

Abstract
Cystatin SN, a specific cysteine protease inhibitor, is thought to be involved in various malignant tumors. Therefore, we evaluated the role of cystatin SN in hepatocellular carcinoma (HCC). Notably, cystatin SN was elevated in tumorous samples and cells. Moreover, overexpression of cystatin SN was correlated with tumor diameter and TNM stage. Cox multivariate analysis displayed that cystatin SN was an independent prognosis indicator and that high cystatin SN level was associated with a dismal prognosis. Moreover, cystatin SN enhancement facilitated the proliferation, migratory, and invasive potential of Huh7 and HCCLM3 cells, whereas cystatin SN knockdown caused the opposite effect. Cystatin SN also modulated the epithelial‐mesenchymal transition progression through the PI3K/AKT pathway. In vivo cystatin SN promoted HCCLM3 cell growth and metastasis in xenograft mice model. Thus, cystatin SN was involved in HCC progression and could be a latent target for HCC treatment.
Keywords: cell proliferation, cystatin SN, epithelial‐mesenchymal transition, hepatocellular carcinoma, prognostic marker
1. INTRODUCTION
The liver tumor is a kind of malignant tumor with high malignancy and death rates. Meanwhile, males have a higher incidence than females (Siegel, Miller, & Jemal, 2017). Globally, the majority of primary liver tumors (70–90%) occurring worldwide are hepatocellular carcinoma (HCC; Zamor, Delemos, & Russo, 2017). Nowadays, HCC is ranked as the fifth frequent malignancy and has a high cancer‐related mortality worldwide; in 2012, global statistics estimated that nearly 800,000 new HCC cases and deaths occurred worldwide, with China alone occupying over 55% of the world's HCC burden (Fitzmaurice et al., 2015; Torre et al., 2015). Hepatitis virus infection becomes the main cause of HCC, mainly caused by the hepatitis B virus (HBV) and hepatitis C virus (HCV; El‐Serag, 2011). Other pathogenic factors cover the consumption of foods containing aflatoxin, Alcoholic cirrhosis transforms into hepatitis and nonalcoholic fatty liver disease an so forth (Singal & Elserag, 2015). Nevertheless, the underlying molecular mechanism of HCC remains unclear. Therefore, it is imperative to further unravel the unknown molecular mechanisms of HCC.
Cystatin SN (CST1) is a specific inhibitor of cysteine proteases. Various research have inferred that CST1 acts as an imperative role in inflammation and tumorigenesis. For instance, Upregulation of CST1 promotes gastric carcinoma cells proliferation and inhibits cathepsin activity (Choi et al., 2009). Moreover, raised CST1 expression promotes malignant progression of breast carcinoma and forecasts a poor prognosis (Dai et al., 2017). In colorectal cancer and pancreatic cancer, CST1 is considered as a new tumor biomarker (Jiang, Liu, Liu, Tan, & Wu, 2015; Kim et al., 2013; Yoneda et al., 2009). Meanwhile, CST1 may be used as an independent prognosis factor for patients with esophageal cancer surgery (Y. F. Chen et al., 2013). However, the roles and mechanisms of CST1 in HCC have not been clarified.
Accordingly, in this study, we used microarray analysis (G. Yang et al., 2016) to verify the expression profile of CST1 in HCC. We then explored the role of CST1 in the promotion of HCC, both clinically and experimentally. Overall, our outcomes displayed that CST1 was upregulation in HCC. High CST1 expression promoted HCC cells proliferation, migration, and invasion, thereby leading to poor prognosis.
2. MATERIALS AND METHODS
2.1. HCC sample collection
The samples of HCC specimens and their matched samples were gathered from surgically resected specimens from patients with HCC between April 2009 and May 2013. All patients signed written informed consent and received ethical approval to use human liver tissue for resection. The diagnosis of HCC was referred to World Health Organization criteria.
2.2. Culture conditions and HCC cells
Human normal hepatic cells, (L02, chang) and HCC cells HepG2, Huh7, SMMC7721, MHCC97L, MHCC97H, Sk‐Hep‐1, and HCCLM3 were obtained from the Chinese Academy of Science (Shanghai, China) and Liver Cancer Institute. SMMC7721 were cultivated in Rosewell Park Memorial Institute (RPMI)‐1640 medium (Thermo Fisher Scientific, Waltham, MA), and the other cell lines were maintained in Dulbecco's modified Eagle medium (DMEM; Thermo Fisher Scientific). All medium contained 10% FBS (Thermo Fisher Scientific). The cells were cultured in an incubator (37°C, 5% CO2).
2.3. Immunoblotting analysis
Immunoblotting assays were executed following our previous study (Han et al., 2017). Briefly, the protein sample was loaded, separated, and transferred to NC membranes, then blocked at 25°C for 1 hr, and incubated with anti‐CST1 antibodies (1:1,000; Novus Biologicals, Littleton, CO) or anti‐glyceraldehyde phosphate dehydrogenase antibodies. Horseradish peroxidase‐conjugated secondary antibodies were used at 25°C. After sufficient washing, protein blots were detected.
2.4. Immunohistochemistry
Immunohistochemistry was executed as previously described (G. Yang et al., 2016). Tissue sections and adjacent tissue and tumorous human microarrays of liver tissue (BC03119a) were incubated with CST1 monoclonal antibodies (1:100; Novus Biologicals) for 12 hrs at 4°C, whereafter added with secondary antibody (1:200; Sigma‐Aldrich, Darmstadt, Germany).
2.5. Polymerase chain reaction (PCR)
PCR was executed as previously described (Zheng et al., 2014). PureLink™ RNA Kit (Thermo Fisher Scientific) was used to extract total RNA from tissues and cells. High Capacity cDNA Reverse Transcription kit (Thermo Fisher Scientific) was applied for reverse transcription.
2.6. Cell counting kit‐8 (CCK8) experiment
CCK8 was executed as previously described (Zheng et al., 2010). Cells in log‐phase growth were inoculated in 96‐well plates at a concentration of 5 × 103 cells/well, and cultured overnight in DMEM for attachment. Cell viability was measured with CCK8 reagent (CK04‐01; Kyushu, Japan).
2.7. Colony formation assay
Cells in log‐phase growth were inoculated in six‐well plates at a concentration of 1,000 cells/well, and cultured for 14 days, then settled in methanol for 20 min, and stained with 1% crystal violet in methanol for 20 min. Finally, the plates were captured and then calculated the colonies.
2.8. Scratched wound assay
Scratched wound assay was executed as previously performed (Li et al., 2016). Cells in log‐phase growth were cultured in six‐well plates and grown to convergence. Next, rinsing the cells with polymerase chain reaction (PBS) three times, and a wound in the monolayer was made by scratching the well with a 10‐μl pipette tip. Pictures were photographed at 0 and 24 hr on a microscope (Nikon, Tokyo, Japan).
2.9. Transwell experiment
Transwell experiment was executed followed by our previous study (J. Wang et al., 2016) using Matrigel‐coated membranes (BD Biosciences, Franklin Lakes, NJ). Cells in log‐phase were inoculated in 24‐well dishes at a concentration of 4 × 104 cells/insert. DMEM with a low concentration of FBS was added and then moved to wells injected with DMEM containing a high concentration of FBS. When the cells were cultured for 24–48 hr. Then the cells that passed through the chamber filter were settled in methanol, then the membrane filter stained with 0.5% crystal violet. Next, the cells were calculated and photographed on a microscope (×100 magnification, five microscopic fields/filter).
2.10. Immunofluorescence assay
Immunofluorescence was executed as previously described (Lan et al., 2018). Cells in log‐phase growth were inoculated on cover glasses and cultured overnight in DMEM for attachment. Then, settled in 4% paraformaldehyde for 15 min. Next, permeabilized the cells for 30 min in 0.5% Triton X‐100 at 25°C for 30 min, then incubated with special primary antibodies at 4°C overnight, whereafter incubated with fluorescent secondary antibody (Thermo Fisher Scientific) for 60 min at 25°C, and rinsed three times. Finally, the cells were counterstained and pictures were photographed.
2.11. Tumor‐bearing mouse model
Male BALB/c nude mice (4–6‐week old) were obtained from the experimental animal center of Shanghai Institute (Shanghai, China). To establish an orthotopic HCC mouse model, 4 × 106 cells dissolved in PBS were inoculated subcutaneously into each flank of the nude mice. After 1 week, the subcutaneous neoplasms were resected and diced into 1 mm3 cubes, then implanted into the livers (left lobes). Tumors were resected after 4 weeks, and their sizes were measured.
To establish a metastasis model 3 × 106 cells were inoculated into the tail veins of each mouse. After 8 weeks, the mice were euthanized, and their lungs were resected. The procedures were executed followed by the protocols authorized by the Institutional Animal Care and Use Committee of Harbin Medical University.
3. RESULTS
3.1. CST1 expression was increased in HCC tissue
First, we evaluated the CST1 expression at message RNA (mRNA) and protein levels in matched tumorous tissues and adjacent tissues. The outcomes demonstrated that CST1 was upregulated in HCC tissues in comparison with that in corresponding normal tissues at mRNA and protein levels (Figure 1a,b). Next, to detect the localization of CST1 in HCC, we used immunohistochemical analysis of a recently prepared tissue microarray. As a result, CST1 expression was dramatically upregulated in HCC specimens than their counterparts and was localized in the cytoplasm (Figure 1c–h). Moreover, when quantitatively evaluating CST1 expression based on intensity of staining as previously reported (Furukawa et al., 2015; Yamamoto et al., 2017), among 75 patients with HCC, 49 (65.33%) patients showed low expression of CST1, and 26 (34.67%) patients had high CST1 expression.
Figure 1.
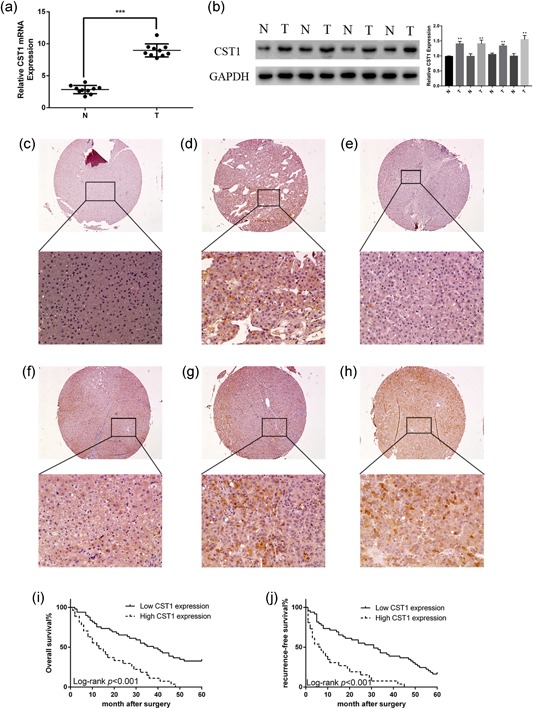
CST1 was overexpressed in human HCC and predicted poor prognosis. (a) mRNA levels of CST1 in different HCC tissues and matched adjacent normal liver tissues. (b) Protein levels of CST1 in different HCC tissues and matched adjacent normal liver tissues (left). Gray value ratio (right). (c) Negative control in normal liver tissue. (d) Positive cytoplasmic expression in liver tissue. (e) Negative control in HCC. (f) Weak positive cytoplasmic expression in HCC. (g) Moderate positive cytoplasmic expression in HCC. (h) Strong positive cytoplasmic expression in HCC. (i) Kaplan–Meier analysis of overall survival in patients with HCC. (j) Kaplan–Meier analysis of recurrence‐free survival in patients with HCC. Scale bar = 100 μm (top) and 500 μm (bottom). Data are presented as means ± standard deviations of three independent experiments. GAPDH: glyceraldehyde 3‐phosphate dehydrogenase; HCC: hepatocellular carcinoma; mRNA: messenger RNA. *p < .05; **p < .01; and ***p < .001 [Color figure can be viewed at wileyonlinelibrary.com]
3.2. Upregulation of CST1 was linked to clinicopathologic parameters and predicted dismal prognosis
To explore the relationship between CST1 and parameters of clinicopathological, we concluded the clinicopathological features of 75 patients with HCC. As shown in Table 1, statistical method, Fisher's exact text, demonstrated that overexpression of CST1 in cancerous samples was related to tumor diameter as well as TNM stage. However, the expression of CST1 was not related to age, sex, HBV infection, liver cirrhosis, or α‐fetoprotein. There was a potential relationship between CST1 expression and lymph node invasion (p = .073). Furthermore, Kaplan–Meier analysis of 75 patients with HCC showed that higher CST1 expression levels in HCC tissues were dramatically related to reduced overall survival (OS; Figure 1i) and recurrence‐free survival (RFS; Figure 1j) for HCC patients after surgery. The univariate and multivariate analysis exposed that high level of CST1 (p = .046), advanced TNM stages (p = .032), and lymph node invasion (p = .030) were independent prognostic factors for overall survival (Table 2). On the whole, these data suggested that CST1 act as a crucial role in HCC progression and might play as a promising prognosis factor.
Table 1.
Clinical characteristics of 75 HCC patient
| Characteristics | CST1 expression | p value | ||||
|---|---|---|---|---|---|---|
| Total (n = 75) | Low (n = 49), n (%) | High (n = 26), n (%) | ||||
| Age (years) | ||||||
| >50 | 31 | 19 (38.78) | 12 (46.15) | .625 | ||
| ≤50 | 44 | 30 (61.22) | 14 (53.85) | |||
| Gender | ||||||
| Male | 60 | 40 (81.63) | 20 (76.92) | .763 | ||
| Female | 15 | 9 (18.37) | 6 (23.08) | |||
| HBV infection | ||||||
| Yes | 58 | 35 (71.43) | 23 (88.46) | .147 | ||
| No | 17 | 14 (28.57) | 3 (11.54) | |||
| Liver cirrhosis | ||||||
| Yes | 68 | 43 (87.76) | 25 (96.15) | .410 | ||
| No | 7 | 6 (12.24) | 1 (3.85) | |||
| Serum AFP (ng/ml) | ||||||
| >20 | 52 | 37 (75.51) | 15 (57.69) | .124 | ||
| ≤20 | 23 | 12 (24.49) | 11 (42.31) | |||
| Tumor diameter (cm) | ||||||
| >5 | 25 | 11 (55.10) | 14 (26.92) | .010 | ||
| ≤5 | 50 | 38 (44.90) | 12 (73.08) | |||
| Lymph node metastasis | ||||||
| Yes | 26 | 13 (26.53) | 13 (50.00) | .073 | ||
| No | 49 | 36 (73.47) | 13 (50.00) | |||
| TNM stage | ||||||
| I‐II | 31 | 25 (51.02) | 6 (23.08) | .027 | ||
| III‐IV | 44 | 24 (48.98) | 20 (76.92) | |||
Abbreviations: AFP: α‐fetoprotein; HBV: hepatitis B virus.
Table 2.
Univariate and multivariate Cox regression analysis for overall survival in patients with HCC
| HR | 95% CI | p value | |
|---|---|---|---|
| Univariate | |||
| Age ≥/<50 | 1.136 | 0.679–1.902 | .626 |
| Gender M/F | 0.931 | 0.483–1.793 | .830 |
| HBV +/− | 1.808 | 0.937–3.489 | .077 |
| AFP ≥/<20 | 1.673 | 0.929–3.021 | .087 |
| Tumor diameter ≥/<5 | 3.3613 | 1.990–6.558 | .000 |
| Lymph node metastasis +/− | 5.313 | 3.015–9.360 | .000 |
| TNM stage I‐II/III‐IV | 11.637 | 5.560–24.395 | .000 |
| CST1 high/low | 3.258 | 1.893–5.607 | .000 |
| Multivariate | |||
| Tumor diameter ≥/<5 | 2.214 | 0.831–5.899 | .112 |
| Lymph node metastasis +/− | 2.975 | 1.111–7.966 | .030 |
| TNM stage I‐II/III‐IV | 3.782 | 1.122–12.758 | .032 |
| CST1 high/low | 1.846 | 1.012–3.369 | .046 |
Abbreviations: AFP: α‐fetoprotein; CI: confidence interval; HBV: hepatitis B virus; HR: hazard ratio.
3.3. Expression of CST1 in HCC cell lines
To confirm the findings from HCC tissues in vitro using HCC cell lines, we performed western blot analysis on Huh7, HepG2, SMMC7721, MHCC97L, MHCC97H, HCCLM3, and Sk‐Hep‐1 cells. Moreover, we detected L02 and chang cells as control. The outcomes demonstrated that CST1 levels were higher in HCC cells than that in L02 and chang cells (Figure 2a). Analogous results were observed from real‐time polymerase chain reaction analysis (Figure 2b). From these findings, Huh7 and HCCLM3 cells were selected to further analyze the role of CST1 in cell motility. Cell lines stably transfected with CST1 plasmid (GeneChem, Shanghai, China) were established using G418 to screen out untransfected cells. Conversely, small interfering RNA targeting the mRNA of human CST1 was used to knockdown CST1. The overexpression and knockdown efficiency of CST1 as shown in Figure 2e,f.
Figure 2.
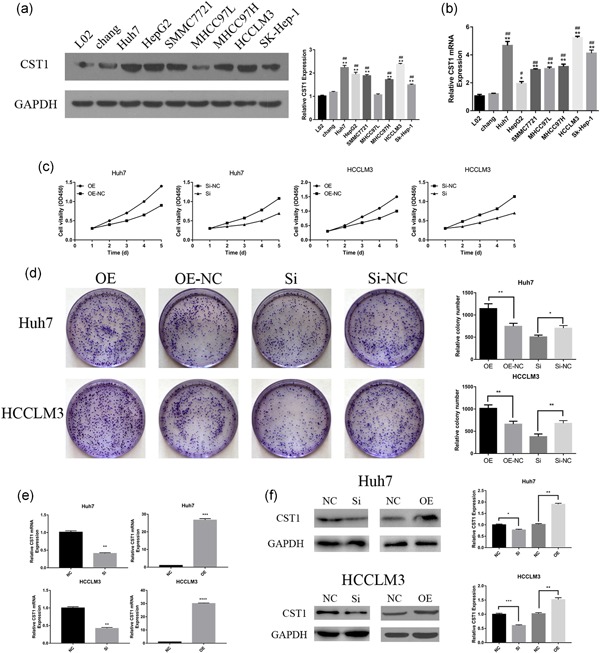
Expression of CST1 in HCC cell lines and CST1 promoted the growth of HCC cells in vitro. (a) CST1 protein expressed in HCC cell lines (left). Gray value ratio (right). (b) CST1 mRNA expressed in HCC cell lines. (c) Proliferation curves were obtained using CCK8 assays. (d) Colony formation assays (left). Number of colonies counted (right). (e) CST1 was efficiently overexpressed and silenced in Huh7 and HCCLM3 on mRNA level. (f) CST1 was efficiently overexpressed and silenced in Huh7 and HCCLM3 on the protein level. Data are presented as means ± standard deviations of three independent experiments. CCK8: cell counting kit‐8; GAPDH: glyceraldehyde 3‐phosphate dehydrogenase; HCC: hepatocellular carcinoma; mRNA: messenger RNA. NC: negative control; Si: small interfering. *Comparison of HCC cells with L02, *p < .05; **p < .01; and ***p < .001. #Comparison of HCC cells with chang, # p < .05; ## p < .01; and ### p < .001 [Color figure can be viewed at wileyonlinelibrary.com]
3.4. CST1 promoted HCC cell proliferation and carcinogenicity
To examine the effects of CST1 on growth and carcinogenicity of HCC cells, CCK8 assays and colony formation assays were applied. As expected, overexpression of CST1 stimulated Huh7 and HCCLM3 cell proliferation (Figure 2c). Inversely, knockdown CST1 had the opposite effects. Colony formation assays showed the same outcomes (Figure 2d).
3.5. CST1 facilitated HCC cell migratory and invasive potential
To examine the effects of CST1 on HCC cell migration and invasion, scratched wound assays and transwell invasion experiments were utilized, for the reason that cell migration and invasion are the initial stages of metastasis. In wound‐healing assays, CST1‐knockdown cells exhibited gentler closure compared with the control group (Figure 3a). Inversely, overexpression of CST1 increased the capacities of the cells to traverse the scratched “wound”. Transwell invasion assays indicated that upregulation of CST1 dramatically elevated the invasive potential of HCC cells. Conversely, silence the expression of CST1 dramatically attenuated cell migratory and invasive capacities (Figure 3b).
Figure 3.
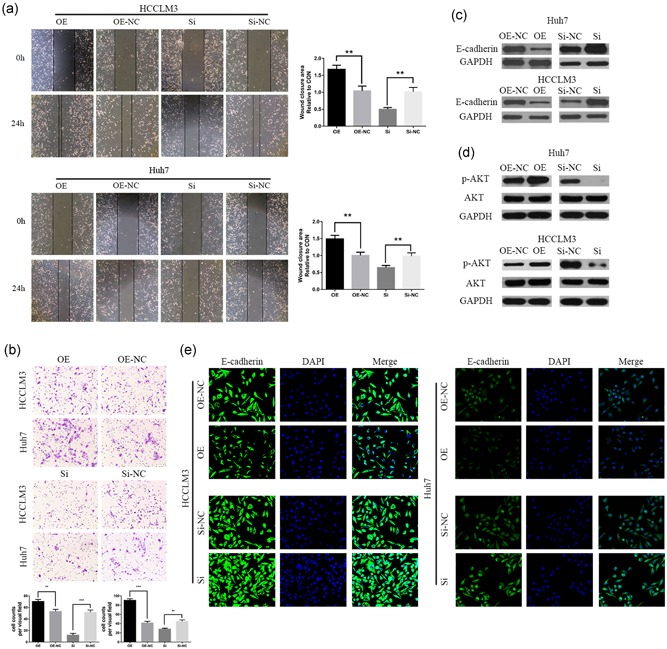
CST1 promoted migration and invasion through the EMT via the PI3K/AKT signaling pathway. (a) Migration capacity was detected by wound‐healing assays. The wound‐closure area was calculated. Scale bar: 100 μm. (b) Invasive capacity was detected by transwell assays. The number of invaded cells was counted. (c) Expression of E‐cadherin was measured by Western blot analysis. (d) Levels of phospho‐AKT and total AKT were measured by Western blot analysis. Scale bar = 100 μm (top) and 200 μm (bottom). (e) Immunofluorescence analysis of E‐cadherin expression in Huh7 and HCCLM3 cells. Data are presented as means ± standard deviations of three independent experiments. EMT: epithelial‐mesenchymal transition; GAPDH: glyceraldehyde 3‐phosphate dehydrogenase; NC: negative control; Si: small interfering *p < .05; **p < .01; and ***p < .001 [Color figure can be viewed at wileyonlinelibrary.com]
The epithelial‐mesenchymal transition (EMT) has a vital function in cancer metastatic properties; hence, we explored the influence of CST1 on EMT. Our outcomes displayed that E‐cadherin expression was inhibited by CST1 overexpression. In contrast, silencing of CST1 expression caused the opposite effect (Figure 3c). Immunofluorescence analysis also confirmed these results (Figure 3e). Many pathways have been reported to be involved in the EMT, such as Wnt/β‐catenin and PI3K/AKT signaling (Lan et al., 2018; C. Wang et al., 2015). Thus, we examined the expression and activation of AKT. Immunoblotting analyses displayed that the expression of phospho‐AKT increased and decreased after CST1 overexpression and silencing, respectively, but that total AKT levels were unchanged (Figure 3d). These results displayed that CST1 could promote HCC metastatic properties through the EMT, and the PI3K/AKT signaling might be involved in this progression.
3.6. PI3K/AKT signaling is involved in CST1‐caused HCC progression
To investigate whether PI3K/AKT signaling was involved in HCC progression. We blocked PI3K activity with LY294002 and observed subsequent cellular behavioral functions, such as proliferation, invasion, and metastasis. As expected, when treated CST1 overexpressed HCCLM3 cell with LY294002, the proliferation, metastasis, and invasion abilities of HCCLM3‐CST1 cell were restored (Figure 5a–d). Similar results were observed in Huh7‐CST1 cell (Figure 5a‐d). Meanwhile, Western blot analysis images documented that the level of phospho‐AKT was also decreased by LY294002 treatment (Figure 5e). Therefore, PI3K/AKT signaling pathway might be involved in CST1‐induced HCC progression and can promote HCC cell lines proliferation, invasion, and migration.
Figure 5.
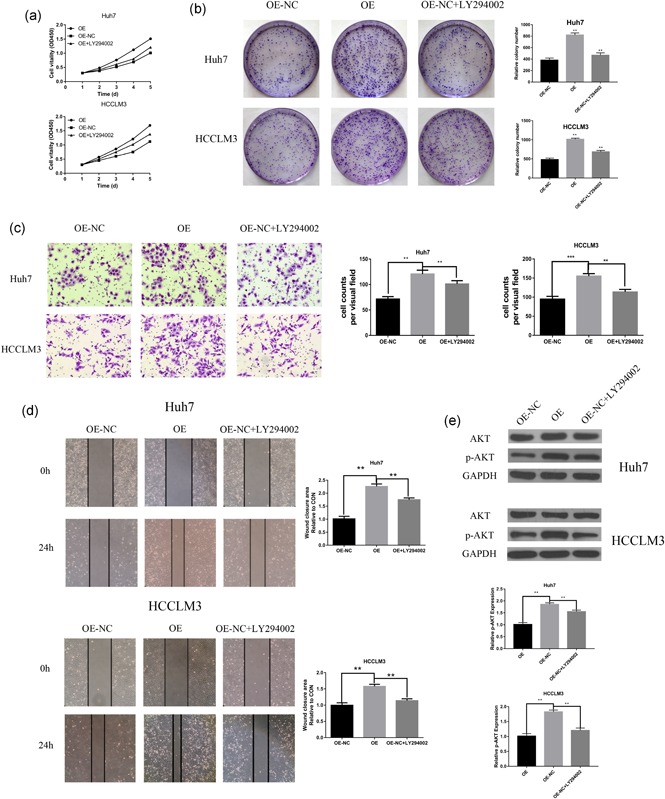
PI3K/AKT signaling pathway involved in CST1‐induced HCC progression. (a) LY294002 decreased the CST1‐induced proliferation curves. (b) Colony formation assays (left) demonstrated that LY294002 could reverse cell proliferation induced by CST1, Number of colonies counted (right). (c) Representative images from the transwell invasive assays, LY294002 decreased the CST1‐induced invasion of Huh7 and HCCLM3 cells. The number of invaded cells was counted. (d) Representative images from the wound‐healing assays, LY294002 decreased the CST1‐induced migration of Huh7 and HCCLM3 cells. The wound‐closure area was calculated. (e) Western blot analysis (left) of p‐AKT expression after LY294002 treatment with Huh7 and HCCLM3 cells. Gray value ratio (right). GAPDH: glyceraldehyde 3‐phosphate dehydrogenase; HCC: hepatocellular carcinoma; NC: negative control. *p < .05; **p < .01; and ***p < .001 [Color figure can be viewed at wileyonlinelibrary.com]
3.7. CST1 expression promoted HCC tumor growth and metastasis
To further explored the influence of CST1 on the HCC tumor growth, we used stable expressed exogenous CST1, HCCLM3‐CST1 cells, to construct an orthotopic tumor‐bearing mouse model. After 4 weeks, the volumes and weights of the tumor were increased in HCCLM3‐CST1 group than those in the control group (Figure 4a,b). Moreover, immunohistochemical analyses showed that the orthotopic tumor tissues expressed higher levels of Ki‐67 in the HCCLM3 overexpression group compared with that in control group (Figure 4c).
Figure 4.
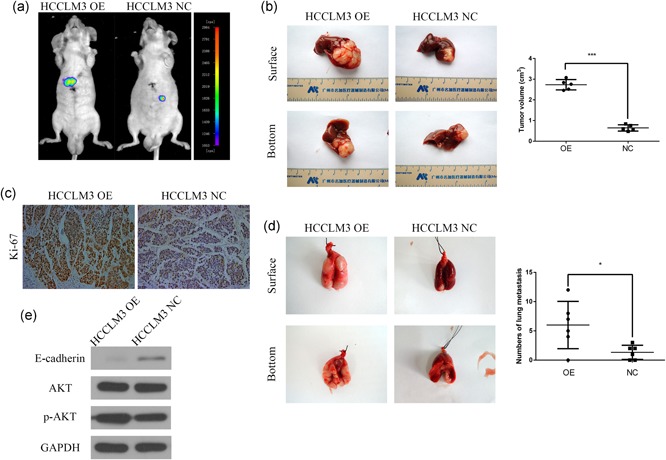
CST1 promoted HCC growth and metastasis in vivo. (a) Images of the orthotopic model were acquired using bioluminescence imaging. (b) CST1 overexpression increased HCCLM3 cell growth in the orthotopic model in nude mice. Tumor volumes were calculated. (c) Images of immunohistochemical detection of Ki‐67 in orthotopic HCC tissues. Scale bar = 500 μm. (d) CST1 overexpression promoted HCCLM3 cell metastasis to the lung. The number of nodules transferred was counted. (e) The levels of E‐cadherin, phospho‐AKT, and total AKT in the lung metastasis model were measured by western blot analysis. Data are presented as means ± standard deviations of three independent experiments. GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; HCC, hepatocellular carcinoma; NC, negative control. *p < .05; **p < .01; and ***p < .001 [Color figure can be viewed at wileyonlinelibrary.com]
Next, we injected the stably transfected HCCLM3 cell line into nude mice through the tail vein so as to examined the effect of CST1 on tumor metastasis in vivo. Necropsies conducted after 8 weeks revealed a significant increase in metastatic modules in CST1‐overexpressing nude mice than that in control mice (Figure 4d). Furthermore, western blot analysis showed that E‐cadherin expression was decreased and phospho‐AKT, but not total AKT, was boosted in the metastatic nodules from the lungs, in the HCCLM3 overexpression group relative to those in the control group (Figure 4e).
4. DISCUSSION
The leading pathogenic factors for HCC vary in different incidence areas. Chronic hepatitis virus infection, HBV and HCV, become the main cause of HCC. In Asia, infection of HBV and consumption of foods containing aflatoxin B1 are the main pathogenic factors. Conversely, infection of HCV, heavy drinking of alcoholic beverages, and many chronic diseases act as an imperative role in low‐incidence areas (McGlynn, Petrick, & London, 2015). Of the two viruses that caused HCC, HBV accounts for nearly 75% of virus‐associated HCC, whereas HCV accounts for 15–25% (Perz, Armstrong, Farrington, Hutin, & Bell, 2006).
Cysteine proteases are proteolytic enzymes. They are widely expressed in human organs and have a variety of functions, including inflammatory tissue lesion, immune response regulation, monocyte induction, and enhanced the migratory capacity of cancer cell et al (Koblinski, Ahram, & Sloane, 2000; Lah, Babnik, Schiffmann, Turk, & Skaleric, 1993). Furthermore, lysosomal cathepsins may also activate several important signaling pathways in cells, resulting in cell death. Under the circumstances, these proteolytic enzymes serve as tumor suppressors (Pišlar, Perišić, & Kos, 2015; Vasiljeva & Turk, 2008). The cystatin superfamily, which including Stefins type 1, CSTs type 2, and kininogen, are cysteine protease inhibitors and have been shown to specifically inhibit the proteolytic activities of cysteine protease (Barrett, 1986). CSTs, type 2 CST superfamily, includes CST1–5, CSTP1, and CSTP2, in which CST1 encodes a secreted protein named cystatin SN (Koblinski et al., 2000). Type 2 CST genes are physically clustered at human chromosome 20p11.2 (Dickinson, Zhao, Thiesse, & Siciliano, 1994; Thiesse, Millar, & Dickinson, 1994), are widely distributed throughout nature (Henskens, Veerman, & Nieuw Amerongen, 1996). The proteins encoded by these genes typically include a secretory leader peptide.
In our research, we explored the role of CST1 in HCC. The data indicated that CST1 was overexpressed in HCC samples and HCC cells than that in paired noncancerous samples and normal liver cells (L02 and chang), respectively. Additionally, immunohistochemistry showed that CST1 was localized in the cytoplasm, and elevated CST1 level was associated with tumor diameter and TNM stage and predicted a poor prognosis. Notably, however, lymph node metastasis only showed a tendency to be correlated, which may be related to the insufficient number of samples, and we believe that future multicenter studies with larger datasets may yield clearer results. Moreover, Kaplan–Meier curves indicated that patients with high CST1 level had worse OS and RFS than those with low CST1 level. Finally, we also found that CST1 acted as a crucial role in many malignant biological behaviors of HCC cells, including HCC cell viability, carcinogenicity, migration, and invasion, both in vivo and in vitro. Therefore, the results supported CST1 as a new prognostic marker for predicting HCC prognosis and latent novel target for HCC treatment.
The mechanism through which CST1 contributes to cell progression remains unclear. The EMT is imperative for many physiological and pathological processes, such as embryogenesis, tissue regeneration, and so forth (Kalluri & Weinberg, 2009; Thiery & Sleeman, 2006; J. Yang & Weinberg, 2008). Importantly, we found that upregulation of CST1 dramatically facilitated cell progression via inducing the EMT through the PI3K/AKT signaling. This pathway is often dysfunctional in cancer and acts as a crucial role in modulating cell processes that are characteristic of carcinoma (Thorpe, Yuzugullu, & Zhao, 2015). Additionally, mutations that activate these enzymes have often been detected in human carcinoma, thereby leading to cancer growth. PI3K converts PIP2 to PIP3 on the plasma membrane and subsequently phosphorylates and activates AKT (Alessi et al., 1997; Stokoe et al., 1997). Following AKT activation, AKT phosphorylates several downstream effectors, a pathway that regulates a variety of biological processes in different types of cancer, and its excessive activation leads to abnormal cell cycle progression, proliferation, migration, invasion, altered adhesion and motility, inhibition of apoptosis, and induction of angiogenesis (Fruman et al., 2017; Manning & Cantley, 2007). Several publications have showed that PI3K/AKT signaling is involved in mediating the progression of HCC (Samarin et al., 2016; R. Y. Wang et al., 2013), and is involved in oncogenesis of various malignancies of the digestive system (Z. Chen et al., 2019; Gao et al., 2017; Y. Wang et al., 2017). In our outcomes, LY294002 was used for rescue experiments. When HCCLM3‐CST1 and Huh7‐CST1 cells treated with LY294002, the growth and metastatic properties of these two cells were partly reversed. This consequence suggests that PI3K/AKT signal is involved in CST1‐induced HCC progression.
In conclusion, we demonstrated the role of CST1 in promoting malignant biological behaviors (proliferation, migration, invasion, etc.) in HCC. However, how does CST1 regulate the progression of HCC through PI3K/AKT signaling and how to induce the occurrence of EMT are a series of problems, which still need further studies to investigate the potential mechanism of CST1's involvement in the occurrence of HCC.
ACKNOWLEDGMENTS
This study was supported by the National Natural Scientific Foundation of China (grant nos. 81272705, 81001081, 81602149, 81301807, 81302060, 81602050, and 81602149) and the Heilongjiang Postdoctoral Fund (LBH‐Z18132).
CONFLICT OF INTERESTS
The authors declare that there are no conflict of interests.
AUTHOR CONTRIBUTIONS
Y. C. and D. S. designed the study and wrote the initial draft of the manuscript. Z. L. and L. L. contributed to analysis and interpretation of data and assisted in the preparation of the manuscript. All other authors have contributed to data collection and interpretation and critically reviewed the manuscript. All authors approved the final version of the manuscript and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Cui Y, Sun D, Song R, et al. Upregulation of cystatin SN promotes hepatocellular carcinoma progression and predicts a poor prognosis. J Cell Physiol. 2019;234:22623–22634. 10.1002/jcp.28828
Contributor Information
Zhaoyang Lu, Email: lzy76772005@163.com.
Lianxin Liu, Email: LiuLX@ems.hrbmu.edu.cn.
References
REFERENCES
- Alessi, D. R. , James, S. R. , Downes, C. P. , Holmes, A. B. , Gaffney, P. R. , Reese, C. B. , & Cohen, P. (1997). Characterization of a 3‐phosphoinositide‐dependent protein kinase which phosphorylates and activates protein kinase Balpha. Current Biology, 7, 261–296. [DOI] [PubMed] [Google Scholar]
- Barrett, A. J. (1986). The cystatins: A diverse superfamily of cysteine peptidase inhibitors. Biomedica Biochimica Acta, 45, 1363. [PubMed] [Google Scholar]
- Chen, Y. F. , Ma, G. , Cao, X. , Luo, R. Z. , He, L. R. , He, J. H. , … Wen, Z. S. (2013). Overexpression of Cystatin SN positively affects survival of patients with surgically resected esophageal squamous cell carcinoma. BMC Surgery, 13, 15 10.1186/1471-2482-13-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Z. , Liu, Z. , Zhang, M. , Huang, W. , Li, Z. , Wang, S. , … Shen, L. (2019). EPHA2 blockade reverses acquired resistance to afatinib induced by EPHA2‐mediated MAPK pathway activation in gastric cancer cells and avatar mice. International Journal of Cancer, 10.1002/ijc.32313 [DOI] [PubMed] [Google Scholar]
- Choi, E. H. , Kim, J. T. , Kim, J. H. , Kim, S. Y. , Song, E. Y. , Kim, J. W. , … Lee, H. G. (2009). Upregulation of the cysteine protease inhibitor, cystatin SN, contributes to cell proliferation and cathepsin inhibition in gastric cancer. Clinica Chimica Acta, 406, 45–51. 10.1016/j.cca.2009.05.008 [DOI] [PubMed] [Google Scholar]
- Dai, D. N. , Li, Y. , Chen, B. , Du, Y. , Li, S. B. , Lu, S. X. , … Wei, W. D. (2017). Elevated expression of CST1 promotes breast cancer progression and predicts a poor prognosis. Journal of Molecular Medicine, 95, 873 10.1007/s00109-017-1537-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson, D. P. , Zhao, Y. , Thiesse, M. , & Siciliano, M. J. (1994). Direct mapping of seven genes encoding human type 2 cystatins to a single site located at 20p11.2. Genomics, 24, 172. [DOI] [PubMed] [Google Scholar]
- El‐Serag, H. B. (2011). Hepatocellular carcinoma. New England Journal of Medicine, 365, 1118–1127. 10.1056/NEJMra1001683 [DOI] [PubMed] [Google Scholar]
- Fitzmaurice, C. , Dicker, D. , Pain, A. , Hamavid, H. , Moradi‐Lakeh, M. , MacIntyre, M. F. , … Naghavi, M. (2015). The global burden of cancer 2013. JAMA Oncology, 1(4), 505–527. 10.1001/jamaoncol.2015.0735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fruman, D. A. , Chiu, H. , Hopkins, B. D. , Bagrodia, S. , Cantley, L. C. , & Abraham, R. T. (2017). The PI3K pathway in human disease. Cell, 170, 605–635. 10.1016/j.cell.2017.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa, K. , Kawamoto, K. , Eguchi, H. , Tanemura, M. , Tanida, T. , Tomimaru, Y. , … Nagano, H. (2015). Clinicopathological significance of leucine‐rich α2‐glycoprotein‐1 in sera of patients with pancreatic cancer. Pancreas, 44, 93 10.1097/MPA.0000000000000205 [DOI] [PubMed] [Google Scholar]
- Gao, Y. , Xiao, X. , Zhang, C. , Yu, W. , Guo, W. , Zhang, Z. , … Deng, W. (2017). Melatonin synergizes the chemotherapeutic effect of 5‐fluorouracil in colon cancer by suppressing PI3K/AKT and NF‐κB/iNOS signaling pathways. Journal of Pineal Research, 62(2), 62 10.1111/jpi.12380 [DOI] [PubMed] [Google Scholar]
- Han, J. , Xie, C. , Pei, T. , Wang, J. , Lan, Y. , Huang, K. , … Liu, L. (2017). Deregulated AJAP1/β‐catenin/ZEB1 signaling promotes hepatocellular carcinoma carcinogenesis and metastasis. Cell Death and Disease, 8, e2736 10.1038/cddis.2017.126 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Henskens, Y. M. , Veerman, E. C. , & Nieuw Amerongen, A. V. (1996). Cystatins in health and disease. Biological Chemistry, 377, 71. [DOI] [PubMed] [Google Scholar]
- Jiang, J. , Liu, H. L. , Liu, Z. H. , Tan, S. W. , & Wu, B. (2015). Identification of cystatin SN as a novel biomarker for pancreatic cancer. Tumor Biology, 36, 3903–3910. 10.1007/s13277-014-3033-3 [DOI] [PubMed] [Google Scholar]
- Kalluri, R. , & Weinberg, R. A. (2009). The basics of epithelial‐mesenchymal transition. Journal of Clinical Investigation, 119, 1420–1428. 10.1172/JCI39104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, J. T. , Lee, S. J. , Kang, M. A. , Park, J. E. , Kim, B. Y. , Yoon, D. Y. , … Lee, H. G. (2013). Cystatin SN neutralizes the inhibitory effect of cystatin C on cathepsin B activity. Cell Death and Disease, 4, e974 10.1038/cddis.2013.485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koblinski, J. E. , Ahram, M. , & Sloane, B. F. (2000). Unraveling the role of proteases in cancer. Clinica Chimica Acta, 291, 113–135. [DOI] [PubMed] [Google Scholar]
- Lah, T. T. , Babnik, J. , Schiffmann, E. , Turk, V. , & Skaleric, U. (1993). Cysteine proteinases and inhibitors in inflammation: Their role in periodontal disease. Journal of Periodontology, 64, 485–491. [PubMed] [Google Scholar]
- Lan, Y. , Han, J. , Wang, Y. , Wang, J. , Yang, G. , Li, K. , … Liu, L. (2018). STK17B promotes carcinogenesis and metastasis via AKT/GSK‐3β/Snail signaling in hepatocellular carcinoma. Cell Death and Disease, 9, 236 10.1038/s41419-018-0262-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, K. , Wang, J. , Han, J. , Lan, Y. , Xie, C. , Pan, S. , & Liu, L. (2016). Overexpression of ZNF703 facilitates tumorigenesis and predicts unfavorable prognosis in patients with cholangiocarcinoma. Oncotarget, 7, 76108–76117. 10.18632/oncotarget.12627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning, B. D. , & Cantley, L. C. (2007). AKT/PKB signaling: Navigating downstream. Cell, 129, 1261–1274. 10.1016/j.cell.2007.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGlynn, K. A. , Petrick, J. L. , & London, W. T. (2015). Global epidemiology of hepatocellular carcinoma: An emphasis on demographic and regional variability. Clinics in Liver Disease, 19, 223–238. 10.1016/j.cld.2015.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perz, J. F. , Armstrong, G. L. , Farrington, L. A. , Hutin, Y. J. , & Bell, B. P. (2006). The contributions of hepatitis B virus and hepatitis C virus infections to cirrhosis and primary liver cancer worldwide. Journal of Hepatology, 45, 529 10.1016/j.jhep.2006.05.013 [DOI] [PubMed] [Google Scholar]
- Pišlar, A. , Perišić, N. M. , & Kos, J. (2015). Lysosomal cysteine peptidases—molecules signaling tumor cell death and survival. Seminars in Cancer Biology, 35, 168–179. 10.1016/j.semcancer.2015.08.001 [DOI] [PubMed] [Google Scholar]
- Samarin, J. , Laketa, V. , Malz, M. , Roessler, S. , Stein, I. , Horwitz, E. , … Breuhahn, K. (2016). PI3K/AKT/mTOR‐dependent stabilization of oncogenic far‐upstream element binding proteins in hepatocellular carcinoma cells. Hepatology, 63, 813–826. 10.1002/hep.28357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel, R. L. , Miller, K. D. , & Jemal, A. (2017). Cancer statistics, 2017. CA: A Cancer Journal for Clinicans, 67, 7–30. 10.3322/caac.21387 [DOI] [PubMed] [Google Scholar]
- Singal, A. G. , & Elserag, H. B. (2015). Hepatocellular carcinoma from epidemiology to prevention: Translating knowledge into practice. Clinical Gastroenterology and Hepatology, 13, 2140 10.1016/j.cgh.2015.08.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokoe, D. , Stephens, L. R. , Copeland, T. , Gaffney, P. R. , Reese, C. B. , Painter, G. F. , … Hawkins, P. T. (1997). Dual role of phosphatidylinositol‐3,4,5‐trisphosphate in the activation of protein kinase B. Science, 277, 567–570. [DOI] [PubMed] [Google Scholar]
- Thiery, J. P. , & Sleeman, J. P. (2006). Complex networks orchestrate epithelial‐mesenchymal transitions. Nature Reviews in Molecular and Cellular Biology, 7, 131–142. 10.1038/nrm1835 [DOI] [PubMed] [Google Scholar]
- Thiesse, M. , Millar, S. J. , & Dickinson, D. P. (1994). The human type 2 cystatin gene family consists of eight to nine members, with at least seven genes clustered at a single locus on human chromosome 20. DNA and Cell Biology, 13, 97–116. 10.1089/dna.1994.13.97 [DOI] [PubMed] [Google Scholar]
- Thorpe, L. M. , Yuzugullu, H. , & Zhao, J. J. (2015). PI3K in cancer: Divergent roles of isoforms, modes of activation and therapeutic targeting. Nature Reviews Cancer, 15, 7–24. 10.1038/nrc3860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torre, L. A. , Bray, F. , Siegel, R. L. , Ferlay, J. , Lortet‐Tieulent, J. , & Jemal, A. (2015). Global cancer statistics, 2012. CA: A Cancer Journal for Clinicians, 65, 87 10.3322/caac.21262 [DOI] [PubMed] [Google Scholar]
- Vasiljeva, O. , & Turk, B. (2008). Dual contrasting roles of cysteine cathepsins in cancer progression: Apoptosis versus tumour invasion. Biochimie, 90, 380–386. 10.1016/j.biochi.2007.10.004 [DOI] [PubMed] [Google Scholar]
- Wang, C. , Wang, X. , Su, Z. , Fei, H. , Liu, X. , & Pan, Q. (2015). miR‐25 promotes hepatocellular carcinoma cell growth, migration and invasion by inhibiting RhoGDI1. Oncotarget, 6, 36231–36244. 10.18632/oncotarget.4740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, J. , Xie, C. , Pan, S. , Liang, Y. , Han, J. , Lan, Y. , … Liu, L. (2016). N‐myc downstream‐regulated gene 2 inhibits human cholangiocarcinoma progression and is regulated by leukemia inhibitory factor/MicroRNA‐181c negative feedback pathway. Hepatology, 64, 1606 10.1002/hep.28781. [DOI] [PubMed] [Google Scholar]
- Wang, R. Y. , Chen, L. , Chen, H. Y. , Hu, L. , Li, L. , Sun, H. Y. , … Wang, H. Y. (2013). MUC15 inhibits dimerization of EGFR and PI3K‐AKT signaling and is associated with aggressive hepatocellular carcinomas in patients. Gastroenterology, 145, 1436–1448. e1‐12. 10.1053/j.gastro.2013.08.009 [DOI] [PubMed] [Google Scholar]
- Wang, Y. , Kuramitsu, Y. , Baron, B. , Kitagawa, T. , Tokuda, K. , Akada, J. , … Nakamura, K. (2017). PI3K inhibitor LY294002, as opposed to wortmannin, enhances AKT phosphorylation in gemcitabine‐resistant pancreatic cancer cells. International Journal of Oncology, 50, 606–612. 10.3892/ijo.2016.3804 [DOI] [PubMed] [Google Scholar]
- Yamamoto, M. , Takahashi, T. , Serada, S. , Sugase, T. , Tanaka, K. , Miyazaki, Y. , … Doki, Y. (2017). Overexpression of leucine‐rich α2‐glycoprotein‐1 is a prognostic marker and enhances tumor migration in gastric cancer. Cancer Science, 108(10), 2052–2060. 10.1111/cas.13329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, G. , Liang, Y. , Zheng, T. , Song, R. , Wang, J. , Shi, H. , … Liu, L. (2016). FCN2 inhibits epithelial‐mesenchymal transition‐induced metastasis of hepatocellular carcinoma via TGF‐beta/Smad signaling. Cancer Letters, 378, 80–86. 10.1016/j.canlet.2016.05.007 [DOI] [PubMed] [Google Scholar]
- Yang, J. , & Weinberg, R. A. (2008). Epithelial‐mesenchymal transition: At the crossroads of development and tumor metastasis. Developmental Cell, 14, 818–829. 10.1016/j.devcel.2008.05.009 [DOI] [PubMed] [Google Scholar]
- Yoneda, K. , Iida, H. H. , Endo, H. , Hosono, K. , Akiyama, T. , Takahashi, H. , … Nakajima, A. (2009). Identification of cystatin SN as a novel tumor marker for colorectal cancer. International Journal of Oncology, 35, 33–40. [PubMed] [Google Scholar]
- Zamor, P. J. , Delemos, A. S. , & Russo, M. W. (2017). Viral hepatitis and hepatocellular carcinoma: Etiology and management. Journal of Gastrointestinal Oncology, 8, 229 10.21037/jgo.2017.03.14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng, T. , Hong, X. , Wang, J. , Pei, T. , Liang, Y. , Yin, D. , … Liu, L. (2014). Gankyrin promotes tumor growth and metastasis through activation of IL‐6/STAT3 signaling in human cholangiocarcinoma. Hepatology, 59, 935–946. 10.1002/hep.26705 [DOI] [PubMed] [Google Scholar]
- Zheng, T. , Wang, J. , Chen, X. , Meng, X. , Song, X. , Lu, Z. , … Liu, L. (2010). Disruption of p73‐MDM2 binding synergizes with gemcitabine to induce apoptosis in HuCCT1 cholangiocarcinoma cell line with p53 mutation. Tumor Biology, 31(4), 287–295. 10.1007/s13277-010-0035-7 [DOI] [PubMed] [Google Scholar]