

Abstract
The study of the bacterial periplasm requires techniques with sufficient spatial resolution and sensitivity to resolve the components and processes within this subcellular compartment. Peroxidase-mediated biotinylation has enabled targeted labeling of proteins within subcellular compartments of mammalian cells. We investigated whether this methodology could be applied to the bacterial periplasm. In this study we demonstrated that peroxidase-mediated biotinylation can be performed in mycobacteria and Escherichia coli. To eliminate detection artifacts from natively biotinylated mycobacterial proteins, we validated two alternative labeling substrates, tyramide azide and tyramide alkyne, which enable biotin-independent detection of labeled proteins. We also targeted peroxidase expression to the periplasm, resulting in compartment-specific labeling of periplasmic versus cytoplasmic proteins in mycobacteria. Finally, we showed that this method can be used to validate protein relocalization to the cytoplasm upon removal of a secretion signal. This novel application of peroxidase-mediated protein labeling will advance efforts to characterize the role of the periplasm in bacterial physiology and pathogenesis.
Keywords: APEX, periplasm, peroxidase-mediated biotinylation, bacteria, CuAAC click chemistry, mycobacteria
Graphical Abstract
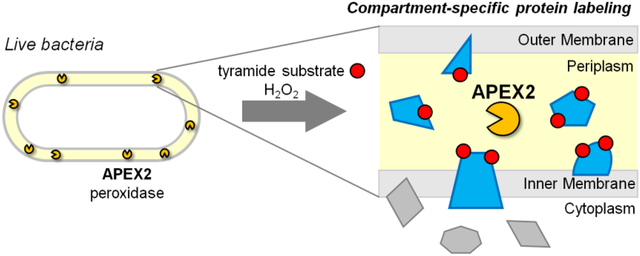
As the physical barrier between bacteria and their environments, the cell envelope is critical for bacterial survival and a major target for antibiotic development. A common feature of many bacterial cell envelopes is the periplasm – the space between the inner and outer membranes. This bacterial subcellular compartment, however, remains poorly characterized. The discovery of novel periplasmic biology requires proper tools for identifying proteins that localize to and perform specialized functions within this compartment.
Periplasmic proteins are typically identified using subcellular fractionation by sedimentation to separate membrane or cell wall constituents away from the cytoplasm, but this method has several critical drawbacks1. The disruptive nature of subcellular fractionation risks cross-contamination, especially by abundant cytoplasmic proteins. Notably, cytoplasmic proteins that peripherally associate with the inner membrane can be enriched in membrane and cell wall fractions. Furthermore, soluble proteins that localize to the periplasm, but do not associate with cell wall material may not be captured by this method. Another experimental approach is to use genetic reporters that are active exclusively in the cytoplasm or the periplasm (e.g., GFP fluorescence in the cytoplasm or alkaline phosphatase activity in the periplasm)2–4. This is a powerful method, particularly when used to screen libraries of reporter fusions or insertions5–6, and has been most recently used to identify exported proteins in Mycobacterium tuberculosis (Mtb) in a mouse infection7. Such studies make the key assumption that insertions and fusions do not alter the export or topology of the encoded protein. An alternative approach that avoids experimental complications is to identify periplasmic proteins by the presence of a secretion signal in the protein’s amino acid sequence8. However, several secreted proteins are known to lack a discernible secretion signal. In Mtb, the channel protein with necrosis-inducing toxin (CpnT) localizes to the outer membrane of the bacterium but has no apparent canonical secretion signal9. Mtb secreted antigen ESAT-6 and superoxide dismutase SodA also have no N-terminal secretion signal but are exported by the ESX-1 and SecA2 secretion systems respectively10–12. A bioinformatics approach based on secretion signals8 would miss these and other periplasmic and secreted proteins. Thus, due to the limitations of present methods for identifying periplasmic proteins, the localization of many bacterial proteins remains ambiguous.
Given the present challenges in validating periplasmic proteins, we explored whether periplasmic proteins could be selectively labeled. Ideally, such protein labeling would occur within live cells to preserve the membrane architecture that defines the periplasm. The labeling method must also function in the conditions found in the periplasm, which is believed to be an oxidizing environment13 and devoid of common enzyme co-factors such as ATP14. (Evidence supporting these statements comes from E. coli and to our knowledge there have been no corresponding studies in mycobacteria, but the analogous cell envelope architecture and presence of enzymes that catalyze disulfide formation15 suggest that the same restrictions apply.) Critically, the labeling method must be compartment-specific and selectively label periplasmic proteins without contamination from cytoplasmic proteins.
Peroxidase-mediated biotinylation is a protein labeling technique that is performed in live cells16–18. In the presence of hydrogen peroxide and biotin-phenol, the peroxidase APEX generates short-lived phenoxyl radicals that covalently biotinylate proximal proteins, typically on surface-exposed tyrosine residues. Biotinylated proteins can then undergo affinity enrichment and identification via mass spectrometry. When APEX was targeted to the mitochondrial matrix, APEX-generated biotin-phenoxyl radicals did not cross lipid membranes and selective labeling of matrix proteins was achieved16. Thus, peroxidase-mediated biotinylation can provide specific labeling of proteins within membrane-bound subcellular compartments. With the increased catalytic activity of the engineered variant APEX2, the sensitivity of peroxidase-mediated biotinylation improved significantly, making this technique highly suitable for subcellular proteomics19.
To evaluate peroxidase-mediated biotinylation in bacteria, we first expressed cytoplasmic APEX2 (Figure 1A) in Mycobacterium smegmatis (Msm) using a riboswitch-based theophylline-inducible expression system20. Msm cultures induced with theophylline had detectable APEX2 expression and coincident peroxidase activity (Figure 1B), indicating expression of active APEX2. We next treated the Msm cultures with biotin-phenol and hydrogen peroxide to initiate biotinylation. We assessed protein biotinylation by streptavidin blot analysis. Mycobacteria, including Msm, express several natively biotinylated proteins (Figure 1B, streptavidin blot, APEX2 uninduced lane)21–22. This endogenous biotinylation is the probable source of signal in uninduced samples. In Msm expressing cytoplasmic APEX2, we detected numerous APEX2-dependent biotinylated protein bands, demonstrating that peroxidase-mediated biotinylation can be performed in mycobacteria (Figure 1B, streptavidin blot, APEX2 induced lane). Optimal labeling was attained with 1 mM biotin-phenol and an incubation time of 30 minutes (Figure S1). We also performed peroxidase-mediated biotinylation in E. coli (Figure S2). Streptavidin blot analysis detected AccB (17 kDa), the only natively biotinylated protein in E. coli (Figure S2, streptavidin blot, APEX2 uninduced lane)23. As observed in Msm, expression of cytoplasmic APEX2 in E. coli also resulted in APEX2-dependent protein biotinylation, demonstrating the broad applicability of peroxidase-mediated biotinylation.
Figure 1. Cytoplasmic and secreted APEX2 have distinct protein biotinylation profiles in Msm.

(A) To localize APEX2 to the periplasmic space, we generated Sec-APEX2 and Tat-APEX2, in which a Sec or Tat secretion pathway signal is fused to APEX2. Red circles denote labeling substrate that reacts covalently with proteins in an APEX2 activity-dependent manner and subsequently serves as an enrichment or detection tag. (B) Msm expressing V5-tagged APEX2 (28 kDa), Sec-APEX2 (31 kDa) or Tat-APEX2 (33 kDa) was grown without or with theophylline and subjected to the labeling protocol with biotin-phenol. Anti-V5 immunoblot analysis of the lysates established expression of APEX2, Sec-APEX2 and Tat-APEX2. Anti-GroEL was used to confirm equal loading. Peroxidase activity was assayed in whole cells using guaiacol. Streptavidin blot analysis was used to detect protein biotinylation. Arrowheads indicate examples of APEX2 expression-dependent bands. Data are representative of >3 biological replicates.
Peroxidase-mediated biotinylation was previously used to inventory the proteome of the mitochondrial matrix by targeting APEX2 to this compartment16. We asked whether APEX2 could similarly be used to label a mycobacterial sub-compartment: the periplasm. To localize APEX2 to the periplasm, we generated Sec-APEX2 and Tat-APEX2, in which a mycobacterial Sec or Tat secretion signal is fused to the N-terminus of APEX2 (Figure 1A). We expressed either Sec-APEX2 or Tat-APEX2 in Msm (Figure 1B) and detected induced protein bands comparable in size to APEX2, indicative of proper secretion and processing of each protein by the respective secretion pathways. We also detected a second, slightly larger Tat-APEX2 product, which likely corresponds to the uncleaved precursor protein and has been observed for several mycobacterial Tat substrates24. Msm expressing either Sec-APEX2 or Tat-APEX2 had detectable peroxidase activity in a colorimetric assay on whole cells, suggesting that APEX2 remains active after secretion to the periplasm (Figure 1B). We noticed that expression of Sec-APEX2 was markedly lower than that of APEX2 or the processed form of Tat-APEX2. Consistent with this observation, Msm expressing Sec-APEX2 had lower peroxidase activity compared to APEX2- and Tat-APEX2-expressing strains. Despite these differences in expression and activity, both Sec-APEX2 and Tat-APEX2 expression yielded similar levels of protein biotinylation. Importantly, the pattern of biotinylation was similar between the two, but distinct from that of cytoplasmic APEX2 (Figure 1B). These results suggest that the cytoplasmic and secreted forms of APEX2 localize to different compartments (the cytoplasm vs. the periplasm) and thus biotinylate distinct subsets of proteins.
As demonstrated in Figure 1B, the discrimination of APEX2-dependent biotinylated proteins in mycobacteria is confounded by the abundance of natively biotinylated proteins in this genus. To address this problem, we synthesized two alternative labeling substrates, tyramide azide and tyramide alkyne (Figures S3 and S4), which can be coupled to detection or enrichment moieties via a Copper-catalyzed Azide/Alkyne Cycloaddition (CuAAC) “click” reaction. Similar to the biotin-phenol labeling protocol, we treated Msm with tyramide azide and hydrogen peroxide to enable APEX2 activity. We then performed CuAAC on total lysates using a fluorescein-conjugated alkyne and detected the labeled proteins by in-gel fluorescence. In the absence of APEX2 expression labeling with tyramide azide resulted in significantly fewer bands than observed with the biotin-phenol substrate (Figures 1B and 2, Figure S5). The tyramide azide labeling patterns of uninduced samples were also weaker compared to those of induced samples. These results are consistent with an absence of detection artifacts from natively biotinylated proteins in the labeling profile. We confirmed that the tyramide azide labeling profile observed by in-gel fluorescence was substrate- and copper-dependent (Figure S6) and that optimal labeling was achieved with 1 mM tyramide azide and an incubation time of 15 minutes (Figure S7). As observed with biotin-phenol, Sec-APEX2 and Tat-APEX2 labeling with tyramide azide generated a unique labeling pattern that differed from that of APEX2, further supporting compartment-specific labeling by cytoplasmic versus secreted APEX2 (Figure 2).
Figure 2. Alternative labeling substrates reduce detection artifacts.

Msm expressing APEX2, Sec-APEX2 or Tat-APEX2 was grown without or with theophylline and subjected to the labeling protocol with either tyramide azide or tyramide alkyne. Lysates underwent a CuAAC reaction with alkyne or azide-conjugated fluorescein. In-gel fluorescence detection of the clicked lysates was used to assess protein labeling. Equal protein loading was confirmed by subsequent Coomassie staining (Figure S5). Data are representative of 3 biological replicates under the specified conditions.
Labeling with tyramide alkyne similarly removed detection artifacts from natively biotinylated proteins (Figure 2, Figure S5). Optimal labeling was attained with 1 mM tyramide alkyne and an incubation time of 15 minutes (Figure S8). The labeling patterns of cytoplasmic and secreted APEX2 with tyramide alkyne matched those observed with tyramide azide. Thus, differential protein labeling by cytoplasmic and secreted APEX2 was observed when either tyramide azide or tyramide alkyne were used. Interestingly, the fluorescence intensity of the tyramide alkyne labeling profiles were consistently lower than those observed with tyramide azide (Figure 2, Figure S5, S9), suggesting differential labeling efficiency by the two substrates. With either tyramide azide or tyramide alkyne, residual labeling occurred in the absence of APEX2 expression (Figure 2, Figure S9). We suspected that this non-specific background labeling was due to the CuAAC conditions used. Indeed, we found that changing the ligand-to-copper ratio from 10:1 to 1:1 and reducing the copper concentration from 1 mM to 200 μM reduced non-specific background while maintaining APEX2-dependent labeling (Figure S10, S11).
To confirm compartment-specific protein biotinylation by cytoplasmic and secreted APEX2, we asked whether these forms of APEX2 differentially biotinylate eGFP, which is expressed in the cytoplasm, and the mycobacterial lipoprotein LprG (MSMEG_3070), which has a predicted secretion signal and localizes to the cell wall fraction25. In Msm we co-expressed eGFP or LprG with either APEX2 or Sec-APEX2 and treated cells with the same biotin-phenol labeling protocol as above (Figure 3A). To probe for biotinylation of either substrate, we first performed avidin enrichment on the lysates to enrich for biotinylated proteins and then probed for FLAG-tagged eGFP or LprG by immunoblot with fluorescence detection to enable quantitative analysis. eGFP was detected at higher levels in the avidin-enriched protein fraction when cells expressed APEX2 (Figure 3A). These results are consistent with APEX2 being localized to the cytoplasm where it can biotinylate eGFP. In contrast, LprG was detected at significantly higher levels after enrichment when co-expressed with Sec-APEX2, consistent with Sec-APEX2 being localized to the periplasm where it can biotinylate LprG.
Figure 3. Protein biotinylation by APEX2 and Sec-APEX2 is compartment-specific.

(A) Msm co-expressing APEX2 or Sec-APEX2 from a single integrated chromosomal copy and eGFP-3XFLAG (31 kDa) or LprG-3XFLAG (27 kDa) from a multi-copy episomal plasmid were grown without or with theophylline and subjected to the labeling protocol with biotin-phenol. Biotinylated proteins were enriched by avidin affinity purification. Anti-FLAG immunoblot analysis with fluorescence detection was used to quantify expression and biotinylation. The fold increase in biotinylation upon induction of APEX2 or Sec-APEX2 (“Ratio”) was calculated by taking the ratio of the + / − theophylline output signals after normalizing to the corresponding inputs. (B) Msm expressing APEX2 or Sec-APEX2 from a multi-copy episomal plasmid were treated as in (A) except that immunoblot analysis was performed with antibody against Mtb Ag85 complex. Purified Mtb Ag85A (34 kDa) was included as a positive control for the antibody. All data are from the same blot; intervening lanes were removed for clarity. All data are representative of ≥ 2 biological replicates. Lane labels I and O indicate input and output for the avidin enrichment.
Since overexpressing certain proteins may result in their mislocalization or in other undesirable effects on bacterial physiology, we further showed the generality of the method by assaying APEX2-dependent biotinylation of native proteins. The Ag85 complex is a well characterized group of mycobacterial cell wall enzymes26. Using Msm strains expressing cytoplasmic APEX2 or Sec-APEX2, we performed the same biotin-phenol labeling protocol and avidin enrichment as above and then probed for the Ag85 complex using a native antibody raised against the Mtb Ag85 complex. This polyclonal antibody cross-reacted with Msm lysates and yielded multiple bands in the input samples (Figure 3B). However, after avidin enrichment, distinct and specific bands in the predicted size range for the Msm Ag85 complex (~28–33 kDa) were detected only upon Sec-APEX2 expression (Figure 3B). Similar to the results with LprG, these observations are consistent with Sec-APEX2 being co-localized with Ag85 in the periplasm. The direct detection of Ag85 in the periplasm is a singular result that contrasts with previous cellular fractionation studies in which the Ag85 complex, which comprises fully soluble proteins, was detected in the supernatant along with cytoplasmic proteins27.
We went on to demonstrate that compartment-specific labeling by APEX2 can validate protein relocalization upon the removal of a putative secretion signal. When the N-terminal region of LprG containing its secretion signal is deleted (NA-LprG), the lipoprotein is presumably no longer secreted to the periplasm and remains in the cytoplasm28. In agreement with cytoplasmic localization, NA-LprG was only detected in the avidin-enriched protein fraction from APEX2-expressing cells (Figure 4). The inability of Sec-APEX2 to biotinylate NA-LprG supports the periplasmic localization of Sec-APEX2. Similar to Sec-APEX2, Tat-APEX2 expression resulted in biotinylation of LprG but not NA-LprG, demonstrating that Tat-APEX2 is also localized to the periplasm (Figure S12). We note that avidin-enriched LprG migrates slightly higher than total LprG detected in the input (Figures 3A, 4, S12). LprG-3XFLAG contains 6 tyrosines (3 native, 3 in the FLAG epitope) and biotin-phenol is somewhat hydrophobic due to the alkyl chain linker between the biotin and phenol moieties. Thus, labeling at multiple sites could lead to altered migration through combined effects on molecular weight and hydrophobicity.
Figure 4. Truncating the predicted secretion signal relocalizes LprG to the cytoplasm.

Msm co-expressing APEX2 or Sec-APEX2 from a multi-copy episomal plasmid and LprG-3XFLAG (27 kDa) or NA-LprG-3XFLAG (24 kDa) from a single integrated chromosomal copy were grown without or with theophylline and subjected to the labeling protocol with biotin-phenol. Biotinylated proteins were enriched by avidin affinity purification. Anti-FLAG immunoblot analysis with chemiluminescence detection of the input and output fractions was used to assess expression and biotinylation of LprG-3XFLAG and NA-LprG-3XFLAG. Data are representative of 3 biological replicates. Lane labels I and O indicate input and output for the avidin enrichment.
In addition, avidin enrichment of LprG even in the absence of APEX2 expression is much more apparent in Figure 3A than Figure 4. The major differences between these experiments that likely led to this result were the relative protein expression levels (higher target protein, lower APEX2 in Figure 3A), amount of lysate used in the enrichment (twice as much in Figure 3A) and the method of detection (fluorescence vs. chemiluminescence). This may have resulted in greater non-specific binding of LprG and more accurate (linear) detection of relative protein levels between samples in Figure 3. Regardless, the conclusions regarding LprG are the same from both experiments and in general fluorescence detection is recommended to enable quantitative comparisons, as discussed further below. Taken together, our findings demonstrate that APEX2 and Sec-APEX2/Tat-APEX2 localize to different subcellular compartments, the cytosol and the periplasm respectively, and each variant biotinylate proteins within their respective compartments.
In the course of applying APEX2-dependent labeling the localization of individual proteins we consistently observed a lower but consistent enrichment above background (ratio >1) when APEX2 and the target were expressed in different compartments (e.g., for eGFP and sec-APEX2 and for LprG and cytoplasmic APEX2; Fig. 3A). The degree of apparent “extra-compartmental” labeling correlated with the expression level of APEX2 (Figure S13), consistent with limited diffusion of the biotin-phenoxyl radical across membranes to yield detectable labeling of proteins outside the targeted compartment. Although no evidence for extra-compartmental protein labeling was detected by microscopy in a previous APEX study16, labeling due to cross-membrane diffusion of biotin-phenol- or biotin-aryl azide-derived radicals has been suggested by other peroxidase-based proteomics studies29–30. Our results are thus consistent with previous observations, but do not preclude the use of APEX2 labeling to localize proteins thanks to the ability to quantitatively compare labeling by cytoplasmic and sec-APEX2. We acknowledge that localizing targets that accumulate appreciably in both compartments will not necessarily be definitive by this method. In the future labeling reagents with lower membrane propensity (e.g., that have a shorter or more hydrophilic linker between the biotin and the phenol) may enable greater compartment specificity.
While labeling of a protein by APEX2 or Sec-APEX2 can determine whether a protein is in the cytoplasm or periplasm respectively, the extent of labeling cannot be used to determine the relative distribution of the protein across these compartments for two key reasons. First, the observed differences in both expression and activity of cytoplasmic and secreted APEX2s will contribute to differences in protein labeling levels. Second, the dramatically smaller size of the periplasm in comparison to the cytoplasm results in a higher effective concentration of Sec-APEX2 versus APEX2, resulting in differential labeling efficiencies in each subcellular compartment. For these reasons, it is not possible to comment on the relative amounts of a given protein that occupy the cytoplasm vs. the periplasm. Also, in general we recommend a low level of APEX2 and target protein expression, such as from a single gene copy when driven from a strong constitutive promoter, for validating localization, as this reduces spurious extra-compartmental labeling due to apparent phenoxyl radical diffusion across the membrane, but maintains robust intra-compartmental labeling (Figures 3A, S13). This may not be a concern for localizing native, rather than overexpressed, proteins due to target concentrations that are likely lower than those observed when using standard mycobacterial expression vectors.
To our knowledge, our work represents the first demonstration of peroxidase-mediated biotinylation in bacteria. Importantly, we show that this technique is compartment-specific, enabling selective labeling of periplasmic versus cytoplasmic proteins in mycobacteria. With our LprG labeling experiments, we show the utility of the described methodology in validating the localization of a protein-of-interest. Using alternative labeling substrates and click chemistry, we also demonstrated detection of APEX2-labeled mycobacterial proteins without artifacts from natively biotinylated proteins. Since detection of natively biotinylated proteins is avoided, these reagents will be especially useful for applications involving proteomics or the localization of specific proteins because they increase the specificity with which labeled proteins can be enriched. Nevertheless, labeling with biotin remains a valid option (Figures 3, 4) and currently the most convenient due to the commercial availability of biotin-phenol. Combined with quantitative proteomics, the described methodology has the potential to improve significantly upon existing approaches for studying periplasmic proteins in bacteria. Better identification and characterization of periplasmic proteins will help elucidate the role of the periplasm in bacterial physiology and virulence.
Methods
Reagents
Unless otherwise stated, reagents were obtained from Sigma-Aldrich. Phosphate buffered saline (PBS) was prepared as 18 mM sodium phosphate, 2.7 mM potassium chloride, 137 mM sodium chloride, 1.47 mM potassium phosphate, pH 7.4. Biotin-phenol (Iris Biotech), tyramide azide and tyramide alkyne were prepared as 50 mM stocks in DMSO and used in cultures at 1 mM final concentration (final DMSO concentration was 2%).
Strains and Culture Conditions
E. coli BL21 (DE3) and Stellar (Clontech) strains were cultured in LB medium and incubated at 37 °C with shaking at 250 rpm. For selection, 50 μg ml−1 kanamycin, 100 μg ml−1 hygromycin B were used where indicated. Msm mc2155 (ATCC 700084) was cultured in Middlebrook 7H9 containing 0.05% (v/v) Tween-80, 0.2% (v/v) glycerol, 0.2% (w/v) glucose, 1% (w/v) casamino acids (Amresco) and incubated at 37 °C with shaking at 250 rpm. For selection 25 μg ml−1 kanamycin or 50 μg ml−1 hygromycin B were used where indicated. Antibiotic concentrations were halved when used together.
Cloning
All vectors were generated using In-Fusion HD Cloning (Clontech), assembly PCR or traditional ligation as described in Tables S1 and S2. N-terminal V5-tagged APEX was subcloned from pcDNA3-mito-APEX (Addgene plasmid # 49386)16 and inserted into pRibo, a theophylline-inducible expression vector20. APEX2 was subsequently generated by site-directed mutagenesis19. Sec-APEX2 and Tat-APEX2 were generated by introducing a secretion signal connected by a two amino acid linker to the N-terminus of V5-tagged APEX2: (Sec/Tat signal)-GS-(V5 tag)-(APEX2). The secretion signals of mpt63 (rv1926c) and blaC (rv2068c) were used as Sec and Tat signals, respectively, and have been previously validated31–32. Vectors expressing C-terminally FLAG-tagged proteins contained a constitutive hsp60 promoter33 and encoded the 3XFLAG sequence: (Protein)-AS-DYKDDDGDYKDHDIDYKDDDDK. NA-LprG was generated by deleting residues 1–25 from the Msm LprG homologue (MSMEG_3070) and mutating the cysteine residue at position 26 to a methionine by site-directed mutagenesis28, 34.
Synthesis of Tyramide Azide and Tyramide Alkyne
For tyramide azide, tyramine hydrochloride (465 mg, 2.675 mmol, 1 equiv.) was combined with 4-azidobutyric acid (0.311 mL, 2.675 mmol, 1 equiv.) and diisopropyl carbodiimide (0.479 mL, 3.211 mmol, 1.2 equiv.) in pyridine (16 mL) in a round bottom flask. The reaction was allowed to proceed overnight at 22 °C. Excess solvent was then evaporated. The solid power remaining was subsequently dissolved in ethyl acetate (3.40 mL) and partially purified by flash column using ethyl acetate as the mobile phase. Collected fractions were pooled and evaporated to dryness. The residue was dissolved in acetonitrile and purified by high-pressure liquid chromatography (HPLC) (60–50% acetonitrile in water with 1% trifluoroacetic acid over 85 min, retention time = 26 min, 35% acetonitrile). 1H-NMR (400 MHz, CDCl3) δ: 7.00 (d, J = 8.4 Hz, 2H), 6.77 (d, J = 8.5 Hz, 2H), 5.57 (s, 1H), 3.47 (m, 2H), 3.28 (t, J = 6.5 Hz, 2H), 2.71 (t, J = 6.9 Hz, 2H), 2.2 (t, J = 7.2 Hz, 2H), 1.86 (m, 2H) (Figure S3). MS (ESI): m/z calculated for C12H17N4O2 [M + H+] 249.1352, observed 249.1346 [M+H]+.
Tyramide alkyne was synthesized and purified in the same manner, except that 5-hexynoic acid (0.295 mL, 2.675 mmol, 1 equiv.) was substituted for 4-azidobutyric acid. For HPLC the retention time was 24 min, 29% acetonitrile. 1H-NMR (400 MHz, CDCl3) δ: 6.99 (d, J = 8.1 Hz, 2H), 6.80 (d, J = 8.0 Hz, 2H), 5.74 (s, 1H), 3.46 (m, 2H), 2.71 (t, J = 7.0 Hz, 2H), 2.26 (t, J = 7.4 Hz, 2H), 2.1 (td, J = 6.9, 2.6 Hz, 2H), 1.94 (t, J = 2.6 Hz, 1H), 1.83 (m, 2H) (Figure S4). MS (ESI): m/z calculated for C14H18NO2 [M + H+] 232.1338, observed 232.1 [M+H]+.
Protein Labeling in Live Mycobacteria
The labeling protocol was adapted from the method described by Rhee and coauthors16. For labeling with biotin-phenol, 15-mL Msm cultures were subcultured from log phase starter cultures at OD600 = 0.25 with or without 2 mM theophylline and incubated at 37 °C for 6 h. Cells were pelleted, resuspended in 1 mL fresh medium and transferred to microcentrifuge tubes. Cell suspensions were incubated with 1 mM biotin-phenol (Iris Biotech) at 37 °C for 30 min. To initiate the labeling reaction, cell suspensions were treated with 1 mM H2O2 for 1 min. The reaction was quenched by adding 1 mL 2X quenching solution containing 20 mM sodium ascorbate, 20 mM sodium azide and 10 mM Trolox in PBS with 0.05% Tween-80 (PBS-Tw80). Cells were washed with 1X quenching solution and then with PBS-Tw80 using a wash volume of 2 mL. The cells were then washed with 1X quenching solution a second time and then with PBS-Tw80 using wash volumes of 1.5 mL before being resuspended in 1.5 mL PBS (no Tween 80). Cells were lysed by bead beating with 0.1 mm zirconia/silica beads four times at 4,500 rpm for 30 s each with samples kept on ice for 5 min between beatings. Beads and cell debris were removed from lysates by centrifugation (10,000 g, 5 min, 4 °C).
Labeling with tyramide azide or tyramide alkyne was performed as described above, but with the following changes: 1 mL cell suspensions were incubated with 1 mM tyramide azide or tyramide alkyne for 15 min. After the second wash with 1X quenching solution, the cells were washed with 1.5 mL 20 mM Tris, pH 7.4 and resuspended in 1.5 mL Click Reaction Lysis Buffer (20 mM Tris, 150 mM NaCl, 1% Triton X-100, 1% SDS, pH 7.4) before lysing.
Protein Labeling in Live E. coli
15-mL E. coli cultures were inoculated at OD600 = 0.1 until cultures reached OD600 = 0.4–0.6. Isopropyl β-D-1-thiogalactopyranoside (IPTG) was added to a final concentration of 0.1 mM and cultures were incubated at 37°C for an additional 1 h. Cells were pelleted, resuspended in 1 mL fresh medium and transferred to microcentrifuge tubes. Samples were then treated with biotin-phenol and H2O2, quenched and lysed as described for mycobacteria above.
Whole Cell Peroxidase Activity
Guaiacol was added to 1% (v/v) final concentration to 500 μL of culture in a microcentrifuge tube. H2O2 was then added to the samples to a final concentration of 1% (v/v). Samples were immediately mixed by vortexing and briefly centrifuged. Cell pellets were examined for the development of a reddish-orange color.
CuAAC Click-Chemistry
Protein lysates from Msm cells labeled with tyramide azide or tyramide alkyne were generated as described above. Click reactions in 500 μL consisted of labeled protein lysates with 5 mM sodium ascorbate, 25 μM 5-fluorescein amidite (FAM) azide (Lumiprobe) or 5-FAM alkyne (Lumiprobe), 100 μM tris(benzyltriazolylmethyl)amine (TBTA) (Thermo Fisher Scientific) and 1 mM CuSO4, added in that order. Reactions were protected from light and incubated at 22 °C with gentle shaking for 1 h after which the reactions were quenched by adding EDTA to a final concentration of 10 mM. SDS loading dye was added to samples before separation on a 10% SDS-PAGE gel and visualization using a Typhoon FLA 9000 scanner (GE Healthcare Life Sciences). Equal protein loading of SDS-PAGE gels was confirmed by Coomassie staining. Optimized click reactions were set up as above, but with the following changes: Reactions were scaled down to 100 μl total volume and used 200 μM CuSO4 and 200 μM TBTA to give a 1:1 ligand-to-copper ratio.
Enrichment of Biotinylated Proteins
Lysates from Msm cells treated with biotin-phenol were generated in PBS as described above. Protein concentration was determined using the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific). NeutrAvidin agarose resin (100 μL; Thermo Fisher Scientific) was pre-equilibrated in microcentrifuge tubes by washing with 500 μl PBS twice. Input samples consisted of 400–1200 μg total protein lysate (normalized across samples) with 1% SDS in a total volume of 500 μl. Input samples were boiled for 5 min and 420 μl (Figure 4) or 960 μL (Figure 3) was then added to the washed resin and incubated at 22 °C with gentle rotation for 1 h. The supernatant (flow-through) was then removed. The resin was washed with 500 μl PBS containing 0.2% SDS at 22 °C with gentle shaking for 10–20 min. The supernatant (wash) was removed and the resin was washed an additional three times with 500 μl PBS (no SDS) each. Proteins (output) were eluted by boiling the resin in 50 μl 2X SDS loading dye for 5 min.
Immunoblots
All antibodies were diluted with Odyssey Blocking Buffer (LI-COR, cat# 927–40000) containing 0.1% SDS and 0.1% Tween-20 unless otherwise noted. For total protein biotinylation profile analysis, 10–20 μg total protein was separated on 10% SDS-PAGE gels and then transferred to nitrocellulose membranes. Membranes were probed with 1:10,000 Streptavidin IRDye®700Dx (Rockland Immunochemicals) and imaged on an Odyssey CLx imaging system (LI-COR). For analysis of APEX2 expression levels, 10–20 μg total protein was separated and transferred as above and then probed with 1:5000 mouse IgG anti-V5 (Invitrogen) or 1:200 mouse anti-GroEL (Abcam) and 1:15,000 goat anti-mouse IgG CW800 (LI-COR) secondary antibody before imaging.
For analysis of eGFP, LprG and NA-LprG biotinylation, input (10–20 μg total protein) and output (20 μL) samples were separated and transferred as above. Membranes were blocked with 5% BSA in PBST (PBS with 0.1% Tween-20) and then probed with 1:1000 mouse monoclonal anti-FLAG M2 (Sigma Aldrich) in 5% BSA in PBST. For fluorescence detection (Figure 3), membranes were probed with 1:15,000 goat anti-mouse IgG CW800 (LI-COR) secondary antibody before imaging. For chemiluminescence detection (Figure 4), membranes were probed with 1:20,000 goat polyclonal anti-mouse IgG-HRP (Abcam) secondary antibody. Millipore Immobilon Western Chemiluminescent HRP Substrate was used for detection of HRP-conjugated antibody on film.
For analysis of Ag85 biotinylation, input (2.5 μg total protein) and output (20 μL) samples were separated and transferred as above. Membranes were blocked with Odyssey Blocking Buffer (LI-COR) and then probed with 1:5000 rabbit polyclonal anti-Mtb Ag85 complex (BEI Resources NR-13800). Membranes were probed with 1:10,000 goat anti-rabbit lgG CW 680 (LI-COR) secondary antibody followed by fluorescence imaging. Mtb Ag85A was expressed and purified as reported35 and 0.1 μg was loaded as a positive control for the antibody. All image processing and analysis was performed using Image Studio Lite (LI-COR)
Supplementary Material
Acknowledgments
The authors thank Julia Joseph and Marissa Alsaloum for technical assistance and to members of the Seeliger Lab for feedback. This work was supported by NIH R21 AI126044 (J.C.S.) and R21 EB01795501 (I.S.C.). The authors declare no conflicts of interest.
Footnotes
- Supplementary Tables S1–S2
- Supplementary Figures S1–S13
References
- 1.Weiner JH; Li L, Proteome of the Escherichia coli envelope and technological challenges in membrane proteome analysis. Biochim Biophys Acta 2008, 1778 (9), 1698–1713. DOI: 10.1016/j.bbamem.2007.07.020. [DOI] [PubMed] [Google Scholar]
- 2.Belardinelli JM; Larrouy-Maumus G; Jones V; Sorio de Carvalho LP; McNeil MR; Jackson M, Biosynthesis and translocation of unsulfated acyltrehaloses in Mycobacterium tuberculosis. J. Biol. Chem 2014, 289 (40), 27952–27965. DOI: 10.1074/jbc.M114.581199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Seeliger JC; Holsclaw CM; Schelle MW; Botyanszki Z; Gilmore SA; Tully SE; Niederweis M; Cravatt BF; Leary JA; Bertozzi CR, Elucidation and chemical modulation of sulfolipid-1 biosynthesis in Mycobacterium tuberculosis. J. Biol. Chem 2012, 287 (11), 7990–8000. DOI: 10.1074/jbc.M111.315473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Touchette MH; Holsclaw CM; Previti ML; Solomon VC; Leary JA; Bertozzi CR; Seeliger JC, The rv1184c locus encodes Chp2, an acyltransferase in Mycobacterium tuberculosis polyacyltrehalose lipid biosynthesis. J. Bacteriol 2015, 197 (1), 201–210. DOI: 10.1128/JB.02015-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McCann JR; McDonough JA; Sullivan JT; Feltcher ME; Braunstein M, Genome-Wide Identification of Mycobacterium tuberculosis Exported Proteins with Roles in Intracellular Growth. J. Bacteriol 2011, 193 (4), 854–861. DOI: 10.1128/jb.01271-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Braunstein M; Griffin TJIV; Kriakov JI; Friedman ST; Grindley NDF; Jacobs WR Jr., Identification of Genes Encoding Exported Mycobacterium tuberculosis Proteins Using a Tn552’phoA In Vitro Transposition System. J. Bacteriol 2000, 182 (10), 2732–2740. DOI: 10.1128/jb.182.10.2732-2740.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Perkowski EF; Zulauf KE; Weerakoon D; Hayden JD; Ioerger TR; Oreper D; Gomez SM; Sacchettini JC; Braunstein M, The EXIT Strategy: an Approach for Identifying Bacterial Proteins Exported during Host Infection. MBio 2017, 8 (2). DOI: 10.1128/mBio.00333-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Petersen TN; Brunak S; von Heijne G; Nielsen H, SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods 2011, 8 (10), 785–786. DOI: 10.1038/nmeth.1701. [DOI] [PubMed] [Google Scholar]
- 9.Danilchanka O; Sun J; Pavlenok M; Maueroder C; Speer A; Siroy A; Marrero J; Trujillo C; Mayhew DL; Doornbos KS; Munoz LE; Herrmann M; Ehrt S; Berens C; Niederweis M, An outer membrane channel protein of Mycobacterium tuberculosis with exotoxin activity. Proc. Natl. Acad. Sci. U. S. A 2014, 111 (18), 6750–6755. DOI: 10.1073/pnas.1400136111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stanley SA; Raghavan S; Hwang WW; Cox JS, Acute infection and macrophage subversion by Mycobacterium tuberculosis require a specialized secretion system. Proc. Natl. Acad. Sci. U. S. A 2003, 100 (22), 13001–13006. DOI: 10.1073/pnas.2235593100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Champion PAD; Stanley SA; Champion MM; Brown EJ; Cox JS, C-Terminal Signal Sequence Promotes Virulence Factor Secretion in Mycobacterium tuberculosis. Science 2006, 313 (5793), 1632–1636. DOI: 10.1126/science.1131167. [DOI] [PubMed] [Google Scholar]
- 12.Braunstein M; Espinosa BJ; Chan J; Belisle JT; Jacobs WR Jr., SecA2 functions in the secretion of superoxide dismutase A and in the virulence of Mycobacterium tuberculosis. Mol. Microbiol 2003, 48 (2), 453–464. [DOI] [PubMed] [Google Scholar]
- 13.Messens J; Collet JF; Van Belle K; Brosens E; Loris R; Wyns L, The oxidase DsbA folds a protein with a nonconsecutive disulfide. J Biol Chem 2007, 282 (43), 31302–31307. DOI: 10.1074/jbc.M705236200. [DOI] [PubMed] [Google Scholar]
- 14.Wulfing C; Pluckthun A, Protein folding in the periplasm of Escherichia coli. Mol. Microbiol 1994, 12 (5), 685–692. [DOI] [PubMed] [Google Scholar]
- 15.Reardon-Robinson ME; Ton-That H, Disulfide-Bond-Forming Pathways in Gram-Positive Bacteria. J Bacteriol 2015, 198 (5), 746–754. DOI: 10.1128/JB.00769-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rhee HW; Zou P; Udeshi ND; Martell JD; Mootha VK; Carr SA; Ting AY, Proteomic mapping of mitochondria in living cells via spatially restricted enzymatic tagging. Science 2013, 339 (6125), 1328–1331. DOI: 10.1126/science.1230593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hung V; Zou P; Rhee HW; Udeshi ND; Cracan V; Svinkina T; Carr SA; Mootha VK; Ting AY, Proteomic mapping of the human mitochondrial intermembrane space in live cells via ratiometric APEX tagging. Mol Cell 2014, 55 (2), 332–341. DOI: 10.1016/j.molcel.2014.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hung V; Udeshi ND; Lam SS; Loh KH; Cox KJ; Pedram K; Carr SA; Ting AY, Spatially resolved proteomic mapping in living cells with the engineered peroxidase APEX2. Nat Protoc 2016, 11 (3), 456–475. DOI: 10.1038/nprot.2016.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lam SS; Martell JD; Kamer KJ; Deerinck TJ; Ellisman MH; Mootha VK; Ting AY, Directed evolution of APEX2 for electron microscopy and proximity labeling. Nat Methods 2015, 12 (1), 51–54. DOI: 10.1038/nmeth.3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Seeliger JC; Topp S; Sogi KM; Previti ML; Gallivan JP; Bertozzi CR, A riboswitch-based inducible gene expression system for mycobacteria. PLoS One 2012, 7 (1), e29266 DOI: 10.1371/journal.pone.0029266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rhee KY; Erdjument-Bromage H; Tempst P; Nathan CF, S-nitroso proteome of Mycobacterium tuberculosis: Enzymes of intermediary metabolism and antioxidant defense. Proc Natl Acad Sci U S A 2005, 102 (2), 467–472. DOI: 10.1073/pnas.0406133102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Duckworth BP; Geders TW; Tiwari D; Boshoff HI; Sibbald PA; Barry CE 3rd; Schnappinger D; Finzel BC; Aldrich CC, Bisubstrate adenylation inhibitors of biotin protein ligase from Mycobacterium tuberculosis. Chem Biol 2011, 18 (11), 1432–1441. DOI: 10.1016/j.chembiol.2011.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cronan JE Jr., Biotination of proteins in vivo. A post-translational modification to label, purify, and study proteins. J Biol Chem 1990, 265 (18), 10327–10333. [PubMed] [Google Scholar]
- 24.McDonough JA; McCann JR; Tekippe EM; Silverman JS; Rigel NW; Braunstein M, Identification of functional Tat signal sequences in Mycobacterium tuberculosis proteins. J Bacteriol 2008, 190 (19), 6428–6438. DOI: 10.1128/JB.00749-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bigi F; Espitia C; Alito A; Zumarraga M; Romano MI; Cravero S; Cataldi A, A novel 27 kDa lipoprotein antigen from Mycobacterium bovis. Microbiology 1997, 143 (Pt 11), 3599–3605. DOI: 10.1099/00221287-143-11-3599. [DOI] [PubMed] [Google Scholar]
- 26.Belisle JT; Vissa VD; Sievert T; Takayama K; Brennan PJ; Besra GS, Role of the major antigen of Mycobacterium tuberculosis in cell wall biogenesis. Science 1997, 276 (5317), 1420–1422. [DOI] [PubMed] [Google Scholar]
- 27.Wells RM; Jones CM; Xi Z; Speer A; Danilchanka O; Doornbos KS; Sun P; Wu F; Tian C; Niederweis M, Discovery of a Siderophore Export System Essential for Virulence of Mycobacterium tuberculosis. PLoS pathogens 2013, 9, e1003120 DOI: 10.1371/journal.ppat.1003120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Drage MG; Tsai HC; Pecora ND; Cheng TY; Arida AR; Shukla S; Rojas RE; Seshadri C; Moody DB; Boom WH; Sacchettini JC; Harding CV, Mycobacterium tuberculosis lipoprotein LprG (Rv1411c) binds triacylated glycolipid agonists of Toll-like receptor 2. Nat Struct Mol Biol 2010, 17 (9), 1088–1095. DOI: 10.1038/nsmb.1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jiang S; Kotani N; Ohnishi T; Miyagawa-Yamguchi A; Tsuda M; Yamashita R; Ishiura Y; Honke K, A proteomics approach to the cell-surface interactome using the enzyme-mediated activation of radical sources reaction. Proteomics 2012, 12 (1), 54–62. DOI: 10.1002/pmic.201100551. [DOI] [PubMed] [Google Scholar]
- 30.Li XW; Rees JS; Xue P; Zhang H; Hamaia SW; Sanderson B; Funk PE; Farndale RW; Lilley KS; Perrett S; Jackson AP, New insights into the DT40 B cell receptor cluster using a proteomic proximity labeling assay. J Biol Chem 2014, 289 (21), 14434–14447. DOI: 10.1074/jbc.M113.529578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McDonough JA; Hacker KE; Flores AR; Pavelka MS Jr.; Braunstein M, The twin-arginine translocation pathway of Mycobacterium smegmatis is functional and required for the export of mycobacterial beta-lactamases. J Bacteriol 2005, 187 (22), 7667–7679. DOI: 10.1128/JB.187.22.7667-7679.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McCann JR; McDonough JA; Pavelka MS; Braunstein M, Beta-lactamase can function as a reporter of bacterial protein export during Mycobacterium tuberculosis infection of host cells. Microbiology 2007, 153 (Pt 10), 3350–3359. DOI: 10.1099/mic.0.2007/008516-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stover CK; de la Cruz VF; Fuerst TR; Burlein JE; Benson LA; Bennett LT; Bansal GP; Young JF; Lee MH; Hatfull GF; et al. , New use of BCG for recombinant vaccines. Nature 1991, 351 (6326), 456–460. DOI: 10.1038/351456a0. [DOI] [PubMed] [Google Scholar]
- 34.Martinot AJ; Farrow M; Bai L; Layre E; Cheng TY; Tsai JH; Iqbal J; Annand JW; Sullivan ZA; Hussain MM; Sacchettini J; Moody DB; Seeliger JC; Rubin EJ, Mycobacterial Metabolic Syndrome: LprG and Rv1410 Regulate Triacylglyceride Levels, Growth Rate and Virulence in Mycobacterium tuberculosis. PLoS Pathog. 2016, 12 (1), e1005351 DOI: 10.1371/journal.ppat.1005351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Backus KM; Dolan MA; Barry CS; Joe M; McPhie P; Boshoff HI; Lowary TL; Davis BG; Barry CE 3rd, The three Mycobacterium tuberculosis antigen 85 isoforms have unique substrates and activities determined by non-active site regions. J. Biol. Chem 2014, 289 (36), 25041–25053. DOI: 10.1074/jbc.M114.581579. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.