|---|
| Cancer | NOD protein | Protective (P)/ detrimental (D) | References | |
| Colorectal | NOD1 | P | NOD1 deficiency alone or together with a mutation in Apc (ApcMin/+) leads to increased risk of tumor formation in the AOM/DSS mouse model of colon cancer. Increased tumor formation is a consequence of increased intestinal epithelial apoptosis as well as intestinal permeability associated with enhanced inflammatory cytokine production and epithelial cell proliferation. | 69 |
| NOD1 | P | NOD1 deficiency in T cells increases risk of tumor formation in mice using AOM/DSS model of colon cancer. NOD1 deficiency in T cells is associated with impaired IFN‐γ production and STAT1 activation. | 70 | |
| NOD2 | P | NOD2 (or RIPK2) deficiency results in increased susceptibility to tumor formation in AOM/DSS mouse model of colon cancer. Absence of NOD2 (or RIPK2) promotes pro‐inflammatory microenvironment in the intestines leading to enhanced epithelial dysplasia following chemically induced injury. | 71 | |
| Gastric | NOD1 | P | Decreased expression level of NOD1 in Helicobacter pylori‐positive GC patients. Stimulation of NOD1 by C12‐iE‐DAP before infection with Helicobacter pylori reduced risk of GC development in gerbils. | 72 |
| NOD1/NOD2 | D | Increased expression level of NOD1 in Helicobacter pylori‐positive and Helicobacter pylori‐negative GC patients. Increased expression level of NOD2 in Helicobacter pylori‐positive GC patients. | 73, 74 | |
| Breast | NOD1 | P | NOD1 deficiency leads to increased tumor growth in mouse model of breast cancer. Stimulation of NOD1 overexpressed breast cancer cells results in caspase 8–mediated apoptosis. | 53, 54 |
| Oral squamous cell carcinoma (OSCC) | NOD1 | P | Decreased expression level of NOD1 in OSCC patients. NOD1 expression decreases along with OSCC progression. | 75 |
| NOD1/NOD2 | D | NOD1 and NOD2 are apparently expressed in YD‐10B and FaDu cell line. Stimulation of NOD1 and NOD2 in YD‐10B cells by Tri‐DAP and MDP, respectively, results in production of IL‐8 and MAPK activation. Stimulation of YD‐10B cells by MDP results in inhibition of the proliferation and induction of apoptosis. | 76 | |
| Head and neck squamous cell carcinoma (HNSCC) | NOD1 | D | Increased expression level of NOD1 in tumor biopsies, Detroit‐562 and FaDu cell line. Stimulation of NOD1 in HNSCC cells by iE‐DAP increases the production of β‐defensin 2, GM‐CSF, G‐CSF, and upregulates ICAM‐1. NOD1 activation by iE‐DAP increases the apoptosis and decreases the number of dead Detroit‐562 cells. | 77 |
| NOD1 | D | Increased expression level of NOD1 (as well as IL‐8 and RIPK2) in tumor biopsies. IL‐8 is a key factor in NOD1‐mediated RIPK2 activation and HNSCC progression. | 78 | |
| Pancreatic | NOD1 | D | Increased expression level of NOD1 in peripheral blood leukocytes of pancreatic cancer. | 79 |
Abbreviations: AOM, azoxymethane; CSF, colony‐stimulating factor; DSS, dextran sulfate sodium; GC, gastric cancer; ICAM‐1, intercellular adhesion molecule‐1; iE‐DAP, d‐glutamyl‐meso‐diaminopimelic acid; IFN, interferon; IL, interleukin; MDP, muramyl dipeptide; NOD, nucleotide‐binding oligomerization domain; RIPK2, receptor‐interacting serine/threonine‐protein kinase 2; STAT1, signal transducer and activator of transcription 1; Tri‐DAP, l‐alanyl‐γ‐d‐glutamyl‐meso‐DAP.
4. TARGETING NOD RECEPTORS IN CANCER IMMUNOTHERAPY
Extensive research has shown that cancer is not just a group of malignant cells but a complex structure within a tumor microenvironment (TME).81, 82, 83 Besides malignant cells, TMEs comprise a variety of immune and nonimmune cell types that, in concert with the many other factors that they secrete, create an effective environment that favors tumor growth and metastatic dissemination.8, 81 Infiltration of TME by immune cells such as macrophages, lymphocytes, natural killer (NK) cells, and DCs in the early stages of tumor development is crucial for an appropriate anticancer immune response. Unfortunately, the beneficial effect produced by these cells is often inhibited by the action of immunosuppressive cells, including regulatory T cells, type 2 (M2) macrophages, and myeloid‐derived suppressor cells (MDSCs) that also infiltrate the TME of developing tumors.83 In such an immunosuppressive environment, cancer cells are able to adapt and remain undetected by host immunosurveillance.83 The overarching goal of cancer immunotherapy is to overcome the immunosuppression in TME, thereby enabling immune cells to effectively eliminate cancer cells without causing intolerable side‐effects.84 To achieve this, various strategies have been used, among which targeting of PRR, including NOD1 and NOD2, constitutes an interesting and novel approach that could be used as an adjunct to current cancer therapies.8 In terms of anticancer activity, NOD agonists can act as (i) immunotherapeutics or (ii) adjuvants in cancer vaccines whereas NOD antagonists have recently proposed to mediate their antitumor activity by preventing the formation of an inflammatory TME.
4.1. NOD agonists as immunotherapeutic agents
When NOD agonists act as immunotherapeutics, they activate the cytotoxic potential of immune cells residing in the TME and, consequently, facilitate their engagement with cancer cells. Such enhancement of anticancer immunity has been investigated in the context of NOD2 agonists which, mainly, activate monocytes and macrophages85 although stimulation of NK cells86, 87 and DCs88, 89, 90 has also been reported. In general, two possible mechanisms on how NOD agonists activate the antitumor activity of macrophages have been suggested. They can either induce tumoricidal macrophages that, in turn, attack cancer cells or stimulate macrophages to mediate anticancer activity indirectly by the release of pro‐inflammatory molecules and other factors. Moreover, macrophages also collaborate with Th1 cells to effectively recognize and eliminate malignant cells. Specifically, type 1 (M1) macrophages and Th1 cells reinforce one another, with M1‐produced IL‐12 maintaining the Th1 phenotype and Th1‐produced IFN‐γ maintaining the M1 phenotype.91 In this Th1‐driven environment, pro‐inflammatory cytokines such as tumor necrosis factor (TNF)‐α, IL‐1, and IL‐6 are also involved in cancer elimination by stimulating various aspects of antitumor immunity, including recruitment of macrophages and T cells from the circulation and stimulation of leukocyte tumoricidal functions.91
4.2. NOD agonists as adjuvants for cancer vaccines
NOD agonists have a potential for use as adjuvants in cancer vaccines and therefore enhance innate as well as adaptive immune responses toward coadministered antigens with insufficient immunostimulatory capabilities.84, 92, 93, 94 In fact, NOD ligands have been involved in vaccination strategies throughout the 20th century, being essential components of Freund's Complete Adjuvant (FCA), one of the most potent and widely used adjuvants in animals. Among them, DAP‐containing peptides have demonstrated favorable adjuvant activity, whereas MDP was recognized to be the minimal structure required for the adjuvanticity of FCA.95, 96, 97 MDP and other muropeptides exert their immune‐enhancing effects through several mechanisms. For example, they increase expression of cell surface markers, which are involved in cell adhesion and presentation of antigens, thereby stimulating phagocytic and antimicrobial activity as well as increasing antibody‐mediated cytotoxicity.98 Moreover, MDP is reported to increase immune responses of other immunomodulatory molecules such as IFN‐γ and to synergize with several cytokines, thereby stimulating differentiation and proliferation of lymphocytes.99, 100
5. NOD LIGANDS AS ANTICANCER AGENTS
5.1. NOD1 agonists
NOD1 senses different DAP‐containing ligands that originate from bacterial PGN. Among them iE‐DAP (1; Figure 3) is recognized as the minimal component sufficient for NOD1 activation. Elongation by an additional alanine resulted in Tri‐DAP (2), which turned out to be an even more potent NOD1 agonist.11, 34, 101 In addition to these DAP‐containing NOD1 agonists released from bacteria, various iE‐DAP analogs with immunostimulatory activities have been designed and synthesized, mostly by introducing lipophilic moieties to the d‐glutamyl (d‐Glu) portion of the iE‐DAP molecule.102 For example, Jakopin et al103 synthesized several iE‐DAP analogs with lauroyl and didodecyl moieties attached to the amino group of the d‐Glu residue. These NOD1 agonists alone (10 µM) or in synergy with lipopolysaccharide (LPS) (1 ng/mL) exhibited significant immunostimulatory effects in human peripheral blood mononuclear cells (PBMCs) resulting in increased cytokine production (TNF‐α, IL‐6, IL‐8, and IL‐10).
Figure 3.
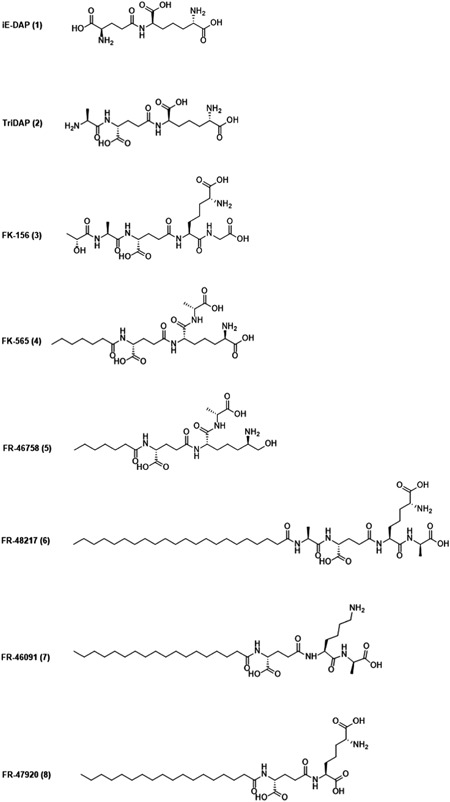
Chemical structures of iE‐DAP, Tri‐DAP, and NOD1 agonists with anticancer activity. iE‐DAP, d‐glutamyl‐meso‐diaminopimelic acid; NOD, nucleotide‐binding oligomerization domain; Tri‐DAP, l‐alanyl‐γ‐d‐glutamyl‐meso‐DAP
In addition to immunostimulation, there is evidence suggesting that NOD1 agonists also possess anticancer activity.104, 105, 106 In the 1980s, researchers from Fujisawa Pharmaceutical Co synthesized a series of structurally related meso‐DAP incorporating analogs that exhibited antitumor activity in vivo.104 For the first screening of compounds, two intratumoral injections of meso‐DAP analog (10‐100 µg/site) were administered in DBA/2 mice with established P388 solid tumors. Among 21 synthetic analogs tested, only six compounds, namely FK‐156 (3), FK‐565 (4), FR‐46758 (5), FR‐48217 (6), FR‐46091 (7), and FR‐47920 (8) showed promising tumor growth inhibition (20%‐50%). Moreover, significant tumor growth inhibition (20%‐40%) was also observed when P388 tumor‐bearing mice received two subcutaneous injections of 3, 4, 5, and 6 (6 mg/kg) or multiple systemic injections of 4 (200 µg/kg). In contrast to the substantial antitumor activity of 3, 4, and 5 observed in vivo, these three compounds at 1 mg/mL concentration demonstrated no toxicity against P388 cells in vitro suggesting that these compounds boost the antitumor activity of immune effector cells such as monocytes, macrophages, or NK cells.104 In subsequent studies, 4 was indeed shown to stimulate NK cells and induced tumoricidal activities of murine macrophages.105, 106 Remarkably, small amounts of 4 (more than 0.1 µg/kg) stimulated NK cell activity and inhibited experimental lung metastasis formation when administered prophylactically 2 or 3 days before inoculation of B16 melanoma cells.106 Conversely, in the study of Inamura et al106 doses as high as 100 µg/kg of 4 were not effective in inhibiting the formation of lung metastasis administered 3 days after B16 tumor cell inoculation. Since NK cells were not effective in controlling pulmonary metastasis when administered 3 days after B16 tumor cell inoculation, they probably destroy only circulating but not extravascular, metastatic cells. Interestingly, repeated intravenous or subcutaneous injections of 4 given at high doses of 1 to 10 mg/kg after B16 tumor cell inoculation significant reduced the number of pulmonary metastases in an established experimental lung metastasis model.106 In our opinion, the difference between the efficacy of prophylactic and therapeutic treatment is probably due to the different mechanisms responsible for the antimetastatic activity. Namely, it is likely that antimetastatic effect observed in the case of therapeutic treatment involves macrophage activation, and not NK cell activation since repeated intraperitoneal injections of 4 at high doses (more than 10 mg/kg) significantly increased the cytotoxicity of murine peritoneal macrophages. Similarly, Schultz et al105 also confirmed the role of macrophages as the primary effectors of 4‐induced antimetastatic activity. Specifically, reduced M109 lung metastasis formation was observed when high doses of 4 (1‐10 mg/kg) were administered prophylactically 2 to 4 days before the inoculation of M109 tumor cells in an experimental metastasis model. This protective activity was, however, abolished by the selective macrophage inhibitor 2‐chloroadenosine, thus further corroborating the notion that macrophage activation is underlying the antimetastatic activity of 4 in this experimental model.105 In spite of the fact that activation of macrophages was proposed to be the primary mechanism of 4‐mediated prophylactic activity against M109 metastasis, further studies are needed to determine if other effector mechanisms, such as NK cell activation, are also involved in the observed antimetastatic activity. Although the exact mechanism of action has not been fully characterized, NOD1 agonists turned out to be effective immunomodulators with promising antimetastatic activity when used as prophylactic treatment. Among all described compounds, 4 efficiently inhibited th growth of lung metastases in vivo while exhibiting minimal toxicity in animal studies, and thus emerged as a prospective immunotherapeutic agent. In fact, compound 4 indeed entered phase I clinical trial for use in cancer therapy but unfortunately, this study was discontinued. Nevertheless, from our perspective, the field of NOD1 agonists still holds a lot of potential. For example, several recently synthesized potent NOD1 agonists102, 103 have yet to be examined for their anticancer activity.
5.2. NOD2 agonists
MDP (9) is the minimal structural component of PGN that activates NOD2. Structurally speaking, MDP is a small molecule composed of an N‐acetylmuramic acid linked to a dipeptide consisting of l‐alanine (l‐Ala) and d‐isoglutamine (d‐isoGln) (Figure 4). Due to its low molecular weight, MDP is highly water soluble and rapidly excreted from the body,107 resulting in weaker in vivo activity.108 To overcome this issue and obtain NOD2 agonists with improved immunostimulatory and anticancer activities, the parent structure of MDP has been (i) incorporated into different nanocarrier delivery systems such as liposomes and nanocapsules or (ii) equipped with various classes of compounds including lipophilic molecules, biomolecules, drugs, and many others. To date, several hundred MDP analogs have been synthesized (reviewed in109, 110). According to the type of modifications in MDP, NOD2 agonists possessing anticancer activities can be classified into four groups namely, lipophilic derivatives, hydrophilic derivatives, conjugates, and desmuramylpeptides.
Figure 4.
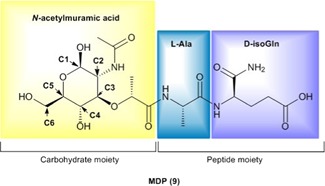
Chemical structure of muramyl dipeptide (MDP). MDP molecule is composed of a carbohydrate moiety represented by N‐acetylmuramic acid, and peptide part consisting of l‐Ala and d‐isoGln. Positions of C atoms at the carbohydrate moiety, which are important for further modifications are marked with arrows [Color figure can be viewed at wileyonlinelibrary.com]
5.2.1. Lipophilic MDP derivatives
As noted previously, lipophilic derivatives with improved immunostimulatory and anticancer activities have been designed to overcome their rapid elimination and consequently weaker stimulation of immune cells with MDP in vivo. To achieve this goal, lipophilic moieties were incorporated into MDP, either at the muramyl moiety at positions C1, C6, or at the C‐terminus of the peptide part. Encapsulation of lipophilic MDP derivatives into liposomes showed even better results in terms of effective immune stimulation since these derivatives are more readily incorporated into and retained within liposomes than MDP, thus leading to increased efficacy. The identification of the optimal liposome‐encapsulated formulations is based on numerous preclinical and animal studies and ensures effective drug delivery into different parts of the body. Such liposome formulations allow for enhanced uptake by monocytes and macrophages, the primary targets of NOD2 agonists via phagocytosis, in turn, they are concentrated in the lysosomal compartment where they are degraded, thus finally releasing lipophilic MDP derivatives.
The group of lipophilic MDP derivatives comprises a variety of molecules such as 6‐O‐acyl derivatives of MDP with mycolic, hydroxyl fatty and quinonylalkanoic acids as well as stearoyl, glycoside, glycerol dipalmitate (GDP), and phosphatidylethanolamine derivatives of MDP. Mifamurtide is certainly the most important representative of this group. Due to its favorable pharmacokinetics, pharmacodynamics, and clinical efficacy, it has already been granted a marketing authorization in Europe for use in combination with chemotherapy in patients with high‐grade osteosarcoma after complete surgical removal of the primary tumor.
Mifamurtide
Muramyl tripeptide phosphatidylethanolamine or MTP‐PE (10; Figure 5) is a fully synthetic lipophilic derivative of MDP that has monocyte‐ and macrophage‐activating properties similar to those of the parent compound, with additional improvements in terms of longer half‐life in plasma and lower toxicity.108 In mifamurtide, 10 is encapsulated into multilamellar liposomes by combining the active substance with phospholipids at a ratio 1:250. This formulation facilitated the delivery of 10 to monocytes and macrophages, especially those in the liver, lungs, and spleen. Following phagocytosis by monocytes and macrophages, liposomes incorporating 10 are degraded and release 10, resulting in activated monocytes and macrophages.111 It has been proposed that the anticancer activity of 10 is associated with its ability to induce tumoricidal monocytes and macrophages that attack cancer cells directly, as well as with the release of pro‐inflammatory molecules such as TNF‐α, IL‐1, IL‐6, IL‐8, and IL‐12.112, 113, 114, 115 In vitro studies showed that human monocytes activated by mifamurtide selectively recognized and killed tumor cells, while no cytotoxic effect toward normal cells was demonstrated.113, 116, 117, 118, 119 This selective cytotoxicity was observed even under cocultivation conditions of the tumor and normal cells.113 Moreover, mifamurtide also synergized with IFN‐γ to increase the tumoricidal activity of human monocytes.117, 118, 120 For example, Sone et al120 demonstrated that monocytes derived from healthy donors exhibited cytotoxicity against human A375 melanoma cells when incubated in vitro with mifamurtide (500 nM) or IFN‐γ (100 U/mL). Interestingly, the cytotoxic effect against A375 tumor cells was significantly enhanced when monocytes were treated with low doses of mifamurtide (50 nM) and IFN‐γ (10 U/mL), indicating that mifamurtide and IFN‐γ–activated monocytes in a synergistic manner. From our point of view, synergistic actions of NOD2 agonists with cytokines are of particular importance, given that the doses used can be drastically reduced resulting in no or fewer undesirable side effects. Furthermore, studies in dogs revealed a beneficial role of mifamurtide in the treatment of spontaneous osteosarcoma, which has many similarities with osteosarcoma in humans. In both, dogs and humans, osteosarcoma arises from long bones and has the same pattern of metastasis, with more than 80% of metastases occurring in the lungs. Specifically, mifamurtide (2 mg/m2, dosed twice weekly for 8 weeks) significantly improved overall survival as compared with placebo in dogs with spontaneous osteosarcoma and splenic hemangiosarcoma when used as part of adjuvant therapy after resection of primary tumor.121, 122, 123, 124 In contrast, mifamurtide was not effective in mice with high tumor burden, or in cats and dogs with mammary metastatic tumors, which suggests that the impact of macrophage activation on controlling tumor growth depends on tumor burden and tumor location.125, 126, 127 Moreover, mifamurtide also demonstrated no strong interactions in terms of enhancing macrophage activation with chemotherapeutics such as doxorubicin (DOX), cisplatin (CDDP), methotrexate (MTX), and ifosfamide (IFO), which are usually used concomitantly with mifamurtide in therapy of osteosarcoma.128, 129, 130 Briefly, Kleinerman et al130 assessed the tumoricidal activity of blood monocytes isolated from osteosarcoma patients receiving CDDP, high‐dose MTX, cyclophosphamide (CTX), or DOX following in vitro activation with mifamurtide (liposomes containing 100 nmol 10) and observed no difference when compared to monocytes isolated from normal donors. Of note, 10 was also studied as an adjuvant in cancer vaccines.131 The study of Bergers et al131 demonstrated that 10 in combination with tumor antigens effectively induced specific protective antitumor immunity against SL2 lymphosarcoma cells. The studied mice received two subcutaneous immunizations (at a 10‐day interval) using liposomal formulations containing tumor antigens (solubilized from crude membranes of SL2 cells) and different immunomodulators (10, lipid A, dimethyl dioctadecyl ethanolamine). Ten days after the second immunization mice were challenged with live SL2 cells. Mice immunized with liposomes containing tumor antigens and 10 (20 µg/dose) provided better protection against the challenge with SL2 cells in comparison with alternative immunomodulators. On the other hand, mice immunized with tumor antigens derived from unrelated P825 tumor and 10 were not able to reject a challenge with SL2 cells evidently highlighting the need to develop specific antitumor immunity. The obtained results also revealed that mouse peritoneal macrophages isolated from immunized mice 5 to 7 days after tumor challenge demonstrated high nonspecific cytotoxicity in vitro (macrophages destroyed the SL2 as well as the nonrelated P815 cells), and that no major cytotoxic lymphocyte activity or substantial cytotoxic antibody titers were detectable. These results indicate that although tumor cells can be destroyed by nonspecific macrophage cytotoxicity, T cells should be involved at least in the induction of tumor immunity due to the specificity in the tumor rejection. Although exact mechanism underlying inducing antitumor immunity due to the specificity in the tumor rejection by liposomal formulations containing tumor antigens and 10 has not been fully characterized, 10 clearly showed the potential to be used as an adjuvant in cancer vaccines.
Figure 5.
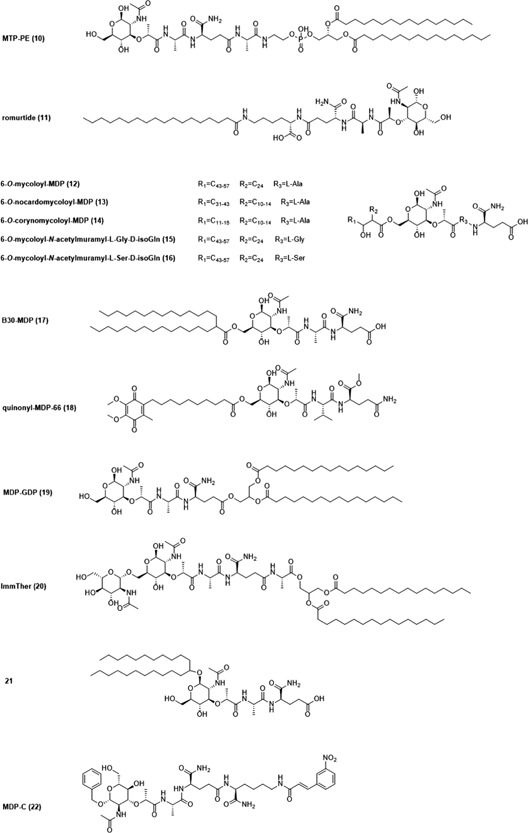
Lipophilic muramyl dipeptide (MDP) derivatives with anticancer activity
The clinical efficacy of mifamurtide has been evaluated in a large number of phase I and II trials.132 Additionally, one large, randomized, prospective, open‐label, multicenter phase III trial (Intergroup Study 0133) has been conducted evaluating the addition of mifamurtide to a three‐drug combination (DOX, CDDP, and high‐dose MTX) and to a four‐drug combination (DOX, CDDP, high‐dose MTX, and IFO) chemotherapy for the treatment of osteosarcoma.132, 133 Briefly, after the resection of the primary tumor, three‐ or four‐drug chemotherapy was given to patients to complete the full course of therapy. Mifamurtide treatment started simultaneously with chemotherapy following surgery. Mifamurtide was administered intravenously at doses 2 mg/m2 twice weekly for 12 weeks, and then twice weekly for an additional 24 weeks (altogether 48 doses in 36 weeks). Importantly, the results demonstrated that addition of mifamurtide was associated with a statistically significant improvement in 6‐year overall survival (but not event‐free survival) in patients with newly diagnosed, high‐grade, nonmetastatic, resectable osteosarcoma in comparison to the patients who did not receive mifamurtide treatment (P = 0.03).132, 133 Currently, mifamurtide holds an orphan drug status in the United States and is marketed in Europe for the treatment, in combination with other chemotherapeutics, of high‐grade, nonmetastatic, resectable osteosarcoma in children, adolescents, and young adults (aged between 2 and 30 years), following complete surgical removal. The recommended regimen of mifamurtide is 2 mg/m2 intravenously administered over 1 hour twice weekly (at least 3 days apart) for an initial 12 weeks, followed by 2 mg/m2 once weekly for additional 24 weeks (it amounts to 48 doses in 36 weeks). In general, mifamurtide therapy is safe and well tolerated. The major adverse events are fever and chills, which are usually transient and associated with initial administration. Most patients rapidly develop tolerance leading to no adverse events with subsequent administration.115, 132 Although the exact mechanism by which mifamurtide improves overall survival in patients has not been fully elucidated, it may eliminate micrometastasis or tumor cells after surgery that are not removed by or are resistant to, chemotherapy.115, 132 In our opinion, it may be possible that mifamurtide could exhibit similar beneficial effects in the therapy of other cancers, especially those which predominantly metastasize to the lungs. Mifamurtide should be further examined to ascertain and potentially harness its potential in the treatment of other cancers.
Romurtide
Romurtide (11), also known under names MDP‐Lys(L18) and muroctasin, is a synthetic stearoyl‐MDP derivative and an effective immunostimulant in vitro and in vivo.134, 135 When injected subcutaneously for 10 consecutive days into healthy cynomolgus monkeys 11 (1 mg/dose) significantly increased the number of peripheral neutrophils, monocytes, and platelets. This effect may be the consequence of the ability of 11 to augment the production of several cytokines by the monocytes including colony‐stimulating factors (CSFs), IL‐1, and IL‐6, which have central roles in the regulation of hematopoiesis.136 Similar results, in terms of increased hematopoiesis, were also obtained in immunosuppressed mice in which multiple injections of 11 (100 µg/dose) effectively restored the white blood cell count, mainly due to an increase in neutrophil counts.137 In both studies (healthy cynomolgus monkeys and mice), the increase in white blood count may be attributable to the augmenting effect of 11 on the production of CSFs, followed by the proliferation and differentiation of stem cells in bone marrow.136, 137 Due to the success in animal studies, 11 was entered into clinical trials in which it demonstrated a restorative effect on leukopenia in cancer patients.138, 139 In 1991, 11 (trade name Nopia) was put in the market in Japan for treating cancer patients with leukopenia induced by chemotherapy or radiotherapy.134, 135 Besides its important role in stimulating hematopoiesis and immune functions, 11 also elicits antitumor immunity against tumors and metastases in vivo.88, 140, 141 Yoo et al141 investigated the antimetastatic effect of 11 in three highly metastatic cancers in mice, namely B16‐BL6 melanoma, colon 26‐M3.1 carcinoma, and L5178Y‐ML25 T lymphoma. A single subcutaneous injection of 11 (100 µg) given 2 or 4 days before tumor cell inoculation caused a significant reduction of lung metastasis of B16‐BL6 melanoma (60%) and colon 26‐M3.1 carcinoma (25%‐40%) as well as liver metastasis of L5178Y‐ML25 T lymphoma (65%‐70%). Although similar treatment was not effective when 11 was administered 1 or 3 days after tumor cell inoculation, five doses of 11 (100 µg/dose) into B16‐BL6 bearing mice after tumor cell inoculation again achieved a significant reduction of experimental and spontaneous lung metastasis. Given the fact in vitro studies demonstrated that 11 increased tumoricidal activity of mouse peritoneal macrophages against B16‐BL6 and that serum of mice treated with 11 inhibited growth of L929 cell line (TNF‐α sensitive cell line) it was suggested that the antimetastatic activity of 11 was associated with enhanced nonspecific immune responses of hosts, including activation of macrophages and induction of cytotoxic factors such as TNF‐α.141 Activation of macrophages by 11 was also proposed as the underlying mechanism of antimetastatic activity in the study of Nitta et al,140 in which free and liposome‐encapsulated 11 inhibited, lung metastasis of transplantable osteosarcoma in hamsters when the 11 was given before or after surgical removal of the primary tumor. Specifically, treatment with free 11 given at a dose 50 µg daily or liposomal 11 given at a dose 20 µg twice a week, which started 3 weeks after tumor transplantation and immediately after primary tumor removal and lasted 4 weeks, resulted in inhibition of lung metastasis for 45% and 40%, respectively, when compared to untreated controls. Despite the fact that both free and liposome‐encapsulated 11 effectively eliminated lung metastases, liposome‐encapsulated 11 exhibited a far greater inhibitory effect than free 11 (40 vs. 350 µg/week), apparently due to the longer retention of the liposomal form in the lung.140 In contrast to the beneficial activity of 11 in terms of suppressing lung metastasis, it exhibited no significant effect on primary tumor mass. The precise reason for that is not clear, but it may be linked to the accelerated growth of transplanted osteosarcoma with time, thus surpassing the tumoricidal activity of activated macrophages.140 Furthermore, the antitumor effect of 11 was also studied in combination with IFN‐β, which has already shown promising results in the treatment of malignant melanoma.88 An in vitro study demonstrated that 11, in combination with IFN‐β, synergistically augmented activation of human monocyte‐derived DCs (MoDCs), resulting in the production of the pro‐inflammatory cytokines TNF‐α, IL‐6, and IL‐12. Moreover, both T cells cocultured with 11 and IFN‐β–treated MoDCs produced significant levels of IFN‐γ, the pivotal cytokine involved in the Th1 response against malignancy. Due to the promising results obtained in vitro, the 11/IFN‐β combination was further studied in murine models of melanoma. It was demonstrated that five intradermal injections of 11 (100 ng‐1 µg) and IFN‐β (10,000 U), given 8 to 10 days after B16‐F10 melanoma tumor cell injection significantly augmented the antitumor effect of IFN‐β in a dose‐dependent manner.88 It should be noted that doses as 11 as low as 100 ng were already sufficient for a statistically significant increase of IFN‐β–mediated antitumor activity against mouse melanoma. However, further investigations are needed to determine whether the therapy with a combination of 11 and IFN‐β could be beneficial for melanoma patients.
6‐O‐acyl‐MDP derivatives
Efforts to develop new potent antitumor agents by modifying the MDP molecule led to the discovery of 6‐O‐acyl derivatives of MDP in which the muramyl moiety was conjugated at the C6 position with various lipophilic molecules. In particular, conjugations of MDP with mycolic, hydroxyl fatty, and quinonylalkanoic acids showed some promising results. Azuma et al142 carried out extensive studies using 6‐O‐acyl derivatives of MDP in which the latter was coupled with natural mycolic acid isolated from bacterial cell walls.143 In the context of antitumor activity, administration of 100 µg 6‐O‐mycoloyl‐MDP (12), 6‐O‐nocardomycoloyl‐MDP (13), 6‐O‐corynomycoloyl‐MDP (14), or 6‐O‐mycoloyl‐N‐acetylmuramyl‐l‐Gly‐d‐isoGln (15) in an oil‐based vehicle suppressed growth of fibrosarcoma in mice while 6‐O‐mycoloyl‐N‐acetylmuramyl‐l‐Ser‐d‐isoGln (16) was active in terms of regression of an established line 10 hepatoma in guinea pigs.142, 143 To avoid the ambiguities concerning the question whether a certain structural variation or the heterogeneity in the natural mycolic acid is required for the antitumor activity, new derivatives were designed, in which natural mycolic acid was substituted by pure synthetic fatty acids of high molecular weight.144 Among them, compound B30‐MDP (17), a mycoloyl‐mimic long‐chain fatty acid derivative of MDP, was recognized as a strong adjuvant capable of inducing antitumor immunity. Kataoka et al145, 146 used a tumor vaccine composed of X‐ray–irradiated line 10 hepatoma cells or acute B cell leukemia cells EN‐L2C in combination with small amounts of 17 (5 µg/dose) to protect guinea pigs against hepatocarcinoma or acute B cell leukemia. Moreover, a similar tumor vaccine formulation also enhanced the activity of cytotoxic killer T cells to inhibit liver metastases of L5178Y‐ML25 lymphoma cells in mice when X‐ray–irradiated L5178Y‐ML25 lymphoma cells and 17 (100 µg/dose) were injected before or after tumor inoculation.147 Since MDP derivatives coupled with mycolic and synthetic long chain fatty acids showed promising antitumor activity, the MDP molecule was further modified by introducing the quinonyl 10‐(2,3‐dimethoxy‐5‐methyl‐1,4‐benzoquinon‐6‐yl)decanoic acid, a highly lipophilic analog of ubiquinones, which resulted in the identification of quinonyl‐MDP‐66 (18).148 In vivo studies demonstrated that a single intradermal injection of 18 (100 µg) administered in a PBS suspension effectively suppressed the growth of Meth A fibrosarcoma in mice.148, 149, 150 In addition, compound 18 was also tested in an oil‐based vehicle following the discovery that oil vehicles including squalene and squalane were required to engage the antitumor activity of some mycobacterial cell‐wall extracts in vivo.151 Experiments in strain‐2 guinea pigs with established line‐10 hepatocarcinoma revealed that 4 intratumoral injections of 18 (0.1‐0.4 mg) incorporated into squalene (or squalane) vehicle oil caused a complete tumor regression resulting in 7 tumor‐free animals out of 7. In contrast, multiple injections of 18 (0.4 mg) in PBS or treatment with squalene (or squalene) vehicle oil alone were less effective in terms of tumor regression resulting in only 2 or even 0 to 1 tumor‐free animals out of 7, respectively.151 It is worth noting that despite squalene (or squalane) vehicle oil alone showed no significant antitumor activity in that tumor model, several squalene‐based oil‐in‐water emulsion such as FM59 have been recognized as efficient adjuvants and are already used in various vaccines.84 Moreover, two injections of 18 (400 µg/dose) also restored the depressed allogeneic cell‐mediated cytotoxicity of spleen cells in 3LL‐bearing mice when administered intraperitoneally, intravenously, or intratumorally.152
MDP‐GDP
Conjugation of MDP to GDP led to the formation of lipophilic MDP‐GDP (19) which was recognized as a very potent immunostimulant, especially when incorporated into liposomes. Specifically, liposomal 19 was able to induce the cytotoxic activity of macrophages in in vitro and in situ studies, as well as antimetastatic activity in vivo.153, 154, 155, 156, 157, 158 Phillips et al153 incorporated 19 into liposomes composed of distearoylphosphatidylcholine (DSPC) and phosphatidylserine (PS) (7:3 molar ratio). They discovered that liposomal 19 was far more efficient in inducing alveolar macrophage cytotoxicity than liposomal MDP or free MDP (10‐ and 7000‐fold, respectively). Moreover, these 19‐containing liposomes efficiently accumulated in the lungs of normal mice and activated murine alveolar macrophages to become cytotoxic against B16‐BL6 tumor cells in vitro. Based on these findings, liposomal 19 was further studied in mice with lung metastases of B16‐BL6 melanoma. Five intravenous injections of liposomal 19 (10 µg) were given to tumor‐bearing mice resulting in significant reduction of lung metastases whereas treatment of tumor‐bearing mice with control liposomes or free MDP (10 µg) had no effect. The similar antimetastatic effect was observed when B16‐BL6 tumor‐bearing mice were treated with liposomes containing 19 or two other MDP‐GDP derivatives, namely GDP derivative of murabutide (hydrophilic, apyrogenic MDP derivative with immunoadjuvant and antitumor activity) and MDP(d,d) (MDP derivative completely devoid of immunoadjuvant activity).154 Mice with lung metastases received five intravenous injections of liposomal 19, murabutide‐GDP, or MDP(d,d)‐GDP resulting in 53%, 36%, and 71% fewer number of lung metastases than control mice, respectively. The order of the ability of liposomal MDP‐GDP derivatives to enhance macrophage activation in vitro and in situ was in fact in good agreement with that observed for the antimetastatic effect. This is particularly interesting since MDP(d,d) itself is completely devoid of immunoadjuvant activity whereas its conjugation with GDP at the terminal amino acid led to a very potent inducer of macrophages and antimetastatic activity. It has been proposed that the difference in the activity is a consequence of its structure. Also MDP(d,d)‐GDP is more resistant to lysosomal enzyme hydrolysis resulting in long‐lived depots within macrophages. In addition to enhancing the cytotoxic potential of alveolar macrophages, 19 incorporated into liposomes also showed promising results in stimulating the tumoricidal activity of macrophages in liver.155, 156, 157, 158 To achieve optimal delivery to the liver, 19 was incorporated into liposomes composed of (i) DSPC and PS (7:0.3 molar ratio)153, 155, 157, 158 or (ii) DSPC and dimyristoylphosphatidylglycerol (DMPG) (10:1 molar ratio).156 Philips et al158 demonstrated that liposomal 19 was 16‐fold more effective than liposomal MDP or 2400‐fold more effective than free MDP in inducing Kupffer cell cytotoxic activity in vitro. Moreover, Kupffer cells were also activated after administration of a single intravenous injection of liposomal 19 (0.1‐1 µg/dose) into healthy mice.158 More importantly, prophylactic or therapeutic treatment with liposomal 19 (0.1‐1 µg/dose), but not free MDP or control liposomes, resulted in a significant reduction in the number of metastasis in B16‐F10 melanoma–bearing mice. Specifically, prophylactic or therapeutic treatment led to a 70% to 90% reduction of liver tumor burden.155 Similar results were obtained by Brodt et al157 who demonstrated that multiple intravenous injections of liposomal 19 (2 µg/dose) administered either as a therapeutic or prophylactic/therapeutic treatment was equally effective in diminishing the number of hepatic metastases in H‐59 lung carcinoma‐bearing mice. On the contrary, liposomal 19 had no therapeutic activity when mice were inoculated with high tumor cell number indicating that immunotherapy with MDP analogs might only be effective when tumor burden is not too extensive. In another study, it was demonstrated that single intravenous injection of 19 (1 µg) incorporated into liposomes as well as free MDP (100 µg) significantly decreased the growth of hepatic metastases in M5076 reticulum cell sarcoma–bearing mice when used as prophylactic treatment, whereas the therapeutic treatment failed to inhibit the metastatic growth.156 The authors speculated that the lack of therapeutic activity was a result of defective Kupffer cell phagocytotic and/or migratory functions.
ImmTher
N‐acetylglucosaminyl‐N‐acetylmuramyl‐l‐Ala‐d‐isoGlu‐l‐Ala‐GDP also known as DTP‐GDP (20) is a lipophilic liposome‐encapsulated disaccharide tripeptide derivative of MDP (known under the brand name ImmTher) and a potent inducer of monocyte‐mediated cytotoxicity in vitro and in vivo.159, 160, 161 For example, in vitro study of Worth et al159 showed that 20 activated human monocytes to produce inflammatory cytokines (TNF‐α, IL‐1, IL‐6, IL‐8, IL‐12, macrophage chemotactic, and activating factor) and inhibit the growth of several human cell lines including Ewing's sarcoma (RD‐ES, SK‐ES‐1, and A4573‐EWS), osteosarcoma (SAOS‐2, MG‐63, and TE‐85), and melanoma (A375). The exact mechanism by which 20‐stimulated monocytes affect tumor cells has not been fully elucidated but could, in addition to direct monocyte‐tumor cell contact, also include indirect activation of T cells and NK cells via an IL‐12–mediated mechanism.159
A phase I clinical trial was initiated to assess the toxicology and biological activity of ImmTher in patients with advanced colon carcinoma. ImmTher proved to be safe in humans up to a single dose of 1.2 mg/m2 and when given weekly for up to 6 months at doses 0.8 to 1 mg/m2. Toxic effects occurred at doses greater than 0.8 mg/m2 and included fever, chills, and hypotension. More importantly, ImmTher caused a regression of lung and liver metastases in three patients with metastatic colon carcinoma.160, 161 Due to the promising anticancer activity observed in preclinical and phase I clinical study ImmTher entered phase II clinical study to assess the 2‐year disease‐free survival of patients with high‐risk Ewing's sarcoma receiving vincristine, DOX, CTX, and dexrazoxane in the presence or absence of 20.162 Since the lungs are common site of metastases in Ewing's sarcoma, the primary goal of 20 is to activate pulmonary macrophages to destroy residual tumor cells not eliminated by systemic chemotherapy (therapy with 20 is initiated after completion of primary therapy, such as surgery and radiotherapy). Nevertheless, 20 was designated as an orphan drug in the United States for indications including pulmonary and hepatic metastases in patients with colorectal adenocarcinoma, Ewing's sarcoma, and osteosarcoma.163
Dialkylmethyl β‐glycosides of MDP
Zemlyakov et al87 identified four lipophilic glycosides of MDP with symmetric secondary aliphatic alcohols as aglycones that demonstrated promising cytotoxic activities in vitro. In fact, the three most lipophilic compounds caused practically complete lysis of human erythroleukemia cells (K‐562) and evident cytotoxicity on blood mononuclear cells when used at their highest tested concentration (200 µg/mL). Furthermore, these three compounds were also strongly cytotoxic when tested on K‐562 and blood mononuclear cells at concentrations ranging from 2 to 20 µg/mL. It has been reported that the cytotoxic effect toward leukemia cells observed in the previous experiment resulted not only as a consequence of the direct cytotoxic activity of the tested compounds but also from their ability to activate NK cells in a blood mononuclear cell population. Moreover, the ability of compounds to stimulate the cytotoxic activity of NK cells correlates well with the increase in the number of aglycone carbon atoms. Thus, compound 21, with the highest lipophilicity of the tested compounds, demonstrated the highest stimulation of NK cells.87
MDP‐C
Recently, MDP‐C (22) was identified as a new MDP analog that mediated its immunostimulating effect through macrophages and DCs.90 In a cell‐based assay, 22 stimulated the cytotoxic activity of murine macrophages against P388 leukemia cells resulting in an inhibitory rate of 71%. In comparison, romurtide (11) used as a positive control demonstrated a much lower inhibitory rate of 45%. Since compound 22 by itself was not cytotoxic to bone marrow–derived DCs, macrophages or P388 cells when tested under the same experimental conditions, it has been suggested that tumor growth inhibition was a result of macrophage activation by compound 22.
5.2.2. MDP conjugates
MDP is rapidly excreted from biological systems, resulting in a weaker stimulation of immune cells in vivo. As described earlier, its activity can be enhanced by introducing lipophilic moieties or by its incorporation into liposomes. Another mode of enhancing the biological activities includes the conjugation of MDP to macromolecules, such as IgG, IgM, BSA, fibronectin, cholesterol, and 10‐mer polyguanylic acid. As in the case of liposomes, biomolecules facilitate the transport of MDP and enable its phagocytosis by target cells, monocytes, and macrophages. Within these cells, molecules are released and then bind to NOD2 resulting in the activation of NOD2 signaling pathway. Alternatively, MDP has also been conjugated to small molecule drugs such as paclitaxel, batracylin, and acridine. In contrast to MDP‐biomolecule conjugates which mainly induce the antitumor activity of monocytes or macrophages, these conjugates induce anticancer activity either by direct cell killing or via the modulation of TME.
Conjugates with biomolecules
The conjugation of MDP‐l‐Ala to cholesterol, an essential structural component of animal cell membranes, resulted in a new lipophilic MDP derivative MDP‐l‐Ala‐3‐O‐cholesterol (23; Figure 6) which showed promising macrophage‐mediated cytotoxicity in vitro when incorporated into liposomes. Philips et al164 demonstrated that 23 (1 µg/mL) incorporated into DSPC:PS (7:3 molar ratio) liposomes efficiently induced the cytotoxic activity of mouse peritoneal macrophages against P815 mastocytoma cells whereas treatment with free MDP at the concentration of 50 µg/mL had no effect. As opposed to the results obtained in mouse peritoneal macrophages, free MDP (10 µg/mL or greater) was capable of activating rat alveolar macrophages against B16‐BL6 melanoma cells, however, this activation was far more extensive when MDP or 23 were encapsulated into DSPC:PS liposomes. Comparison of relative activities of MDP, liposomal MDP and liposomal 23 in stimulating rat macrophage‐mediated cytotoxicity revealed that liposomal MDP and liposomal 23 were 880‐ and 7400‐fold more effective than free MDP, respectively.164 Results confirmed that encapsulation of MDP into liposomes indeed enhances macrophage‐mediated cytotoxicity when compared with that of free MDP. However, the retention of hydrosoluble MDP in liposomes is still poor resulting in loss of 90% of MDP.164 Conjugation to cholesterol improved the lipophilic character of 23 and facilitated its incorporation into phospholipid bilayers resulting in stable 23‐containing liposomes with greatly improved macrophage‐activating properties as free or liposomal MDP. In addition to liposomal formulations, the effect of 23 incorporated into polymeric carrier systems such as nanocapsules was also studied, given the greater stability as well as improved characteristics following oral delivery.165 These studies showed that 23 incorporated into nanocapsules exhibited antimetastatic activity in a murine model of liver metastasis (histiocytosarcoma M5076 bearing mice), but only when administered as a prophylactic treatment.165, 166 Treatment of mice with nanocapsulated 23 given intravenously (5 µg/dose) or orally (50 µg/dose) twice a week beginning 2 days before M5076 tumor cell injection significantly decreased the number of metastases in the liver. Specifically, systemic and oral administrations resulted in 52% and 23% inhibition of metastasis in comparison to untreated controls, respectively. In contrast, the inhibition of metastasis was not observed in the absence of pretreatment.165 Results suggested that administration of 23 before tumor inoculation allows the circulating tumor cells to encounter activated macrophages. However, after the establishment of metastases in liver parenchyma, access by the activated liver macrophages is more restrained. In an attempt to enhance the antimetastatic activity of 23 in the liver, nanocapsulated 23 was tested in combination with nanocapsules containing indomethacin, a nonsteroidal anti‐inflammatory drug, based on the finding that liposomal indomethacin demonstrated antimetastatic activity in mice bearing 3LL Lewis lung carcinoma.166, 167 Two separate injections of nanoencapsulated 23 (5 µg/dose) and nanoencapsulated indomethacin (100 µg/dose) beginning 2 days before tumor cell injection resulted in enhanced antimetastatic activity which seemed to be additive.166 The exact mechanism of additive antimetastatic activity has yet to be elucidated but may, at least in the case of 23, include activation of monocytes and macrophages. Attempts have also been made to improve the anticancer activity of MDP by binding it to various protein carriers, namely neoglycoproteins,168 antibodies,169 maleylated bovine serum albumin (MBSA),170 and gelatin,171 as well as to the nonproteinaceous carrier polyguanylic acid (MDP‐PolyG).172 Roche et al168 demonstrated that conjugation of MDP to neoglycoproteins enhanced the tumoricidal activity of macrophages, both in vitro and in vivo, thereby protecting the mice against metastatic growth. Moreover, they also found that MDP bound to IgM mAbs specific for L1210 leukemic cells (F2‐10‐23‐IgM) and for Lewis lung carcinoma 3LL cells (6B6‐IgM), activated thioglycolate‐elicited mouse peritoneal macrophages, in turn leading to a growth inhibitory effect in target cancer cells.169 Specifically, the coating of L1210 tumor cells with MDP‐F2‐10‐23‐IgM (10 µg/mL MDP bound to 200 µg/mL F2‐10‐23‐IgM) and incubated with macrophages resulted in 80% growth inhibition of L1210 tumor cells whereas comparable concentrations of free MDP resulted only in 5% to 10% tumor growth inhibition. Moreover, 3LL cells coated with MDP‐6B6‐IgM were even more efficient in activating macrophages. Only 5 µg/mL of MDP bound to 200 µg/mL of 6B6‐IgM resulted in 70% growth inhibition of 3LL cells.169 Results obtained in that study indicate the potential of mAbs to efficiently enhance the macrophage‐mediated cytotoxicity of MDP against cancer cells. Using this approach only macrophages inside or around the tumor would be activated leading to more specific antitumor activity and also less systemic activation of macrophages. Furthermore, Tabata et al171 studied four conjugates of MDP, namely MDP‐gelatin, MDP‐IgG, MDP‐fibronectin, and MDP‐BSA, in terms of their macrophage uptake as well as their ability to induce tumoricidal macrophages. To examine macrophage‐mediated cytotoxicity against Meth A fibrosarcoma (R1) cells, macrophages were isolated from the peritoneal cavity of mice who received one intraperitoneal injection of each MDP conjugate (containing 10 µg of MDP) or free MDP (10 µg). The order of the ability of conjugates to enhance macrophage activation was in good agreement with that for macrophage uptake and was highest in the case of MDP‐gelatin, followed by MDP‐IgG and MDP‐fibronectin, whereas MDP‐BSA, as well as free MDP, showed no activity.171 The highest efficacy, shown by the MDP‐gelatin conjugate, could be a consequence of the high specific affinity of gelatin for macrophages, whereas the absence of activity in the case of MDP‐BSA could be explained by the fact that it is difficult for BSA to be ingested by macrophages.171 Further experiments in mice bearing Meth A fibrosarcoma (R1) confirmed these findings. Administration of four intraperitoneal injections of MDP‐gelatin conjugate (containing 10 µg of MDP) into mice every third day after R1 tumor cell inoculation strongly suppressed the growth of R1 cells whereas free MDP (10 µg) showed no inhibitory activity under the same experimental conditions.171 More recently, Srividya et al170, 172 described scavenger receptor‐mediated endocytosis as a new option for delivery of MDP into macrophages and consequently treatment of cancer. MDP conjugated to MBSA or to polyguanylic acid (PolyG) was internalized by macrophages through scavenger receptor‐mediated endocytosis, which resulted in a 50‐ or 20‐fold higher cytotoxic activity against tumor cells, in comparison to that elicited by free MDP. It has been proposed that this type of MDP delivery activates tumoricidal activity of macrophages by triggering the secretion of cytokines (IL‐1, IL‐6, and TNF‐α) and other soluble mediators leading to final eradication of cancer cells.
Figure 6.
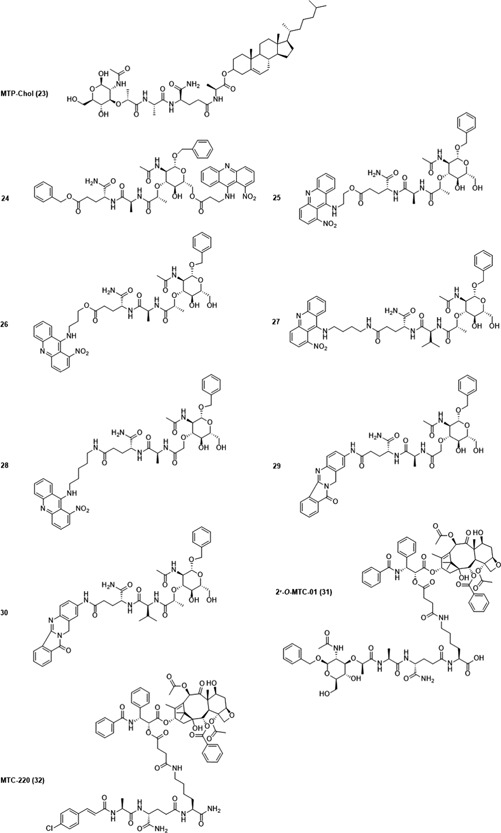
Muramyl dipeptide (MDP) conjugates with anticancer activity
Conjugates with small molecule drugs
Natural and synthetic acridines/acridones are known as potent cytotoxic agents but their clinical application is limited or has even been discontinued due to their severe side effects.173 Dzierzbicka et al86, 174 synthesized several series of conjugates of (nor)MDP with acridine and acridone derivatives to yield compounds with anticancer activity and improved pharmacological properties. Of all the synthesized compounds, the derivatives 24, 25, 26, 27, 28, 29, and 30 showed some promising results. Compound 24 stimulated the cytotoxic activity of NK cells derived from healthy and Ab melanoma–bearing animals, while compounds 25, 26, 27, and 28 exhibited potent cytotoxic activity against several human cell lines.86, 174 Moreover, compounds 25, 27, and 28 were also active in vivo in the hollow fiber assay and compound 26 showed in vivo activity against UACC‐62 melanoma in mice.86, 174 Dzierbicka et al175 prepared a series of (nor)MDP analogues conjugated to a heterocyclic aryl amine batracylin at the carboxylic group of d‐isoGln at the C terminus of the peptide residue. Compounds 29 and 30 inhibited the proliferation in vitro of Ab melanoma cells, as well as demonstrating a prominent proapoptotic effect in WEHI 164 fibrosarcoma cells in the presence of immune cells.176 Furthermore, MDP analogs were also covalently linked to paclitaxel (PTX), one of the most widely used chemotherapeutic agent for treatment of various types of cancer. Several analogs (3′‐N‐MTC‐01, 2′‐O‐MTC‐01, and 7‐O‐MTC‐01) were prepared by coupling MDP analogs to 3′‐amino, 2′‐hydroxyl, or 7‐hydroxyl group of PTX. Among them, compound 2′‐O‐MTC‐01 (31) showed the most potent antitumor as well as immunostimulatory activity in vitro. Although compound 31 by itself demonstrated growth inhibitory effect against a panel of human cell lines with IC50 in the nM concentration range (1.3‐320 nM, 72 hours treatment), the tumor growth inhibition was not as efficient as that of PTX alone (0.3‐280 nM, 72 hours treatment). Further experiments in murine peritoneal macrophages demonstrated that compound 31 increased the expression and production of TNF‐α and IL‐12, particularly at a concentration of 5 µM or higher in a dose‐dependent manner. Interestingly, the ability of 31 to induce expression and production of TNF‐α and IL‐12 even surpassed that of PTX at a concentration of 5 µM.177 The ensuing in vivo experiments on mice bearing metastasis of LLC, however, showed that 31 was completely devoid of antimetastatic activity.178 To obtain analogs of MDP with antitumor and antimetastatic activities, compound 31 was further modified by replacing the muramic acid moiety by various aromatic groups, leading to the discovery of MTC‐220 (32).178 The results obtained by National Cancer Institute‐60 Human Tumor Cell Lines Screen showed that 32 inhibited growth of various tumor cell lines, with a mean GI50 (concentration of drug to cause 50% reduction in proliferation of cancer cells) of 22 nM. Its effectiveness was further confirmed in mouse xenograft models, where 32 effectively inhibited the growth of human breast (MDA‐MB‐231, MCF‐7) and lung cancers (H460, A549, and H1975). Moreover, 32 also exhibited antimetastatic activity in spontaneous metastasis model of LLC and highly invasive and metastatic 4T1 mammary carcinoma model. Specifically, multiple injections of 32 (10 mg/kg) administered daily for 15 days into mice with established LLC tumors resulted in statistically significant inhibition of tumor growth (33%) and lung metastasis (47%) in comparison to control group. Although similar tumor growth inhibition (26%) was observed when LLC bearing mice were treated with 6 mg/kg of PTX (equimolar dose of 32), PTX had no effect on lung metastasis numbers in comparison to control group. The results obtained in a 4T1 mammary carcinoma model further confirmed the antitumor and antimetastatic activity of 32. The significant tumor growth inhibition (approximately 30%) was observed when 4T1 bearing mice received multiple injections of 32 (5 mg/kg) or PTX (3 mg/kg) given daily for 28 days. As in the case of LLC, only 32 significantly reduced the number of lung metastases in a 4T1 mouse model of lung metastasis when compared with the control group. Detailed mechanistic studies suggested the connection between the antimetastatic activity of 32 and its ability to modulate inflammatory TME. It has been discovered that 32 suppressed the accumulation MDSCs in the spleen and bone marrow of 4T1 tumor‐bearing mice, while also repressing the expression of several metastasis‐promoting factors including TNF‐α, chemokine ligand 2 (CCL2), transforming growth factor (TGF) β, and matrix metalloproteinase (MMP) 9 in tumor tissue.178 These results are very encouraging since MDSCs play an important role in tumor progression and metastasis formation. Namely, MDSCs can increase production of MMP9 (involved in tumor angiogenesis promotion) or enhance tumor cell invasion and migration through the TGFβ pathway. The fact that 32 reduced the expression of TNF‐α is of particular interest since, in the majority of studies, MDP derivatives increased TNF‐α expression, in that way contributing to the anticancer effect. There are, however, reports of an ambiguous role of TNF‐α in cancer progression. Besides its anticancer activity, TNF‐α is also involved in the development of the tissue architecture necessary for tumor growth and metastatic dissemination, as well as in the induction of other cytokines, angiogenic factors, and MMPs, thus leading to the increased growth and survival of tumor cells.179 Although 32 exerts its antimetastatic activity through modulation of TME, there is still a lot unknown about the exact mechanism of this modulation. It has been speculated that 32 could also inhibit the TLR4 signaling pathway in cancer cells given the fact that the structure of 32 contains the PTX motif, which has been shown to bind to TLR4 receptors.
5.2.3. Hydrophilic MDP derivatives
GMDP
GMDP (33; Figure 7) is a hydrophilic MDP derivative with an N‐acetylglucosamine residue attached to N‐acetylmuramic acid by a β(1,4)‐glycosidic bond. It is an effective immunomodulator, already marketed in Russia as Likopid, for combined treatment of various infectious diseases.180, 181 In addition to its immunomodulatory activities, 33 also mediates antitumor activity against several murine tumors including adenocarcinoma, LLC, melanoma, and sarcoma. The GMDP‐mediated growth inhibition of these tumors was, however, less than 60%.182 Furthermore, 33 also exhibits antimetastatic activity when given as prophylactic treatment. Experiments in LLC bearing mice showed that 33 reduced, 4.4‐ to 5.6‐fold, the number of metastases as well as their size (7‐10‐fold), as a result of GMDP‐mediated activation of NK cells.183 Since several studies revealed the ability of muramyl peptides to potentiate anticancer activity of therapeutic cytokines, the anticancer potential of 33 has been studied in combination with TNF‐α as well as other compounds includingTLR4 agonists (LPS, Lipid A analogs) and anticancer drugs. Shimizu et al studied the antitumor activity of 33 in a combination with low doses of LPS and its synthetic lipid A analogs A‐103 and 506, since these TLR4 agonists showed promising antitumor effect in several mouse tumor models. Two intravenous injections of A‐103 (50 µg) or 33 (10 µg) exhibited 43% or 52% inhibition of tumor growth rate, whereas a concomitant administration of A‐103 (100 µg) and 33 (10 µg) induced a significant 69% tumor growth inhibition when administered into Meth A fibrosarcoma bearing mice. In the same study it has also been shown that combinations of synthetic lipid A analogs (A‐103 and 506) (50 µg) or LPS (1‐10 µg) with 33 (10 µg) exhibited stronger inhibition of the tumor growth rate than when 33 was replaced by MDP (10 µg). Moreover, concomitant treatment with LPS and 33 resulted in three to four tumor‐free mice out of five.184 The exact mechanism underlying antitumor activity was not identified in this study, but could, at least in part, be attributed to macrophage activation and induction of TNF‐α secretion, as was observed in an experiment on mouse peritoneal exudate macrophages.184 Furthermore, 33 potentiated cytotoxic activity of TNF‐α against L929 murine fibrosarcoma cells. The synergistic effect was dose‐dependent and statistically significant when cells were treated for 24 hours with a combination of TNF‐α at a concentration range of 5 to 5000 U/mL and 33 at a concentration range of 0.014 to 140 µM. Moreover, treatment of L929 cells with a combination of GMDP (1.4 µM) and TNF‐α (10 U/mL) resulted in a greater number of apoptotic cells (43%) than that observed with TNF‐α alone (35%), indicating that 33 accelerated the TNF‐α–induced apoptosis.185 Furthermore, even better results in terms of cytotoxicity were observed when 33 was used in combination with TNF‐α and anticancer drugs such as actinomycin D (ActD) and CDDP in vitro.185, 186 For example, treatment of L929 cells with a TNF‐α in combination with ActD (4 µg/mL) resulted in 100% dead cells at 250 U/mL concentration, whereas in the presence of 33 (1.4 µM), a similar effect was observed even when TNF‐α and ActD were used at much lower 50 U/mL and 1 µg/L concentrations, respectively.185 In addition, 33 (1 µg/mL) also significantly potentiated the cytotoxic effect of TNF‐α (500 U/mL) and CDDP (3‐6 µM) against several other murine and human cell lines (L929, EAT, U‐93, and MCF‐7).186 Besides antitumor activity in vitro, 33 also exhibited antitumor activity in an in vivo setting. Specifically, it augmented the antitumor activity of TNF‐α and CDDP against Ehrlich ascites carcinoma and against melanoma B16 bearing mice.187 More importantly, treatment of mice with 33 at a dose as low as 0.05 µg/mouse decreased the toxicity of CDDP (40 µg/mouse)/TNF‐α (500 U/mouse) combination and normalized changes in hematological parameters (decreased lymphocytes, increased monocytes, and neutrophils) attributed to CDDP/TNF‐α treatment.187
Figure 7.
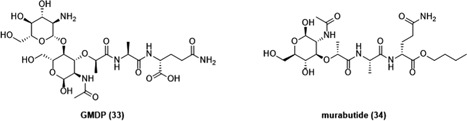
Hydrophilic muramyl dipeptide (MDP) derivatives with anticancer activity
Results obtained in these studies indicated that 33 synergizes with TNF‐α (and anticancer drugs) as well as augments anticancer effect of TNF‐α, one of the essential cytokines involved in regulation of cancer. Despite promising anticancer activity, the clinical application of TNF‐α is limited due to its high toxicity and deleterious side effects.188 Combination of 33 with TNF‐α and anticancer drugs reduced therapeutic doses of TNF‐α and anticancer drugs as well as augmented their therapeutic effect. This is particularly important since chemotherapeutics used in therapeutic doses, besides cancer cells, kill also normal rapid‐dividing cells such as the cells of bone marrow. The utilization of drugs characterized by synergistic effects, therefore, enables the reduction of therapeutically efficient doses, thereby decreasing the toxicity against normal cells.
Murabutide
Replacement of d‐isoGln with a d‐Gln‐n‐butyl‐ester residue in the peptide part of MDP afforded murabutide (34), another hydrophilic MDP derivative, that has similar adjuvant activity but lacks the pyrogenicity and toxicity of MDP.189, 190, 191, 192 Besides its adjuvant activity, 34 has also been shown to activate the immune system to fight cancer. Although in vast majority of studies MDP derivatives have been shown only to enhance anticancer activity of monocytes and macrophages, a study of Vidal et al89 showed that 34 triggered maturation and activation of monocyte‐derived immature DCs. 34‐induced maturation of DCs was found to be beneficial since, in tumors, DCs are often found in an immature, immunosuppressive state and therefore unable to mount a proper immune response. Similarly, it has been proposed that the profile of cytokines secreted by 34‐stimulated DCs including MIP‐1β, TNF‐α, IL‐10, and GM‐CSF, could be useful in mounting a strong immune response against tumors. In accordance with these results 34 at 10 µg/mL final concentration indeed significantly augmented the cytostatic activity of immature DCs against THP‐1 cancer cells.89 Moreover, treatment of DCs with 34 also resulted in an enhanced stimulatory capacity of DCs for both allogeneic and autologous T cells. These results revealed a great potential of 34 to be used as an adjuvant in DC‐based cancer vaccines.
Although 34 by itself has been shown to activate immune cells to fight cancer, an optimal activation was observed when 34 was used in combination with therapeutic cytokines IL‐2 and IFN‐α/β. In these studies, 34 synergized with IL‐2 or IFN‐α/β in vitro and in vivo as well as enhanced their biological activities. Specifically, in vitro experiments on human PBMCs revealed the synergistic activity of 34 and IL‐2 in inducing IL‐1β, IL‐12, IFN‐y, and CSFs. Moreover, in the same study it was demonstrated that the weak antitumor effect of IL‐2 in Meth A fibrosarcoma bearing mice was enhanced by concomitant administration of 34 (10 mg/kg, 5 or 3 times per week) and IL‐2 (5 × 106 U/kg, 5 or 3 times per week) following 2 weeks of treatment. In fact, complete tumor regression was achieved in nearly 70% tumor‐bearing mice when they were treated with both compounds together.193 Importantly, concomitant treatment with both compounds was well‐tolerated since the net gain in body weight was not significantly different from that observed in the control group. The exact mechanism on how 34 potentiates antitumor activity of IL‐2 has not been elucidated in this study. On the basis of results of cytokine profile induced by a combination of 34 and IL‐2 in vitro, it was suggested that the antitumor effect may be due to induction of the Th1 cytokines IL‐12 and IFN‐y.193 In addition, stimulation of other effector mechanisms such as activation of NK cells killing cell activities could also be involved in potentiated antitumor effect. Similarly, synergistic antitumor activity was also observed when Meth A fibrosarcoma bearing mice were treated with 34 in combination with IFN‐α/β.194 Multiple injections of 34 (10 mg/kg) and IFN‐α/β (1.25 × 106 U/kg), both given three times per week following 2 weeks of treatment into Meth A fibrosarcoma bearing mice resulted in almost 50% tumor‐free mice. In sharp contrast, treatment of tumor‐bearing mice with IFN‐α/β or 34 by themselves did not bring about a significant regression of tumor size.194 Collectively, the conducted studies show a limited efficacy and dose‐dependent toxicity of therapeutic cytokines in anticancer therapy. In our opinion, the utilization of safe immunomodulators such as compound 34, capable of potentiating the anticancer activities of cytokines could, therefore, represent a significant advantage in the therapy of cancer. By employing this approach lower doses of therapeutic cytokines are needed for anticancer activity resulting in reduced toxicity, typically associated with high‐dose and long‐term administrations.
5.2.4. Desmuramylpeptides
The finding that the presence of the N‐acetylmuramyl moiety is not necessary for the immunomodulatory properties of MDP led to the design and synthesis of a new class of MDP derivatives, termed desmuramylpeptides.195, 196, 197, 198, 199, 200, 201, 202 Desmuramylpeptides are devoid of the N‐acetylmuramyl moiety and have therefore more lipophilic character than MDP. This class contains compounds that are able to enhance host defense against microbial infections as well as exhibit strong adjuvant activity and, even, remarkable antitumor potency.202 The latter was extensively studied using two nor‐MDP analogs, namely LK‐409 (35; Figure 8) and LK‐410 (36), in which the N‐acetylmuramyl moiety was replaced by the N‐(7‐oxooctanoyl) and N‐trans‐2‐((2′‐(acetylamino)cyclohexyl)oxy)acetyl groups, respectively.203, 204 It was demonstrated that treatment of SA‐1 fibrosarcoma bearing mice with multiple intraperitoneal injections of 35 (2.5 or 25 µg/dose) or 36 (25 µg/dose) administered 5 consecutive days after tumors achieved 35 mm3 in size resulted in a moderate but statistically significant inhibition of tumor growth measured as tumor growth delay (time required for the tumor to achieve a volume of 150 mm3). An antitumor effect was substantially augmented when either 35 or 36 were administered together with TNF‐α analog TNFNv3. The most significant tumor growth delay was seen when mice with established SA‐1 tumors received 2.5 µg of 36 (five injections in 5 consecutive days) and 5 × 105 U of TNFNv3 (three injections given every second day), and was prolonged to 9.2 days when compared to untreated controls and 3.2 days when compared to TNFNv3 (5 × 105 U/dose) treated mice. Remarkably, 36 also reduced the side effects of TNFNv3 in mice, resulting in lower body weight loss and better general conditions.202 Results indicate that desmuramylpeptides effectively potentiated the antitumor activity of TNF‐α analog, which is especially desirable since lower doses of compounds, in comparison to both compounds by themselves, are needed to achieve a similar antitumor effect. The exact mechanism on how these nor‐MDP analogs mediated antitumor activity has yet to be elucidated, but it most probably includes activation of macrophages. Moreover, 35 and 36 also demonstrated pronounced immunorestorative effects in vivo.203, 204 The effectiveness of 35 even surpassed that of romurtide (11) in restoring the activity of the immune response in tumor‐bearing mice and in immunocompromised animals evaluated in numerous in vitro and in vivo experiments.204 On the other hand, multiple injections of 36 at doses 10 and 100 mg/kg significantly increased the survival of mice suppressed by CTX and challenged with a suspension of Candida albicans.203 To evaluate the mechanism underlying its immunorestorative activity, 36 was further evaluated in several immunopharmacological models. It has been shown that 36 stimulated maturation of B cells as well as increasing the activity of B cells, T cells, and macrophages, but had no effect on cell counts.203 Since desmuramylpeptides have shown promising antitumor activity when used in combination with cytokines in addition to their ability to restore immune cell functions, in our opinion they could represent an excellent adjunct to current cancer therapy. Also, based on the observation that desmuramylpeptides can potentiate the antitumor activity of cytokines, it might be interesting to also test them in combination with other chemotherapeutics at low doses, potentially resulting in fewer side effects. Finally, desmuramylpeptides with immunorestorative activity could also restore the immune cell functions often impaired by chemotherapeutics, which is of particular importance, given the fact that the patients with the impaired immune system are more vulnerable to infection.
Figure 8.
Desmuramylpeptides with antitumor and immunorestorative activity
5.2.5. SAR of MDP
The structure of MDP in correlation with its adjuvant and antitumor activities has been widely studied. Investigations of their structure‐activity relationship (SAR) are summarized in Figure 9.
Figure 9.
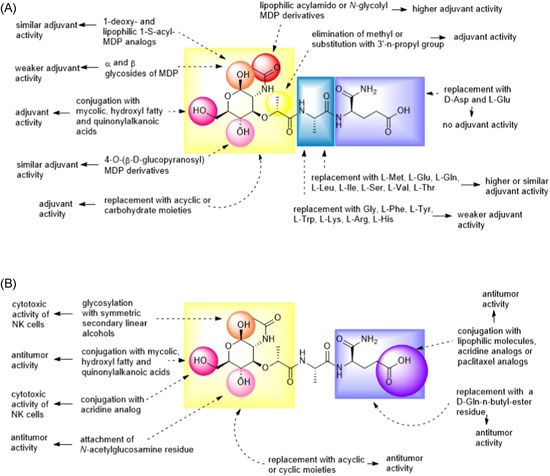
SAR of MDP. A, Modifications in MDP molecule in correlation with adjuvant activity and B, modifications in MDP molecule in correlation with antitumor activity. MDP, muramyl dipeptide; SAR, structure‐activity relationship [Color figure can be viewed at wileyonlinelibrary.com]
5.2.5.1. Modifications of the peptide moiety
The l‐configuration of the first amino acid (l‐Ala) and the d‐configuration of the glutamic acid residue of the second amino acid (d‐isoGln) are essential for retaining or increasing the adjuvant activity of MDP.205 Substitution of l‐Ala with other amino acids generated MDP analogs with (i) similar or higher adjuvant activities (l‐methionine (l‐Met), l‐glutamic acid (l‐Glu), l‐glutamine (l‐Gln), l‐leucine (l‐Leu), l‐isoleucine (l‐Ile), l‐serine (l‐Ser), l‐valine (l‐Val), l‐threonine (l‐Thr)) or (ii) weaker (glycine (Gly), l‐phenylalanine (l‐Phe), l‐tyrosine (l‐Tyr), l‐tryptophan (l‐Trp), l‐lysine (l‐Lys), l‐arginine (l‐Arg), l‐histidine (l‐His)) than that of MDP.206, 207, 208 d‐isoGln (α‐amide) is an essential part of MDP, since its substitution with l‐Glu (γ‐amide) or d‐aspartic acid (d‐Asp) (side chain shortened by one methylene group) produces analogs devoid of adjuvant activity.209 In the context of the antitumor activity, conjugation of MDP at the C‐terminal end of the peptide part with lipophilic molecules such as stearic acid, dipalmitoyl phosphatidylethanolamine, GDP, cholesterol, m‐nitrocinnamic acid, acridine analogs, and paclitaxel analogs resulted in analogs with antitumor activity.
5.2.5.2. Modifications of the carbohydrate moiety
Besides substitutions in the peptide portion, modifications can also be introduced into the carbohydrate moiety of MDP. The hydroxyl group at the C1 position can be eliminated, replaced by thiol, or substituted by α‐ or β‐glycosides without loss of adjuvant activity.210 1‐deoxy and lipophilic 1‐S‐acyl analogs of MDP demonstrated strong adjuvant activity, closely similar to that of MDP,211, 212 while α‐ and β‐methyl glycosides showed weaker adjuvant activity.213 Moreover, dialkylmethyl β‐glycosides of MDP were found to stimulate cytotoxicity of NK cells.87 Furthermore, the acetamide group at the C2 position can be replaced by a hydroxyl, amino, methylamino, N‐methylacetamide or N‐glycolyl group.210, 214 Introduction of lipophilic acylamido or N‐glycolyl moieties into the structure of MDP led to analogs with enhanced adjuvant activity.210, 214 The chiral center of the lactic acid moiety at the C3 position appears to have a minimal effect on the biological activities of MDP. Elimination of the methyl group at the chiral center, or its substitution with 3′‐n‐propyl group, gives MDP analogs with adjuvant activity but lower toxicity than MDP.210 Substitution of the hydroxyl group at the C4 position is not that common, although some 4‐O‐(β‐d‐glucopyranosyl) derivatives of MDP demonstrated strong adjuvant activity, comparable to that of MDP.215, 216 Moreover, attachment of an N‐acetylglucosamine residue to the hydroxyl group at the C4 position gives 33, a hydrophilic analog of MDP with antitumor activity. Substitution of the hydroxyl group at the C6 position by mycolic, hydroxyl fatty or quinonylalkanoic acids produced 6‐O‐acyl derivatives of MDP possessing significant adjuvant and antitumor activities.142, 143 Similarly, the 6‐O‐acyl derivative of MDP with acridine, compound 24, stimulated the cytotoxic activity of NK cells derived from healthy and Ab melanoma–bearing animals.86 Moreover, some studies also showed that replacement of the N‐acetylmuramic acid in MDP with certain acyclic, cyclic, and carbohydrate moieties led to the retained immunomodulatory activity of MDP and, in some cases, even exhibited antitumor activities.195, 196, 197, 198, 199, 200, 201, 202
5.3. NOD1 and NOD2 antagonists
NOD antagonists have recently been recognized as potential anticancer agents with a unique mechanism of action. In sharp contrast to NOD agonists, which generally produce a pro‐inflammatory TME and enhance the ability of immune cells to fight cancer cells, it has been proposed that NOD antagonists mainly mediate their antitumor activity by preventing the formation of an inflammatory TME. Several compounds have so far been synthesized, among which DY‐16‐43 (37; Figure 10), 38, MDC‐405 (39) and salutaxel (40) exhibit the most effective anticancer activity.
Figure 10.
NOD1 and NOD2 antagonists with anticancer activity. NOD, nucleotide‐binding oligomerization domain
An NOD2 antagonist 37 (a noncleavable analog of 32) markedly increased the therapeutic efficacy of PTX. It has been shown that concomitant treatment of LLC tumor‐bearing mice with multiple injections of PTX (12 mg/kg) and 37 (30 mg/kg) resulted, not only in the reduction of tumor weight but also in prevention of tumor metastasis. Surprisingly, treatment with 37 alone demonstrated no significant improvement in inhibition of tumor growth and prevention of metastasis. It was therefore proposed that, in mice, NOD2 was activated mainly by DAMPs generated as a result of the treatment with PTX and, in turn, resulted in TME remodeling, chemoresistance, and metastasis. Furthermore, it was suggested that 37 prevented the establishment of an inflammatory TME by blocking DAMPs and therefore sensitized the chemotherapeutic response of PTX.217 Recently, Wang et al218 identified a heterocyclic 1,4‐benzodiazepine‐2,5‐dione derivative 38 as a dual NOD1/NOD2 antagonist that, remarkably, enhanced the antitumor efficacy of PTX. Concomitant administration of multiple injections of 38 (20 mg/kg) and PTX (12 mg/kg) into mice beginning one day after LLC tumor cell inoculation resulted in a substantial tumor weight inhibition (67%) whereas treatment with PTX (34%) or 38 (10%) alone exerted no significant beneficial effect. Moreover, it has been shown that compound 38 could also decrease the mRNA levels of NOD1/2‐mediated IL‐6 and TNF‐α in human PBMC‐derived macrophages; further, it potently inhibited the NF‐κB and MAPK pathways. The exact mechanism underlying the observed antitumor activity of 38 has not so far been elucidated; however, it most probably involves the remodeling of TME as in the case of NOD2 antagonist 37. Due to the promising results obtained by conjugates of PTX and MDP analogs, MDP analogs were further conjugated to docetaxel (DTX), another chemotherapeutic drug used in treating many cancers.219 Of all the synthesized DTX conjugates, compound 39 showed excellent results in preventing 4T1 breast tumor growth and metastasis in mice. Moreover, the beneficial effects of 39 have been associated with its ability to inhibit NOD1 signaling.219 Despite promising antitumor and antimetastatic activities in vivo, 39 has some unfavorable physicochemical characteristics such as low solubility in water. It has, therefore, been modified by replacing the amide in the C‐terminus with a carboxylic acid group, affording a new NOD1 antagonist 40.219 In a preliminary screening, 40 inhibited growth of 14 human cancer cell lines of different origin with a mean GI50 of 16.3 nM after 72 hours treatment. Its effectiveness was further confirmed in mouse xenograft models, where 40 effectively inhibited the growth of human breast (MDA‐MB‐231), colon (HTC116), and lung (H1975, A549/T) cancers. Moreover, 40 also exhibited antimetastatic activity in a highly invasive and metastatic 4T1 mammary carcinoma model. Specifically, multiple injections of 40 (5, 10, or 20 mg/kg) given once per week for 15 days into mice with established LLC tumors demonstrated a substantial tumor growth inhibition in a dose‐dependent manner (12%, 37%, or 57%, respectively). Under the same experimental conditions, 5.7 mg/kg of DTX inhibited tumor growth only by 19%. Moreover, compound 40 also exhibited a significant antimetastatic effect at all doses used (5, 10, or 20 mg/kg) and was also significantly superior to DTX in the prevention of tumor metastasis when administered at doses of 10 or 20 mg/kg. In addition, compound 40 (10 mg/kg) was also tested in combination with DOX (4 mg/kg) and exhibited superior antitumor and antimetastatic effect in 4T1 carcinoma model in comparison to DTX (5.7 mg/kg)/DOX combination. Importantly, no significant body weight loss in mice was observed under the experimental conditions indicating that compounds were well tolerated at all tested concentrations. Mechanistically, 40 suppressed the accumulation of MDSC in the spleen of tumor‐bearing mice and decreased levels of several pro‐inflammatory molecules.219 As noted previously, MDSCs infiltrate the TME of developing tumors and promote their invasion and metastasis, most probably through the release of MMPs.220 Compound 40 was reported to significantly suppress MMP9 expression in the serum, spleen, and lungs of tumor‐bearing mice as well as to decrease the serum levels of tissue inhibitor of metalloproteinase (TIMP) 1 whose activity is also associated with the progression of several cancer types.221 Moreover, lung tissue derived from 4T1 tumor‐bearing mice treated with compound 40 demonstrated a significant decrease in the mRNA levels of prokineticin (PROK) 2, MMP8, S100 calcium‐binding protein A8 (S100A8) and S100A9, which might be also critically involved in metastasis formation.219 In addition, compound 40 also suppressed accumulation of neutrophils in the blood of 4T1 tumor‐bearing mice. This effect could be advantageous, since several studies have shown a correlation between elevated blood neutrophil count and poor clinical outcome in many cancers.222, 223 Moreover, it has been shown that dysfunctional neutrophils express reduced levels of NOD1 and that treatments that blocked the NOD1/NF‐κB pathway resulted in inhibition of neutrophil migration and their phagocytic killing capacity.224 Due to these findings, it has been proposed that compound 40 inhibits neutrophil recruitment by blocking the NOD1 pathway.
6. CONCLUSION
To date, a plethora of NOD ligands have been synthesized among which several compounds showed potent antitumor and even antimetastatic activity in numerous in vitro and in vivo studies (Supporting Information Table S1) as well as in clinical trials. Specifically, NOD2 agonist mifamurtide certainly represents the most important compound to have been granted with marketing authorization by the European Medicine Agency for treating osteosarcoma in combination with other chemotherapeutics, following complete surgical removal of a primary tumor. For a long time, only NOD agonists were considered as promising antitumor and antimetastatic compounds. Recently, a shift in this paradigm has occurred with the emerging knowledge that NOD antagonists could also facilitate the elimination of some cancers. Incidentally, the NOD1 antagonist salutaxel currently holds the status of an investigational new drug for cancer therapy. Furthermore, preclinical studies have also shown the great potential of NOD ligands for fighting cancer in a synergistic manner, in combination with chemotherapeutics, TLR ligands, cytokines, or when used as adjuvants in cancer vaccines.
To conclude, the innate immune receptors NOD1 and NOD2 constitute promising targets in cancer immunotherapy. It is, however, unlikely that NOD ligands alone could be sufficient for complete elimination of cancer, but their tumor‐suppressing capacities could be harnessed by introducing them as adjuncts to already existing cancer immunotherapies or to traditional cancer therapies such as chemotherapy and radiation.
CONFLICTS OF INTEREST
The authors declare that there are no conflicts of interest.
Supporting information
Supplementary information
ACKNOWLEDGEMENTS
This study was supported by the Slovenian Research Agency grant (No. P1‐0208). We thank Roger Pain for proofreading the manuscript.
Biographies
Sanja Nabergoj obtained her degree in Pharmacy in 2014 at University of Ljubljana, Slovenia (Faculty of Pharmacy). She performed experimental part of her master thesis at the University of Strasbourg, France (Faculty of Pharmacy). At present she is an assistant of Toxicology at the Faculty of Pharmacy, University of Ljubljana. She started her graduate studies in 2016 and is currently in pursuit of her PhD under mentorship of Assoc Prof Dr Žiga Jakopin and Prof Dr Irena Mlinarič‐Raščan. Her main research focus is on identification of programmed cell death modulators (NOD and EP4 agonists) in chronic lymphocytic leukemia cells.
Irena Mlinarič‐Raščan is a full professor in pharmacogenomics, immunology, and cell biology, National director of EATRIS.SI and dean at the Faculty of Pharmacy, University Ljubljana, Slovenia. She had completed two postdoctoral fellowships at the Mount Sinai Hospital, Toronto and at the Tokyo University, and was a guest professor at the University of Bern. Her research work involves pharmacogenomics approaches in individualization of leukemia and lymphoma therapies, focusing on thiopurine‐based ALL therapy and identification of novel target molecules including prostaglandin receptor 4, proteasome, and immunoproteasome, which further serve for lead design and optimization.
Assoc Prof Dr Žiga Jakopin received his degree in Pharmacy in 2005 and his PhD in Pharmaceutical Sciences in 2010 from the University of Ljubljana (Faculty of Pharmacy, Ljubljana, Slovenia). At present he is Associate Professor of Medicinal Chemistry at the Faculty of Pharmacy (University of Ljubljana). He continued his training abroad and worked as a visiting researcher (in 2009) and as a postdoctoral researcher (in 2017) in the laboratory of Prof Dr Emanuela Corsini at the Universitá degli studi di Milano, Italy, where he supplemented his background with cell‐based assays. His primary research interests concern the design, synthesis, and evaluation of peptidomimetics and immunomodulatory compounds and in vitro immunotoxicology assays. His research focus at present is on the development of new ligands of NOD receptors. He has authored over 25 publications in prominent scientific journals, is a recipient of several national and international fellowships and grants, has participated in EU‐funded and national projects, and is currently involved in MSCA‐ITN‐EJD project PhD4GlycoDrug. He has also served on several international and national organizing committees.
Nabergoj S, Mlinarič‐Raščan I, Jakopin Ž. Harnessing the untapped potential of nucleotide‐binding oligomerization domain ligands for cancer immunotherapy. Med Res Rev. 2019;39:1447‐1484. 10.1002/med.21557
References
REFERENCES
- 1. Virchow R. Die Krankhaften Geschwülste. Berlin: Verlag von August Hirschwald; 1863. [Google Scholar]
- 2. Coley WB. II. Contribution to the knowledge of sarcoma. Ann Surg. 1891;14(3):199‐220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Morales A, Eidinger D, Bruce AW. Intracavitary Bacillus Calmette‐Guerin in the treatment of superficial bladder tumors. J Urol. 1976;116(2):180‐183. [DOI] [PubMed] [Google Scholar]
- 4. Zheng YQ, Naguib YW, Dong Y, Shi YC, Bou S, Cui Z. Applications of bacillus Calmette‐Guerin and recombinant bacillus Calmette‐Guerin in vaccine development and tumor immunotherapy. Expert Rev Vaccines. 2015;14(9):1255‐1275. [PMC free article] [PubMed] [Google Scholar]
- 5. Emens LA, Ascierto PA, Darcy PK, et al. Cancer immunotherapy: opportunities and challenges in the rapidly evolving clinical landscape. Eur J Cancer Oxf Engl 1990. 2017;81:116‐129. [DOI] [PubMed] [Google Scholar]
- 6. Galluzzi L, Vacchelli E, Bravo‐San Pedro JM, et al. Classification of current anticancer immunotherapies. Oncotarget. 2014;5(24):12472‐12508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Papaioannou NE, Beniata OV, Vitsos P, Tsitsilonis O, Samara P. Harnessing the immune system to improve cancer therapy. Ann Transl Med. 2016;4(14):261‐261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Shekarian T, Valsesia‐Wittmann S, Brody J, et al. Pattern recognition receptors: immune targets to enhance cancer immunotherapy. Ann Oncol. 2017;28(8):1756‐1766. [DOI] [PubMed] [Google Scholar]
- 9. Cui J, Chen Y, Wang HY, Wang R‐F. Mechanisms and pathways of innate immune activation and regulation in health and cancer. Hum Vaccines Immunother. 2014;10(11):3270‐3285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kim YK, Shin JS, Nahm MH. NOD‐like receptors in infection, immunity, and diseases. Yonsei Med J. 2016;57(1):5‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chamaillard M, Hashimoto M, Horie Y, et al. An essential role for NOD1 in host recognition of bacterial peptidoglycan containing diaminopimelic acid. Nat Immunol. 2003;4(7):702‐707. [DOI] [PubMed] [Google Scholar]
- 12. Girardin SE, Boneca IG, Carneiro LA, et al. Nod1 detects a unique muropeptide from gram‐negative bacterial peptidoglycan. Science. 2003;300(5625):1584‐1587. [DOI] [PubMed] [Google Scholar]
- 13. Hasegawa M, Yang K, Hashimoto M, et al. Differential release and distribution of Nod1 and Nod2 immunostimulatory molecules among bacterial species and environments. J Biol Chem. 2006;281(39):29054‐29063. [DOI] [PubMed] [Google Scholar]
- 14. Inohara N, Nuñez G. NODs: intracellular proteins involved in inflammation and apoptosis. Nat Rev Immunol. 2003;3(5):371‐382. [DOI] [PubMed] [Google Scholar]
- 15. Correa RG, Milutinovic S, Reed JC. Roles of NOD1 (NLRC1) and NOD2 (NLRC2) in innate immunity and inflammatory diseases. Biosci Rep. 2012;32(6):597‐608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kanazawa N, Okafuji I, Kambe N, et al. Early‐onset sarcoidosis and CARD15 mutations with constitutive nuclear factor‐kappaB activation: common genetic etiology with Blau syndrome. Blood. 2005;105(3):1195‐1197. [DOI] [PubMed] [Google Scholar]
- 17. Miceli‐Richard C, Lesage S, Rybojad M, et al. CARD15 mutations in Blau syndrome. Nat Genet. 2001;29(1):19‐20. [DOI] [PubMed] [Google Scholar]
- 18. Ogura Y, Bonen DK, Inohara N, et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn's disease. Nature. 2001;411(6837):603‐606. [DOI] [PubMed] [Google Scholar]
- 19. Caruso R, Warner N, Inohara N, Núñez G. NOD1 and NOD2: signaling, host defense, and inflammatory disease. Immunity. 2014;41(6):898‐908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Saxena M, Yeretssian G. NOD‐like receptors: master regulators of inflammation and cancer. Front Immunol. 2014;5:327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kutikhin AG. Role of NOD1/CARD4 and NOD2/CARD15 gene polymorphisms in cancer etiology. Hum Immunol. 2011;72(10):955‐968. [DOI] [PubMed] [Google Scholar]
- 22. Strober W, Murray PJ, Kitani A, Watanabe T. Signalling pathways and molecular interactions of NOD1 and NOD2. Nat Rev Immunol. 2006;6(1):9‐20. [DOI] [PubMed] [Google Scholar]
- 23. Barnich N, Aguirre JE, Reinecker H‐C, Xavier R, Podolsky DK. Membrane recruitment of NOD2 in intestinal epithelial cells is essential for nuclear factor‐{kappa}B activation in muramyl dipeptide recognition. J Cell Biol. 2005;170(1):21‐26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kufer TA, Kremmer E, Adam AC, Philpott DJ, Sansonetti PJ. The pattern‐recognition molecule Nod1 is localized at the plasma membrane at sites of bacterial interaction. Cell Microbiol. 2008;10(2):477‐486. [DOI] [PubMed] [Google Scholar]
- 25. Ogura Y, Inohara N, Benito A, Chen FF, Yamaoka S, Núñez G. Nod2, a Nod1/Apaf‐1 family member that is restricted to monocytes and activates NF‐kappaB. J Biol Chem. 2001;276(7):4812‐4818. [DOI] [PubMed] [Google Scholar]
- 26. Tada H, Aiba S, Shibata K‐I, Ohteki T, Takada H. Synergistic effect of Nod1 and Nod2 agonists with toll‐like receptor agonists on human dendritic cells to generate interleukin‐12 and T helper type 1 cells. Infect Immun. 2005;73(12):7967‐7976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ogura Y, Lala S, Xin W, et al. Expression of NOD2 in Paneth cells: a possible link to Crohn's ileitis. Gut. 2003;52(11):1591‐1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Marriott I, Rati DM, McCall SH, Tranguch SL. Induction of Nod1 and Nod2 intracellular pattern recognition receptors in murine osteoblasts following bacterial challenge. Infect Immun. 2005;73(5):2967‐2973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Voss E, Wehkamp J, Wehkamp K, Stange EF, Schröder JM, Harder J. NOD2/CARD15 mediates induction of the antimicrobial peptide human beta‐defensin‐2. J Biol Chem. 2006;281(4):2005‐2011. [DOI] [PubMed] [Google Scholar]
- 30. Nigro G, Rossi R, Commere P‐H, Jay P, Sansonetti PJ. The cytosolic bacterial peptidoglycan sensor Nod2 affords stem cell protection and links microbes to gut epithelial regeneration. Cell Host Microbe. 2014;15(6):792‐798. [DOI] [PubMed] [Google Scholar]
- 31. Hisamatsu T, Suzuki M, Reinecker H‐C, Nadeau WJ, McCormick BA, Podolsky DK. CARD15/NOD2 functions as an antibacterial factor in human intestinal epithelial cells. Gastroenterology. 2003;124(4):993‐1000. [DOI] [PubMed] [Google Scholar]
- 32. Sugawara Y, Uehara A, Fujimoto Y, et al. Toll‐like receptors, NOD1, and NOD2 in oral epithelial cells. J Dent Res. 2006;85(6):524‐529. [DOI] [PubMed] [Google Scholar]
- 33. Uehara A, Fujimoto Y, Fukase K, Takada H. Various human epithelial cells express functional Toll‐like receptors, NOD1 and NOD2 to produce anti‐microbial peptides, but not proinflammatory cytokines. Mol Immunol. 2007;44(12):3100‐3111. [DOI] [PubMed] [Google Scholar]
- 34. Jakopin Ž. Nucleotide‐binding oligomerization domain (NOD) inhibitors: a rational approach toward inhibition of NOD signaling pathway. J Med Chem. 2014;57(16):6897‐6918. [DOI] [PubMed] [Google Scholar]
- 35. Moreira LO, Zamboni DS. NOD1 and NOD2 signaling in infection and inflammation. Front Immunol. 2012;3:328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Philpott DJ, Sorbara MT, Robertson SJ, Croitoru K, Girardin SE. NOD proteins: regulators of inflammation in health and disease. Nat Rev Immunol. 2014;14(1):9‐23. [DOI] [PubMed] [Google Scholar]
- 37. Inohara N, Koseki T, del Peso L, et al. Nod1, an Apaf‐1‐like activator of caspase‐9 and nuclear factor‐kappaB. J Biol Chem. 1999;274(21):14560‐14567. [DOI] [PubMed] [Google Scholar]
- 38. Lechtenberg BC, Mace PD, Riedl SJ. Structural mechanisms in NLR inflammasome signaling. Curr Opin Struct Biol. 2014;29:17‐25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Riedl SJ, Li W, Chao Y, Schwarzenbacher R, Shi Y. Structure of the apoptotic protease‐activating factor 1 bound to ADP. Nature. 2005;434(7035):926‐933. [DOI] [PubMed] [Google Scholar]
- 40. Bertrand MJ, Doiron K, Labbé K, Korneluk RG, Barker PA, Saleh M. Cellular inhibitors of apoptosis cIAP1 and cIAP2 are required for innate immunity signaling by the pattern recognition receptors NOD1 and NOD2. Immunity. 2009;30(6):789‐801. [DOI] [PubMed] [Google Scholar]
- 41. Damgaard RB, Nachbur U, Yabal M, et al. The ubiquitin ligase XIAP recruits LUBAC for NOD2 signaling in inflammation and innate immunity. Mol Cell. 2012;46(6):746‐758. [DOI] [PubMed] [Google Scholar]
- 42. Hasegawa M, Fujimoto Y, Lucas PC, et al. A critical role of RICK/RIP2 polyubiquitination in Nod‐induced NF‐kappaB activation. EMBO J. 2008;27(2):373‐383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Krieg A, Correa RG, Garrison JB, et al. XIAP mediates NOD signaling via interaction with RIP2. Proc Natl Acad Sci USA. 2009;106(34):14524‐14529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tigno‐Aranjuez JT, Asara JM, Abbott DW. Inhibition of RIP2's tyrosine kinase activity limits NOD2‐driven cytokine responses. Genes Dev. 2010;24(23):2666‐2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yang Y, Yin C, Pandey A, Abbott D, Sassetti C, Kelliher MA. NOD2 pathway activation by MDP or Mycobacterium tuberculosis infection involves the stable polyubiquitination of Rip2. J Biol Chem. 2007;282(50):36223‐36229. [DOI] [PubMed] [Google Scholar]
- 46. Abbott DW, Yang Y, Hutti JE, Madhavarapu S, Kelliher MA, Cantley LC. Coordinated regulation of Toll‐like receptor and NOD2 signaling by K63‐linked polyubiquitin chains. Mol Cell Biol. 2007;27(17):6012‐6025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Abbott DW, Wilkins A, Asara JM, Cantley LC. The Crohn's disease protein, NOD2, requires RIP2 in order to induce ubiquitinylation of a novel site on NEMO. Curr Biol CB. 2004;14(24):2217‐2227. [DOI] [PubMed] [Google Scholar]
- 48. Wang C, Deng L, Hong M, Akkaraju GR, Inoue J, Chen ZJ. TAK1 is a ubiquitin‐dependent kinase of MKK and IKK. Nature. 2001;412(6844):346‐351. [DOI] [PubMed] [Google Scholar]
- 49. Yeretssian G. Effector functions of NLRs in the intestine: innate sensing, cell death, and disease. Immunol Res. 2012;54(1‐3):25‐36. [DOI] [PubMed] [Google Scholar]
- 50. Navas TA, Baldwin DT, Stewart TA. RIP2 is a Raf1‐activated mitogen‐activated protein kinase kinase. J Biol Chem. 1999;274(47):33684‐33690. [DOI] [PubMed] [Google Scholar]
- 51. Askari N, Correa RG, Zhai D, Reed JC. Expression, purification, and characterization of recombinant NOD1 (NLRC1): a NLR family member. J Biotechnol. 2012;157(1):75‐81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yeretssian G, Correa RG, Doiron K, et al. Non‐apoptotic role of BID in inflammation and innate immunity. Nature. 2011;474(7349):96‐99. [DOI] [PubMed] [Google Scholar]
- 53. da Silva Correia J, Miranda Y, Austin‐Brown N, et al. Nod1‐dependent control of tumor growth. Proc Natl Acad Sci USA. 2006;103(6):1840‐1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. da Silva Correia J, Miranda Y, Leonard N, Hsu J, Ulevitch RJ. Regulation of Nod1‐mediated signaling pathways. Cell Death Differ. 2007;14(4):830‐839. [DOI] [PubMed] [Google Scholar]
- 55. Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol. 2013;13(10):722‐737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Knodler LA, Celli J. Eating the strangers within: host control of intracellular bacteria via xenophagy. Cell Microbiol. 2011;13(9):1319‐1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Levine B, Deretic V. Unveiling the roles of autophagy in innate and adaptive immunity. Nat Rev Immunol. 2007;7(10):767‐777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Puleston DJ, Simon AK. Autophagy in the immune system. Immunology. 2014;141(1):1‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Cooney R, Baker J, Brain O, et al. NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat Med. 2010;16(1):90‐97. [DOI] [PubMed] [Google Scholar]
- 60. Homer CR, Richmond AL, Rebert NA, Achkar J‐P, McDonald C. ATG16L1 and NOD2 interact in an autophagy‐dependent antibacterial pathway implicated in Crohn's disease pathogenesis. Gastroenterology. 2010;139(5):1630‐1641. 1641.e1‐2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Travassos LH, Carneiro LA, Ramjeet M, et al. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol. 2010;11(1):55‐62. [DOI] [PubMed] [Google Scholar]
- 62. Hsu L‐C, Ali SR, McGillivray S, et al. A NOD2‐NALP1 complex mediates caspase‐1‐dependent IL‐1beta secretion in response to Bacillus anthracis infection and muramyl dipeptide. Proc Natl Acad Sci U S A. 2008;105(22):7803‐7808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Pan Q, Mathison J, Fearns C, et al. MDP‐induced interleukin‐1beta processing requires Nod2 and CIAS1/NALP3. J Leukoc Biol. 2007;82(1):177‐183. [DOI] [PubMed] [Google Scholar]
- 64. Yoo NJ, Park WS, Kim SY, et al. Nod1, a CARD protein, enhances pro‐interleukin‐1beta processing through the interaction with pro‐caspase‐1. Biochem Biophys Res Commun. 2002;299(4):652‐658. [DOI] [PubMed] [Google Scholar]
- 65. Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol. 2013;13(6):397‐411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Lupfer C, Thomas PG, Kanneganti T‐D. Nucleotide oligomerization and binding domain 2‐dependent dendritic cell activation is necessary for innate immunity and optimal CD8 + T cell responses to influenza A virus infection. J Virol. 2014;88(16):8946‐8955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Sabbah A, Chang TH, Harnack R, et al. Activation of innate immune antiviral responses by Nod2. Nat Immunol. 2009;10(10):1073‐1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Kent A, Blander JM. Nod‐like receptors: key molecular switches in the conundrum of cancer. Front Immunol. 2014;5:185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Chen GY, Shaw MH, Redondo G, Nunez G. The innate immune receptor Nod1 protects the intestine from inflammation‐induced tumorigenesis. Cancer Res. 2008;68(24):10060‐10067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Zhan Y, Seregin SS, Chen J, Chen GY. Nod1 limits colitis‐associated tumorigenesis by regulating IFN‐γ production. J Immunol Baltim Md 1950. 2016;196(12):5121‐5129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Couturier‐Maillard A, Secher T, Rehman A, et al. NOD2‐mediated dysbiosis predisposes mice to transmissible colitis and colorectal cancer. J Clin Invest. 2013;123(2):700‐711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Suarez G, Romero‐Gallo J, Piazuelo MB, et al. Modification of Helicobacter pylori peptidoglycan enhances NOD1 activation and promotes cancer of the stomach. Cancer Res. 2015;75(8):1749‐1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Allison CC, Ferrand J, McLeod L, et al. Nucleotide oligomerization domain 1 enhances IFN‐γ signaling in gastric epithelial cells during Helicobacter pylori infection and exacerbates disease severity. J Immunol Baltim Md 1950. 2013;190(7):3706‐3715. [DOI] [PubMed] [Google Scholar]
- 74. Mohammadian Amiri R, Tehrani M, Taghizadeh S, Shokri‐Shirvani J, Fakheri H, Ajami A. Association of nucleotide‐binding oligomerization domain receptors with peptic ulcer and gastric cancer. Iran J Allergy Asthma Immunol. 2016;15(5):355‐362. [PubMed] [Google Scholar]
- 75. Wang X, Jiang W, Duan N, et al. NOD1, RIP2 and Caspase12 are potentially novel biomarkers for oral squamous cell carcinoma development and progression. Int J Clin Exp Pathol. 2014;7(4):1677‐1686. [PMC free article] [PubMed] [Google Scholar]
- 76. Yoon H‐E, Ahn M‐Y, Kwon S‐M, Kim D‐J, Lee J, Yoon J‐H. Nucleotide‐binding oligomerization domain 2 (NOD2) activation induces apoptosis of human oral squamous cell carcinoma cells. J Oral Pathol Med Off Publ Int Assoc Oral Pathol Am Acad Oral Pathol. 2016;45(4):262‐267. [DOI] [PubMed] [Google Scholar]
- 77. Millrud CR, Kvarnhammar AM, Tajti J, Munck‐Wikland E, Uddman R, Cardell LO. Nod‐like receptors in head and neck squamous cell carcinoma. Acta Otolaryngol (Stockh). 2013;133(12):1333‐1344. [DOI] [PubMed] [Google Scholar]
- 78. Chan L‐P, Wang L‐F, Chiang F‐Y, Lee K‐W, Kuo P‐L, Liang C‐H. IL‐8 promotes HNSCC progression on CXCR1/2‐mediated NOD1/RIP2 signaling pathway. Oncotarget. 2016;7(38):61820‐61831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Słotwiński R, Dąbrowska A, Lech G, Słodkowski M, Słotwińska SM. Gene expression disorders of innate antibacterial signaling pathway in pancreatic cancer patients: implications for leukocyte dysfunction and tumor progression. Cent‐Eur J Immunol. 2014;39(4):498‐507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Zaki H. The emerging role of NOD‐like receptors in colorectal cancer. J Neoplasm. 2016;1(1 [Google Scholar]
- 81. Balkwill FR, Capasso M, Hagemann T. The tumor microenvironment at a glance. J Cell Sci. 2012;125(Pt 23):5591‐5596. [DOI] [PubMed] [Google Scholar]
- 82. Egeblad M, Nakasone ES, Werb Z. Tumors as organs: complex tissues that interface with the entire organism. Dev Cell. 2010;18(6):884‐901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Pitt JM, Marabelle A, Eggermont A, Soria J‐C, Kroemer G, Zitvogel L. Targeting the tumor microenvironment: removing obstruction to anticancer immune responses and immunotherapy. Ann Oncol Off J Eur Soc Med Oncol. 2016;27(8):1482‐1492. [DOI] [PubMed] [Google Scholar]
- 84. Temizoz B, Kuroda E, Ishii KJ. Vaccine adjuvants as potential cancer immunotherapeutics. Int Immunol. 2016;28(7):329‐338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Long KB, Beatty GL. Harnessing the antitumor potential of macrophages for cancer immunotherapy. Oncoimmunology. 2013;2(12):e26860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Dzierzbicka K, Kołodziejczyk AM, Wysocka‐Skrzela B, Myśliwski A, Sosnowska D. Synthesis and antitumor activity of conjugates of muramyldipeptide, normuramyldipeptide, and desmuramylpeptides with acridine/acridone derivatives. J Med Chem. 2001;44(22):3606‐3615. [DOI] [PubMed] [Google Scholar]
- 87. Zemlyakov AE, Tsikalova VN, Tsikalov VV, et al. Dialkylmethyl β‐glycosides of N‐acetylmuramyl‐l‐alanyl‐d‐isoglutamine: synthesis and protective antiinfection and cytotoxic activities. Russ J Bioorganic Chem. 2008;34(1):103‐109. [PubMed] [Google Scholar]
- 88. Fujimura T, Yamasaki K, Hidaka T, Ito Y, Aiba S. A synthetic NOD2 agonist, muramyl dipeptide (MDP)‐Lys (L18) and IFN‐β synergistically induce dendritic cell maturation with augmented IL‐12 production and suppress melanoma growth. J Dermatol Sci. 2011;62(2):107‐115. [DOI] [PubMed] [Google Scholar]
- 89. Vidal V, Dewulf J, Bahr GM. Enhanced maturation and functional capacity of monocyte‐derived immature dendritic cells by the synthetic immunomodulator murabutide. Immunology. 2001;103(4):479‐487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Yang H‐Z, Xu S, Liao X‐Y, et al. A novel immunostimulator, N2‐[α‐O‐Benzyl‐N‐(acetylmuramyl)‐l‐alanyl‐d‐ isoglutaminyl]‐N6‐trans‐(m‐nitrocinnamoyl)‐l‐lysine, and its adjuvancy on the hepatitis B surface antigen. J Med Chem. 2005;48(16):5112‐5122. [DOI] [PubMed] [Google Scholar]
- 91. Haabeth OAW, Bogen B, Corthay A. A model for cancer‐suppressive inflammation. Oncoimmunology. 2012;1(7):1146‐1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Pasquale A, Preiss S, Silva F, Garçon N. Vaccine adjuvants: from 1920 to 2015 and beyond. Vaccines. 2015;3(2):320‐343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Geddes K, Magalhães JG, Girardin SE. Unleashing the therapeutic potential of NOD‐like receptors. Nat Rev Drug Discov. 2009;8(6):465‐479. [DOI] [PubMed] [Google Scholar]
- 94. Maisonneuve C, Bertholet S, Philpott DJ, De Gregorio E. Unleashing the potential of NOD‐ and Toll‐like agonists as vaccine adjuvants. Proc Natl Acad Sci USA. 2014;111(34):12294‐12299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Adam A, Ellouz F, Ciorbaru R, Petit JF, Lederer E. Peptidoglycan adjuvants: minimal structure required for activity. Z Immunitatsforsch Exp Klin Immunol. 1975;149(2‐4):341‐348. [PubMed] [Google Scholar]
- 96. Ellouz F, Adam A, Ciorbaru R, Lederer E. Minimal structural requirements for adjuvant activity of bacterial peptidoglycan derivatives. Biochem Biophys Res Commun. 1974;59(4):1317‐1325. [DOI] [PubMed] [Google Scholar]
- 97. Kotani S, Watanabe Y, Shimono T, Narita T, Kato K. Immunoadjuvant activities of cell walls, their water‐soluble fractions and peptidoglycan subunits, prepared from various gram‐positive bacteria, and of synthetic N‐acetylmuramyl peptides. Z Immunitatsforsch Exp Klin Immunol. 1975;149(2‐4):302‐319. [PubMed] [Google Scholar]
- 98. Traub S, von Aulock S, Hartung T, Hermann C. MDP and other muropeptides—direct and synergistic effects on the immune system. J Endotoxin Res. 2006;12(2):69‐85. [DOI] [PubMed] [Google Scholar]
- 99. Saiki I, Fidler IJ. Synergistic activation by recombinant mouse interferon‐gamma and muramyl dipeptide of tumoricidal properties in mouse macrophages. J Immunol Baltim Md 1950. 1985;135(1):684‐688. [PubMed] [Google Scholar]
- 100. Souvannavong V, Brown S, Adam A. Muramyl dipeptide (MDP) synergizes with interleukin 2 and interleukin 4 to stimulate, respectively, the differentiation and proliferation of B cells. Cell Immunol. 1990;126(1):106‐116. [DOI] [PubMed] [Google Scholar]
- 101. Laroui H, Yan Y, Narui Y, et al. l‐Ala‐γ‐d‐Glu‐meso‐diaminopimelic acid (DAP) interacts directly with leucine‐rich region domain of nucleotide‐binding oligomerization domain 1, increasing phosphorylation activity of receptor‐interacting serine/threonine‐protein kinase 2 and its interaction with nucleotide‐binding oligomerization domain 1. J Biol Chem. 2011;286(35):31003‐31013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Fujimoto Y, Fukase K. Structures, synthesis, and human Nod1 stimulation of immunostimulatory bacterial peptidoglycan fragments in the environment. J Nat Prod. 2011;74(3):518‐525. [DOI] [PubMed] [Google Scholar]
- 103. Jakopin Ž, Gobec M, Kodela J, Hazdovac T, Mlinarič‐Raščan I, Sollner Dolenc M. Synthesis of conformationally constrained γ‐d‐glutamyl‐meso‐diaminopimelic acid derivatives as ligands of nucleotide‐binding oligomerization domain protein 1 (Nod1). Eur J Med Chem. 2013;69:232‐243. [DOI] [PubMed] [Google Scholar]
- 104. Izumi S, Nakahara K, Gotoh T, et al. Antitumor effects of novel immunoactive peptides, FK‐156 and its synthetic derivatives. J Antibiot (Tokyo). 1983;36(5):566‐574. [DOI] [PubMed] [Google Scholar]
- 105. Schultz RM, Altom MG. Macrophage involvement in the antitumor activity of a synthetic acyltripeptide (FK‐565) against experimental lung carcinoma metastases. J Immunopharmacol. 1986;8(4):515‐528. [DOI] [PubMed] [Google Scholar]
- 106. Inamura N, Nakahara K, Kino T, et al. Activation of tumoricidal properties in macrophages and inhibition of experimentally‐induced murine metastases by a new synthetic acyltripeptide, FK‐565. J Biol Response Mod. 1985;4(4):408‐417. [PubMed] [Google Scholar]
- 107. Parant M, Parant F, Chedid L, Yapo A, Petit JF, Lederer E. Fate of the synthetic immunoadjuvant, muramyl dipeptide (14C‐labelled) in the mouse. Int J Immunopharmacol. 1979;1(1):35‐41. [DOI] [PubMed] [Google Scholar]
- 108. Fogler WE, Wade R, Brundish DE, Fidler IJ. Distribution and fate of free and liposome‐encapsulated [3H]nor‐muramyl dipeptide and [3H]muramyl tripeptide phosphatidylethanolamine in mice. J Immunol Baltim Md 1950. 1985;135(2):1372‐1377. [PubMed] [Google Scholar]
- 109. Ogawa C, Liu Y‐J, S. kobayashi K. Muramyl dipeptide and its derivatives: peptide adjuvant in immunological disorders and cancer therapy. Curr Bioact Compd. 2011;7(3):180‐197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Pashenkov MV, Dagil YA, Pinegin BV. NOD1 and NOD2: molecular targets in prevention and treatment of infectious diseases. Int Immunopharmacol. 2018;54:385‐400. [DOI] [PubMed] [Google Scholar]
- 111. Nardin A, Lefebvre M, Labroquere K, Faure O, Abastado J. Liposomal muramyl tripeptide phosphatidylethanolamine: targeting and activating macrophages for adjuvant treatment of osteosarcoma. Curr Cancer Drug Targets. 2006;6(2):123‐133. [DOI] [PubMed] [Google Scholar]
- 112. Asano T, McWatters A, An T, Matsushima K, Kleinerman ES. Liposomal muramyl tripeptide up‐regulates interleukin‐1 alpha, interleukin‐1 beta, tumor necrosis factor‐alpha, interleukin‐6 and interleukin‐8 gene expression in human monocytes. J Pharmacol Exp Ther. 1994;268(2):1032‐1039. [PubMed] [Google Scholar]
- 113. Kleinerman ES, Erickson KL, Schroit AJ, Fogler WE, Fidler IJ. Activation of tumoricidal properties in human blood monocytes by liposomes containing lipophilic muramyl tripeptide. Cancer Res. 1983;43(5):2010‐2014. [PubMed] [Google Scholar]
- 114. Maeda M, Knowles RD, Kleinerman ES. Muramyl tripeptide phosphatidylethanolamine encapsulated in liposomes stimulates monocyte production of tumor necrosis factor and interleukin‐1 in vitro. Cancer Commun. 1991;3(10‐11):313‐321. [DOI] [PubMed] [Google Scholar]
- 115. European Medicines Agency . Mepact 4 mg powder for suspension for infusion—Summary of Product Characteristics (SmPC). http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_‐_Product_Information/human/000802/WC500026565.pdf Accessed June 1, 2018.
- 116. Bucana CD, Hoyer LC, Schroit AJ, Kleinerman E, Fidler IJ. Ultrastructural studies of the interaction between liposome‐activated human blood monocytes and allogeneic tumor cells in vitro. Am J Pathol. 1983;112(1):101‐111. [PMC free article] [PubMed] [Google Scholar]
- 117. Galligioni E, Quaia M, Spada A, et al. Activation of cytolytic activity in peripheral blood monocytes of renal cancer patients against non‐cultured autologous tumor cells. Int J Cancer. 1993;55(3):380‐385. [DOI] [PubMed] [Google Scholar]
- 118. Galligioni E, Santarosa M, Favaro D, Spada A, Talamini R, Quaia M. In vitro synergic effect of interferon gamma combined with liposomes containing muramyl tripeptide on human monocyte cytotoxicity against fresh allogeneic and autologous tumor cells. Tumori. 1994;80(5):385‐391. [DOI] [PubMed] [Google Scholar]
- 119. Sone S, Utsugi T, Tandon P, Yanagawa H, Okubo A, Ogura T. Tumor cytotoxicity and interleukin 1 production of blood monocytes of lung cancer patients. Cancer Immunol Immunother CII. 1990;30(6):357‐362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Sone S, Tandon P, Utsugi T, et al. Synergism of recombinant human interferon gamma with liposome‐encapsulated muramyl tripeptide in activation of the tumoricidal properties of human monocytes. Int J Cancer. 1986;38(4):495‐500. [DOI] [PubMed] [Google Scholar]
- 121. Kurzman ID, MacEwen EG, Rosenthal RC, et al. Adjuvant therapy for osteosarcoma in dogs: results of randomized clinical trials using combined liposome‐encapsulated muramyl tripeptide and cisplatin. Clin Cancer Res. 1995;1(12):1595‐1601. [PubMed] [Google Scholar]
- 122. MacEwen EG, Kurzman ID, Rosenthal RC, et al. Therapy for osteosarcoma in dogs with intravenous injection of liposome‐encapsulated muramyl tripeptide. J Natl Cancer Inst. 1989;81(12):935‐938. [DOI] [PubMed] [Google Scholar]
- 123. MacEwen EG, Kurzman ID, Helfand S, et al. Current studies of liposome muramyl tripeptide (CGP 19835A lipid) therapy for metastasis in spontaneous tumors: a progress review. J Drug Target. 1994;2(5):391‐396. [DOI] [PubMed] [Google Scholar]
- 124. Vail DM, MacEwen EG, Kurzman ID, et al. Liposome‐encapsulated muramyl tripeptide phosphatidylethanolamine adjuvant immunotherapy for splenic hemangiosarcoma in the dog: a randomized multi‐institutional clinical trial. Clin Cancer Res. 1995;1(10):1165‐1170. [PubMed] [Google Scholar]
- 125. Fidler I. Optimization and limitations of systemic treatment of murine melanoma metastases with liposomes containing muramyl tripeptide phosphatidylethanolamine. Cancer Immunol Immunother CII. 1986;21(3):169‐173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Fox LE, MacEwen EG, Kurzman RD, et al. Liposome‐encapsulated muramyl tripeptide phosphatidylethanolamine for the treatment of feline mammary adenocarcinoma—a multicenter randomized double‐blind study. Cancer Biother. 1995;10(2):125‐130. [DOI] [PubMed] [Google Scholar]
- 127. Teske E, Rutteman GR, vd Ingh TS, van Noort R, Misdorp W. Liposome‐encapsulated muramyl tripeptide phosphatidylethanolamine (L‐MTP‐PE): a randomized clinical trial in dogs with mammary carcinoma. Anticancer Res. 1998;18(2A):1015‐1019. [PubMed] [Google Scholar]
- 128. Hudson MM, Snyder JS, Jaffe N, Kleinerman ES. In vitro and in vivo effect of adriamycin therapy on monocyte activation by liposome‐encapsulated immunomodulators. Cancer Res. 1988;48:5256‐5263. [PubMed] [Google Scholar]
- 129. Kleinerman ES, Meyers PA, Raymond AK, Gano JB, Jia SF, Jaffe N. Combination therapy with ifosfamide and liposome‐encapsulated muramyl tripeptide: tolerability, toxicity, and immune stimulation. J Immunother Emphasis Tumor Immunol. 1995;17(3):181‐193. [DOI] [PubMed] [Google Scholar]
- 130. Kleinerman ES, Snyder JS, Jaffe N. Influence of chemotherapy administration on monocyte activation by liposomal muramyl tripeptide phosphatidylethanolamine in children with osteosarcoma. J Clin Oncol. 1991;9(2):259‐267. [DOI] [PubMed] [Google Scholar]
- 131. Bergers JJ, Den Otter W, Dullens HF, et al. Effect of immunomodulators on specific tumor immunity induced by liposome‐encapsulated tumor‐associated antigens. Int J Cancer. 1994;56(5):721‐726. [DOI] [PubMed] [Google Scholar]
- 132. Meyers PA. Muramyl tripeptide (mifamurtide) for the treatment of osteosarcoma. Expert Rev Anticancer Ther. 2009;9(8):1035‐1049. [DOI] [PubMed] [Google Scholar]
- 133. Frampton JE. Mifamurtide: a review of its use in the treatment of osteosarcoma. Paediatr Drugs. 2010;12(3):141‐153. [DOI] [PubMed] [Google Scholar]
- 134. Azuma I. Development of the cytokine inducer romurtide: experimental studies and clinical application. Trends Pharmacol Sci. 1992;13(12):425‐428. [DOI] [PubMed] [Google Scholar]
- 135. Azuma I. Review: inducer of cytokines in vivo: overview of field and romurtide experience. Int J Immunopharmacol. 1992;14(3):487‐496. [DOI] [PubMed] [Google Scholar]
- 136. Nakajima R, Yshida Y, Akahane K, Sekiguchi M, Osada Y. Stimulatory effect of romurtide on hematopoiesis in monkeys. Arzneimittelforschung. 1991;41(1):60‐65. [PubMed] [Google Scholar]
- 137. Nakajima R, Ishida Y, Yamaguchi F, et al. Beneficial effect of muroctasin on experimental leukopenia induced by cyclophosphamide or irradiation in mice. Arzneimittelforschung. 1988;38(7A):986‐992. [PubMed] [Google Scholar]
- 138. Furuse K, Sakuma A. Activation of the cytokine network by muroctasin as a remedy for leukopenia and thrombopenia. Arzneimittelforschung. 1989;39(8):915‐917. [PubMed] [Google Scholar]
- 139. Tsubura E, Nomura T, Niitani H, et al. Restorative activity of muroctasin on leukopenia associated with anticancer treatment. Arzneimittelforschung. 1988;38(7A):1070‐1074. [PubMed] [Google Scholar]
- 140. Nitta Y, Sugita T, Ikuta Y, Murakami T. Inhibitory effect of liposomal MDP‐Lys on lung metastasis of transplantable osteosarcoma in hamster. Oncol Res. 2000;12(1):25‐31. [DOI] [PubMed] [Google Scholar]
- 141. Yoo YC, Saiki I, Sato K, Azuma I. MDP‐Lys(L18), a lipophilic derivative of muramyl dipeptide, inhibits the metastasis of haematogenous and non‐haematogenous tumours in mice. Vaccine. 1994;12(2):175‐160. [DOI] [PubMed] [Google Scholar]
- 142. Azuma I, Sugimura K, Yamawaki M, et al. Adjuvant activity of synthetic 6‐O‐"mycoloyl"‐N‐ acetylmuramyl‐l‐alanyl‐d‐isoglutamine and related compounds. Infect Immun. 1978;20(3):600‐607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Uemiya M, Sugimura K, Kusama T, et al. Adjuvant activity of 6‐O‐mycoloyl derivatives of N‐acetylmuramyl‐l‐seryl‐d‐isoglutamine and related compounds in mice and guinea pigs. Infect Immun. 1979;24(1):83‐89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Kusumoto S, Inage M, Shiba T, Azuma I, Yamamura Y. Synthesis of long chain fatty acid esters of N‐acetylmuramyl‐l‐alanyl‐d‐isoglutamine in relation to antitumor activity. Tetrahedron Lett. 1978;19(49):4899‐4902. [Google Scholar]
- 145. Kataoka T, Tokunaga T. A synthetic adjuvant effective in inducing antitumor immunity. Jpn J Cancer Res Gann. 1988;79(7):817‐820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Kataoka T, Kinomoto M, Takegawa M, Tokunaga T. Effect of a synthetic adjuvant for inducing anti‐tumour immunity. Vaccine. 1991;9(5):300‐302. [DOI] [PubMed] [Google Scholar]
- 147. Yung choon yoo Y, Saiki I, Sato K, Azuma I. B30‐MDP, a synthetic muramyl dipeptide derivative for tumour vaccination to enhance antitumour immunity and antimetastatic effect in mice. Vaccine. 1992;10(11):792‐797. [DOI] [PubMed] [Google Scholar]
- 148. Kobayashi S, Fukuda T, Imada I, Fujino M, Azuma I, Yamamura Y. Novel quinonyl derivatives of muramyl dipeptide possessing potent antitumor activity. Chem Pharm Bull (Tokyo). 1979;27(12):3193‐3196. [DOI] [PubMed] [Google Scholar]
- 149. Azuma I, Yamawaki M, Uemiya M, et al. Adjuvant and antitumor activities of quinonyl‐N‐acetylmuramyldipeptides. Gan. 1979;70(6):847‐848. [PubMed] [Google Scholar]
- 150. Saiki I, Tanio Y, Yamawaki M, et al. Adjuvant activities of quinonyl‐N‐acetyl muramyl dipeptides in mice and guinea pigs. Infect Immun. 1981;31(1):114‐121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Tanio Y, Souma H, Tokushima Y, Yamamura Y, Azuma I. Regression of line‐10 hepatocarcinoma with synthetic quinonyl muramyl dipeptide in strain‐2 guinea pigs. Gan. 1983;74(2):192‐195. [PubMed] [Google Scholar]
- 152. Saiki I, Tanio Y, Yamamoto KI, Yamamura Y, Azuma I. Effect of quinonyl‐N‐acetyl muramyl dipeptide on immune responses in tumor‐bearing mice. Infect Immun. 1983;39(1):137‐141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Phillips NC, Moras ML, Chedid L, Lefrancier P, Bernard JM. Activation of alveolar macrophage tumoricidal activity and eradication of experimental metastases by freeze‐dried liposomes containing a new lipophilic muramyl dipeptide derivative. Cancer Res. 1985;45:128‐134. [PubMed] [Google Scholar]
- 154. Phillips NC, Chedid L, Bernard JM, Level M, Lefrancier P. Induction of murine macrophage tumoricidal activity and treatment of experimental pulmonary metastases by liposomes containing lipophilic muramyl dipeptide analogs. J Biol Response Mod. 1987;6(6):678‐691. [PubMed] [Google Scholar]
- 155. Phillips NC, Tsao MS. Inhibition of experimental liver tumor growth in mice by liposomes containing a lipophilic muramyl dipeptide derivative. Cancer Res. 1989;49(4):936‐939. [PubMed] [Google Scholar]
- 156. Phillips NC, Tsao MS. Liposomal muramyl dipeptide therapy of experimental M5076 liver metastases in mice. Cancer Immunol Immunother CII. 1991;33(2):85‐90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Brodt P, Blore J, Phillips N, Munzer JS, Rioux J. Inhibition of murine hepatic tumor growth by liposomes containing a lipophilic muramyl dipeptide. Cancer Immunol Immunother CII. 1989;28(1):54‐58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Phillips NC, Rioux J, Tsao MS. Activation of murine Kupffer cell tumoricidal activity by liposomes containing lipophilic muramyl dipeptide. Hepatol Baltim Md. 1988;8(5):1046‐1050. [DOI] [PubMed] [Google Scholar]
- 159. Worth LL, Jia SF, An T, Kleinerman ES. ImmTher, a lipophilic disaccharide derivative of muramyl dipeptide, up‐regulates specific monocyte cytokine genes and activates monocyte‐mediated tumoricidal activity. Cancer Immunol Immunother CII. 1999;48(6):312‐320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Vosika GJ, Cornelius DA, Bennek JA, Sadlik JR, Gilbert CW. Immunologic and toxicologic study of disaccharide tripeptide glycerol dipalmitoyl: a new lipophilic immunomodulator. Mol Biother. 1990;2(1):50‐56. [PubMed] [Google Scholar]
- 161. Vosika GJ, Cornelius DA, Gilbert CW, et al. Phase I trial of ImmTher, a new liposome‐incorporated lipophilic disaccharide tripeptide. J Immunother. 1991;10(4):256‐266. [DOI] [PubMed] [Google Scholar]
- 162. U. S. National Library of Medicine . Vincristine, Doxorubicin, Cyclophosphamide and Dexrazoxane (VACdxr) in High Risk Ewing's Sarcoma Patients. https://clinicaltrials.gov/ct2/show/NCT00038142 Accessed June 15, 2018.
- 163. U. S. Food and Drug Administration . Search Orphan Drug Designations and Approvals. https://www.accessdata.fda.gov/scripts/opdlisting/oopd/index.cfm Accessed June 15, 2018.
- 164. Phillips NC, Moras ML, Chedid L, et al. Activation of macrophage cytostatic and cytotoxic activity in vitro by liposomes containing a new lipophilic muramyl peptide derivative, MDP‐l‐alanyl‐cholesterol (MTP‐CHOL). J Biol Response Mod. 1985;4(5):464‐474. [PubMed] [Google Scholar]
- 165. Yu WP, Barratt GM, Devissaguet JP, Puisieux F. Anti‐metastatic activity in vivo of MDP‐l‐alanyl‐cholesterol (MTP‐Chol) entrapped in nanocapsules. Int J Immunopharmacol. 1991;13(2‐3):167‐173. [DOI] [PubMed] [Google Scholar]
- 166. Barratt G, Puisieux F, Yu WP, Foucher C, Fessi H, Devissaguet JP. Anti‐metastatic activity of MDP‐l‐alanyl‐cholesterol incorporated into various types of nanocapsules. Int J Immunopharmacol. 1994;16(5‐6):457‐461. [DOI] [PubMed] [Google Scholar]
- 167. Aliño SF, Iruarrizaga A, Alfaro J, Almena A, Lejarreta M, Unda FJ. Antimetastatic effects of liposome entrapped indomethacin. Life Sci. 1991;48(2):149‐154. [DOI] [PubMed] [Google Scholar]
- 168. Roche AC, Bailly P, Monsigny M. Macrophage activation by MDP bound to neoglycoproteins: metastasis eradication in mice. Invasion Metastasis. 1985;5(4):218‐232. [PubMed] [Google Scholar]
- 169. Roche AC, Bailly P, Midoux P, Monsigny M. Selective macrophage activation by muramyldipeptide bound to monoclonal antibodies specific for mouse tumor cells. Cancer Immunol Immunother CII. 1984;18(3):155‐159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Srividya S, Roy RP, Basu SK, Mukhopadhyay A. Scavenger receptor‐mediated delivery of muramyl dipeptide activates antitumor efficacy of macrophages by enhanced secretion of tumor‐suppressive cytokines. J Leukoc Biol. 2000;67(5):683‐690. [DOI] [PubMed] [Google Scholar]
- 171. Tabata Y, Ikada Y. Targeting of muramyl dipeptide to macrophages by gelatin conjugation to enhance their in vivo antitumor activity. J Controlled Release. 1993;27(1):79‐88. [Google Scholar]
- 172. Srividya S, Roy RP, Basu SK, Mukhopadhyay A. Selective activation of antitumor activity of macrophages by the delivery of muramyl dipeptide using a novel polynucleotide‐based carrier recognized by scavenger receptors. Biochem Biophys Res Commun. 2000;268(3):772‐777. [DOI] [PubMed] [Google Scholar]
- 173. Cholewiński G, Dzierzbicka K, Kołodziejczyk AM. Natural and synthetic acridines/acridones as antitumor agents: their biological activities and methods of synthesis. Pharmacol Rep. 2011;63(2):305‐336. [DOI] [PubMed] [Google Scholar]
- 174. Dzierzbicka K, Kołodziejczyk AM. Synthesis and antitumor activity of conjugates of muramyldipeptide or normuramyldipeptide with hydroxyacridine/acridone derivatives. J Med Chem. 2003;46(1):183‐189. [DOI] [PubMed] [Google Scholar]
- 175. Dzierzbicka K, Trzonkowski P, Sewerynek P, Myśliwski A. Synthesis and cytotoxic activity of conjugates of muramyl and normuramyl dipeptides with batracylin derivatives. J Med Chem. 2003;46(6):978‐986. [DOI] [PubMed] [Google Scholar]
- 176. Trzonkowski P, Dzierzbicka K, Bociewicz J, Szmit E, Myśliwski A. Biological activity of conjugates of muramyl dipeptides with batracylin derivatives. Int Immunopharmacol. 2005;5(2):241‐251. [DOI] [PubMed] [Google Scholar]
- 177. Li X, Yu J, Xu S, et al. Chemical conjugation of muramyl dipeptide and paclitaxel to explore the combination of immunotherapy and chemotherapy for cancer. Glycoconj J. 2008;25(5):415‐425. [DOI] [PubMed] [Google Scholar]
- 178. Ma Y, Zhao N, Liu G. Conjugate (MTC‐220) of muramyl dipeptide analogue and paclitaxel prevents both tumor growth and metastasis in mice. J Med Chem. 2011;54(8):2767‐2777. [DOI] [PubMed] [Google Scholar]
- 179. Wu Y, Zhou BP. TNF‐alpha/NF‐kappaB/Snail pathway in cancer cell migration and invasion. Br J Cancer. 2010;102(4):639‐644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Buniatian AA, Inviiaeva EV, Nikoda VV, Vinnitskiĭ LI. [Immunocorrectors in the complex treatment of postoperative suppurative‐inflammatory complications in surgical patients and monitoring of immunological parameters]. Anesteziol Reanimatol. 2004;5:79‐83. [PubMed] [Google Scholar]
- 181. Svistunova AS, Pinegin BV, Selitskaia RP, et al. [The use of immunomodulator likopid in the combined treatment pulmonary tuberculosis]. Probl Tuberk. 2002;3:21‐25. [PubMed] [Google Scholar]
- 182. Andronova T, Ivanov V, Petrov R, Mikhailova A. The Structure and Immunomodulating Function of Glucosaminylmuramyl Peptides/the Thymus‐ and Bone Marrow‐Derived Immunoregulatory Peptides. London: Harwood Academic; 1992. [Google Scholar]
- 183. Akhmatova NK, Semenova IB, Donenko FV, Kiselevskiĭ MV, Kurbatova EA, Egorova NB. [Immunomodulators of microbial origin enhance cytotoxicity of human mononuclear leukocytes and reduce metastatic progression of Lewis lung carcinoma in mice]. Zh Mikrobiol Epidemiol Immunobiol. 2006;6:35‐40. [PubMed] [Google Scholar]
- 184. Shimizu T, Iwamoto Y, Yanagihara Y, Ikeda K, Achiwa K. Combined effects of synthetic lipid A analogs or bacterial lipopolysaccharide with glucosaminylmuramyl dipeptide on antitumor activity against Meth A fibrosarcoma in mice. Int J Immunopharmacol. 1992;14(8):1415‐1420. [DOI] [PubMed] [Google Scholar]
- 185. Petrova EE, Valyakina TI, Simonova MA, et al. Muramyl peptides augment cytotoxic effect of tumor necrosis factor‐alpha in combination with cytotoxic drugs on tumor cells. Int Immunopharmacol. 2006;6(9):1377‐1386. [DOI] [PubMed] [Google Scholar]
- 186. Petrova EE, Valyakina TI, Komaleva RL, Simonova MA, Nesmeyanov VA. Glucosaminylmuramyl dipeptide potentiates the effects of tumor necrosis factor‐alpha and cisplatin on transformed cells in vitro. Bull Exp Biol Med. 2007;143(2):251‐254. [DOI] [PubMed] [Google Scholar]
- 187. Petrova EE, Simonova MA, Komaleva RL, et al. GMDP augments antitumor action of the CP/TNFα combination in vivo. Biomed Pharmacother. 2010;64(4):240‐248. [DOI] [PubMed] [Google Scholar]
- 188. Wang X, Lin Y. Tumor necrosis factor and cancer, buddies or foes? Acta Pharmacol Sin. 2008;29(11):1275‐1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Bahr GM, Darcissac E, Bevec D, Dukor P, Chedid L. Immunopharmacological activities and clinical development of muramyl peptides with particular emphasis on murabutide. Int J Immunopharmacol. 1995;17(2):117‐131. [DOI] [PubMed] [Google Scholar]
- 190. Jakopin Z. Murabutide revisited: a review of its pleiotropic biological effects. Curr Med Chem. 2013;20(16):2068‐2079. [DOI] [PubMed] [Google Scholar]
- 191. Riveau G. Central pyrogenic activity of muramyl dipeptide. J Exp Med. 1980;152(4):869‐877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Telzak E, Wolff SM, Dinarello CA, et al. Clinical evaluation of the immunoadjuvant murabutide, a derivative of MDP, administered with a tetanus toxoid vaccine. J Infect Dis. 1986;153(3):628‐633. [DOI] [PubMed] [Google Scholar]
- 193. Bahr GM, Darcissac E, Pouillart PR, Chedid LA. Synergistic effects between recombinant interleukin‐2 and the synthetic immunomodulator murabutide: selective enhancement of cytokine release and potentiation of antitumor activity. J Interferon Cytokine Res. 1996;16(2):169‐178. [DOI] [PubMed] [Google Scholar]
- 194. Bahr GM, Pouillart PR, Chedid LA. Enhancement in vivo of the antiinflammatory and antitumor activities of type I interferon by association with the synthetic immunomodulator murabutide. J Interferon Cytokine Res. 1996;16(4):297‐306. [DOI] [PubMed] [Google Scholar]
- 195. Azuma I, Okumura H, Saiki I, et al. Adjuvant activity of carbohydrate analogs of N‐acetylmuramyl‐L‐alanyl‐D‐isoglutamine on the induction of delayed‐type hypersensitivity to azobenzenearsonate‐N‐acetyl‐l‐tyrosine in guinea pigs. Infect Immun. 1981;33(3):834‐839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Danklmaier J, Hünig H. Synthesis of acyclic analogs of N‐acetylmuramyl‐l‐alanyl‐d‐isoglutamine (MDP). Liebigs Ann Chem. 1990;1990:145‐150. [Google Scholar]
- 197. Gobec M, Mlinarič‐Raščan I, Dolenc MS, Jakopin Ž. Structural requirements of acylated Gly‐l‐Ala‐d‐Glu analogs for activation of the innate immune receptor NOD2. Eur J Med Chem. 2016;116:1‐12. [DOI] [PubMed] [Google Scholar]
- 198. Gobec M, Tomašič T, Štimac A, et al. Discovery of nanomolar desmuramylpeptide agonists of the innate immune receptor nucleotide‐binding oligomerization domain‐containing protein 2 (NOD2) possessing immunostimulatory properties. J Med Chem. 2018;61(7):2707‐2724. [DOI] [PubMed] [Google Scholar]
- 199. Jakopin Ž, Corsini E, Gobec M, Mlinarič‐Raščan I, Dolenc MS. Design, synthesis and biological evaluation of novel desmuramyldipeptide analogs. Eur J Med Chem. 2011;46(9):3762‐3777. [DOI] [PubMed] [Google Scholar]
- 200. Jakopin Ž, Gobec M, Mlinarič‐Raščan I, Sollner Dolenc M. Immunomodulatory properties of novel nucleotide oligomerization domain 2 (NOD2) agonistic desmuramyldipeptides. J Med Chem. 2012;55(14):6478‐6488. [DOI] [PubMed] [Google Scholar]
- 201. Migliore‐Samour D, Bouchaudon J, Floc'h F, et al. A short lipopeptide, representative of a new family of immunological adjuvants devoid of sugar. Life Sci. 1980;26(11):883‐888. [DOI] [PubMed] [Google Scholar]
- 202. Sersa G, Novakovic S, Stalc A. Antitumor effect of recombinant human tumor necrosis factor‐alpha analog combined with desmuramyl dipeptides LK‐409 or LK‐410 on sarcoma in mice. Mol Biother. 1992;4(4):188‐192. [PubMed] [Google Scholar]
- 203. Kikelj D, Pečar S, Kotnik V, et al. N‐{trans‐2‐[[2′‐(Acetylamino)cyclohexyl]oxy]acetyl}‐l‐alanyl‐d‐glutamic acid: a novel immunologically active carbocyclic muramyl dipeptide analogue. J Med Chem. 1998;41(4):530‐539. [DOI] [PubMed] [Google Scholar]
- 204. Kotnik V, Štalc A. Potential therapeutic indications for synthetic desmuramyl MDP analogue (LK‐409). Pflugers Arch. 1996;431(6 Suppl 2):R235‐R236. [DOI] [PubMed] [Google Scholar]
- 205. Pabst MJ, Beranova‐Giorgianni S, Krueger JM. Effects of muramyl peptides on macrophages, monokines, and sleep. Neuroimmunomodulation. 1999;6(4):261‐283. [DOI] [PubMed] [Google Scholar]
- 206. Adam A, Devys M, Souvannavong V, Lefrancier P, Choay J, Lederer E. Correlation of structure and adjuvant activity of N‐acetyl muramyl‐l‐alanyl‐d‐isoglutamine (MDP), its derivatives and analogues. Anti‐adjuvant and competition properties of stereoisomers. Biochem Biophys Res Commun. 1976;72(1):339‐346. [DOI] [PubMed] [Google Scholar]
- 207. Kamisango K, Saiki I, Tanio Y, et al. Chemical synthesis and adjuvant activity of N‐acetylmuramyl‐l‐alanyl‐d‐isoglutamine (MDP) analogs 1, 2. Chem Pharm Bull (Tokyo). 1981;29(6):1644‐1654. [DOI] [PubMed] [Google Scholar]
- 208. Kobayashi S, Fukuda T, Yukimasa H, Fujino M, Azuma I, Yamamura Y. Synthesis of muramyl dipeptide analogs with enhanced adjuvant activity. Bull Chem Soc Jpn. 1980;53(9):2570‐2577. [Google Scholar]
- 209. Adam A, Lederer E. Muramyl peptides: immunomodulators, sleep factors, and vitamins. Med Res Rev. 1984;4(2):111‐152. [DOI] [PubMed] [Google Scholar]
- 210. Lefrancier P, Lederer E. Muramyl‐peptides. Pure Appl Chem. 1987;59(3):449‐454. [Google Scholar]
- 211. Hasegawa A, Hioki Y, Kiso M, Okumura H, Azuma I. Synthesis and biological activities of N‐acetyl‐1‐thiomuramoyl‐l‐alanyl‐d‐isoglutamine and some of its lipophilic derivatives. Carbohydr Res. 1983;123(2):183‐199. [DOI] [PubMed] [Google Scholar]
- 212. Hasegawa A, Hioki Y, Seki E, Kiso M, Azuma I. Synthesis of N‐[2‐O‐(2‐acetamido‐1, 5‐anhydro‐2, 3‐dideoxy‐d‐glucitol‐3‐yl)‐d‐lactoyl]‐l‐alanyl‐d‐isoglutamine (1‐deoxy‐MDP) and some of its lipophilic analogs, and their immunoadjuvant activities. Agric Biol Chem. 1986;50(7):1873‐1878. [Google Scholar]
- 213. Nagai Y, Akiyama K, Kotani S, et al. Structural specificity of synthetic peptide adjuvant for induction of experimental allergic encephalomyelitis. Cell Immunol. 1978;35(1):168‐172. [DOI] [PubMed] [Google Scholar]
- 214. Coulombe F, Divangahi M, Veyrier F, et al. Increased NOD2‐mediated recognition of N‐glycolyl muramyl dipeptide. J Exp Med. 2009;206(8):1709‐1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Farkaš J, Ledvina M, Brokeš J, Ježek J, Zajíček J, Zaoral M. The synthesis of O‐(2‐acetamido‐2‐deoxy‐β‐d‐glucopyranosyl)‐(1→4)‐N‐acetylnormuramoyl‐l‐α‐aminobutanoyl‐d‐isoglutamine. Carbohydr Res. 1987;163(1):63‐72. [DOI] [PubMed] [Google Scholar]
- 216. Hasegawa A, Kaneda Y, Amano M, Kiso M, Azuma I. A facile synthesis of N‐acetyl‐muramyl‐l‐alanyl‐d‐isoglutamine and its carbohydrate analogs, and their immunoadjuvant activities. Agric Biol Chem. 1978;42(11):2187‐2189. [Google Scholar]
- 217. Dong Y, Wang S, Wang C, Li Z, Ma Y, Liu G. Antagonizing NOD2 signaling with conjugates of paclitaxel and muramyl dipeptide derivatives sensitizes paclitaxel therapy and significantly prevents tumor metastasis. J Med Chem. 2017;60(3):1219‐1224. [DOI] [PubMed] [Google Scholar]
- 218. Wang S, Yang J, Li X, et al. Discovery of 1,4‐benzodiazepine‐2,5‐dione (BZD) derivatives as dual nucleotide binding oligomerization domain containing 1/2 (NOD1/NOD2) antagonists sensitizing paclitaxel (PTX) to suppress Lewis lung carcinoma (LLC) growth in vivo. J Med Chem. 2017;60(12):5162‐5192. [DOI] [PubMed] [Google Scholar]
- 219. Wen X, Zheng P, Ma Y, et al. Salutaxel, a conjugate of docetaxel and a muramyl dipeptide (MDP) analogue, acts as multifunctional prodrug that inhibits tumor growth and metastasis. J Med Chem. 2018;61(4):1519‐1540. [DOI] [PubMed] [Google Scholar]
- 220. Umansky V, Blattner C, Gebhardt C, Utikal J. The role of myeloid‐derived suppressor cells (MDSC) in cancer progression. Vaccines. 2016;4(4):36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Ries C. Cytokine functions of TIMP‐1. Cell Mol Life Sci. 2014;71(4):659‐672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Donskov F. Immunomonitoring and prognostic relevance of neutrophils in clinical trials. Semin Cancer Biol. 2013;23(3):200‐207. [DOI] [PubMed] [Google Scholar]
- 223. Han Y, Yu Z, Wen S, Zhang B, Cao X, Wang X. Prognostic value of chemotherapy‐induced neutropenia in early‐stage breast cancer. Breast Cancer Res Treat. 2012;131(2):483‐490. [DOI] [PubMed] [Google Scholar]
- 224. Wei L‐J, Tan X, Fan G‐J, Jiang Y‐N, Shah QA. Role of the NOD1/NF‐κB pathway on bovine neutrophil responses to crude lipopolysaccharide. Vet J Lond Engl 1997. 2016;214:24‐31. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary information