

Abstract
Nicotine activates nicotinic acetylcholine receptors and improves cognitive and sensory function, in part by its actions in cortical regions. Physiological studies show that nicotine amplifies stimulus‐evoked responses in sensory cortex, potentially contributing to enhancement of sensory processing. However, the role of specific cell types and circuits in the nicotinic modulation of sensory cortex remains unclear. Here, we performed whole‐cell recordings from pyramidal (Pyr) neurons and inhibitory interneurons expressing parvalbumin (PV), somatostatin (SOM), and vasoactive intestinal peptide (VIP) in mouse auditory cortex, in vitro. Bath application of nicotine strongly depolarized and excited VIP neurons, weakly depolarized Pyr neurons, and had no effect on the membrane potential of SOM or PV neurons. The use of receptor antagonists showed that nicotine's effects on VIP and Pyr neurons were direct and indirect, respectively. Nicotine also enhanced the frequency of spontaneous inhibitory postsynaptic currents (sIPSCs) in Pyr, VIP, and SOM, but not PV, cells. Using Designer Receptors Exclusively Activated by Designer Drugs (DREADDs), we show that chemogenetic inhibition of VIP neurons prevents nicotine's effects on Pyr neurons. Since VIP cells preferentially contact other inhibitory interneurons, we suggest that nicotine drives VIP cell firing to disinhibit Pyr cell somata, potentially making Pyr cells more responsive to auditory stimuli. In parallel, activation of VIP cells also directly inhibits Pyr neurons, likely altering integration of other synaptic inputs. These cellular and synaptic mechanisms likely contribute to nicotine's beneficial effects on cognitive and sensory function.
Keywords: interneuron, nicotine, nicotinic acetylcholine receptor, parvalbumin, pyramidal neuron, somatostatin, VIP
1. INTRODUCTION
Nicotine is known to enhance cognitive function and has been shown to improve performance on a variety of attentional, memory, and sensory tasks (Lawrence, Ross, & Stein, 2002; Levin, McClernon, & Rezvani, 2006; Rezvani & Levin, 2001; Swan & Lessov‐Schlaggar, 2007). Consequently, nicotine may be a promising therapeutic drug for some cognitive or sensory disorders (Gil & Metherate, 2019; Kumari & Postma, 2005; Newhouse et al., 2012). Nicotine acts via nicotinic acetylcholine receptors (nAChR), which are distributed throughout the brain (Clarke, Schwartz, Paul, Pert, & Pert, 1985; Dani & Bertrand, 2007). However, neocortex in particular has been implicated as a region critical to the performance enhancement observed with nicotine, and nAChR activation increases neuronal responsiveness in cortical areas associated with attention and sensory processing (Disney, Aoki, & Hawken, 2007; Lawrence et al., 2002; Sun et al., 2017).
Nicotinic acetylcholine receptors are not equally distributed across cell types in cortex, and this selectivity may be key to understanding the mechanism of nicotine's pro‐cognitive effects (Arroyo, Bennett, & Hestrin, 2014; Gulledge, Park, Kawaguchi, & Stuart, 2007; Porter et al., 1999). Functional nAChRs in pyramidal (Pyr) neurons appear to be sparse and mostly in deeper layers, while inhibitory interneurons appear to express more nAChRs and are more responsive to nAChR agonist (Disney et al., 2007; Poorthuis et al., 2013; Porter et al., 1999; Zolles, Wagner, Lampert, & Sutor, 2009). Among the main classes of cortical inhibitory interneurons, most parvalbumin (PV) and many somatostatin (SOM)‐expressing neurons are not sensitive to nAChR activation (Gulledge et al., 2007; Porter et al., 1999), whereas vasoactive intestinal peptide (VIP), cholecystokinin, and calretinin‐expressing neurons are more sensitive (Gulledge et al., 2007; Porter et al., 1999). Of the latter cell types, VIP interneurons are of special interest since they are increasingly implicated in cortical disinhibition; that is, VIP cells preferentially innervate other interneurons, especially SOM cells, that in turn inhibit Pyr cells (Lee, Kruglikov, Huang, Fishell, & Rudy, 2013; Pfeffer, Xue, He, Huang, & Scanziani, 2013). VIP cell‐mediated disinhibition occurs during locomotion and task performance, thereby increasing Pyr cell excitability and firing (Fu et al., 2014; Jackson, Ayzenshtat, Karnani, & Yuste, 2016; Pi et al., 2013). Thus, if VIP interneurons express nAChRs, nicotine might similarly activate a disinhibitory microcircuit.
Here, we performed whole‐cell recordings from Pyr, VIP, SOM, and PV cells in acute brain slices containing mouse auditory cortex to examine the cell‐type specificity of nicotinic modulation. We found that nicotine weakly depolarizes Pyr cells, while potently depolarizing and exciting VIP cells. Additionally, using Designer Receptors Activated by Designer Drugs (DREADDs) to silence VIP cell activity, we found that VIP neurons mediate nicotinic effects on Pyr cells. Thus, nicotine‐induced, VIP‐mediated disinhibition of Pyr neurons likely leads to the increased responsiveness observed in other studies, providing a probable mechanism for nicotine's beneficial effects on cognitive and sensory function.
2. MATERIALS AND METHODS
2.1. Animals
Male and female mice, 25–50 days old, were used for all experiments. The care and use of mice were approved by the University of California, Irvine Institutional Animal Care and Use Committee. To identify interneuron subtypes for recording, we used three different mouse lines that expressed the fluorescent protein tdTomato under interneuron‐specific promoters. For VIP, SOM, and PV cells, we crossed the respective homozygous mice VIP‐ires‐cre (VIPtm1(cre)Zjh), SOM‐ires‐cre (Ssttm2.1(cre)Zjh), or PV‐ires‐cre (Pvalbtm1(cre)Arb) with the homozygous tdTomato reporter mouse Ai9 (B6.Cg‐Gt(ROSA)26Sortm9(CAG‐tdTomato)/Hze). All mice were obtained from The Jackson Laboratory. In one animal, immunohistochemistry was used to confirm near‐complete overlap between tdTomato fluorescence and anti‐VIP antibodies for neurons in auditory cortex (VIP antibody H‐6, sc‐25347, Santa Cruz Biotech). To generate mice for injection of DREADDs, we crossed homozygous VIP‐ires‐cre mice with FVB mice. Recordings from Pyr cells were performed in either FVB mice or the offspring of VIP‐ires‐cre/FVB mice.
2.2. Slice preparation
Mice were anesthetized with isoflurane and decapitated. Brains were quickly removed into cold ACSF containing 125 mM NaCl, 2.5 mM KCl, 25 mM NaHCO3, 1.25 KH2PO4, 1.2 mM MgSO4, 2.0 mM CaCl2, and 10 mM dextrose, bubbled with 95% O2/5% CO2. Auditory thalamocortical slices (∼400 μm thick for electrical stimulation experiments and ~250 μm for all other experiments) were prepared using a vibroslicer (Leica VT1000) as described previously (Cruikshank, Rose, & Metherate, 2002). Slices were placed in a holding chamber containing oxygenated ACSF at room temperature for ~1 hr before recording.
2.3. Electrophysiology
Slices were transferred to a submersion chamber for recording and maintained in continuous bath flow of ACSF (~2.5–3 ml/min) at room temperature. Whole‐cell recordings were obtained with patch pipettes (1.5–5 MΩ) filled with either a K+‐based solution (for current‐clamp recordings) containing (in mM) 135 K‐gluconate, 1 KCl, 2 MgCl2, 1 Na‐ATP, 0.5 Na‐GTP, 1 EGTA, 10 HEPES, or a Cs+‐based solution (for voltage‐clamp recordings) containing (in mM) 135 CsMeSO4, 5 CsCl, 2 MgCl2, 1 Na‐ATP, 0.5 Na‐GTP, 1 EGTA, 10 HEPES (pH 7.3 and 270 mOsm). Responses were acquired in voltage‐clamp or current‐clamp mode with a MultiClamp 700B amplifier (Molecular Devices) and AxoGraph software. Signals were amplified and low‐pass filtered at 2 kHz and digitally sampled at 10 kHz. Series resistance (6–15 MΩ) was continuously monitored, and data were discarded if the resistances changed more than 30%. Voltages were not adjusted to compensate for the liquid junction potential (~10 mV). Neurons were visualized using infrared differential interference contrast (IR‐DIC) and fluorescence (Zeiss Axioskop 2). The recording location in auditory cortex was based on previous studies in the mouse thalamocortical slice (Cruikshank et al., 2002) and confirmed in some recordings by a short‐latency response to stimulation of the thalamocortical pathway.
All drugs were added to ACSF and bath‐applied to the slice: 1 μM nicotine (Sigma), 50–100 μM picrotoxin (PTX, Sigma), 10–20 μM 6‐cyano‐7‐nitroquinoxaline‐2,3‐dione (CNQX, Sigma), 10 μM D‐2‐amino‐5‐phosphonovalerate (AP5, Tocris), 100 nM clozapine N‐oxide (CNO, abcam), 10 nM methyllycaconitine citrate (MLA, Sigma), 0.5–1 μM dihydro‐β‐erythroidine (DHβE, Sigma). To stimulate thalamocortical afferents, a bipolar concentric electrode (125 μm outer diameter, Frederick Haer) was placed in visually identified superior thalamic radiation (STR) in the thalamocortical pathway. Stimulus pulses (100–400 μA) were given every 10 s and evoked responses were averaged from 5 to 10 repetitions.
For current‐clamp recordings, neurons were selected only if the resting membrane potential was negative to −50 mV and experiments were conducted at resting membrane potential. For voltage‐clamp recordings of isolated inhibitory postsynaptic currents (IPSCs), the reversal potential for excitatory postsynaptic currents (EPSC) was presumed to be around 0 mV. Recordings at 0 mV contained small negative amplitude spontaneous currents in addition to the large positive amplitude currents; these negative currents are likely spontaneous EPSCs and confirm that the positive amplitude currents are exclusively IPSCs. To isolate EPSCs, we first estimated stimulus‐evoked IPSC reversal potential in 1 mV steps in a subset of neurons and obtained a value of ~52 mV; this clamp potential was then used for EPSC measurements.
2.4. Viral infusion
Three‐week old male and female hemizygous VIP‐Cre mice received 2 x 0.5‐μL unilateral infusions to auditory cortex (from bregma: M/L + 4.0 mm, A/P −2.55/−2.85 mm; from cortical surface D/V −1.1/−0.8 mm) of either AAV2.8‐hSyn‐DIO‐mCherry or AAV2.8‐hSyn‐DIO‐HM4D‐mCherry. Viruses were infused at a rate of 6 μL/h using a 30‐gauge Neuros Hamilton syringe (product #65456‐01) mounted on a Leica Biosystems Nanoinjector Motorized f/Stereotaxics pump (Product #39462901). All infusions used the Leica Microsystem Angle Two Stereotaxic System. For all experiments, animals were allowed to recover for a minimum of 3 weeks before tissue harvesting. All viruses were purchased from UNC Vector Core (mCherry Lot: AV4981CD 2014; HM4D AV4980B) or Addgene (mCherry #44362, Lot: v4330; HM4D #44362, Lot: v4331). Viral purity was confirmed via Sanger Sequencing (Genewiz) as previously described (Lopez et al., 2019).
2.5. Immunohistochemistry
Mice were anesthetized with 50 mg/kg sodium pentobarbital and perfused with ice‐cold 0.1 M PBS and 4% paraformaldehyde. Brains were harvested, soaked in 4% paraformaldehyde for 24 hr at 4°C, and cryoprotected in 30% sucrose at 4°C until completely submerged. Tissue was then flash frozen in dry ice‐chilled isopentane and 40 μm histological sections containing auditory cortex were collected using a Leica CM 1850 cryostat at −20°C. To confirm expression of Cre‐dependent vector in VIP + neurons of the auditory cortex, free‐floating sections were washed three times for 5 min in 0.1 M PBS. Slices were then blocked in blocking serum (10% Normal Goat Serum, 0.5% Triton X‐100, in 0.1 M PBS; 1 hr) and incubated at 4°C overnight in primary solution (10% Normal Goat Serum, 0.5% Triton X‐100; anti‐DsRed [1:500], Clontech #1408015; anti‐VIP [1:500], Santa Cruz #sc‐25347). Slices were washed in 0.1% PBS‐Tween 20 and incubated in secondary solution (10% Normal Goat Serum, 0.5% Triton X‐100; DsRed/mCherry, Alexa Fluor goat anti‐rabbit 555 [1:1000]; VIP, Alexa Fluor 488 goat anti‐mouse [1:1000], in PBS). Following secondary incubation, tissue was washed in 0.1% PBS‐Tween20 and incubated in DAPI [1:15000] in 0.1 M PBS. Sections were then slide mounted and cover slipped using VectaShield Mounting Medium (product #H‐1000).
2.6. Experimental design and statistical analysis
All recordings were analyzed in AxoGraph and all statistical tests were performed in GraphPad Prism (α = 0.05). Data are expressed as the mean ± standard error of the mean. The laminar location of each recorded neuron was determined as a percent of full cortical width, and the layer estimated based on previous studies in mouse auditory cortex (Cruikshank, Killackey, & Metherate, 2001): L1 0%–13%, L2/3 14%–33%, L4 34%–49%, L5 50%–72%, L6 73%–100%.
Membrane potential, spontaneous IPSC/EPSC frequency and amplitude, and firing rate were all determined from a 1‐min recording span; pre‐nicotine data were measured from the 1 min immediately prior to nicotine application and nicotine data were measured from 4 to 5 min after the start of nicotine application. Paired statistical tests (e.g., paired t tests) were used to compare pre‐nicotine to nicotine data, with the exact test along with number of cells and number of animals reported for each comparison in the Results. Membrane potential was the average over the 1‐min period. Nicotinic effects on membrane potential were expressed in the figures as “Depolarization”, i.e., nicotine membrane potential—pre‐nicotine membrane potential. IPSC/EPSC frequency (in Hertz) was determined from the number of events during the 1 min. Amplitude for spontaneous IPSCs/EPSCs was determined by measuring peak amplitude for each event, then averaging all event amplitudes over the 1 min. The amplitude for evoked IPSCs/EPSCs was measured as peak amplitude over the period 0–100 ms after stimulation, averaged from 5 to 10 repetitions.
3. RESULTS
Nicotine selectively modulates specific cell types, a feature that is likely critical to understanding the neural basis of nicotinic effects (Gulledge et al., 2007; Porter et al., 1999). To determine the specificity of nicotine in auditory cortex, we obtained whole‐cell recordings from four nonoverlapping classes of cells that constitute the majority of cortical neurons: Pyr, VIP, SOM, and PV neurons (Rudy, Fishell, Lee, & Hjerling‐Leffler, 2010). Pyr cells were identified by their pyramidal‐shaped soma and prominent apical dendrite. VIP, SOM, and PV cells were identified by crossing VIP‐Cre, SOM‐Cre, or PV‐Cre mice with the Cre reporter mouse Ai9, thus conferring tdTomato fluorescence to one cell type in each experiment.
Slices were taken from 25‐ to 50 day‐old mice and although P25‐P50 represents a time of continuing cortical and nAChR development (Kawai, Kang, & Metherate, 2011; Slotkin, 2002), we found no correlation of major nicotine effects with age (details below). All recordings were performed in the auditory cortex in a thalamocortical slice preparation, and each data set includes cells from all cortical layers; nicotine was bath‐applied (1 μΜ).
3.1. Nicotine selectively depolarizes VIP and Pyr neurons
First, we examined how nicotine alters the membrane potential of each cell type using current‐clamp whole‐cell recordings and a K+‐based pipette solution. We found that nicotine weakly but consistently depolarized Pyr cells an average of 1.39 ± 0.22 mV (Figure 1a,b; paired t test: n = 46 cells, 22 mice, t (45) = 6.17, p < 0.0001). This effect was observed in each cortical layer containing Pyr cell somata (Figure 1c; paired t test: L2/3 n = 18 cells, 11 mice, t (17) = 2.71, p = 0.015; L4 n = 15 cells, 11 mice, t (14) = 4.60, p = 0.0004; L5 n = 6 cells, 5 mice, t (5) = 3.14, p = 0.026; L6 n = 6 cells, 5 mice, t (5) = 2.61, p = 0.048), although it was stronger in deeper layers (Figure 1c; one‐way ANOVA: F (3,41) = 4.002, p = 0.014, with Tukey's post hoc test L2/3 versus L5 p = 0.024).
Figure 1.
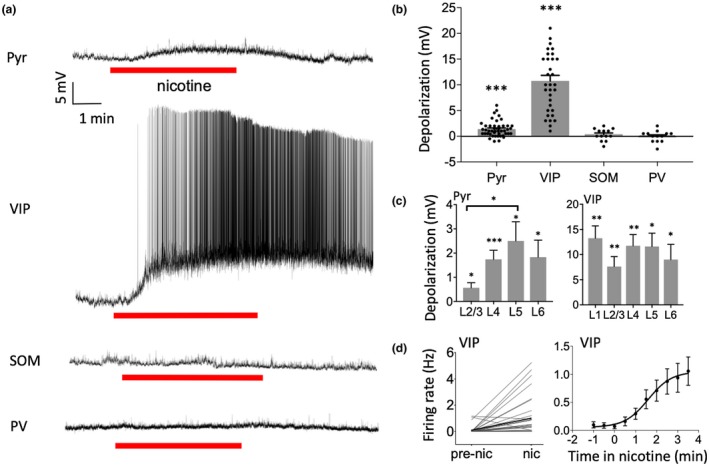
Nicotine selectively depolarized Pyr and VIP neurons. (a) Example recordings showing effect of nicotine (1 µM in bath) on membrane potential for each cell type. (b) Group data demonstrating nicotine‐induced depolarization in Pyr and VIP cells and no effect on SOM or PV cells. Data points represent individual cells. (c) Nicotinic depolarization by layer for Pyr (left) and VIP (right) neurons. Depolarization was stronger in deeper layers for Pyr neurons. (d) Nicotine increased action potential firing rate in VIP cells (left) and firing appears to peak after several minutes (right, firing rate examined in 30 s bins). For statistical comparisons in this and subsequent figures, asterisks indicate: *p < 0.05, **p < 0.01, ***p < 0.001
Furthermore, nicotine strongly depolarized VIP interneurons an average of 10.76 ± 1.08 mV (Figure 1a,b; paired t test: n = 33 cells, 14 mice, t (32) = 9.94, p < 0.0001). This potent response caused a majority (22/33) of VIP cells to fire action potentials, as reflected in spike frequency measures before and after nicotine application (Figure 1d, left; paired t test: n = 33 cells, 14 mice, t (32) = 3.64, p = 0.0009). Depolarization and spiking began soon after bath application of nicotine and took several minutes to reach peak firing with no evidence of adaptation (Figure 1d, right; one‐way ANOVA comparing firing rate across time bins: F (32,288) = 9.84, p < 0.0001, with Tukey's post hoc test comparing bins vs. −1 min: −0.5 p = 0.96, 0 p = 0.91, 0.5 p = 0.97, 1 p = 0.14, 1.5 p = 0.048, 2 p = 0.015, 2.5 p = 0.012, 3 p = 0.020, 3.5 p = 0.005).
Nicotine's depolarization of VIP cells also occurred in all layers (Figure 1c; paired t test: L1 n = 8 cells, 4 mice, t (7) = 5.38, p = 0.001. L2/3 n = 8 cells, 7 mice, t (7) = 3.87, p = 0.0062. L4 n = 8 cells, 6 mice, t (7) = 5.28, p = 0.0012. L5 n = 5 cells, 4 mice, t (4) = 4.37, p = 0.012. L6 n = 4 cells, 2 mice, t (3) = 2.97, p = 0.048). The degree of nicotinic depolarization in Pyr and VIP cells did not correlate with age (not shown; Pearson correlation for age [days] versus depolarization: Pyr n = 46 cells, 22 mice, r 2 = 0.015, p = 0.42; VIP n = 33 cells, 14 mice, r 2 = 0.010, p = 0.58).
In contrast to its effects on Pyr and VIP cells, nicotine did not alter the membrane potential of SOM or PV neurons (Figure 1a,b; paired t test: SOM n = 13 cells, 5 mice, t (12) = 0.95, p = 0.36; PV n = 13 cells, 5 mice, t (12) = 0.12, p = 0.90). Therefore, nicotine selectively depolarizes VIP and Pyr cells across cortical layers, with the most powerful effect being on VIP cells.
3.2. Nicotine directly depolarizes VIP neurons via β2‐containing nAChRs and indirectly depolarizes Pyr cells
To determine if the depolarization of Pyr and VIP cells resulted from direct activation of nAChRs located on these cell types, we applied nicotine after blocking synaptic activity. We bath‐applied 10 μΜ CNQX and 50 μΜ PTX for 7–10 min prior to nicotine, to block AMPA and GABA‐A receptors, respectively. CNQX and PTX prevented nicotinic depolarization of Pyr cells (Figure 2a; paired t test: n = 7 cells, 2 mice, t (6) = 1.99, p = 0.09), suggesting that nicotine's effects occurred indirectly. However, nicotinic depolarization of VIP cells persisted in the presence of CNQX and PTX, implying direct nAChR activation (Figure 2a; paired t test: n = 12 cells, 6 mice, t (11) = 6.21, p < 0.0001).
Figure 2.
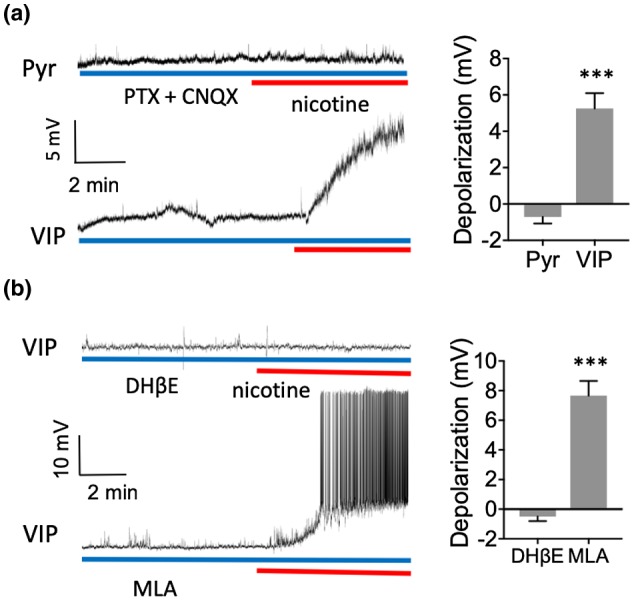
Nicotine directly depolarized VIP neurons via β2‐containing nAChRs and indirectly depolarized Pyr neurons. (a) Example recordings demonstrating nicotine's effect on membrane potential in the presence of 50 µM PTX and 10 µM CNQX (left). Group data (right) showing that PTX and CNQX prevented nicotinic depolarization in Pyr cells but not VIP cells. (b) Example recordings in VIP cells in the presence of 1 µM DHβE (top left) or 10 nM MLA (bottom left). DHβE prevented the nicotinic depolarization of VIP cells while MLA did not
We then sought to identify the receptor subtype mediating activation of VIP cells. The two main types of nAChRs in cortex are homomeric α7 and heteromeric α4β2 receptors that can be antagonized by MLA and DhβE, respectively (Arroyo et al., 2014; Dani & Bertrand, 2007; Xiao & Kellar, 2004). Nicotine's depolarization of VIP cells was blocked by DHβE (0.5–1 μΜ; Figure 2b, paired t test: n = 10 cells, 4 mice, t (9) = 1.63, p = 0.14), but not by MLA (10 nM; Figure 2b; paired t test: n = 6 cells, 2 mice, t (5) = 7.75, p = 0.0006), demonstrating that nicotine's actions on VIP cells are mediated by β2‐containing nAChRs. The DHβE results indicate the likely involvement of α4β2 receptors due to their predominance in cortex, but do not preclude the involvement of relatively sparse α subunits, such as α2 or α5 (Kleeman et al., 2016; Koukouli et al., 2017). Overall, the results indicate that nicotine indirectly depolarizes Pyr cells and directly depolarizes VIP neurons via β2‐containing, but not α7, nAChRs.
3.3. Nicotine enhances sIPSCs in Pyr, VIP, and SOM neurons
Given nicotine's potent excitation of VIP interneurons, we next assessed potential downstream effects by recording spontaneous inhibitory postsynaptic currents (sIPSC). Recordings were performed in voltage clamp with a Cs+‐based pipette solution and cells were clamped at 0 mV to isolate sIPSCs. We found that nicotine weakly, but consistently, increased the frequency of sIPSCs in Pyr cells (Figure 3a,b; paired t test: n = 26 cells, 10 mice, t (25) = 7.44, p < 0.0001) and that this enhancement occurred in each cortical layer (Figure 3d; paired t test: L2/3 n = 8 cells, 4 mice, t (7) = 3.96, p = 0.006; L4 n = 9 cells, 6 mice, t (8) = 6.41, p = 0.0002; L5/6 n = 9 cells, 5 mice, t (8) = 3.13, p = 0.014). Nicotine also weakly, but consistently, increased the frequency of sIPSCs in VIP cells (Figure 3a,b; paired t test: n = 10 cells, 3 mice, t (9) = 3.94, p = 0.0034), but had no effect on sIPSC frequency in PV cells (Figure 3a,b; paired t test: n = 8 cells, 2 mice, t (7) = 0.11, p = 0.91).
Figure 3.

Nicotine increased the frequency of sIPSCs in Pyr, SOM, and VIP neurons. (a) Example recordings showing nicotine's effects on sIPSCs for each cell type (0 mV holding potential). (b) Group data showing that nicotine increased the frequency of sIPSCs in Pyr, VIP, and SOM cells with no effect on PV cells (left). Nicotine did not alter the mean amplitude of sIPSCs in any cell type (right). (c) Same data as in B but for individual cells, to show the consistency of nicotine's effects. (d) Nicotine enhanced the frequency of sIPSCs in Pyr cells in all layers
More notable was nicotine's effect on SOM cells. Prior to nicotine application, SOM neurons had few sIPSCs, e.g., relative to Pyr cells (Figure 3a,b; unpaired t test comparing frequency: t (30) = 3.42, p = 0.0019). However, nicotine strongly increased sIPSC frequency in SOM cells (Figure 3a,b; paired t test: n = 6 cells, 3 mice, t (5) = 4.19, p = 0.0086). Although the degree of nicotinic effects on sIPSCs varied considerably among Pyr, VIP, and SOM cells, the enhancement occurred in almost all of these cells, as seen in individual plots (Figure 3c). The time course of nicotine effects was slow, on the order of minutes, similar to that of VIP cell depolarization and spiking (Figure 1). Finally, nicotine had no effect on the mean amplitude of sIPSCs in any cell type (Figure 3b; paired t test: Pyr t (25) = 1.09, p = 0.29; VIP t (9) = 1.39, p = 0.20; SOM t (5) = 0.46, p = 0.67; PV t (7) = 0.24, p = 0.82), although we did observe in some cells that the largest amplitude responses appeared only with nicotine application.
We additionally examined spontaneous excitatory postsynaptic currents (sEPSC) in Pyr cells by clamping the membrane potential at −52 mV, the observed reversal potential of stimulus‐evoked IPSCs (see Methods). Nicotine had no effect on frequency or amplitude of sEPSCs (Figure 4a,b; paired t test: n = 9 cells, 5 mice, frequency t (8) = 1.23, p = 0.25, amplitude t (8) = 0.96, p = 0.37).
Figure 4.
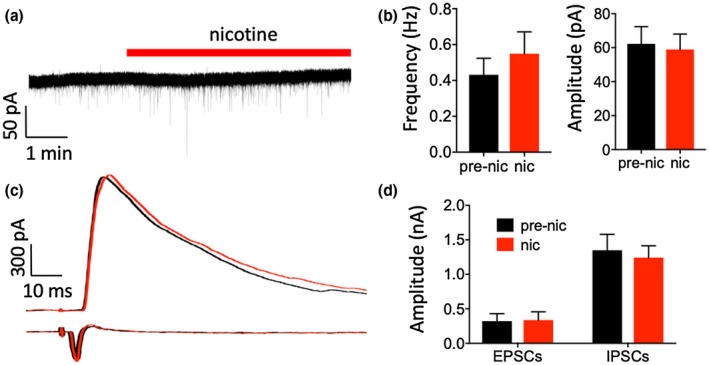
Nicotine had no effect on EPSCs or evoked IPSCs in Pyr neurons. (a) Example recording of sEPSCs in a Pyr neuron (holding potential, −52 mV, see Results). (b) Group data showing that nicotine had no effect on the frequency (left) or amplitude (right) of sEPSCs. (c) Example recordings of evoked IPSCs (top, holding potential 0 mV) and evoked EPSCs (bottom, holding potential −52 mV) in two separate Pyr neurons. (d) Group data showing that nicotine had no effect on the peak amplitude of evoked EPSCs or evoked IPSCs
Finally, we recorded thalamic afferent‐evoked EPSCs and IPSCs by placing a stimulating electrode in the thalamocortical pathway and stimulating with above‐minimal intensities. As with spontaneous events, evoked IPSCs were recorded at 0 mV and evoked EPSCs at −52 mV. Nicotine did not alter the amplitude of evoked IPSCs or evoked EPSCs (Figure 4c,d; paired t test: IPSCs n = 10 cells, 6 mice, t (9) = 1.07, p = 0.31; EPSCs n = 6 cells, 4 mice, t (5) = 0.85, p = 0.43). Thus, nicotine appears to modulate sIPSCs, likely due to excitation of VIP neurons, but not sEPSCs or thalamic afferent‐evoked synaptic responses.
3.4. Nicotine disinhibits Pyr neurons via VIP interneurons
Recent studies have shown that VIP interneurons preferentially inhibit other inhibitory interneurons that, in turn, inhibit Pyr cells. Consequently, VIP cell activation results in the disinhibition of Pyr cells (Lee et al., 2013; Pfeffer et al., 2013). Since our results show that nicotine directly activates VIP cells and indirectly depolarizes Pyr cells, it is possible that nicotinic depolarization of Pyr cells depends on activation of VIP interneurons. To address this, we silenced VIP interneurons using inhibitory DREADDs that primarily prevent synaptic release of neurotransmitter from HM4D‐expressing cells (Amat et al., 2017; Lichtenberg et al., 2017; Stachniak, Ghosh, & Sternson, 2014). Cre‐inducible AAV hM4D viruses were injected into the auditory cortex of VIP‐Cre mice (Figure 5a), and inhibitory DREADDs expressed in VIP neurons were activated by the agonist CNO (100 nM). We initially used higher concentrations of CNO (1–10 μM) as in prior electrophysiology studies (Alexander et al., 2009; Krashes et al., 2011; Urban et al., 2016), but found in control studies that these higher concentrations depolarized about half of pyramidal cells 1–2 mV (data not shown).
Figure 5.

Nicotine depolarized and enhanced sIPSC frequency in Pyr neurons via VIP neurons. (a) Coronal section with immunohistochemistry against DAPI (blue), mCherry from the HM4D construct (red), and VIP (green) in HM4D‐transduced auditory cortex. Inset shows co‐labeling of mCherry and VIP in an example cell. (b) Example recordings from Pyr neurons in HM4D‐expressing mice demonstrating that CNO application prevents nicotine's effects on membrane potential (top) and sIPSCs (bottom). (c) Group data from Pyr cells in HM4D‐expressing mice; nicotine depolarized Pyr cells and CNO prevented the nicotinic depolarization of Pyr cells. (d) Group data from Pyr cells in HM4D‐expressing mice; nicotine enhanced the frequency of sIPSCs and CNO prevented the nicotinic enhancement of sIPSC frequency
To ensure that HM4D expression per se did not alter nicotinic effects in Pyr cells, we first confirmed that nicotine still depolarized Pyr cells and enhanced sIPSCs (Figure 5c,d; paired t test: depolarization n = 5 cells, 2 mice, t (4) = 4.71, p = 0.0093; sIPSC frequency n = 7 cells, 3 mice, t (6) = 3.32, p = 0.02). Similarly, we confirmed that 100 nM CNO in the absence of HM4D did not affect Pyr cells (not shown; paired t test: depolarization n = 5 cells, 2 mice, t (4) = 0.18, p = 0.87; sIPSC frequency n = 6 cells, 2 mice, t (5) = 0.97, p = 0.38).
In VIP‐Cre/HM4D mice, however, CNO prevented the nicotine‐induced depolarization and enhancement of sIPSC frequency (Figure 5b,c,d; paired t test: depolarization n = 9 cells, 3 mice, t (8) = 0.60, p = 0.56; sIPSC frequency n = 7 cells, 3 mice, t (6) = 0.76, p = 0.47). Thus, silencing VIP neurons prevents nicotine's effects in Pyr cells and provides evidence that nicotinic regulation of Pyr neurons depends on excitation of VIP neurons.
4. DISCUSSION
In this study, we examined the cell‐type specificity of nicotine's effects in auditory cortex to reveal three key findings: (a) Nicotine depolarizes cell types selectively; i.e., nicotine weakly depolarizes Pyr cells, strongly depolarizes and excites VIP cells, and does not alter membrane potential of SOM or PV cells. (b) Nicotine enhances the frequency of sIPSCs selectively; i.e., weakly in Pyr and VIP cells, strongly in SOM cells and not at all in PV cells. (c) Nicotine‐induced depolarization and enhanced sIPSC frequency in Pyr cells require activation of VIP neurons, implicating nicotinic activation of a disinhibitory neural circuit as well as direct VIP neuron projections to Pyr neurons.
It is important to note that in our study nicotine bath application lasts several minutes, whereas many studies use rapid and brief application to avoid desensitizing nAChRs. Thus, our results—sustained activation of VIP cells and elevation of sIPSC frequency—reflect weakly desensitizing or non‐desensitizing effects of nicotine that are relevant to understanding the effects of in vivo systemic administration of nicotine (which is our rationale for using bath application). Conversely, endogenous ACh activation of nAChRs presents a more complex picture, with both phasic and tonic actions contributing to effects (Klinkenberg, Sambeth, & Blokland, 2011; Sarter, Parikh, & Howe, 2009).
4.1. Nicotine's effects on Pyr cells
We observed that nicotine weakly and indirectly depolarizes Pyr cells in cortical layers 2–6. Other studies similarly find little evidence of direct nAChR activation on Pyr cells (Christophe et al., 2002; Disney et al., 2007; Gulledge et al., 2007). A few exceptions include direct nAChR activation of L5 and L6 Pyr cells, although these studies used higher concentrations of nicotine (10 μM–1 mM) or ACh (1 mM) and were performed in regions other than auditory cortex (Kassam, Herman, Goodfellow, Alves, & Lambe, 2008; Zolles et al., 2009). It is possible that some Pyr cells in auditory cortex, especially in deeper layers, express nAChRs that may respond to higher concentrations or rapid application of nicotine.
We also found that nicotine enhances the frequency of sIPSCs in Pyr cells, consistent with prior studies (Couey et al., 2007). Our experiments additionally show that this enhancement is mediated through VIP cells, presumably by the weak but direct VIP projection to Pyr cells seen in previous studies (Lee et al., 2013; Pfeffer et al., 2013).
Although we saw no change in the amplitude of thalamic afferent‐evoked responses under voltage clamp, this doesn't preclude the ability of nicotine to modify cortical responses to sensory stimuli in vivo, as seen previously (Askew, Intskirveli, & Metherate, 2017; Intskirveli & Metherate, 2012). Rather, when not under voltage‐clamp control, nicotinic depolarization of Pyr cells would move the membrane potential closer to spike threshold, resulting in heightened responsiveness and potentially contributing to the increased gain of acoustic responses observed previously. Also, the nicotinic effects on neural circuit dynamics observed here and in prior in vitro studies will likely have complex outcomes on intracortical processing of sensory stimuli, potentially contributing to the increased gain within narrowed acoustic receptive fields observed previously (Askew et al., 2017; Intskirveli & Metherate, 2012).
4.2. Nicotine's effects on interneurons
We found that nicotine has distinct effects on specific interneuron types. Nicotine strongly depolarizes and excites VIP cells via β2‐containing nAChRs, while having no effect on SOM or PV cell membrane potential. Also, nicotine potently enhanced the frequency of sIPSCs in SOM neurons, weakly enhanced the frequency of sIPSCs in VIP cells, and had no effect on sIPSCs in PV cells.
Several prior studies support direct nAChR activation of VIP cells. A majority of cells responsive to nicotinic agonists express VIP, and ACh activates VIP neurons via α4β2 receptors (Bell, Bell, & McQuiston, 2015; Lee, Hjerling‐Leffler, Zagha, Fishell, & Rudy, 2010; Porter et al., 1999). VIP cell activation increases evoked responses in visual cortex (Fu et al., 2014) and in frontal cortex improves behavioral performance in a memory‐dependent task (Kamigaki & Dan, 2017), so nicotinic activation of VIP cells may have similar systems‐level effects. Our experiments extend these results by demonstrating direct and potent VIP cell depolarization and sustained spiking by low concentrations of nicotine relevant to therapeutic administration (Newhouse et al., 2012; Rezvani & Levin, 2001), as well as revealing functional consequences for neural circuitry.
PV and SOM generally do not seem to express nAChRs, although there is some indication that subpopulations within these groups may contain nAChRs (Gulledge et al., 2007; Porter et al., 1999). One study demonstrated L2/3 SOM neurons with functional nAChRs and fast‐spiking (presumably PV) neurons expressing α7 receptors (Poorthuis et al., 2013). However, as mentioned above, rapid desensitization of α7 nAChRs would preclude observation of effects in the present study with bath application of nicotine. Moreover, in hippocampus, a subpopulation of putative SOM interneurons exhibit a non‐desensitizing α2 nAChR‐mediated response to bath application of 1 μM nicotine (Jia, Yamazaki, Nakauchi, & Sumikawa, 2009), though in our study such depolarization might be masked by counteracting enhanced inhibition. Further studies of specific interneuron subtypes may be needed to resolve these discrepancies.
There is substantial evidence that VIP neurons preferentially innervate and inhibit SOM cells, consistent with the powerful, nicotine‐induced enhancement of sIPSC frequency that we observed in SOM cells (Lee et al., 2013; Pfeffer et al., 2013; Pi et al., 2013). Although we did not directly demonstrate VIP involvement, it appears likely that the nicotinic excitation of VIP cells causes the sIPSC enhancement in SOM neurons.
4.3. VIP interneuron‐mediated inhibitory mechanisms
Nicotine increased the frequency of sIPSCs in three cell types, yet we found no evidence of a corresponding hyperpolarization that might be expected with enhanced inhibitory input. This could be due to the space‐clamp limitations of our current‐clamp recordings; i.e., cortical cells are known to have extensive dendritic processes, which may not be accurately recorded from by somatic recordings, especially with K+‐based internal solutions. If VIP cells primarily innervate distal dendrites of their postsynaptic target, the inhibition and hyperpolarization evoked by VIP cells might remain localized to this cellular compartment. Some SOM neurons are characterized by this type of specificity and their preferential inhibition of Pyr cell distal dendrites is thought to alter the balance of synaptic integration (Di Cristo et al., 2004). VIP cells may similarly target dendritic processes in select cell types.
In fact, in voltage‐clamp recordings (using a Cs+‐based solution that blocks potassium channels and reduces space‐clamp error) nicotine does appear to alter the baseline holding current in SOM cells (see example in Figure 3a). This change in baseline holding current may reflect a small hyperpolarization that cannot be seen with the K+‐based solution, suggesting that alterations in the membrane potential of SOM neurons may occur distant from the soma. On the other hand, previous studies found that VIP cells target both the dendrites and soma of Pyr cells (Kawaguchi & Kubota, 1996, 1997). Even though direct VIP inhibition of Pyr cells is weak, it is possible that it still results in the hyperpolarization of Pyr cells. Yet it appears in our experiments and in other studies that the predominate effect of exciting VIP interneurons is that of disinhibition (Fu et al., 2014; Lee et al., 2013; Pfeffer et al., 2013; Pi et al., 2013), thus any direct hyperpolarization of Pyr cells may be overridden by the counteracting depolarization.
4.4. Disinhibition of Pyr cells by VIP neurons
We also observed that nicotine depolarizes Pyr cells via activation of VIP interneurons. This finding supports growing evidence that activating VIP neurons exerts a disinhibitory effect on Pyr cells. In the auditory cortex of awake mice, optogenetic activation of VIP neurons suppresses SOM cells and increases tone‐evoked responses in principal neurons (Pi et al., 2013). A similar effect occurs in visual cortex, where VIP cell activation also enhances sensory‐evoked responses (Fu et al., 2014). Given that VIP cells strongly inhibit SOM cells (Lee et al., 2013; Pfeffer et al., 2013; Pi et al., 2013), it is probable that the nicotinic disinhibition of Pyr cells involves SOM cells that tonically inhibit Pyr cells (Gentet et al., 2012). That is, in our experiments, nicotine activates VIP cells which then inhibit SOM cells, causing a release from tonic inhibition of Pyr cells.
4.5. Conclusions and broader implications
We conclude that non‐desensitizing (or weakly desensitizing) effects of nicotine selectively excite VIP neurons in auditory cortex to directly inhibit VIP, SOM, and Pyr neurons, and indirectly disinhibit Pyr cells. VIP cell excitation may alter cortical processing by making Pyr cells more responsive to inputs near the soma, i.e., the site of nicotine‐induced depolarization, and less responsive to inputs near the site of direct inhibition. Presumed direct VIP neuron projections to interneurons and Pyr cells produced sIPSCs but not somatic hyperpolarization, suggesting that the projections are to distal dendrites (particularly in SOM neurons; in VIP and Pyr neurons somatic inhibition may have been masked by depolarization). It is not clear how inhibition of distal dendrites would alter intracortical processing, but the overall result of nicotine's actions may contribute to increased Pyr neuron responsiveness and selectivity to acoustic inputs (Askew et al., 2017; Intskirveli & Metherate, 2012). Given the complexity of nAChR‐mediated cellular actions in this and prior studies, a full understanding of nicotinic regulation will require integrating the contributions of diverse nAChRs with varying subunit composition, cellular distribution, and response to agonist (Gil & Metherate, 2019; Poorthuis et al., 2013). Overall, nicotinic modulation may serve as a preparatory mechanism for incoming input, resulting in improved cortical processing. Although these experiments were performed in auditory cortex, other cortical regions may contain similar networks and nAChR functionality. Thus, these data provide insight into potential mechanisms underlying the pro‐cognitive and sensory processing effects of nicotine in multiple cortical regions.
CONFLICT OF INTEREST
The authors declare no competing financial interests. The data that support the findings of this study are available from the corresponding author upon reasonable request.
ACKNOWLEDGMENTS
Supported by the National Institutes of Health (R01 DC013200, R01 DA025922, P30 DC08369, T32 DC010775, F99 NS105217) and the National Science Foundation (DGE‐1321846).
Askew CE, Lopez AJ, Wood MA, Metherate R. Nicotine excites VIP interneurons to disinhibit pyramidal neurons in auditory cortex. Synapse. 2019;73:e22116 10.1002/syn.22116
REFERENCES
- Alexander, G. M. , Rogan, S. C. , Abbas, A. I. , Armbruster, B. N. , Pei, Y. , Allen, J. A. , … Roth, B. L. (2009). Remote control of neuronal activity in transgenic mice expressing evolved G protein‐coupled receptors. Neuron, 63(1), 27–39. 10.1016/j.neuron.2009.06.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amat, S. B. , Rowan, M. J. M. , Gaffield, M. A. , Bonnan, A. , Kikuchi, C. , Taniguchi, H. , & Christie, J. M. (2017). Using c‐kit to genetically target cerebellar molecular layer interneurons in adult mice. PLoS One, 12(6), e0179347 10.1371/journal.pone.0179347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arroyo, S. , Bennett, C. , & Hestrin, S. (2014). Nicotinic modulation of cortical circuits. Frontiers in Neural Circuits, 8, 30 10.3389/fncir.2014.00030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Askew, C. , Intskirveli, I. , & Metherate, R. (2017). Systemic nicotine increases gain and narrows receptive fields in A1 via integrated cortical and subcortical actions. eNeuro, 4(3). 10.1523/ENEURO.0192-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell, L. A. , Bell, K. A. , & McQuiston, A. R. (2015). Acetylcholine release in mouse hippocampal CA1 preferentially activates inhibitory‐selective interneurons via alpha4beta2* nicotinic receptor activation. Frontiers in Cellular Neuroscience, 9, 115 10.3389/fncel.2015.00115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christophe, E. , Roebuck, A. , Staiger, J. F. , Lavery, D. J. , Charpak, S. , & Audinat, E. (2002). Two types of nicotinic receptors mediate an excitation of neocortical layer I interneurons. Journal of Neurophysiology, 88(3), 1318–1327. 10.1152/jn.2002.88.3.1318 [DOI] [PubMed] [Google Scholar]
- Clarke, P. B. , Schwartz, R. D. , Paul, S. M. , Pert, C. B. , & Pert, A. (1985). Nicotinic binding in rat brain: Autoradiographic comparison of [3H]acetylcholine, [3H]nicotine, and [125I]‐alpha‐bungarotoxin. Journal of Neuroscience, 5(5), 1307–1315. 10.1523/JNEUROSCI.05-05-01307.1985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couey, J. J. , Meredith, R. M. , Spijker, S. , Poorthuis, R. B. , Smit, A. B. , Brussaard, A. B. , & Mansvelder, H. D. (2007). Distributed network actions by nicotine increase the threshold for spike‐timing‐dependent plasticity in prefrontal cortex. Neuron, 54(1), 73–87. 10.1016/j.neuron.2007.03.006 [DOI] [PubMed] [Google Scholar]
- Cruikshank, S. J. , Killackey, H. P. , & Metherate, R. (2001). Parvalbumin and calbindin are differentially distributed within primary and secondary subregions of mouse auditory forebrain. Neuroscience, 105, 553–569. [DOI] [PubMed] [Google Scholar]
- Cruikshank, S. J. , Rose, H. J. , & Metherate, R. (2002). Auditory thalamocortical synaptic transmission in vitro. Journal of Neurophysiology, 87(1), 361–384. 10.1152/jn.00549.2001 [DOI] [PubMed] [Google Scholar]
- Dani, J. A. , & Bertrand, D. (2007). Nicotinic acetylcholine receptors and nicotinic cholinergic mechanisms of the central nervous system. Annual Review of Pharmacology and Toxicology, 47, 699–729. 10.1146/annurev.pharmtox.47.120505.105214 [DOI] [PubMed] [Google Scholar]
- Di Cristo, G. D. , Wu, C. , Chattopadhyaya, B. , Ango, F. , Knott, G. , Welker, E. , … Huang, Z. J. (2004). Subcellular domain‐restricted GABAergic innervation in primary visual cortex in the absence of sensory and thalamic inputs. Nature Neuroscience, 7(11), 1184–1186. 10.1038/nn1334 [DOI] [PubMed] [Google Scholar]
- Disney, A. A. , Aoki, C. , & Hawken, M. J. (2007). Gain modulation by nicotine in macaque v1. Neuron, 56(4), 701–713. 10.1016/j.neuron.2007.09.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, Y. U. , Tucciarone, J. M. , Espinosa, J. S. , Sheng, N. , Darcy, D. P. , Nicoll, R. A. , … Stryker, M. P. (2014). A cortical circuit for gain control by behavioral state. Cell, 156(6), 1139–1152. 10.1016/j.cell.2014.01.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentet, L. J. , Kremer, Y. , Taniguchi, H. , Huang, Z. J. , Staiger, J. F. , & Petersen, C. C. (2012). Unique functional properties of somatostatin‐expressing GABAergic neurons in mouse barrel cortex. Nature Neuroscience, 15(4), 607–612. 10.1038/nn.3051nn.3051 [DOI] [PubMed] [Google Scholar]
- Gil, S. M. , & Metherate, R. (2019). Enhanced sensory‐cognitive processing by activation of nicotinic acetylcholine receptors. Nicotine & Tobacco Research, 21(3), 377–382. 10.1093/ntr/nty134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulledge, A. T. , Park, S. B. , Kawaguchi, Y. , & Stuart, G. J. (2007). Heterogeneity of phasic cholinergic signaling in neocortical neurons. Journal of Neurophysiology, 97(3), 2215–2229. 10.1152/jn.00493.2006 [DOI] [PubMed] [Google Scholar]
- Intskirveli, I. , & Metherate, R. (2012). Nicotinic neuromodulation in auditory cortex requires MAPK activation in thalamocortical and intracortical circuits. Journal of Neurophysiology, 107(10), 2782–2793. 10.1152/jn.01129.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson, J. , Ayzenshtat, I. , Karnani, M. M. , & Yuste, R. (2016). VIP+ interneurons control neocortical activity across brain states. Journal of Neurophysiology, 115(6), 3008–3017. 10.1152/jn.01124.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia, Y. , Yamazaki, Y. , Nakauchi, S. , & Sumikawa, K. (2009). Alpha2 nicotine receptors function as a molecular switch to continuously excite a subset of interneurons in rat hippocampal circuits. European Journal of Neuroscience, 29(8), 1588–1603. 10.1111/j.1460-9568.2009.06706.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamigaki, T. , & Dan, Y. (2017). Delay activity of specific prefrontal interneuron subtypes modulates memory‐guided behavior. Nature Neuroscience, 20(6), 854–863. 10.1038/nn.4554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassam, S. M. , Herman, P. M. , Goodfellow, N. M. , Alves, N. C. , & Lambe, E. K. (2008). Developmental excitation of corticothalamic neurons by nicotinic acetylcholine receptors. Journal of Neuroscience, 28(35), 8756–8764. 10.1523/JNEUROSCI.2645-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi, Y. , & Kubota, Y. (1996). Physiological and morphological identification of somatostatin‐ or vasoactive intestinal polypeptide‐containing cells among GABAergic cell subtypes in rat frontal cortex. Journal of Neuroscience, 16, 2701–2715. 10.1523/JNEUROSCI.16-08-02701.1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi, Y. , & Kubota, Y. (1997). GABAergic cell subtypes and their synaptic connections in rat frontal cortex. Cerebral Cortex, 7, 476–486. 10.1093/cercor/7.6.476 [DOI] [PubMed] [Google Scholar]
- Kawai, H. D. , Kang, H. A. , & Metherate, R. (2011). Heightened nicotinic regulation of auditory cortex during adolescence. Journal of Neuroscience, 31(40), 14367–14377. 10.1523/JNEUROSCI.1705-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleeman, E. , Nakauchi, S. , Su, H. , Dang, R. , Wood, M. A. , & Sumikawa, K. (2016). Impaired function of alpha2‐containing nicotinic acetylcholine receptors on oriens‐lacunosum moleculare cells causes hippocampus‐dependent memory impairments. Neurobiology of Learning and Memory, 136, 13–20. 10.1016/j.nlm.2016.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinkenberg, I. , Sambeth, A. , & Blokland, A. (2011). Acetylcholine and attention. Behavioral Brain Research, 221(2), 430–442. 10.1016/j.bbr.2010.11.033 [DOI] [PubMed] [Google Scholar]
- Koukouli, F. , Rooy, M. , Tziotis, D. , Sailor, K. A. , O'Neill, H. C. , Levenga, J. , … Maskos, U. (2017). Nicotine reverses hypofrontality in animal models of addiction and schizophrenia. Nature Medicine, 23(3), 347–354. 10.1038/nm.4274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krashes, M. J. , Koda, S. , Ye, C. P. , Rogan, S. C. , Adams, A. C. , Cusher, D. S. , … Lowell, B. B. (2011). Rapid, reversible activation of AgRP neurons drives feeding behavior in mice. Journal of Clinical Investigation, 121(4), 1424–1428. 10.1172/JCI46229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumari, V. , & Postma, P. (2005). Nicotine use in schizophrenia: The self medication hypotheses. Neuroscience and Biobehavioral Reviews, 29(6), 1021–1034. 10.1016/j.neubiorev.2005.02.006 [DOI] [PubMed] [Google Scholar]
- Lawrence, N. S. , Ross, T. J. , & Stein, E. A. (2002). Cognitive mechanisms of nicotine on visual attention. Neuron, 36(3), 539–548. 10.1016/S0896-6273(02)01004-8 [DOI] [PubMed] [Google Scholar]
- Lee, S. , Hjerling‐Leffler, J. , Zagha, E. , Fishell, G. , & Rudy, B. (2010). The largest group of superficial neocortical GABAergic interneurons expresses ionotropic serotonin receptors. Journal of Neuroscience, 30(50), 16796–16808. 10.1523/JNEUROSCI.1869-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, S. , Kruglikov, I. , Huang, Z. J. , Fishell, G. , & Rudy, B. (2013). A disinhibitory circuit mediates motor integration in the somatosensory cortex. Nature Neuroscience, 16(11), 1662–1670. 10.1038/nn.3544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin, E. D. , McClernon, F. J. , & Rezvani, A. H. (2006). Nicotinic effects on cognitive function: Behavioral characterization, pharmacological specification, and anatomic localization. Psychopharmacology (Berl), 184(3–4), 523–539. 10.1007/s00213-005-0164-7 [DOI] [PubMed] [Google Scholar]
- Lichtenberg, N. T. , Pennington, Z. T. , Holley, S. M. , Greenfield, V. Y. , Cepeda, C. , Levine, M. S. , & Wassum, K. M. (2017). Basolateral amygdala to orbitofrontal cortex projections enable cue‐triggered reward expectations. Journal of Neuroscience, 37(35), 8374–8384. 10.1523/JNEUROSCI.0486-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- López, A. J. , Jia, Y. , White, A. O. , Kwapis, J. L. , Espinoza, M. , Hwang, P. , … Wood, M. A. (2019). Medial habenula cholinergic signaling regulates cocaine‐associated relapse‐like behavior. Addiction Biology, 24(3), 403–413. 10.1111/adb.12605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newhouse, P. , Kellar, K. , Aisen, P. , White, H. , Wesnes, K. , Coderre, E. , … Levin, E. D. (2012). Nicotine treatment of mild cognitive impairment: A 6‐month double‐blind pilot clinical trial. Neurology, 78(2), 91–101. 10.1212/WNL.0b013e31823efcbb [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeffer, C. K. , Xue, M. , He, M. , Huang, Z. J. , & Scanziani, M. (2013). Inhibition of inhibition in visual cortex: The logic of connections between molecularly distinct interneurons. Nature Neuroscience, 16(8), 1068–1076. 10.1038/nn.3446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pi, H. J. , Hangya, B. , Kvitsiani, D. , Sanders, J. I. , Huang, Z. J. , & Kepecs, A. (2013). Cortical interneurons that specialize in disinhibitory control. Nature, 503(7477), 521–524. 10.1038/nature12676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poorthuis, R. B. , Bloem, B. , Schak, B. , Wester, J. , de Kock, C. P. , & Mansvelder, H. D. (2013). Layer‐specific modulation of the prefrontal cortex by nicotinic acetylcholine receptors. Cerebral Cortex, 23(1), 148–161. 10.1093/cercor/bhr390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter, J. T. , Cauli, B. , Tsuzuki, K. , Lambolez, B. , Rossier, J. , & Audinat, E. (1999). Selective excitation of subtypes of neocortical interneurons by nicotinic receptors. Journal of Neuroscience, 19(13), 5228–5235. 10.1523/JNEUROSCI.19-13-05228.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezvani, A. H. , & Levin, E. D. (2001). Cognitive effects of nicotine. Biological Psychiatry, 49(3), 258–267. 10.1016/S0006-3223(00)01094-5 [DOI] [PubMed] [Google Scholar]
- Rudy, B. , Fishell, G. , Lee, S. , & Hjerling‐Leffler, J. (2010). Three groups of interneurons account for nearly 100% of neocortical GABAergic neurons. Developmental Neurobiology, 105(3), 553–569. 10.1002/dneu.20853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarter, M. , Parikh, V. , & Howe, W. M. (2009). Phasic acetylcholine release and the volume transmission hypothesis: Time to move on. Nature Reviews Neuroscience, 10(5), 383–390. 10.1038/nrn2635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slotkin, T. A. (2002). Nicotine and the adolescent brain: Insights from an animal model. Neurotoxicology and Teratology, 24(3), 369–384. 10.1016/S0892-0362(02)00199-X [DOI] [PubMed] [Google Scholar]
- Stachniak, T. J. , Ghosh, A. , & Sternson, S. M. (2014). Chemogenetic synaptic silencing of neural circuits localizes a hypothalamus–>midbrain pathway for feeding behavior. Neuron, 82(4), 797–808. 10.1016/j.neuron.2014.04.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, Y. , Yang, Y. , Galvin, V. C. , Yang, S. , Arnsten, A. F. , & Wang, M. (2017). Nicotinic alpha4beta2 cholinergic receptor influences on dorsolateral prefrontal cortical neuronal firing during a working memory task. Journal of Neuroscience, 37(21), 5366–5377. 10.1523/JNEUROSCI.0364-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swan, G. E. , & Lessov‐Schlaggar, C. N. (2007). The effects of tobacco smoke and nicotine on cognition and the brain. Neuropsychology Review, 17(3), 259–273. 10.1007/s11065-007-9035-9 [DOI] [PubMed] [Google Scholar]
- Urban, D. J. , Zhu, H. U. , Marcinkiewcz, C. A. , Michaelides, M. , Oshibuchi, H. , Rhea, D. , … Roth, B. L. (2016). Elucidation of the behavioral program and neuronal network encoded by dorsal raphe serotonergic neurons. Neuropsychopharmacology, 41(5), 1404–1415. 10.1038/npp.2015.293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao, Y. , & Kellar, K. J. (2004). The comparative pharmacology and up‐regulation of rat neuronal nicotinic receptor subtype binding sites stably expressed in transfected mammalian cells. Journal of Pharmacology and Experimental Therapeutics, 310(1), 98–107. 10.1124/jpet.104.066787 [DOI] [PubMed] [Google Scholar]
- Zolles, G. , Wagner, E. , Lampert, A. , & Sutor, B. (2009). Functional expression of nicotinic acetylcholine receptors in rat neocortical layer 5 pyramidal cells. Cerebral Cortex, 19(5), 1079–1091. 10.1093/cercor/bhn158 [DOI] [PubMed] [Google Scholar]