

Abstract
Fructose is a commonly ingested dietary sugar which has been implicated in playing a particularly harmful role in the development of metabolic disease. Fructose is primarily metabolised by the liver in humans, and increases rates of hepatic de novo lipogenesis. Fructose increases hepatic de novo lipogenesis via numerous mechanisms: by altering transcriptional and allosteric regulation, interfering with cellular energy sensing, and disrupting the balance between lipid synthesis and lipid oxidation. Hepatic de novo lipogenesis is also upregulated by the inability to synthesise glycogen, either when storage is inhibited in knock‐down animal models or storage is saturated in glycogen storage disease. Considering that fructose has the capacity to upregulate hepatic glycogen storage, and replenish these stores more readily following glycogen depleting exercise, the idea that hepatic glycogen storage and hepatic de novo lipogenesis are linked is an attractive prospect. We propose that hepatic glycogen stores may be a key factor in determining the metabolic responses to fructose ingestion, and saturation of hepatic glycogen stores could exacerbate the negative metabolic effects of excessive fructose intake. Since physical activity potently modulates glycogen metabolism, this provides a rationale for considering nutrient–physical activity interactions in metabolic health.
Keywords: fructose, liver, hepatic, glycogen, de novo lipogenesis, metabolism
Introduction
Fructose is a hexose with an identical chemical formula to glucose (C6H12O6), but with a keto group in position two of its carbon chain instead of an aldehyde group in position one of the carbon chain (Tappy & Lê, 2010). Whilst some fructose can be endogenously produced (Hwang et al. 2017), most fructose becomes available to humans from the diet. In Europe, it is estimated that two‐thirds of dietary fructose is consumed as sucrose (a glucose–fructose disaccharide, commonly known as ‘table sugar’) and around one‐third is ingested as free fructose (Sluik et al. 2015), although even when fructose is consumed in the free form, it is rarely consumed without the co‐ingestion of glucose (either as free glucose or glucose polymers). Common food sources of fructose intake in Europe include soft drinks (sugar sweetened beverages), fruit juices, fruits, cakes and dairy products (Sluik et al. 2015). Reported intake of free sugars, including fructose, is quantitatively important, ranging from ∼40 to 100 g per day across developed nations (roughly 7–20% of total energy intake) (Wittekind & Walton, 2014). The role of fructose in the human diet could be viewed as contentious, since some would argue that fructose is a uniquely harmful sugar for metabolic health and should be essentially avoided by all (Lustig et al. 2012), whereas others would argue that certain populations have exquisite metabolic health in the presence of extremely high fructose intakes (Pontzer et al. 2018), and there are even some recommendations for fructose‐containing carbohydrates to optimise performance and recovery during competition and intensive training in athletes (Gonzalez et al. 2016, 2017). In an attempt to solve this conflict on the role(s) of dietary fructose, this symposium review will aim to unify the related symposium reviews (Fuchs et al. 2019; Pinnick & Hodson, 2019; Tappy & Rosset, 2019; von Holstein‐Rathlou, 2019) by demonstrating how moderating physiological factors are important to consider when assessing the impact of fructose ingestion on metabolic health. Since fructose is a key contributor to disorders of fat metabolism, the role of fructose in hepatic lipogenesis will be a key focus. We will present the hypothesis that hepatic glycogen stores may regulate metabolic responses to fructose ingestion and could therefore be a target to prevent or mitigate the negative metabolic effects of fructose intake.
Dietary carbohydrate intake
Dietary carbohydrates reportedly comprise ∼46% (224 g per day) of energy intake in the UK, with ‘free sugars’ comprising ∼11% (57 g per day) (Roberts et al. 2018). Dietary carbohydrates are commonly classified as mono‐/disaccharides composed of one/two monomers (e.g. glucose, fructose, galactose), oligosaccharides composed of typically 3–10 monomers (e.g. maltodextrins, raffinose), or polysaccharides composed of many monomers (e.g. amylose, amylopectin) (Scientific Advisory Committee on Nutrition, 2015). With regards to hepatic metabolism of carbohydrate, ingestion of glucose in either free form, or as the various polymers such as maltose, maltodextrin and (amylose) starch can be considered physiologically similar stimuli because hydrolysis of glucose polymers is not thought to be rate‐limiting to intestinal absorption (Gonzalez et al. 2017). Furthermore, free glucose is rarely ingested alone as a sugar and, for this reason, it has been proposed that the ingestion of glucose alone is more reflective of non‐sugar intake from a physiological perspective (Tappy, 2018). In other words, when referring to sugar intake, we are typically referring to the co‐ingestion of fructose and glucose, and not the ingestion of free glucose alone. With this in mind, public health strategies that aim to reduce the intake of free sugars (Scientific Advisory Committee on Nutrition, 2015), such as the Soft Drinks Industry Levy in the UK (Barber, 2017) will, if successful, essentially reduce the intakes of fructose‐containing carbohydrates.
Metabolic health and postprandial lipid metabolism
Metabolic health is an umbrella term which can be defined as the ability to maintain homeostasis of substrates in response to challenging stimuli (including exercise and nutrition). It can be inferred at many levels, from molecular to whole body. Metabolic health is often characterised as the ability to maintain blood glucose or lipid concentrations within a range that does not increase risk of disease (Edinburgh et al. 2017). This is important for cardiovascular disease, for example, because fasting and postprandial hyperglycaemia and hyperlipaemia are associated with the development of cardiovascular disease (Edinburgh et al. 2017). Fructose intake has been implicated to play a role in many facets of metabolic health (Tappy & Lê, 2010) but, due to the notion that fructose is a key contributor towards disorders of fat metabolism (Softic et al. 2016), this review will focus on lipid handling.
In healthy humans, in the overnight fasted state, circulating blood lipid concentrations are composed of non‐esterified fatty acids (NEFAs) and triacylglycerols. When humans ingest a meal containing fat, exogenous fat is digested and broken down into NEFAs and monoglycerides before undergoing re‐esterification in the intestine and appearing in the circulation packaged in chylomicrons. Blood triacylglycerol concentrations increase steadily following ingestion of a meal and peak ∼5 h following ingestion (Chavez‐Jauregui et al. 2010), whereas NEFA concentrations typically decrease to negligible circulating concentrations within the first hour of ingestion, especially if the meal elicits an insulin response (Vega‐Lopez et al. 2013). Adipose tissue, skeletal muscle and the liver function integratively to manage postprandial lipaemia and most lipids are stored as triglycerides in adipose tissue (Frayn et al. 2006). The usual site of lipid storage is the adipose tissue, whereas lipid handling in non‐adipose tissues – including the liver – can cause a burden to these organs.
Compared to glucose ingestion, fructose ingestion (at a dose of 0.75 g⋅kg body mass−1) can increase the postprandial lipaemic response to the first meal of the day in healthy individuals (Abraha et al. 1998). This is predominantly due to a lower insulinaemic response following fructose ingestion compared with glucose ingestion, whereby lipoprotein lipase (LPL) activity in adipose tissue is activated less and clearance of dietary triacylglyceride (TAG) into adipose tissue is reduced (Chong et al. 2007). This dose of fructose equates to 52.5 g of fructose for a 70 kg individual; for context, a 330 ml can of Coca‐Cola contains ∼35 g of sucrose, so this is a high dose of fructose which would not usually be consumed in bolus outside the laboratory. Meta‐analysis of studies where the diet has been supplemented with fructose shows an elevation of postprandial lipaemia only in individuals with overweight or obesity and not in otherwise healthy participants (Wang et al. 2014). This appears to be driven by a disproportional increase in de novo lipogenesis (DNL) and liver fat accumulation with more prolonged (i.e. 3 weeks) fructose overfeeding (Sevastianova et al. 2012), and liver fat correlates strongly with hepatic very‐low‐density lipoprotein (VLDL) production and serum TAG concentrations (Adiels et al. 2006). Thus, the insulin response appears to be the main factor dictating acute responses to fructose ingestion, but longer‐term detriments are characterised by increased hepatic lipogenesis. This suggests that dietary fructose ingestion may be particularly harmful for health in certain contexts, for example when humans are in a positive energy balance and/or low energy turnover.
Hepatic lipid and carbohydrate handling, hepatic de novo lipogenesis, and interactions with fructose
The mechanisms that drive the differences in fructose‐induced hypertriglyceridaemia under various levels of energy balance and energy turnover are likely to be mediated by hepatic metabolism of fructose and lipids. Whilst the intestine can metabolise some fructose (Jang et al. 2018), the liver is the primary site of fructose metabolism in humans (Tappy & Lê, 2010; Gonzalez & Betts, 2018). In the postabsorptive state, NEFA is the primary substrate for hepatic fat oxidation and acts as a precursor for TAG synthesis (Havel et al. 1970). The liver contributes to handling postprandial lipaemia by taking up remnant lipoprotein particles driven by the enzyme hepatic lipase. The liver also has capacity for DNL (Sanders & Griffin, 2016), which is the formation of new lipids from non‐lipid precursors. Therefore, hepatic lipid content is dependent on the rates of hepatic fatty acid uptake and synthesis on the one hand, and rates of fatty acid oxidation and secretion (as VLDL) on the other. Excessive lipid accumulation in the liver or in the circulation are each thought to be detrimental to health. Thus, notwithstanding the intrinsic links between hepatic fatty acid oxidation, DNL and VLDL secretion (Nguyen et al. 2008), perhaps the ultimate fate of net lipid synthesis (VLDL secretion or hepatic TAG storage) is less important than the processes of hepatic lipid uptake, synthesis, and oxidation. In other words, the mechanisms of hepatic lipid uptake, synthesis, and oxidation are likely the most important targets to regulate metabolic health in relation to hepatic lipid metabolism, as the downstream partitioning to VLDL secretion or hepatic TAG storage are both detrimental to metabolic health when excessively stimulated.
At rest, the majority of the fructose (when ingested alone) taken up by the liver contributes to gluconeogenesis and some is converted to liver glycogen (Tappy & Lê, 2010). However, fructose also influences how the liver responds to glucose; infusing fructose into the portal vein of dogs in a stepwise manner results in an incremental rise in net hepatic glucose uptake (Shiota et al. 1998), and in humans, fructose infusion markedly upregulates hepatic glycogen synthesis (Petersen et al. 2001). Some fructose will be metabolised to pyruvate, which can be converted to lactate and enter the systemic circulation, thereby providing substrate for oxidation and/or contributing to glycogen storage in liver and muscle (Fig. 1) (Tappy & Lê, 2010). The quantity of fructose directly contributing to DNL in the liver is low (Tappy & Lê, 2010). However, tracing the fate of fructose carbons alone (e.g. via carbon‐labelled fructose) does not necessarily provide full insight into DNL from all precursors. For example, in addition to serving as a precursor, fructose availability (directly or indirectly via hepatic glycogen) could stimulate the process of DNL and thus increase the conversion of precursors such as glucose, lactate and fructose to triglycerides. In this regard, the use of deuterium oxide to determine DNL (Pinnick et al. 2019) has advantages over labelled fructose.
Figure 1. Pathways of glucose and fructose metabolism in the liver.
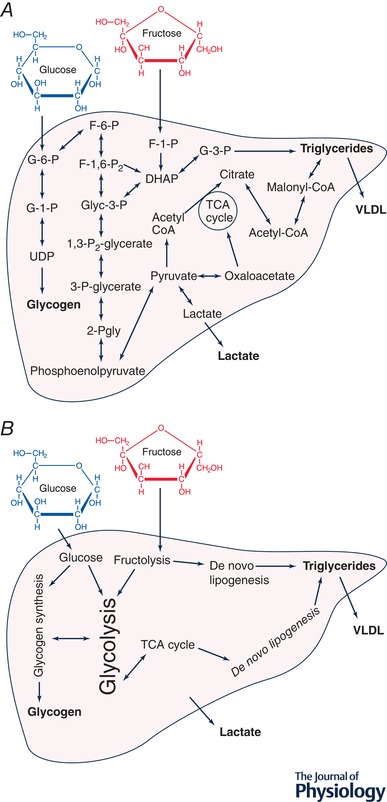
A, major pathways of hepatic glycogen synthesis and de novo lipogenesis. Hepatic glucose uptake is tightly regulated, whereas hepatic fructose uptake occurs in an ‘unregulated’ manner, without negative feedback, driven by the highly affinitive hepatic fructokinase. DNL can occur from glucose and fructose as precursors via multiple pathways: either via increased glycolytic production of acetyl coenzyme A, or via accumulation of dihydroxyacetone phosphate from glycolytic (fructose‐1,6‐bisphosphate) or directly from fructolytic (fructose‐1‐phosphate) intermediates. B, the major processes involved in hepatic glycogen synthesis and de novo lipogenesis which summarise the multiple enzymatic steps highlighted in A. G‐6‐P, glucose‐6‐phosphate; G‐1‐P, glucose‐1‐phosphate; UDP, uridine diphosphate glucose; F‐1‐P, fructose‐1‐phosphate; F‐6‐P, fructose‐6‐phosphate; F‐1,6‐P2, fructose‐1,6‐bisphosphate; Glyc‐3‐P, glyceraldehyde‐3‐phosphate; 1,3‐P2‐glycerate, 1,3‐bisphosphoglycerate; G‐3‐P, glycerol‐3‐phosphate; 3‐Pgly, 3‐phosphoglycerate; 2‐Pgly, 2‐phosphoglycerate; Acetyl‐CoA, acetyl coenzyme A; Malonyl‐CoA, malonyl coenzyme A; DHAP, dihydroxyacetone phosphate; VLDL, very‐low‐density lipoprotein.
Acute co‐ingestion of fructose with glucose results in greater hepatic DNL than from glucose ingestion alone (Parks et al. 2008), and whilst fasting DNL is not upregulated following 10 weeks of fructose feeding in overweight humans (Stanhope et al. 2009), postprandial DNL is massively upregulated (>7‐fold) following overfeeding with fructose compared with glucose for 10 weeks (Stanhope et al. 2009). The regulation of DNL in the presence of fructose is complex, with many contributing processes (Fig. 1). Whereas hepatic glucose uptake is tightly regulated by a combination of hyperglycaemia, hyperinsulinaemia and delivery of glucose via the portal vein (McGuinness & Cherrington, 2003), hepatic fructose uptake occurs in an ‘unregulated’ manner, without negative feedback, driven by hepatic fructokinase with high affinity for fructose (Fig. 2 A) (Adelman et al. 1967). DNL can occur from glucose, fructose and lactate as precursors via multiple pathways (Sanders & Griffin, 2016): either via increased glycolytic production of acetyl coenzyme A (acetyl‐CoA), or via accumulation of dihydroxyacetone phosphate from glycolytic (fructose‐1,6‐bisphosphate) or directly from fructolytic (fructose‐1‐phosphate) intermediates (Fig. 1). The role of fructose in liver metabolism has been reviewed in more detail previously (McGuinness & Cherrington, 2003; Bizeau & Pagliassotti, 2005; Geidl‐Flueck & Gerber, 2017); however, to understand how fructose influences hepatic DNL, we will focus on the role of transcriptional and allosteric regulation, the role of energy sensing and AMP‐activated protein kinase (AMPK), and the interplay between β‐oxidation and lipogenesis.
Figure 2. Highlighting some of the important pathways in hepatic carbohydrate handling and de novo lipogenesis.
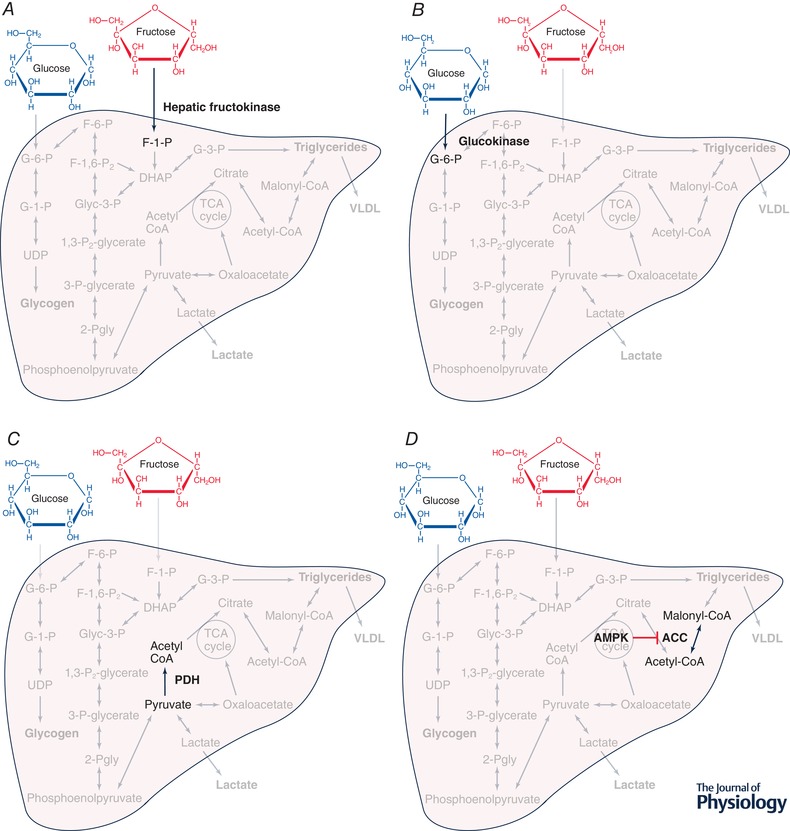
A, fructose is converted into fructose‐1‐phosphate via enzyme hepatic fructokinase. B, glucose is converted into glucose‐6‐phosphate via enzyme glucokinase. C, pyruvate is converted into acetyl‐coenzyme A via enzyme pyruvate dehydrogenase. D, acetyl coenzyme A is converted into malonyl coenzyme A via enzyme acetyl coenzyme A carboxylase, an enzyme which is negatively regulated by AMP‐activated protein kinase. F‐1‐P, fructose‐1‐phosphate; 6‐G‐P, glucose‐6‐phosphate; Acetyl‐CoA, acetyl coenzyme A; PDH, pyruvate dehydrogenase; ACC, acetyl CoA carboxylase; AMPK, AMP‐activated protein kinase; Malonyl‐CoA, malonyl coenzyme A.
Transcriptional and allosteric regulation
Fructose interacts with various transcriptional and allosteric (enzymatic) processes along the pathways of glycogen synthesis, glycolysis and DNL within the liver. Transcriptional regulation translates into long‐term mechanisms of regulation while allosteric (enzymatic) processes will be responsible for acute/immediate mechanisms of regulation.
Sterol regulatory element binding proteins (SREBPs) are transcription factors with three isoforms (SREBP‐1a, ‐1c, ‐2) (Brown & Goldstein, 1997). The SREBP‐1c isoform activates transcription of numerous genes encoding lipogenic enzymes (Horton, 2002). Insulin stimulates transcription of the gene coding for SREBP‐1c and glucagon inhibits this transcription (Goldstein et al. 2006). SREBP‐1c is thought to be the primary mediator of insulin‐induced hepatic lipogenesis because hepatic SREBP‐1c transcription decreases in the liver of rats treated with streptozotocin (which ablates insulin secretion) and rises back to normal levels following insulin treatment (Shimomura et al. 1999). However, insulin‐independent hepatic SREBP‐1c activation can be achieved in rats by refeeding with glucose, sucrose and fructose following streptozotocin administration (Matsuzaka et al. 2004), suggesting that both nutrient availability and insulin concentrations play a role in SREBP‐1c‐stimulated hepatic lipogenesis. Fructose feeding in rats also stimulates peroxisome proliferator‐activated receptor γ coactivator‐1 β (PGC‐1β) (Nagai et al. 2009), which is a transcriptional coactivator of SREBP‐1c. With regards to SREBP‐1c, fructose may be of secondary importance to hepatic lipogenesis compared with the presence of high insulin concentrations, but clearly plays a role in this pathway.
Carbohydrate response element binding protein (ChREBP) is a transcription factor which is activated by carbohydrate feeding and is expressed in the liver of rats (Yamashita et al. 2001) and humans (Hurtado del Pozo et al. 2011), and which also targets numerous lipogenic enzymes (Ma et al. 2006). It has been hypothesised that fructose may activate hepatic ChREBP to a greater extent than glucose due to the unrestricted nature of hepatic fructose uptake (Ter Horst & Serlie, 2017). Interestingly, in ChREBP knockout mice, a high‐fructose diet does not lead to liver fat accumulation but instead accelerates fibrosis (Zhang et al. 2017b), suggesting that ChREBP activation is a necessary signal to allow fructose‐induced hepatic lipogenesis to occur.
Glucokinase is the enzyme which phosphorylates glucose to glucose‐6‐phosphate (Fig. 2 B) and therefore is responsible for facilitating hepatic glucose uptake. Glucokinase is tightly regulated by negative feedback loops, mainly via the glucokinase regulatory protein (GKRP) which binds to, and inhibits activity of, glucokinase (Van Schaftingen, 1989). Fructose‐6‐phosphate promotes GKRP to bind to glucokinase, inhibiting its activity, whereas fructose‐1‐phosphate has the opposite effect on GKRP, facilitating its dissociation from the glucokinase, which increases glucokinase activity (Vandercammen & Van Schaftingen, 1990; Vandercammen et al. 1992). Therefore, greater fructose‐1‐phosphate concentrations result in greater hepatic glucose uptake via glucokinase (Davies et al. 1990). This has been demonstrated in vivo in dogs, where small doses of fructose infused to the portal vein result in increased hepatic glucose uptake, hepatic glycogen synthesis, and hepatic glycolysis (Shiota et al. 2002). The extent to which hepatic fructokinase phosphorylates fructose to fructose‐1‐phosphate is not regulated and instead is driven primarily by the availability of fructose to the liver (Tappy & Lê, 2010). This provides mechanistic evidence for how hepatic fructose uptake potentiates hepatic glucose uptake, which supports evidence in humans that glucose–fructose co‐ingestion approximately doubles hepatic glycogen repletion rates compared with glucose ingestion alone (Gonzalez et al. 2017).
Pyruvate dehydrogenase (PDH) is a tightly regulated protein complex in mitochondria which catalyses the decarboxylation of pyruvate to acetyl‐CoA (Fig. 2 C) (Harris et al. 2002), which is an irreversible step in hepatic lipogenesis. PDH is activated by insulin and Ca2+ via activation of pyruvate dehydrogenase phosphatase (Harris et al. 2002). PDH is inactivated by acetyl‐CoA and NADH, via activation of pyruvate dehydrogenase kinase (Harris et al. 2002). In rats, prolonged fructose ingestion stimulates hepatic PDH activity and increases hepatic DNL compared with prolonged glucose ingestion. However, a direct role of PDH activity in hepatic lipogenesis is unclear, since knockout and pharmacological inhibition of hepatic pyruvate dehydrogenase kinases both suppress hepatic ChREBP‐mediated lipogenesis (Wu et al. 2018). Furthermore, the extent to which PDH activity is quantitatively important for human hepatic lipid metabolism is currently unclear.
Acetyl‐CoA carboxylase (ACC) is an enzyme responsible for catalysing acetyl‐CoA to malonyl coenzyme A (malonyl‐CoA) in the first ‘committed’ step to lipogenesis (Fig. 2 D) (Hardie, 1989). In rat hepatocytes, ACC is stimulated by both glucose and insulin (Katz & Ick, 1981) and is inhibited by phosphorylation with AMP‐activated protein kinase (AMPK) (Carling et al. 1987). In rodents, high‐fructose feeding upregulates ACC activity (Winder et al. 1975), and pharmacological inhibition of ACC can decrease hepatic DNL and hepatic steatosis (Goedeke et al. 2018). This is, however, at the expense of hypertriglyceridaemia (Goedeke et al. 2018). In human‐derived HEPG2 cells, addition of fructose to glucose does not further upregulate ACC expression (Hirahatake et al. 2011), so it is unclear in humans how an acute physiological dose of fructose directly influences hepatic ACC activity. Altered ACC activity from fructose intake may therefore require sustained increases in fructose intake and may therefore interact with energy sensing pathways as the energy and glycogen status of the hepatocytes are changed.
Energy sensing and AMP‐activated protein kinase signalling
The AMP/ATP ratio within eukaryotes determines the principal energy status of the cell, and the major, most widely conserved indicator of this ratio is AMPK (Herzig & Shaw, 2018). AMPK is activated by metabolic stressors including nutrient deprivation, for example depriving hepatocytes or pancreatic β‐cells of glucose markedly upregulates AMPK activity (Salt et al. 1998; Zhang et al. 2013). It is now well established that AMPK is activated under conditions of low‐energy status and that this inhibits anabolic pathways and promotes catabolic pathways. As mentioned previously, this is mediated via the phosphorylation of ACC with an inhibitory effect (Fig. 2 D) (Carling et al. 1987). Refeeding rats with carbohydrate following fasting results in a reduction of hepatic AMPK activity whilst concurrently increasing hepatic ACC activity (Assifi et al. 2005), and AMPK activation via a gain‐of‐function mouse model has been shown to inhibit hepatic DNL without influencing lipid oxidation (Woods et al. 2017).
Fructose stimulates hepatic DNL from multiple pathways (Fig. 1), which is potentially because hepatic fructokinase is not negatively regulated by ATP levels (Adelman et al. 1967), so fructolysis occurs independent of cellular energy status. It has recently been shown that fructose‐1,6‐bisphosphate, an intermediate of fructose metabolism (Fig. 1), impairs activation of AMPK during glucose starvation (Zhang et al. 2017a), suggesting that the presence of fructose metabolites per se accelerates DNL by reducing AMPK activity and increasing ACC activity. Furthermore, the phosphorylation of fructose to fructose‐6‐phosphate results in conversion of ATP to ADP (Hallfrisch, 1990), and this results in uric acid production (Nakagawa et al. 2006). This has been shown to increase lipogenesis and the expression of lipogenic genes in hepatocytes (Lanaspa et al. 2012), providing an additional mechanism related to energy sensing by which fructose increases DNL.
In addition to being regulated by the energy status of the cell via the ATP:AMP ratio, AMPK activity is also regulated by glycogen concentrations, independent of AMP. Glycogen binds to the β‐subunit of AMPK and this inhibits AMPK activity (McBride et al. 2009), at least in skeletal muscle. For example, increasing skeletal muscle glycogen content of rodents by ∼3‐fold decreases basal AMPK‐α2 activity almost proportionally to glycogen concentration. Furthermore, whilst AMPK‐α2 activity is still responsive to stimulation by the AMP analogue, 5‐aminoimidazole‐4‐carboxamide ribonucleotide (AICAR), this stimulated response is drastically blunted in the presence of high versus low muscle glycogen concentrations (Wojtaszewski et al. 2002). Therefore, across a range of AMP concentrations, AMPK activity is modulated by glycogen concentrations. Assuming similar AMPK regulation occurs in the liver, then high glycogen concentrations may inhibit AMPK activity, thus alleviating the inhibitory phosphorylation of ACC, allowing greater DNL to occur.
Interplay between β‐oxidation and lipogenesis
Net hepatic lipid production is the summation of either reduced β‐oxidation, increased lipogenesis, or a combination of both processes. Evidence discussed so far indicates that fructose upregulates lipogenic processes in the liver, but there is also evidence that β‐oxidation is reduced, thereby further contributing to net lipid production. Peroxisome proliferator‐activated receptor α (PPARα) is the master regulator of hepatic lipid metabolism and expression is induced by fasting (Kersten & Stienstra, 2017). In human muscle, glucose and insulin inhibit fat oxidation by reducing the rate of fatty acid entry to the mitochondria (Sidossis et al. 1996). The mechanism of inhibition in rat hepatocytes is an increase in cellular malonyl‐coenzyme A (McGarry et al. 1977). Fructose has been shown to inhibit β‐oxidation in rat and human hepatocytes (Rebollo et al. 2014), and chronic fructose ingestion in rats leads to an increase in hepatic lipogenic genes as well as a concurrent decrease in hepatic carnitine palmitoyltransferase 1 (CPT1) (Teofilovic et al. 2016). This suggests that fructose increases net hepatic lipid production from multiple mechanisms, not solely an increase in lipogenesis.
A role for hepatic glycogen in regulating de novo lipogenesis?
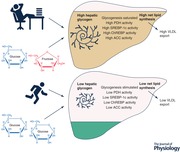
Hepatic glycogen content reflects the balance between glycogen synthesis (via direct and indirect pathways), and glycogenolysis. As mentioned in the previous section, fructose markedly upregulates hepatic glycogen synthesis as well as upregulating hepatic DNL. Whilst this suggests that fructose has the potential to enhance the recovery of athletic performance (Fuchs et al. 2019), it also kindles the idea that hepatic glycogen content could be a key regulator of hepatic lipid metabolism.
Mice with the gene coding for hepatic glycogen synthase (Gys2) knocked down display markedly increased hepatic DNL, which leads to hepatic insulin resistance and steatosis (Irimia et al. 2017), which suggests that the inability to synthesise hepatic glycogen is a driver of hepatic DNL. In the alternative scenario, in glycogen storage disease type 1a, where glycogen stores are saturated (Cori & Cori, 1952), hepatic DNL is also markedly upregulated (Bandsma et al. 2008). Due to the stimulatory effects of fructose on hepatic glycogen synthesis discussed in the previous section, liver glycogen stores may become ‘saturated’ in scenarios where fructose is ingested in large quantities, when glycogen is not utilised at a high rate (i.e. during sedentary conditions), or a combination of these two conditions, and thus, excess carbohydrate is converted into lipid (Fig. 3).
Figure 3. The role of glycogen status on hepatic de novo lipogenesis and VLDL export.
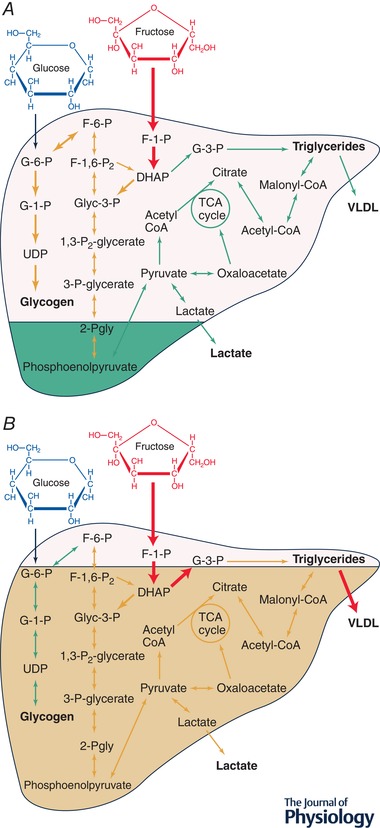
The traffic light system demonstrates flux through a given pathway, where red (thickest lines in black and white version) represents high flux, orange (medium thickness lines in black and white version) represents medium flux, and green (thinnest lines in black and white version) represents low flux. A, low hepatic glycogen status enables ingested fructose to stimulate hepatic glycogen synthesis, allowing flux through lipogenic pathways to remain low. B, high hepatic glycogen status determines that ingested fructose is shunted away from glycogen synthesis and towards lipogenic pathways, which leads to greater VLDL export. G‐6‐P, glucose‐6‐phosphate; G‐1‐P, glucose‐1‐phosphate; UDP, uridine diphosphate glucose; F‐1‐P, fructose‐1‐phosphate; F‐6‐P, fructose‐6‐phosphate; F‐1,6‐P2, fructose‐1,6‐bisphosphate; Glyc‐3‐P, glyceraldehyde‐3‐phosphate; 1,3‐P2‐glycerate, 1,3‐bisphosphoglycerate; G‐3‐P, glycerol‐3‐phosphate; 3‐Pgly, 3‐phosphoglycerate; 2‐Pgly, 2‐phosphoglycerate; Acetyl‐CoA, acetyl coenzyme A; Malonyl‐CoA, malonyl coenzyme A; DHAP, dihydroxyacetone phosphate; VLDL, very‐low‐density lipoprotein.
In scenarios where hepatic glycogen stores are already ‘full’, hepatic glucose and fructose uptake may be shunted more towards DNL, whereas when hepatic glycogen stores are low, glucose and fructose are potentially shifted more towards glycogen synthesis rather than DNL. This is supported by evidence that hepatic glycogen stores undergo autoregulation, whereby low glycogen concentrations stimulate glycogen synthesis (Fleig et al. 1987) and inhibit glycogenolysis (Roden et al. 2001). Furthermore, overriding this autoregulation by overexpressing hepatic glucokinase to accelerate hepatic glugose uptake in an unregulated fashion results in markedly increased hepatic glycogen stores, but at the expense of hypertriglyceridaemia (potentially from DNL) (O'Doherty et al. 1999). However, if glycogen synthesis is stimulated directly – by overexpressing hepatic protein targeting to glycogen – there are no changes in triglyceridaemia (O'Doherty et al. 2000). This suggests that the mechanism binding glycogen stores to fructose‐induced increases in hepatic DNL could be the metabolic fate of hepatic carbohydrate uptake: directed more to glycogen when glycogen stores are low.
Assuming the hepatic glycogen hypothesis is correct, the ingestion of fructose when hepatic glycogen stores are already saturated would stimulate lipogenesis to a greater extent than when hepatic glycogen stores are low. If glycogen is being utilised at a rate high enough to overcome net glycogen synthesis, despite fructose intake, then theoretically this should reduce hepatic DNL and VLDL output. Whilst there are no data in humans, in rats daily exercise can mediate the initial increase in hepatic lipogenic gene expression after 3 days of high‐fructose feeding (Winder et al. 1975). It will be intriguing to explore whether the capacity to store liver glycogen correlates with the lipogenic response to fructose ingestion. With this in mind, from a practical standpoint, to mitigate negative metabolic effects of fructose intake via modulating hepatic glycogen stores, perhaps the advice should be to ‘deplete before you eat’? In support of this, a single bout of exercise, which lowers glycogen content in liver and skeletal muscle, has been shown to potently downregulate postprandial hepatic DNL and liver triglyceride storage in humans with insulin resistance (Rabøl et al. 2011).
Considering nutrient–physical activity interactions
Physiology is fundamentally the study of life, and nutrients are essential for life to be maintained, but it is an oversimplification to view the intake of any nutrient as a sole determinant of one of the many processes of life. Instead, physiology is dictated by interactions between nutrients and many other stimuli, and it is this integrated view that we should strive towards in order to understand the physiological effects of nutritional habits. In a world where obesity and associated metabolic complications are rising (NCD Risk Factor Collaboration, 2016), understanding the role of nutrients in energy balance and metabolism is more pertinent than ever. Considering evidence relevant to fructose, diets high in sucrose have been correlated with higher energy intake (Johnson et al. 2009), and adding sucrose to or removing it from the diet leads to a modest increase or decrease in weight, respectively, over time (Te Morenga et al. 2013). However, sucrose is unlikely to have a role solely on energy intake independent from any other component of energy balance, and instead could influence other facets of energy intake and/or energy expenditure, including physical activity energy expenditure.
At the extremes of physiology, ‘simple carbohydrate’ (sugars) intake of ∼460 g (∼1720 kilocalories), and total energy intake of ∼5800 kilocalories, per day are achieved in Tour de France cyclists (Saris et al. 1989), yet they do not develop metabolic disorders or hepatic steatosis. Similarly, some hunter gatherer populations are reported to consume as much as 50% of energy intake from honey, but also appear to have very low prevalence of metabolic disease (Pontzer et al. 2018). In non‐exercising humans, similar nutrient intake results in metabolic detriment and high rates of net DNL within days (Acheson et al. 1988). Thus, these two similarly extreme nutrient intakes exert quite contrasting physiological effects depending on the physiological state of the individual, and hepatic glycogen content could be a key factor that dictates these responses.
The possibility that hepatic glycogen stores are important for mediating fructose‐driven hepatic DNL is a good example of why nutrient–physical activity interactions are important to consider. Acute endurance exercise increases hepatic glycogenolysis, and is largely dictated by the exercise intensity – especially in untrained individuals – (Gonzalez et al. 2016). However, even in untrained individuals, it would take ∼2 h exercising at 80% for liver glycogen to be depleted to very low levels (>70%) (Gonzalez et al. 2016). Whilst intensity is clearly important for hepatic glycogen stores, this suggests that total volume and patterns of physical activity are also important considerations. Where possible, researchers should strive to understand the effects of fructose ingestion (or any other nutrient) in the context of mediating factors. Measuring physical activity energy expenditure, type, timing and intensity are all useful towards understanding the physiological effects of nutrient ingestion.
Conclusions
Dietary fructose plays an important role in hepatic glycogen and lipid metabolism, with potential consequences for metabolic health. Ingestion of fructose increases rates of hepatic de novo lipogenesis and, in the context of a positive energy balance, can lead to greater VLDL export and hypertriglyceridaemia. An inability to further synthesise glycogen upregulates hepatic DNL and, considering that fructose ingestion increases both DNL and hepatic glycogen synthesis, glycogen stores may play a key role in determining the metabolic responses to fructose ingestion. The corollary is that negative metabolic effects of fructose intake are most likely to manifest when hepatic glycogen stores are saturated. This hypothesis provides a rationale for striving to consider nutrient–physical activity interactions in physiology research, and to target turnover and/or utilisation of hepatic glycogen stores to improve metabolic health. Future research should strive to take an integrated approach towards understanding physiological responses to nutrients.
Additional information
Competing interests
None declared.
Author contributions
All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
The present work was not supported by any specific funding source. A.H. is completing a PhD studentship with financial support from The Rank Prize Funds and the University of Bath. Additional funding for consumables is provided by Kenniscentrum Suiker & Voeding. F.K. has received funding from the Medical Research Council, Diabetes UK and The British Heart Foundation in the past. She is currently funded by the Medical Research Council (MR/P002927/1). J.T.G. has received funding from The European Society for Clinical Nutrition and Metabolism (ESPEN), The Rank Prize Funds, Kenniscentrum Suiker & Voeding, Arla Foods Ingredients, the Medical Research Council, and the Biotechnology and Biological Sciences Research Council. J.T.G. has also acted as a consultant to PepsiCo and Lucozade Ribena Suntory.
Biographies
Aaron Hengist is a PhD student in the Department for Health at the University of Bath. His research focuses on the role of dietary sugars and carbohydrates in aspects of energy balance and metabolic health, with a particular focus on physical activity.

Francoise Koumanov is a Lecturer (Assistant Professor) in the Department for Health at the University of Bath. Her research focuses on molecular signalling processes of glucose and insulin in physiological and pathophysiological states. Her research aims to elucidate the roles of exercise and GLUT4 trafficking in the pathophysiology of peripheral insulin resistance.
Javier Gonzalez is a Senior Lecturer (Associate Professor) in the Department for Health at the University of Bath. His research focuses on interactions between nutrition and exercise in health and disease. His research aims to explore the role of carbohydrate availability in the regulation of energy balance, metabolic health and sports performance.
Edited by: Ole Petersen & Yasuhiko Minokoshi
This review was presented at the symposium ‘Fructose in physiology: friend or foe?’, which took place at QEII Centre, London, UK, 14‐16 September 2018.
References
- Abraha A, Humphreys SM, Clark ML, Matthews DR & Frayn KN (1998). Acute effect of fructose on postprandial lipaemia in diabetic and non‐diabetic subjects. Br J Nutr 80, 169–175. [PubMed] [Google Scholar]
- Acheson KJ, Schutz Y, Bessard T, Anantharaman K, Flatt JP & Jequier E (1988). Glycogen storage capacity and de novo lipogenesis during massive carbohydrate overfeeding in man. Am J Clin Nutr 48, 240–247. [DOI] [PubMed] [Google Scholar]
- Adelman RC, Ballard FJ & Weinhouse S (1967). Purification and properties of rat liver fructokinase. J Biol Chem 242, 3360–3365. [PubMed] [Google Scholar]
- Adiels M, Taskinen MR, Packard C, Caslake MJ, Soro‐Paavonen A, Westerbacka J, Vehkavaara S, Hakkinen A, Olofsson SO, Yki‐Jarvinen H & Boren J (2006). Overproduction of large VLDL particles is driven by increased liver fat content in man. Diabetologia 49, 755–765. [DOI] [PubMed] [Google Scholar]
- Assifi MM, Suchankova G, Constant S, Prentki M, Saha AK & Ruderman NB (2005). AMP‐activated protein kinase and coordination of hepatic fatty acid metabolism of starved/carbohydrate‐refed rats. Am J Physiol Endocrinol Metab 289, E794–E800. [DOI] [PubMed] [Google Scholar]
- Bandsma RHJ, Prinsen BH, de Sain‐van der Velden M, Rake J‐P, Boer T, Smit GPA, Reijngoud D‐J & Kuipers F (2008). Increased de novo lipogenesis and delayed conversion of large VLDL into intermediate density lipoprotein particles contribute to hyperlipidemia in glycogen storage disease type 1a. Pediatr Res 63, 702–707. [DOI] [PubMed] [Google Scholar]
- Barber S (2017). The Soft Drinks Industry Levy. House of Commons Library Briefing Paper, No. 7876.
- Bizeau ME & Pagliassotti MJ (2005). Hepatic adaptations to sucrose and fructose. Metab Clin Exp 54, 1189–1201. [DOI] [PubMed] [Google Scholar]
- Brown MS & Goldstein JL (1997). The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane‐bound transcription factor. Cell 89, 331–340. [DOI] [PubMed] [Google Scholar]
- Carling D, Zammit VA & Hardie DG (1987). A common bicyclic protein kinase cascade inactivates the regulatory enzymes of fatty acid and cholesterol biosynthesis. FEBS Lett 223, 217–222. [DOI] [PubMed] [Google Scholar]
- Chavez‐Jauregui RN, Mattes RD & Parks EJ (2010). Dynamics of fat absorption and effect of sham feeding on postprandial lipema. Gastroenterology 139, 1538–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong MF, Fielding BA & Frayn KN (2007). Mechanisms for the acute effect of fructose on postprandial lipemia. Am J Clin Nutr 85, 1511–1520. [DOI] [PubMed] [Google Scholar]
- Cori GT & Cori CF (1952). Glucose‐6‐phosphatase of the liver in glycogen storage disease. J Biol Chem 199, 661–667. [PubMed] [Google Scholar]
- Davies DR, Detheux M & Van Schaftingen E (1990). Fructose 1‐phosphate and the regulation of glucokinase activity in isolated hepatocytes. Eur J Biochem 192, 283–289. [DOI] [PubMed] [Google Scholar]
- Edinburgh RM, Betts JA, Burns SF & Gonzalez JT (2017). Concordant and divergent strategies to improve postprandial glucose and lipid metabolism. Nutr Bull 42, 113–122. [Google Scholar]
- Fleig WE, Enderle D, Steudter S, Nother‐Fleig G & Ditschuneit H (1987). Regulation of basal and insulin‐stimulated glycogen synthesis in cultured hepatocytes. Inverse relationship to glycogen content. J Biol Chem 262, 1155–1160. [PubMed] [Google Scholar]
- Frayn KN, Arner P & Yki‐Jarvinen H (2006). Fatty acid metabolism in adipose tissue, muscle and liver in health and disease. Essays Biochem 42, 89–103. [DOI] [PubMed] [Google Scholar]
- Fuchs CJ, Gonzalez JT & van Loon LJC (2019). Fructose co‐ingestion to increase carbohydrate availability in athletes. J Physiol 597, 3549–3560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geidl‐Flueck B & Gerber PA (2017). Insights into the hexose liver metabolism – Glucose versus fructose. Nutrients 9, 1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedeke L, Bates J, Vatner DF, Perry RJ, Wang T, Ramirez R, Li L, Ellis MW, Zhang D, Wong KE, Beysen C, Cline GW, Ray AS & Shulman GI (2018). Acetyl‐CoA carboxylase inhibition reverses NAFLD and hepatic insulin resistance but promotes hypertriglyceridemia in rodents. Hepatology 68, 2197–2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein JL, DeBose‐Boyd RA & Brown MS (2006). Protein sensors for membrane sterols. Cell 124, 35–46. [DOI] [PubMed] [Google Scholar]
- Gonzalez JT & Betts JA (2018). Dietary fructose metabolism by splanchnic organs: size matters. Cell Metab 27, 483–485. [DOI] [PubMed] [Google Scholar]
- Gonzalez JT, Fuchs CJ, Betts JA & van Loon LJ (2016). Liver glycogen metabolism during and after prolonged endurance‐type exercise. Am J Physiol Endocrinol Metab 311, E543–E553. [DOI] [PubMed] [Google Scholar]
- Gonzalez JT, Fuchs CJ, Betts JA & van Loon LJ (2017). Glucose plus fructose ingestion for post‐exercise recovery – Greater than the sum of its parts? Nutrients 9, 344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallfrisch J (1990). Metabolic effects of dietary fructose. FASEB J 4, 2652–2660. [DOI] [PubMed] [Google Scholar]
- Hardie DG (1989). Regulation of fatty acid synthesis via phosphorylation of acetyl‐CoA carboxylase. Prog Lipid Res 28, 117–146. [DOI] [PubMed] [Google Scholar]
- Harris RA, Bowker‐Kinley MM, Huang B & Wu P (2002). Regulation of the activity of the pyruvate dehydrogenase complex. Adv Enzyme Regul 42, 249–259. [DOI] [PubMed] [Google Scholar]
- Havel RJ, Kane JP, Balasse EO, Segel N & Basso LV (1970). Splanchnic metabolism of free fatty acids and production of triglycerides of very low density lipoproteins in normotriglyceridemic and hypertriglyceridemic humans. J Clin Invest 49, 2017–2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzig S & Shaw RJ (2018). AMPK: guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol 19, 121–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirahatake KM, Meissen JK, Fiehn O & Adams SH (2011). Comparative effects of fructose and glucose on lipogenic gene expression and intermediary metabolism in HepG2 liver cells. PLoS One 6, e26583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton JD (2002). Sterol regulatory element‐binding proteins: transcriptional activators of lipid synthesis. Biochem Soc Trans 30, 1091–1095. [DOI] [PubMed] [Google Scholar]
- Hurtado del Pozo C, Vesperinas‐Garcia G, Rubio MA, Corripio‐Sanchez R, Torres‐Garcia AJ, Obregon MJ & Calvo RM (2011). ChREBP expression in the liver, adipose tissue and differentiated preadipocytes in human obesity. Biochim Biophys Acta 1811, 1194–1200. [DOI] [PubMed] [Google Scholar]
- Hwang JJ, Jiang L, Hamza M, Dai F, Belfort‐DeAguiar R, Cline G, Rothman DL, Mason G & Sherwin RS (2017). The human brain produces fructose from glucose. JCI Insight 2, e90508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irimia JM, Meyer CM, Segvich DM, Surendran S, DePaoli‐Roach AA, Morral N & Roach PJ (2017). Lack of liver glycogen causes hepatic insulin resistance and steatosis in mice. J Biol Chem 292, 10455–10464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang C, Hui S, Lu W, Cowan AJ, Morscher RJ, Lee G, Liu W, Tesz GJ, Birnbaum MJ & Rabinowitz JD (2018). The small intestine converts dietary fructose into glucose and organic acids. Cell Metab 27, 351–361.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson RK, Appel LJ, Brands M, Howard BV, Lefevre M, Lustig RH, Sacks F, Steffen LM & Wylie‐Rosett J; American Heart Association Nutrition Committee of the Council on Nutrition, Physical Activity, and Metabolism and the Council on Epidemiology and Prevention (2009). Dietary sugars intake and cardiovascular health: a scientific statement from the American Heart Association. Circulation 120, 1011–1020. [DOI] [PubMed] [Google Scholar]
- Katz NR & Ick M (1981). Induction of acetyl‐CoA carboxylase in primary rat hepatocyte cultures by glucose and insulin. Biochem Biophys Res Commun 100, 703–709. [DOI] [PubMed] [Google Scholar]
- Kersten S & Stienstra R (2017). The role and regulation of the peroxisome proliferator activated receptor alpha in human liver. Biochimie 136, 75–84. [DOI] [PubMed] [Google Scholar]
- Lanaspa MA, Sanchez‐Lozada LG, Cicerchi C, Li N, Roncal‐Jimenez CA, Ishimoto T, Le M, Garcia GE, Thomas JB, Rivard CJ, Andres‐Hernando A, Hunter B, Schreiner G, Rodriguez‐Iturbe B, Sautin YY & Johnson RJ (2012). Uric acid stimulates fructokinase and accelerates fructose metabolism in the development of fatty liver. PLoS One 7, e47948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lustig RH, Schmidt LA & Brindis CD (2012). Public health: The toxic truth about sugar. Nature 482, 27–29. [DOI] [PubMed] [Google Scholar]
- Ma L, Robinson LN & Towle HC (2006). ChREBP·Mlx is the principal mediator of glucose‐induced gene expression in the liver. J Biol Chem 281, 28721–28730. [DOI] [PubMed] [Google Scholar]
- McBride A, Ghilagaber S, Nikolaev A & Hardie DG (2009). The glycogen‐binding domain on the AMPK beta subunit allows the kinase to act as a glycogen sensor. Cell Metab 9, 23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGarry JD, Mannaerts GP & Foster DW (1977). A possible role for malonyl‐CoA in the regulation of hepatic fatty acid oxidation and ketogenesis. J Clin Invest 60, 265–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuinness OP & Cherrington AD (2003). Effects of fructose on hepatic glucose metabolism. Curr Opin Clin Nutr Metab Care 6, 441–448. [DOI] [PubMed] [Google Scholar]
- Matsuzaka T, Shimano H, Yahagi N, Amemiya‐Kudo M, Okazaki H, Tamura Y, Iizuka Y, Ohashi K, Tomita S, Sekiya M, Hasty A, Nakagawa Y, Sone H, Toyoshima H, Ishibashi S, Osuga J & Yamada N (2004). Insulin‐independent induction of sterol regulatory element‐binding protein‐1c expression in the livers of streptozotocin‐treated mice. Diabetes 53, 560–569. [DOI] [PubMed] [Google Scholar]
- Nagai Y, Yonemitsu S, Erion DM, Iwasaki T, Stark R, Weismann D, Dong J, Zhang D, Jurczak MJ, Loffler MG, Cresswell J, Yu XX, Murray SF, Bhanot S, Monia BP, Bogan JS, Samuel V & Shulman GI (2009). The role of peroxisome proliferator‐activated receptor γ coactivator‐1 β in the pathogenesis of fructose‐induced insulin resistance. Cell Metab 9, 252–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa T, Hu H, Zharikov S, Tuttle KR, Short RA, Glushakova O, Ouyang X, Feig DI, Block ER, Herrera‐Acosta J, Patel JM & Johnson RJ (2006). A causal role for uric acid in fructose‐induced metabolic syndrome. Am J Physiol Renal Physiol 290, F625–F631. [DOI] [PubMed] [Google Scholar]
- NCD Risk Factor Collaboration (NCD‐RisC) (2016). Trends in adult body‐mass index in 200 countries from 1975 to 2014: a pooled analysis of 1698 population‐based measurement studies with 19.2 million participants. Lancet 387, 1377–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen P, Leray V, Diez M, Serisier S, Le Bloc'h J, Siliart B & Dumon H (2008). Liver lipid metabolism. J Anim Physiol Anim Nutr (Berl) 92, 272–283. [DOI] [PubMed] [Google Scholar]
- O'Doherty RM, Jensen PB, Anderson P, Jones JG, Berman HK, Kearney D & Newgard CB (2000). Activation of direct and indirect pathways of glycogen synthesis by hepatic overexpression of protein targeting to glycogen. J Clin Invest 105, 479–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Doherty RM, Lehman DL, Telemaque‐Potts S & Newgard CB (1999). Metabolic impact of glucokinase overexpression in liver: lowering of blood glucose in fed rats is accompanied by hyperlipidemia. Diabetes 48, 2022–2027. [DOI] [PubMed] [Google Scholar]
- Parks EJ, Skokan LE, Timlin MT & Dingfelder CS (2008). Dietary sugars stimulate fatty acid synthesis in adults. J Nutr 138, 1039–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen KF, Laurent D, Yu C, Cline GW & Shulman GI (2001). Stimulating effects of low‐dose fructose on insulin‐stimulated hepatic glycogen synthesis in humans. Diabetes 50, 1263–1268. [DOI] [PubMed] [Google Scholar]
- Pinnick KE, Gunn PJ & Hodson L (2019). Measuring human lipid metabolism using deuterium labeling: In vivo and in vitro protocols. Methods Mol Biol 1862, 83–96. [DOI] [PubMed] [Google Scholar]
- Pinnick KE & Hodson L (2019). Challenging metabolic tissues with fructose: tissue‐specific and sex‐specific responses. J Physiol 597, 3527–3537. [DOI] [PubMed] [Google Scholar]
- Pontzer H, Wood BM & Raichlen DA (2018). Hunter‐gatherers as models in public health. Obes Rev 19 (Suppl. 1), 24–35. [DOI] [PubMed] [Google Scholar]
- Scientific Advisory Committee on Nutrition (2015). Carbohydrates and Health. TSO, London. [Google Scholar]
- Rabøl R, Petersen KF, Dufour S, Flannery C & Shulman GI (2011). Reversal of muscle insulin resistance with exercise reduces postprandial hepatic de novo lipogenesis in insulin resistant individuals. Proc Natl Acad Sci U S A 108, 13705–13709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebollo A, Roglans N, Baena M, Sanchez RM, Merlos M, Alegret M & Laguna JC (2014). Liquid fructose downregulates Sirt1 expression and activity and impairs the oxidation of fatty acids in rat and human liver cells. Biochim Biophys Acta 1841, 514–524. [DOI] [PubMed] [Google Scholar]
- Roberts C, Steer T, Maplethorpe N, Cox L, Meadows S, Nicholson S, Page P & Swan G (2018). National Diet and Nutrition Survey: Results from Years 7 and 8 (combined) of the Rolling Programme (2014/2015 to 2015/2016). PHE publications, London: [Google Scholar]
- Roden M, Petersen KF & Shulman GI (2001). Nuclear magnetic resonance studies of hepatic glucose metabolism in humans. Recent Prog Horm Res 56, 219–237. [DOI] [PubMed] [Google Scholar]
- Salt IP, Johnson G, Ashcroft SJ & Hardie DG (1998). AMP‐activated protein kinase is activated by low glucose in cell lines derived from pancreatic beta cells, and may regulate insulin release. Biochem J 335, 533–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders FW & Griffin JL (2016). De novo lipogenesis in the liver in health and disease: more than just a shunting yard for glucose. Biol Rev Camb Philos Soc 91, 452–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saris WH, van Erp‐Baart MA, Brouns F, Westerterp KR & ten Hoor F (1989). Study on food intake and energy expenditure during extreme sustained exercise: the Tour de France. Int J Sports Med 10 (Suppl. 1), S26–S31. [DOI] [PubMed] [Google Scholar]
- Sevastianova K, Santos A, Kotronen A, Hakkarainen A, Makkonen J, Silander K, Peltonen M, Romeo S, Lundbom J, Lundbom N, Olkkonen VM, Gylling H, Fielding BA, Rissanen A & Yki‐Jarvinen H (2012). Effect of short‐term carbohydrate overfeeding and long‐term weight loss on liver fat in overweight humans. Am J Clin Nutr 96, 727–734. [DOI] [PubMed] [Google Scholar]
- Shimomura I, Bashmakov Y, Ikemoto S, Horton JD, Brown MS & Goldstein JL (1999). Insulin selectively increases SREBP‐1c mRNA in the livers of rats with streptozotocin‐induced diabetes. Proc Natl Acad Sci U S A 96, 13656–13661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiota M, Galassetti P, Monohan M, Neal DW & Cherrington AD (1998). Small amounts of fructose markedly augment net hepatic glucose uptake in the conscious dog. Diabetes 47, 867–873. [DOI] [PubMed] [Google Scholar]
- Shiota M, Moore MC, Galassetti P, Monohan M, Neal DW, Shulman GI & Cherrington AD (2002). Inclusion of low amounts of fructose with an intraduodenal glucose load markedly reduces postprandial hyperglycemia and hyperinsulinemia in the conscious dog. Diabetes 51, 469–478. [DOI] [PubMed] [Google Scholar]
- Sidossis LS, Stuart CA, Shulman GI, Lopaschuk GD & Wolfe RR (1996). Glucose plus insulin regulate fat oxidation by controlling the rate of fatty acid entry into the mitochondria. J Clin Invest 98, 2244–2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sluik D, Engelen AI & Feskens EJ (2015). Fructose consumption in the Netherlands: the Dutch National Food Consumption Survey 2007–2010. Eur J Clin Nutr 69, 475–481. [DOI] [PubMed] [Google Scholar]
- Softic S, Cohen DE & Kahn CR (2016). Role of dietary fructose and hepatic de novo lipogenesis in fatty liver disease. Dig Dis Sci 61, 1282–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanhope KL, Schwarz JM, Keim NL, Griffen SC, Bremer AA, Graham JL, Hatcher B, Cox CL, Dyachenko A, Zhang W, McGahan JP, Seibert A, Krauss RM, Chiu S, Schaefer EJ, Ai M, Otokozawa S, Nakajima K, Nakano T, Beysen C, Hellerstein MK, Berglund L & Havel PJ (2009). Consuming fructose‐sweetened, not glucose‐sweetened, beverages increases visceral adiposity and lipids and decreases insulin sensitivity in overweight/obese humans. J Clin Invest 119, 1322–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tappy L (2018). Fructose‐containing caloric sweeteners as a cause of obesity and metabolic disorders. J Exp Biol 221, jeb164202. [DOI] [PubMed] [Google Scholar]
- Tappy L & Lê KA (2010). Metabolic effects of fructose and the worldwide increase in obesity. Physiol Rev 90, 23–46. [DOI] [PubMed] [Google Scholar]
- Tappy L & Rosset R (2019). Health outcomes of a high fructose intake: the importance of physical activity. J Physiol 597, 3561–3571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Te Morenga L, Mallard S & Mann J (2013). Dietary sugars and body weight: systematic review and meta‐analyses of randomised controlled trials and cohort studies. BMJ 346, e7492. [DOI] [PubMed] [Google Scholar]
- Teofilovic A, Bursac B, Djordjevic A, Vojnovic Milutinovic D, Matic G & Velickovic N (2016). High dietary fructose load aggravates lipid metabolism in the liver of Wistar rats through imbalance between lipogenesis and fatty acid oxidation. Turk J Biol 40, 1235–1242. [Google Scholar]
- Ter Horst KW & Serlie MJ (2017). Fructose consumption, lipogenesis, and non‐alcoholic fatty liver disease. Nutrients 9, 981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandercammen A, Detheux M & Van Schaftingen E (1992). Binding of sorbitol 6‐phosphate and of fructose 1‐phosphate to the regulatory protein of liver glucokinase. Biochem J 286, 253–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandercammen A & Van Schaftingen E (1990). The mechanism by which rat liver glucokinase is inhibited by the regulatory protein. Eur J Biochem 191, 483–489. [DOI] [PubMed] [Google Scholar]
- Van Schaftingen E (1989). A protein from rat liver confers to glucokinase the property of being antagonistically regulated by fructose 6‐phosphate and fructose 1‐phosphate. Eur J Biochem 179, 179–184. [DOI] [PubMed] [Google Scholar]
- Vega‐Lopez S, Ausman LM, Matthan NR & Lichtenstein AH (2013). Postprandial lipid responses to standard carbohydrates used to determine glycaemic index values. Br J Nutr 110, 1782–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Holstein‐Rathlou S & Gillum MP (2019). Fibroblast growth factor 21: an endocrine inhibitor of sugar and alcohol appetite. J Physiol 597, 3539–3548. [DOI] [PubMed] [Google Scholar]
- Wang DD, Sievenpiper JL, de Souza RJ, Cozma AI, Chiavaroli L, Ha V, Mirrahimi A, Carleton AJ, Di Buono M, Jenkins AL, Leiter LA, Wolever TM, Beyene J, Kendall CW & Jenkins DJ (2014). Effect of fructose on postprandial triglycerides: a systematic review and meta‐analysis of controlled feeding trials. Atherosclerosis 232, 125–133. [DOI] [PubMed] [Google Scholar]
- Winder WW, Booth FW, Fitts RH & Holloszy JO (1975). Effect of exercise on response of liver lipogenic enzymes to a high fructose diet. Proc Soc Exp Biol Med 148, 1150–1154. [DOI] [PubMed] [Google Scholar]
- Wittekind A & Walton J (2014). Worldwide trends in dietary sugars intake. Nutr Res Rev 27, 330–345. [DOI] [PubMed] [Google Scholar]
- Wojtaszewski JF, Jorgensen SB, Hellsten Y, Hardie DG & Richter EA (2002). Glycogen‐dependent effects of 5‐aminoimidazole‐4‐carboxamide (AICA)‐riboside on AMP‐activated protein kinase and glycogen synthase activities in rat skeletal muscle. Diabetes 51, 284–292. [DOI] [PubMed] [Google Scholar]
- Woods A, Williams JR, Muckett PJ, Mayer FV, Liljevald M, Bohlooly YM & Carling D (2017). Liver‐specific activation of AMPK prevents steatosis on a high‐fructose diet. Cell Rep 18, 3043–3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C‐H, Tso S‐C, Chuang JL, Gui WJ, Lou M, Sharma G, Khemtong C, Qi X, Wynn RM & Chuang DT (2018). Targeting hepatic pyruvate dehydrogenase kinases restores insulin signaling and mitigates ChREBP‐mediated lipogenesis in diet‐induced obese mice. Mol Metab 12, 12–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita H, Takenoshita M, Sakurai M, Bruick RK, Henzel WJ, Shillinglaw W, Arnot D & Uyeda K (2001). A glucose‐responsive transcription factor that regulates carbohydrate metabolism in the liver. Proc Natl Acad Sci U S A 98, 9116–9121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang CS, Hawley SA, Zong Y, Li M, Wang Z, Gray A, Ma T, Cui J, Feng JW, Zhu M, Wu YQ, Li TY, Ye Z, Lin SY, Yin H, Piao HL, Hardie DG & Lin SC (2017a). Fructose‐1,6‐bisphosphate and aldolase mediate glucose sensing by AMPK. Nature 548, 112–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Tong X, VanDommelen K, Gupta N, Stamper K, Brady GF, Meng Z, Lin J, Rui L, Omary MB & Yin L (2017b). Lipogenic transcription factor ChREBP mediates fructose‐induced metabolic adaptations to prevent hepatotoxicity. J Clin Invest 127, 2855–2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YL, Guo H, Zhang CS, Lin SY, Yin Z, Peng Y, Luo H, Shi Y, Lian G, Zhang C, Li M, Ye Z, Ye J, Han J, Li P, Wu JW & Lin SC (2013). AMP as a low‐energy charge signal autonomously initiates assembly of AXIN‐AMPK‐LKB1 complex for AMPK activation. Cell Metab 18, 546–555. [DOI] [PubMed] [Google Scholar]