

Abstract
BACKGROUND
High mobility group box-1 (HMGB1), recognized as a representative of damage-associated molecular patterns, is released during cell injury/death, triggering the inflammatory response and ultimately resulting in tissue damage. Dozens of studies have shown that HMGB1 is involved in certain diseases, but the details on how injured hepatocytes release HMGB1 need to be elicited.
AIM
To reveal HMGB1 release mechanism in hepatocytes undergoing oxidative stress.
METHODS
C57BL6/J male mice were fed a high-fat diet for 12 wk plus a single binge of ethanol to induce severe steatohepatitis. Hepatocytes treated with H2O2 were used to establish an in vitro model. Serum alanine aminotransferase, liver H2O2 content and catalase activity, lactate dehydrogenase and 8-hydroxy-2-deoxyguanosine content, nicotinamide adenine dinucleotide (NAD+) levels, and Sirtuin 1 (Sirt1) activity were detected by spectrophotometry. HMGB1 release was measured by enzyme linked immunosorbent assay. HMGB1 translocation was observed by immunohistochemistry/immunofluorescence or Western blot. Relative mRNA levels were assayed by qPCR and protein expression was detected by Western blot. Acetylated HMGB1 and poly(ADP-ribose)polymerase 1 (Parp1) were analyzed by Immunoprecipitation.
RESULTS
When hepatocytes were damaged, HMGB1 translocated from the nucleus to the cytoplasm because of its hyperacetylation and was passively released outside both in vivo and in vitro. After treatment with Sirt1-siRNA or Sirt1 inhibitor (EX527), the hyperacetylated HMGB1 in hepatocytes increased, and Sirt1 activity inhibited by H2O2 could be reversed by Parp1 inhibitor (DIQ). Parp1 and Sirt1 are two NAD+-dependent enzymes which play major roles in the decision of a cell to live or die in the context of stress . We showed that NAD+ depletion attributed to Parp1 activation after DNA damage was caused by oxidative stress in hepatocytes and resulted in Sirt1 activity inhibition. On the contrary, Sirt1 suppressed Parp1 by negatively regulating its gene expression and deacetylation.
CONCLUSION
The functional inhibition between Parp1 and Sirt1 leads to HMGB1 hyperacetylation, which leads to its translocation from the nucleus to the cytoplasm and finally outside the cell.
Keywords: Sirtuin1, Poly ADP-ribose polymerase 1, High mobility group box-1, Hepatocytes, Hydrogen peroxide
Core tip: High mobility group box 1 (HMGB1) is a nuclear protein that non-specifically binds to the minor grooves in DNA. Once released passively by necrotic and damaged cells, HMGB1 will become a damage-associated molecular pattern molecule triggering the inflammatory response and ultimately results in tissue damage. In the present study, we found that HMGB1 is released from H2O2-injured hepatocytes due to Sirt1 functional inhibition.
INTRODUCTION
High mobility group box 1 (HMGB1), a member of a subfamily of the HMG proteins, non-specifically binds to the minor grooves in DNA[1], and is released passively by necrotic and damaged cells upon various stimulation[2]. Once released in the extracellular space, HMGB1 acts as a damage-associated molecular pattern molecule, triggering the inflammatory response and ultimately results in tissue damage. Therefore, HMGB1 released by injured cells is a key driver of disease progression in both acute and chronic diseases[3,4]. During the past decade, studies in both patients and animal models have established that HMGB1 represents a potential biomarker and novel therapeutic target suggesting that blocking HMGB1 release is a potential therapeutic strategy for certain diseases.
The nuclear localization of HMGB1 and its affinity for DNA are regulated through phosphorylation and acetylation, and have been found to have a dynamic relationship with chromatin. When HMGB1 is not acetylated, it is located in the nucleus and is not secreted or released outside the cell, reducing its inflammatory effects as seen during cellular injury/death[5]. HMGB1 is a novel deacetylation target of nicotinamide adenine dinucleotide (NAD+)-dependent deacetylase Sirtuin1 (Sirt1)[6], which is grouped as a class III histone deacetylase, and is a predominantly nuclear protein which can shuttle to the cytoplasm under conditions of cell stress. Sirt1 and poly ADP-ribose polymerase 1 (Parp1) are two enzymes functionally connected due to their common use of an NAD+ substrate[7] and functional interplay in regulating lipopolysaccharide-mediated HMGB1 secretion[8].
Chromatin relaxation at DNA lesions is one of the earliest cellular responses to DNA damage and is regulated by Parp1 enzymatic activity[9]. Under oxidative stress caused by H2O2, Parp1 is generated at high levels leading to hepatic cell damage and death in CCl4-induced liver injury[10]. Additionally, there was significantly greater DNA damage and excessive Parp1 activation, exhausting NAD+ stores in hepatocyte-specific HMGB1 knockout mice (HMGB1-HC-KO)[11]. On the other hand, Sirt1 plays a critical role in preventing DNA damage caused by H2O2 in human embryonic stem cells and regulates stress responses, genomic stability, and cell survival[12]. H2O2 also evokes injury of cardiomyocytes by upregulating HMGB1[13]. However, the relationship among Parp1, -Sirt1, cell death,and HMGB1 release still needs to be elucidated.
In the present study, we hypothesized that HMGB1 release by H2O2-injured hepatocytes is regulated by DNA-damage-mediated Parp1 activation, which causes NAD+ over-depletion followed by Sirt1 activity suppression, leading to HMGB1 hyperacetylation and finally release. Our findings demonstrated that DNA damage triggers the cascade reaction of Parp1–Sirt1–HMGB1 hyperacetylation and finally HMGB1 release from hepatocytes. The results support our hypothesis and identify HMGB1 as an important biomarker for liver injury induced with a high-fat diet/ethyl alcohol (HFD/etOH).
MATERIALS AND METHODS
Animals, diets and experiments
Twenty-four male C57BL/6J mice, weighing 23 ± 2 g, were obtained from the Shanghai Institute of Materia Medica [license number: SCXK (Shanghai) 2012-0002]. The experimental protocol was approved by the Animal Ethics Committee of Shanghai University of Traditional Chinese Medicine, and the study was performed in accordance with the approved guidelines. The animals were randomly divided into two groups: Control and HFD plus etOH. The HFD group was fed an HFD (Research Diets, United States, D12492), while the control group was given a control diet (Research Diets, United States, D12451). After 12 wk of treatment, on the last day, mice fed an HFD were given 31.25% ethanol solution in water at a single dose of 5 g/kg body weight by oral gavage, while the mice in the control group were given saline. After 9 h, all mice were sacrificed, and the samples were collected for subsequent experiments.
Measurement of serum alanine aminotransferase (ALT) and hepatic H2O2, and catalase
Serum ALT activity was detected using a commercial kits according to the manufacturer’s instructions (Nanjing Jiancheng Biology Co., Ltd, China). The liver tissues were homogenized with a homogenizer in cold saline (1:10, w/v), and then centrifuged at 3000 rpm for 15 min for detecting the levels of H2O2 and catalase using commercial kits (Nanjing Jiancheng Biology Co., Ltd).
Hematoxylin-eosin staining and immunohistochemical assay
Liver tissues were fixed in 10% formalin, embedded in paraffin, cut into 4-μm-thick sections, and stained with hematoxylin-eosin. For immunohistochemical assay, sections were immersed in anti-HMGB1 antibody solution in a humidified chamber at 4 °C overnight, followed by incubation in a biotin–labeled secondary antibody solution at 37 °C for 1 h. Then, the slices were incubated with SABC solution at 37 °C for 1 h. All images were observed by microscopy (ZEISS, German).
Cell culture and treatments
The mouse embryonic hepatocyte cell line BNL.CL2, purchased from the cell bank of the Shanghai Institutes for Biological Sciences (Shanghai, China), was cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco, United States) supplemented with 10% fetal calf serum (Gibco, United States), 100 U/mL penicillin and 100 U/mL streptomycin, at 37 °C in a 5% (v/v) CO2 humidified atmosphere. When cells were grown to 70%-80% confluence, they were treated with H2O2 (Kaiji Biology Co. Ltd, China) at different concentrations for the same duration or at the same concentration for different durations.
Lactate dehydrogenase (LDH) cytotoxicity assay
The LDH cytotoxicity assay was performed with a kit (Cytotoxicity LDH Assay Kit-WST, Dojindo Molecular Technologies, Shanghai) according to the manufacturer’s guidelines. In summary, 1 × 104 cells cultured in 96-well plates in the presence or absence of H2O2 were incubated with 100 µL of working solution for 30 min at room temperature, and the 96-well plates were protected from light. Next, 50 µL of stop solution was added, and the absorbance of each sample was read at 490 nm using a Synergy 2 plate reader (Bio-Tek Ltd, United States). The cytotoxicity of each sample was normalized using the following formula: Cytotoxicity (%) = [(ODsample - ODblank) / (ODpostive - ODblank)] (%).
ELISA for HMGB1
The levels of HMGB1 in the culture medium were measured by ELISA kit (Chondrex Co. Ltd, Redmond, United States) according to the manufacturer’s instructions.
DNA damage assay
The 8-hydroxy-2-deoxyguanosine (8-OHdG) levels were measured using a kit according to the manufacturer's protocol (8-OHdG assay kit, Genmed Inc., Shanghai, China). The kit provided 8-OHdG antibody and peroxidase labeled secondary antibody. TMB (3,3’,5,5’-tetramethylbenzidine) was used as a peroxidase substrate and formed blue precipitates at pH 4.9. The absorbance was read at 650 nm using a Synergy 2 plate reader.
Quantification of NAD+ levels
The NAD+ levels in cultured cells or liver tissues were determined using an enzymatic method according to the manufacturer’s instructions (EnzyChrom, BioAssays Systems, CA, United States). Typically, 20 mg of liver tissue or 1 × 105 cells were homogenized in a 1.5 mL Eppendorf tube with 100 µL of NAD+ extraction buffer preheated for 5 min at 60 °C, and then 20 µL assay buffer was added along with 100 µL of NADH extraction buffer to neutralize the extracts. The extracts were spun down at 14000 rpm for 5 min, and the supernatant was transferred into a clear flat-bottom 96-well plate for NAD+ concentration assays. After the working buffer was added, the absorbance for time “zero” (A0) was read at 565 nm and A15 after a 15-min incubation at room temperature of each well was recorded with a Synergy 2 plate reader (Bio-Tek Ltd, United States). Finally, according to the standard curve, NAD+ levels were normalized using the following formula[14]: NAD+ levels (μM) = (∆Asample-∆Ablank)/slope × n, where ∆A = A15-A0, n equaling 5 was the dilution ratio of samples, and the slope was calculated from the standard curve.
Sirt1 deacetylase activity assay
Sirt1 deacetylase activity was measured using a Sirt1 direct fluorescent screening assay kit (GENMED Inc., Shanghai, China) following the manufacturer’s protocol. The Sirt1 substrate in the kit is an acetylated peptide fragment derived from p53 that was prelabeled with 7-amino-4-methylcoumarin (AMC). After deacetylation by Sirt1, an amino peptidase cleaved the deacetylated substrate, which generated a highly fluorescent molecule group of AMC. The fluorescence intensity was monitored each 5 min for 1 h using a fluorescence plate reader (Bio-Tek FL×800) at an excitation wavelength of 360 nm and an emission wavelength of 460 nm. Values are expressed as the rate of reaction for the first 30 min. The results were normalized by the total protein concentration, determined using a BCA assay.
Isolation of cytoplasmic and nuclear proteins
Cytoplasmic and nuclear protein fractions from BNL.Cl2 cells or liver tissues were extracted with a commercial protein isolation kit from Yeasen Co., Ltd. (Shanghai, China) according to the manufacturer’s instructions. Briefly, 1 × 107 BNL.CL2 cells or 50 mg liver tissue with reagent A were collected into 1.5 mL microcentrifuge tubes and vortexed for 5 s. Afterwards, samples were put on ice for 10 min and centrifuged for an additional 5 min at 12000 rpm and 4 °C. The supernatants were cytoplasmic proteins. The precipitates with Reagent C were vortexed for 25 s per 3 min interval, total 30 min. Finally, samples were centrifuged for 5 min at 12000 rpm and 4 °C, and the supernatants were nuclear proteins.
Western blot analysis
Protein concentration was determined by BCA assay (Beyotime, Suzhou, China). The 4%-20% SDS-PAGE-Resolved protein bands were transferred to PVDF membranes (Millipore, Bedford, MA, United States) with a Trans-Blot Turbo transfer system (Bio-Rad). Primary antibodies (Abcam Co., Ltd, United States) and compatible horseradish peroxidase-conjugated secondary antibodies were used. Proteins were detected using chemiluminescence-plus reagents from Millipore Co., Ltd. (Bedford, MA, United States). The blot image was captured with a Protein Simple multispectral imaging system with a Chemi HR camera and analyzed using AlphaView SA (version 3.4.0; Protein Simple, United States).
Immunoprecipitation (IP) analysis
Total lysates (300 μg)were immunoprecipitated with specific rabbit anti-HMGB1 (1:75) or Parp1 antibody (1:50), while the control lysates were immunoprecipitated with rabbit anti-immunoglobulin antibody on a Labnet Mini LabRoller Rotator (Labnet, Edison, NJ, United States) at 4 °C overnight, followed by the incubation with 30 μL of protein A/G agarose beads to extract the immunocomplexes for 3 h at room temperature. Thereafter, beads were washed with IP lysis buffer and the immunocomplexes were extracted in SDS loading buffer at 97 °C, followed by Western blot analysis.
Fluorescence confocal laser microscopy
Cells (1 × 104) were seeded on cover-glass bottom dishes and treated with either 500 μmol/L H2O2 for 0.5 h, 2 h, 8 h or 24 h, or with 100 μmol/L, 500 μmol/L, or 1000 μmol/L H2O2 for 8 h. Following treatment, cells were fixed with 4% paraformaldehyde for 30 min, and then incubated in phosphate buffered saline (PBS) containing 0.2% Triton-X-100 for 30 min on ice. Thereafter, cells were blocked with 5% Goat serum for 1h at room temperature, followed by incubation in a 5% BSA solution containing HMGB1 primary antibody (1:100) overnight at 4 °C. The next day, after a triple wash in Tween-20 phosphate buffer saline, the cells were incubated in PBS containing Alexa Fluor 594 conjugated secondary antibody (1:300) using a 1% BSA dilution for 1h at room temperature. Lastly, the nucleus were stained nuclei with DAPI, and the dishes were mounted and observed using a ZEISS confocal laser fluorescence microscope (ZEISS, German).
Real-time reverse transcription-polymerase chain reaction assay
After purity determination, each amount of total RNA sample extracted from liver tissues or cells was reverse transcribedinto cDNA using a TransScript® All-in-One First-Strand cDNA Synthesis SuperMix for qPCR Kit (One-Step gDNA Removal) according to the manufacturer’s protocol. The mRNA levels were quantification with a SYBR Green qPCR SuperMix kit. The relative amount of target mRNA was calculated by the 2-ΔΔCt. Primer sequences for the genes used are as follows: Beta-actin (NM_007393.5): forward (5’–3’): ACTGCTCTGGCTCCTAGCAC, and reverse (5’–3’): ACATCTGCTGGAAGGTGGAC; SIRT1 (NM_019812.3): forward (5’–3’): GAACCACCAAAGCGGAAA, and reverse (5’–3’): TCCCACAGGAGACAGAAACC; PARP1 (NM_007415.2): forward Primer (5’–3’): ACCACTTCTCCTGCTTCTGG, and reverse Primer (5’–3’): GCCGTCTTCTTGACCTTCTG.
Sirt1 shRNA transfection
Cells were seeded in six-well plates at a density of 2 × 105 cells, cultured until approximately 70% confluence, and then transfected with control or Sirt1-shRNA-pLKO.1-EGFP (Sirt1-F: CCG GGC CAT GTT TGA TAT TGAGT ATC TCG AGA TAC TCA ATA TCA AAC ATG GCT TTTTG; Sirt1-R: AAT TCA AAA AGC CAT GTT TGA TAT TGA GTA TCT CGA GAT ACT CAA TAT CAA ACA TGGC) lentiviral vector (USEN Biological Technology Co., Ltd, Shanghai, China) (MOI:4-8) according to the manufacturer’s instructions. Briefly, the vectors were dissolved in no fetal calf serum DMEM. Before infection, the cells were treated with 5 μg/mL polybrene for 4 h. After 24 h of infection, the cells were incubated for 48 h in 2% fetal calf serum DMEM including 10 μg/mL Puromycin dihydrochloride. Then, the cells were treated with or without H2O2 (500 μM) for 8 h. The mRNA levels of Sirt1 and Parp1 were determined by Real-time PCR, and protein levels of Sirt1, Parp1, Ace-Parp1, Ace-HMGB1, and HMGB1 were determined as above.
Parp1 over expression
The BNL.CL2 cells were plated in six-well plates at a density of 2 × 105 cells/mL, incubated until approximately 50% confluence, and then transfected with control or Parp1 using synergistic activation mediator CRISPR Activation Plasmid according to the manufacturer’s instructions. In summary, 20 μL of Parp1 plasmid DNA (sc-419018-ACT, Santa Cruz, United States) or negative control (sc-437275, Santa Cruz, United States) plasmid was dissolved in DMEM as solution A. Meanwhile, 10μL of UltraCruz® Transfection Reagent (sc-395739, Santa Cruz, United States) was also dissolved in DMEM to obtained solution B and equilibrated for 5 min at room temperature. Next, solutions A and B were mixed, and equilibrated for 20 min at room temperature to form a mixture of Parp1 plasmid DNA-UltraCruz® Transfection. The cells were transfected with Parp1 plasmid DNA or negative control plasmid. At 48 h after transfection, the medium was changed to fresh medium containing 10 μg/mL Puromycin dihydrochloride (sc-108071), 200 μg/mL Hygromycin B (sc-29067) and 2 μg/mL Blasticidin S HCl (sc-495389) for antibiotic selection, and then the parp1 expression level was detected by realtime-PCR.
Statistical analysis
Data were analyzed by one-way analysis of variance (ANOVA) or Welch ANOVA followed by the Student’s t-test using the GraphPad 8.0 software and are expressed as the mean ± standard deviation (SD), P < 0.05 was considered statistically significant. All experiments at least three independents were performed.
RESULTS
Fatty liver causes HMGB1 release from damaged hepatocytes
To study the release of HMGB1 in the livers following development of oxidative stress damage, C57BL6/J mice were fed a high fat diet for 12 wk and a single binge ethanol feeding before sacrifice. The HFD/etOH mice had higher serum ALT activity (Figure 1A) which is an enzyme in the cytoplasm of normal hepatocytes, but released from the livers into serum during hepatocellular injury[15]. Additionally, there was significantly increased H2O2 content and catalase activity in the livers (Figure 1A), and markedly increased hepatic steatosis (Figure 1B). The results of im-munohistochemistry indicated that HMGB1 was translocated from the nucleus to the cytoplasm in hepatocyte (black arrow in Figure 1C). Also, HMGB1 levels obviously rose in cytoplasm and decreased in the nucleus in hepatocyte of HFD/etOH mice with hyperacetylation (Figure 1D). Mice with fatty liver disease had significantly increased levels of Parp1, followed by decreased content of NAD+ and inhibited Sirt1 (Figure 1E and F).
Figure 1.

Hepatocellular injury in mice treated with a high-fat diet plus single binge alcohol. A: Serum levels of alanine aminotransferase and the levels of H2O2 and catalase in C57BL/6 mice fed a control diet for 12 wk or a high-fat diet plus single binge alcohol (n = 12); B: Hematoxylin-eosin stained images (scale bar = 10 μm); C: High mobility group box-1 (HMGB1) staining (scale bar = 20 μm). The black arrow shows HMGB1 translocation; D and E: Western blot analysis for Parp1, Sirt1, and HMGB1 in whole tissue and HMGB1 in the cytoplasm or in nucleus. HMGB1 acetylation was detected by immunoprecipitation; F: The mRNA expression of Parp1 and Sirt1 by real-time PCR and liver NAD+ content. All data were presented as mean ± SD. Statistical analysis was done by the Student’s t-test. aP < 0.05 vs control, bP < 0.01 vs control, cP < 0.01 vs control. HFD/etOH: High-fat diet plus single binge alcohol; ALT: Alanine aminotransferase; HMGB1: High mobility group box-1; IP: Immunoprecipitation.
H2O2-induced HMGB1 release from injured hepatocytes
Hepatocytes are the most abundant and most vulnerable cell types to be attacked in the liver. To further investigate whether HMGB1 is released during injury/death, hepatocytes were treated with 500 μmol/L H2O2. Progressively elevated LDH release was observed, which peaked at 8h and then decreased gradually to nearly normal levels at 24 h (Figure 2A), and LDH was released from hepatocytes treated with various concentrations of H2O2 (0, 100, 500 and 1000 μmol/L) in a dose-dependent manner (Figure 2B). H2O2 triggered DNA injury by reactive oxygen species (ROS) which produced 8-OHdG, a marker of oxidative stress to DNA[16], therefore, 8-OHdG levels in H2O2-treated hepatocytes were measured. 8-OHdG levels were significantly increased by H2O2, but were suppressed by N-acetylcysteine (NAC), an ROS scavenger[17] (Figure 2C). In recent years, accumulating data have demonstrated that HMGB1 is released passively during various kinds of cellular injury and death[18,19]. In accordance with these reports, our results revealed that the HMGB1 content in medium was over 30-fold at 2 h and reached a 76-fold increase at 8 h, and was still 13-fold at 24 h (Figure 2D) after hepatocytes were exposed to 500 μmol/L H2O2 for different durations of time. Also, the HMGB1 content was decreased in a dose-dependent manner by NAC treatment (Figure 2E).
Figure 2.

Cytotoxicity increase and high mobility group box-1 release induced by H2O2 in BNL CL cells. A and B: BNL CL2 cells cultured with H2O2 for 0, 0.5, 2, 8, or 24 h or at different concentrations (0, 100, 500, and 1000 μmol/L); C: 8-hydroxy-2-deoxyguanosine levels in cells treated with 500 μmol/L H2O2 or N-acetylcysteine (NAC); D: High mobility group box-1 (HMGB1) content in medium detected by ELISA; E: Cells were treated with 500 μM H2O2 for different time intervals HMGB1 content in medium detected by ELISA. Cells were pre-treated with NAC for 24 h, and then incubated with or without H2O2 (500 μmol/L) for 8 h. All data are presented as the mean ± SD. Statistical analysis was done by the Student’s t-test. aP < 0.05 vs 0 group, bP < 0.01 vs 0 group, dP < 0.01 vs H2O2 group. 8-HOdG: 8-hydroxy-2-deoxyguanosine; NAC: N-acetylcysteine; HMGB1: High mobility group box-1.
Nuclear HMGB1 continuously moving across the nuclear membrane into the cytoplasm through specific nuclear pores is a key stage of its extracellular release. Our immunohistochemical results showed that HMGB1 appeared in the cytoplasm after hepatocytes were treated with 500 μmol/L H2O2 for 2 h and 8 h, but not 0.5 h, meanwhile, it appeared in the cytoplasm in all groups of hepatocytes treated with 100, 500, and 1000 μmol/L H2O2 (Figure 3A). Consistent with these results, Western blot showed an increase of HMGB1 in the cytoplasm and decrease in the nucleus in a H2O2 time- and dose-dependent manner (Figure 3B and C). The accumulation of HMGB1 in the cytoplasm is determined by the acetylation status of specific lysine residues located in the two nuclear localization sequences[20]. We found that for the different lengths of time, acetylated (Ac) HMGB1 levels in cell lysates reached 1.2-fold even at 0.5 h and 2-fold peak at 8 h and decreased at 24 h after hepatocytes were incubated with H2O2 (Figure 3D). Likewise, the acetylated (Ac) HMGB1 levels also displayed an increase in a dose-dependent manner with H2O2 treatment (Figure 3E). Together, these findings suggest that as a consequence of injury to hepatocytes by H2O2 , HMGB1 is translocated from the nucleus to the cytoplasm and then released to medium with the concomitantly elevated proportion of the acetylated form.
Figure 3.

H2O2 induced high mobility group box-1 translocation and acetylation. A: Confocal microscopy analysis of cells treated with H2O2 either for different durations or at different concentrations. (scale bar = 40 μm). The white arrow shows the translocation of high mobility group box-1 (HMGB1) to the cytoplasm; B and C: HMGB1 expression determined by Western blot; D and E: Acetylated HMGB1 determined by immunoprecipitation. All data are presented as the mean ± SD. Statistical analysis was done by the Student’s t-test. aP < 0.05 vs 0 group, bP < 0.01 vs 0 group. HMGB1: High mobility group box-1.
H2O2-induced Sirt1 decrease leads to HMGB1 release
Sirt1, a mammalian ortholog of yeast silent information regulator 2, is a NAD+-dependent class III protein deacetylase that is involved in the deacetylation of HMGB1. Our previous findings indicated that hepatocytes exposed to oxidative stress in vitro release hyperacetylated HMGB1[21] and have a H2O2-induced Sirt1 levels[22], which prompted us to examine whether H2O2-induced HMGB1 was release by decreasing Sirt1 expression. As expected, the decrease of Sirt1 protein level was obvious in a H2O2 time- and dose-dependent manner (Figure 4A and B). Furthermore, the Sirt1 activity in the control group was 3.28 ± 0.14 nmoL/mg/min, but was 3.06 ± 0.13 nmoL/mg/min, 3.12 ± 0.02 nmoL/mg/min and 085 ± 0.05 nmoL/mg/min in groups treated with different concentrations of H2O2, respectively (Figure 4E). After cells were treated with 0.1 μmol/L SRT1720 (an activator of Sirt1) for 4 h, the HMGB1 contents in medium significantly decreased to 15.6 ± 2.87 ng/mL compared with 61.09 ± 9.86 ng/mL in cells treated with H2O2 alone (Figure 4F). After cells were treated with EX527, an inhibitor of Sirt1, Sirt1 levels decreased and acetylated HMGB1 increased, and as a consequence, HMGB1 was translocated from the nuclues to the cytoplasm (Figure 5A and B). To further investigate the effects of Sirt1 on HMGB1 acetylation and release, Sirt1 knockdown hepatocytes were employed (Figure 5C) and they strongly enhanced HMGB1 acetylation, accompanied by HMGB1 translocation from the nuclues to the cytoplasm and release to medium (Figure 5D and E). Because the decreased Sirt1 was companied with increased HMGB1 hyperacetylation and its release in H2O2 injured cells, we conclude that the Sirt1 suppression by H2O2 causes hyperacetylated HMGB1 and finally its release to medium.
Figure 4.

H2O2 treatment inhibits Sirt1 expression and activity. A and B: BNL CL2 cells were treated with H2O2 for 0-24 h at 500 μmol/L. Sirt1 levels determined by Western blot; C and D: BNL CL2 cells were treated with H2O2 at 0-1000 μmol/L for 8 h. Sirt1 levels determined by Western blot; E: Sirt1 deacetylase activity detected by fluorescence spectrophotometry; F: BNL CL2 cells were treated with H2O2 (500 μmol/L) for 4 h, and then SRT1720, a Sirt1 activator, was added into the medium. The supernatants were collected to determinate high mobility group box-1 content by ELISA. All data are presented as the mean ± SD. Statistical analysis was done by the Student’s t-test. aP < 0.05 vs 0 group, bP < 0.01 vs 0 group or negative control, dP < 0.01 vs H2O2 group. HMGB1: High mobility group box-1.
Figure 5.

Sirt1 inhibits high mobility group box-1 translocation and release. A: Acetylated HMGB1 levels determined by immunoprecipitation. EX527, a Sirt1 inhibitor inhibited sirt1 level and promoted HMGB1 acetylation; B: High mobility group box-1 (HMGB1) expression in the nucleus or cytoplasm determined by Western blot; C: The mRNA and protein levels of Sirt1. BNL CL2 cells were transfected with control lentiviral vector or Sirt1 shRNA lentiviral vector for 48 h. The mRNA and protein levels of Sirt1 were detected by real-time PCR and Western blot. Multiplicity of infection, refers to the number of lent virus adsorption on the cells and the ratio of the number of cells in culture; D: HMGB1 translocation; E: HMGB1 content in medium determined by ELISA. All data are presented as the mean ± SD. Statistical analysis was done by the Student’s t-test. bP < 0.01 vs 0 group or negative control, fP < 0.01 vs control lent viral group. MOI: Multiplicity of infection; HMGB1: High mobility group box-1.
H2O2-induced NAD+ depletion by Parp1 leads to suppressed Sirt1 activity
Because the same substrate of NAD+ is shared, Parp1 and Sirt1 are functionally connected. We first determined the effect of H2O2 and DIQ (1,5-dihydroxyisoquinoline), a competitive inhibitor of Parp1, on NAD+ levels. The NAD+ levels decreased from 0.5 h and reached approximately 50% inhibition at 8 h, then restored 24 h after cells were treated with H2O2 (Figure 6A). Likewise, the NAD+ levels were also inhibited in a dose-dependent manner after H2O2 treatment (Figure 6B). Furthermore, we examined the effect of DIQ on NAD+ levels. As expected, the NAD+ levels decreased approximately 50% from the control after H2O2 treatment at 8 h, however, when the cells were pretreated with 100 μmol/L DIQ, the NAD+ levels were restored to approximately 70% of control, and pretreatment with 300 μmol/L DIQ reversed the NAD+ levels to over 90% of control (Figure 6C). Next, the effect of Parp1 inhibition on Sirt1 activity was assessed. Cells pretreated with DIQ demonstrated an increase in Sirt1 activity as compared with cells treated with H2O2 alone (Figure 6D). To further investigate whether the effect of Parp1 inhibition on Sirt1 activity is related with its dependence on NAD+ concentration, NAM, a noncompetitive inhibitor of Sirt1, was employed. Cells pretreated with DIQ and then exposed to H2O2 demonstrated an increase in Sirt1 activity, while cells pretreated with both DIQ and NAM (5 mmol/L and 10 mmol/L, respectively) failed to exhibit an increase in Sirt1 activity (Figure 6D). These findings verified the importance of NAD+ in mediating sirt1 activity through Parp1 inhibition.
Figure 6.

Sirt1 activity is inhibited by H2O2 through Parp1-NAD+. A and B: NAD+ contents in cells treated with H2O2 for different durations or at different concentrations; C: NAD+ contents in cells treated with DIQ, a Parp1 inhibitor; D: Sirt1 activity in cells treated with DIQ and NAM, an NAD+ inhibitor; E and F: High mobility group box-1 (HMGB1) acetylation in cells treated with DIQ; G: BNL CL2 cells were treated with DIQ or H2O2. And then HMGB1 contents in medium determined by ELISA; H: HMGB1 contents in medium determined by ELISA in cells treated with CP or Parp1+ transfection; I: Sirt1 activity of cells with Parp1+ transfection. All data are presented as the mean ± SD. Statistical analysis was done by the Student’s t-test. bP < 0.01 vs 0 group or negative control, dP < 0.01 vs H2O2 group, hP < 0.01 vs H2O2 + DIQ group, iP < 0.01 vs CP group. CP: Control Plasmid.
Hyperacetylation status of HMGB1 is a vital step prior to its release. In our experiments, the immunoprecipitated nuclear lysates were incubated with antibodies against HMGB1, and then immunoblotted with acetyl lysine antibodies. As shown in Figure 6E, DIQ pretreatment alone had no appreciable effect (Figure 6E, panel 2). Cells incubated with H2O2 demonstrated an increase in acetylated HMGB1 compared with untreated cells. Moreover, cells incubated with both DIQ and H2O2 demonstrated a significant inhibition of acetylation of HMGB1 (Figure 6E and F). Then, when we investigated the effect of Parp1 inhibition on H2O2-induced HMGB1 release, the results showed that HMGB1 release was significantly increased when cells were treated with H2O2, however, cells pretreated with DIQ (100 μmol/L, or 300 μmol/L) had a significant decrease (Figure 6G). In contrast, we overexpressed Parp1 in cells to investigate Sirt1 enzyme activities and HMGB1 release. As shown in Figure 6H and I, Parp1 overexpression not only suppressed Sirt1 activities, but also promoted HMGB1 release, which is similar to the results with H2O2 treatment.
Sirt1 negatively regulates Parp1
Parp1 is a nuclear protein involved in oxidative stress-induced DNA repair and chromatin remodeling. To examine whether Parp1 is responsive to oxidative stress in hepatocytes, we subjected cells to H2O2 for different times and concentrations, and the results showed that both parp1 and acetylated parp1 protein levels increased in a time- and dose-dependent manner (Figure 7A-D). To preserve cellular NAD+ levels and thus retain its own function, Sirt1 may deactivate Parp1 by deacetylation of Parp1. Besides this, Sirt1 is able to negatively regulate the PARP1 promoter[23]. We therefore hypothesized that a decrease in Sirt1 in cells with H2O2-induced DNA damage would increase Parp1 and its acetylation levels. To test this hypothesis, we examined the mRNA and protein levels of Parp1 levels and its acetylation status in Sirt1 knockdown cells. Coinciding with our expectation, both the mRNA and protein levels of Parp1 levels and its acetylation status were significantly increased, which are in keeping with the results of H2O2 treatment (Figure 7E-G). To further confirm these findings, cells were exposed to a Sirt1 activator, SRT1720. We also found that Parp1 protein levels and its acetylation status were decreased after incubation with SRT1720 alone or SRT1720 plus H2O2 (Figure 7H and I).
Figure 7.

Sirt1 inhibits Parp1 expression and acetylation in BNL CL2 cells. A-D: Parp1 expression and acetylation in cells treated with H2O2 for different durarions or at different concentrations; E: The mRNA levels of Parp1 in Sirt1 knockdown cells treated with H2O2; F and G: Parp1 expression and acetylation in Sirt1 knockdown cells treated with H2O2; H and I: Parp1 expression and acetylation in cells treated with SRT1720, a Sirt1 activator. All data are presented as the mean ± SD. Statistical analysis was done by the Student’s t-test. aP < 0.05 vs 0 group or negative control, bP < 0.01 or negative control, dP < 0.01 vs H2O2 group, fP < 0.01 vs CLv group, jP < 0.05 vs shLv group, kP < 0.01 vs shLv group.
DISCUSSION
Oxidative stress is one of the main mechanisms of either HFD or ethanol-induced liver injury[24,25]. Alcohol and HFD consumption are known to synergistically induce hepatocytes damage and steatohepatitis[26]. Our in vivo experiments verified that hepatocytes injury and steatosis accompanied with oxidative stress appeared after feeding mice with an HFD for 3 mo plus a single binge of ethanol. HMGB1 released outside the cell was highly acetylated[20] and was related with exposure of hepatocytes to oxidative stress[27]. ROS plays a critical role in the induction of HMGB1 release[28]. The similar finding from this study was that both mice fed an HFD/etOH and H2O2-treated hepatocytes exacerbated the release of HMGB1 that was highly acetylated.
Posttranslational modifications play a role in HMGB1 localization. Our previous study found that hyperphosphorylation of HMGB1 has been implicated in its nuclear-to-cytoplasmic translocation[29]. Recent studies on HMGB1 release identified that the decreased activity of SIRT1, an NAD+-dependent histone deacetylase, which is especially predominant in the nucleus, is critical for HMGB1 hyperacetylation[6]. SIRT1 is also involved in both non-alcoholic fatty liver diseases and alcoholic fatty liver disease[30]. In the present study, we observed that HMGB1 translocation from the nucleus to the cytoplasm and the subsequent release were accompanied by time- and dose-dependent hyperacetylation in H2O2-induced hepatocytes. Similarly, the Sirt1 protein levels and activity also acted in a time- and dose-dependent manner. Furthermore, we demonstrated that either SIRT1 shRNA or EX527, a specific SIRT1 inhibitor, promoted HMGB1 hyperacetylation and subsequent release. On the contrary, SRT1720, a SIRT1 activator suppressed HMGB1 release. A parallel change of Sirt1 decrease and HMGB1 hyperacetylation and subsequent translocation from the nucleus to cytoplasm to outside the cell in either mice fed an HFD/etOH or hepatocytes treated with H2O2 confirmed that Sirt1 plays a key role in HMGB1 release.
Sirt1 and Parp1 are two enzymes functionally connected due to their use of the common substrate NAD+[7]. Parp1 was shown to have a greater effect on NAD+-depletion than Sirt1[31]. Rapid depletion of NAD+ levels by Parp1 inhibits Sirt1 activity and the capability of Sirt1 to deacetylate its targets responding to genotoxic stress[9]. We demonstrated that NAD+ content in H2O2 treated cells decreased in a time- and dose-dependent manner. Additionally, this phenomenon was observed with Parp1. Furthermore, DIQ, a specific Parp1 inhibitor, has antagonism for H2O2 and caused a rescue of NAD+levels and increased Sirt1 activity, followed by a reduction of HMGB1 release. On the contrary, overexpression of Parp1 has resulted in a decrease in Sirt1 activity and HMGB1 release. We verified that Sirt1 activity inhibition in H2O2-treated cells was due to overconsuming NAD+ by Parp1. Parp1 is a DNA damage sensor protein which binds to DNA strand, breaks efficiently, and uses NAD+ as a substrate to facilitate DNA repair[32,33]. 8-OHdG, a marker of oxidative stress to DNA, was observed to increase in our study, which supports our hypothesis that NAD+ may be overconsumed, in part, due to Parp1 activation caused by DNA damage led to sirt1 activity suppression.
In addition that Parp1 negatively regulates Sirt1, Sirt1 may deactivate Parp1 to preserve cellular NAD+ levels and thus retain its own function. Sirt1 is able to negatively regulate the Parp1 promoter[34] and can deacetylate Parp1[35]. In accordance with these reports, our results obtained with the use of shRNA of Sirt1 indicated that the levels of both Parp1 mRNA and protein increased due to Parp1 hyperacetylation. The contrary results were revealed in cells with Sirt1 activator and SRT1720 treatment. In summary, our present study demonstrates that functional inhibition of Parp1 and Sirt1 in H2O2-induced hepatocytes is crucial for HMGB1 release outside the cell, thereby uncovering a new mechanism for HFD/etOH caused hepatic steatosis (Figure 8).
Figure 8.
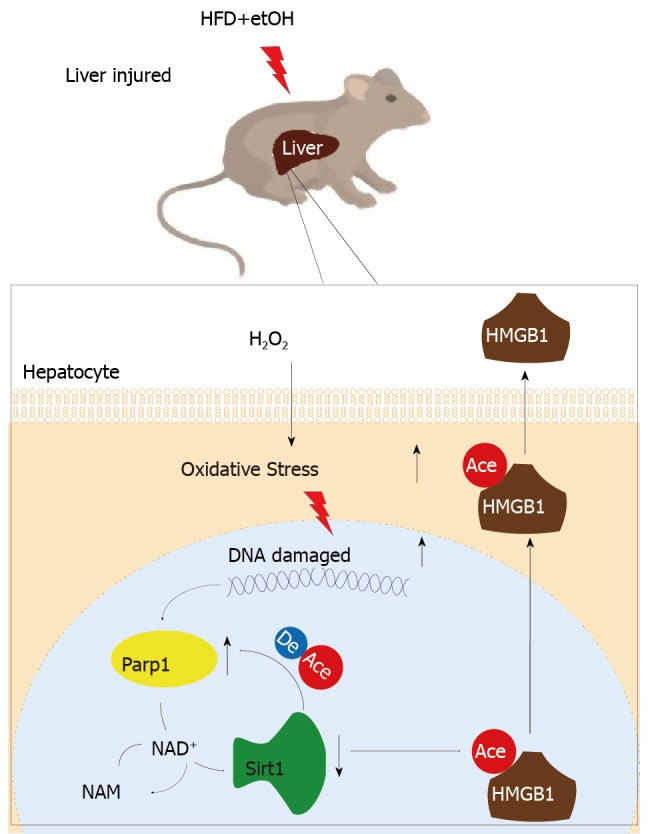
Imbalance of mutual inhibition between Parp1 and Sirt1 causes high mobility group box-1 translocation in hepatocytes. HFD+etOH: High-fat diet plus ethyl alcohol; HMGB1: High mobility group box-1.
ARTICLE HIGHLIGHTS
Research background
High mobility group box-1 (HMGB1) released by injured/dying cells becomes a key of damage-associated molecular patterns molecule to trigger inflammation responses leading to a series of tissue and organ damage, even diseases. But the details on how the injured hepatocytes released HMGB1 are to be elicited.
Research motivation
Hepatocytes are the most abundant cell type in the liver and the most easily attacked target, and loss of hepatocytes would contribute to various hepatic diseases. So, it is important to investigate the mechanism of HMGB1 release during hepatocyte injury or death.
Research objectives
To elicit HMGB1 release regulated by Sirt1 in injured/ dying hepatocytes, male C57BL/6J mice fed a high-fat diet (HFD) plus ethyl alcohol (etOH) were employed to assess the HMGB1 release and translocation from the nuclues to cytoplasm in injured hepatocytes. In vitro, mouse embryonic hepatocyte cell line BNL.CL2 incubated with H2O2 was further investigate the regulatory effect of Sirt1 on HMGB1 release. The main aim ofthis study was to understand HMGB1 release and its regulatory factors during hepatocyte injury/death, which would be a potential target for clinical therapy.
Research methods
Male C57BL/6J mice were fed an HFD plus etOH to establish a model of hepatocytes injury/death. Meanwhile, mouse embryonic hepatocyte cell line BNL.CL2 was cultured in vitro and treated with H2O2. Serum ALT, liver H2O2 content and catalase activity, lactate dehydrogenase and 8-hydroxy-2-deoxyguanosine content, NAD+ levels, Sirt1 activity were detected by spectrophotometry. HMGB1 release was measured by ELISA and its translocation was detected by immunohistochemistry/immunofluorescence or Western blot. The mRNA and protein levels were assayed by qPCR and Western blot, respectively. Acetylated HMGB1 and Parp1 were analyzed by immunoprecipitation.
Research results
After treatment with an HFD plus etOH in vivo or H2O2 in vitro toinduce hepatocyte injury, HMGB1 was translocated from the nucleus to the cytoplasm and passively released outside. This process occured because HMGB1 was hyperacetylated owing to both Sirt1 protein and activity suppression. After treatment with Sirt1-siRNA or Sirt1 inhibitor EX527, hyperacetylated HMGB1 increased (P < 0.01). Further, Sirt1 activity was suppressed by H2O2, which could be reversed by the Parp1 inhibitor DIQ (P < 0.01).
Research conclusions
When hepatocytes injured/die, HMGB1 is translocated from the nucleus to the cytoplasm and finally released, which is related with hyperacetylated HMGB1 due to Sirt1 activity inhibition by NAD+ depletion caused by Parp1 overactivation.
Research perspectives
The future research will focus on herbs affecting this process to investigate the recovery of injured hepatocyte.
Footnotes
Institutional review board statement: All procedures involving animals were approved by the Animal Ethics Committee of Shanghai University of Traditional Chinese Medicine.
Institutional animal care and use committee statement: All procedures involving animals were approved by the Animal Ethics Committee of Shanghai University of Traditional Chinese Medicine. (protocol number: PZSHUTCM190419001).
Conflict-of-interest statement: The authors declared that they have no conflict of interest.
ARRIVE guidelines statement: The authors have read the ARRIVE guidelines, and the manuscript was prepared and revised according to the ARRIVE guidelines.
Manuscript source: Unsolicited manuscript
Peer-review started: April 26, 2019
First decision: May 24, 2019
Article in press: August 19, 2019
Specialty type: Gastroenterology and hepatology
Country of origin: China
Peer-review report classification
Grade A (Excellent): 0
Grade B (Very good): 0
Grade C (Good): C, C
Grade D (Fair): D
Grade E (Poor): 0
P-Reviewer: Demonacos C, Karamouzis MV, Konishi H S-Editor: Yan JP L-Editor: Wang TQE-Editor: Qi LL
Contributor Information
Ting-Jie Ye, Department of Biology, School of Basic Medical Sciences, Shanghai University of Traditional Chinese Medicine, Shanghai 201203, China.
Yan-Lin Lu, Department of Biology, School of Basic Medical Sciences, Shanghai University of Traditional Chinese Medicine, Shanghai 201203, China; Department of Oncology and Institute of Traditional Chinese Medicine in Oncology, Longhua Hospital, Shanghai University of Traditional Chinese Medicine, Shanghai 200032, China.
Xiao-Feng Yan, Department of Biology, School of Basic Medical Sciences, Shanghai University of Traditional Chinese Medicine, Shanghai 201203, China.
Xu-Dong Hu, Department of Biology, School of Basic Medical Sciences, Shanghai University of Traditional Chinese Medicine, Shanghai 201203, China.
Xiao-Ling Wang, Department of Biology, School of Basic Medical Sciences, Shanghai University of Traditional Chinese Medicine, Shanghai 201203, China. shengwuwang12@126.com.
References
- 1.Malarkey CS, Churchill ME. The high mobility group box: The ultimate utility player of a cell. Trends Biochem Sci. 2012;37:553–562. doi: 10.1016/j.tibs.2012.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bertheloot D, Latz E. HMGB1, IL-1α, IL-33 and S100 proteins: Dual-function alarmins. Cell Mol Immunol. 2017;14:43–64. doi: 10.1038/cmi.2016.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zamora R, Barclay D, Yin J, Alonso EM, Leonis MA, Mi Q, Billiar TR, Simmons RL, Squires RH, Vodovotz Y. HMGB1 is a Central Driver of Dynamic Pro-inflammatory Networks in Pediatric Acute Liver Failure induced by Acetaminophen. Sci Rep. 2019;9:5971. doi: 10.1038/s41598-019-42564-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huebener P, Pradere JP, Hernandez C, Gwak GY, Caviglia JM, Mu X, Loike JD, Jenkins RE, Antoine DJ, Schwabe RF. The HMGB1/RAGE axis triggers neutrophil-mediated injury amplification following necrosis. J Clin Invest. 2019;130:1802. doi: 10.1172/JCI126975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bonaldi T, Talamo F, Scaffidi P, Ferrera D, Porto A, Bachi A, Rubartelli A, Agresti A, Bianchi ME. Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. EMBO J. 2003;22:5551–5560. doi: 10.1093/emboj/cdg516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rabadi MM, Xavier S, Vasko R, Kaur K, Goligorksy MS, Ratliff BB. High-mobility group box 1 is a novel deacetylation target of Sirtuin1. Kidney Int. 2015;87:95–108. doi: 10.1038/ki.2014.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Luna A, Aladjem MI, Kohn KW. SIRT1/PARP1 crosstalk: Connecting DNA damage and metabolism. Genome Integr. 2013;4:6. doi: 10.1186/2041-9414-4-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Walko TD, 3rd, Di Caro V, Piganelli J, Billiar TR, Clark RS, Aneja RK. Poly(ADP-ribose) polymerase 1-sirtuin 1 functional interplay regulates LPS-mediated high mobility group box 1 secretion. Mol Med. 2015;20:612–624. doi: 10.2119/molmed.2014.00156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pillai JB, Isbatan A, Imai S, Gupta MP. Poly(ADP-ribose) polymerase-1-dependent cardiac myocyte cell death during heart failure is mediated by NAD+ depletion and reduced Sir2alpha deacetylase activity. J Biol Chem. 2005;280:43121–43130. doi: 10.1074/jbc.M506162200. [DOI] [PubMed] [Google Scholar]
- 10.Wang C, Zhang F, Wang L, Zhang Y, Li X, Huang K, Du M, Liu F, Huang S, Guan Y, Huang D, Huang K. Poly(ADP-ribose) polymerase 1 promotes oxidative-stress-induced liver cell death via suppressing farnesoid X receptor α. Mol Cell Biol. 2013;33:4492–4503. doi: 10.1128/MCB.00160-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang H, Nace GW, McDonald KA, Tai S, Klune JR, Rosborough BR, Ding Q, Loughran P, Zhu X, Beer-Stolz D, Chang EB, Billiar T, Tsung A. Hepatocyte-specific high-mobility group box 1 deletion worsens the injury in liver ischemia/reperfusion: A role for intracellular high-mobility group box 1 in cellular protection. Hepatology. 2014;59:1984–1997. doi: 10.1002/hep.26976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jang J, Huh YJ, Cho HJ, Lee B, Park J, Hwang DY, Kim DW. SIRT1 Enhances the Survival of Human Embryonic Stem Cells by Promoting DNA Repair. Stem Cell Reports. 2017;9:629–641. doi: 10.1016/j.stemcr.2017.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xie J, Zhou X, Hu X, Jiang H. H2O2 evokes injury of cardiomyocytes through upregulating HMGB1. Hellenic J Cardiol. 2014;55:101–106. [PubMed] [Google Scholar]
- 14.Vazquez EJ, Berthiaume JM, Kamath V, Achike O, Buchanan E, Montano MM, Chandler MP, Miyagi M, Rosca MG. Mitochondrial complex I defect and increased fatty acid oxidation enhance protein lysine acetylation in the diabetic heart. Cardiovasc Res. 2015;107:453–465. doi: 10.1093/cvr/cvv183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zeng T, Guo FF, Zhang CL, Zhao S, Dou DD, Gao XC, Xie KQ. The anti-fatty liver effects of garlic oil on acute ethanol-exposed mice. Chem Biol Interact. 2008;176:234–242. doi: 10.1016/j.cbi.2008.07.004. [DOI] [PubMed] [Google Scholar]
- 16.Dąbrowska N, Wiczkowski A. Analytics of oxidative stress markers in the early diagnosis of oxygen DNA damage. Adv Clin Exp Med. 2017;26:155–166. doi: 10.17219/acem/43272. [DOI] [PubMed] [Google Scholar]
- 17.Samuni Y, Goldstein S, Dean OM, Berk M. The chemistry and biological activities of N-acetylcysteine. Biochim Biophys Acta. 2013;1830:4117–4129. doi: 10.1016/j.bbagen.2013.04.016. [DOI] [PubMed] [Google Scholar]
- 18.Ding J, Cui X, Liu Q. Emerging role of HMGB1 in lung diseases: Friend or foe. J Cell Mol Med. 2017;21:1046–1057. doi: 10.1111/jcmm.13048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gao E, Jiang Y, Li Z, Xue D, Zhang W. Association between high mobility group box‑1 protein expression and cell death in acute pancreatitis. Mol Med Rep. 2017;15:4021–4026. doi: 10.3892/mmr.2017.6496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang H, Antoine DJ, Andersson U, Tracey KJ. The many faces of HMGB1: Molecular structure-functional activity in inflammation, apoptosis, and chemotaxis. J Leukoc Biol. 2013;93:865–873. doi: 10.1189/jlb.1212662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tsung A, Klune JR, Zhang X, Jeyabalan G, Cao Z, Peng X, Stolz DB, Geller DA, Rosengart MR, Billiar TR. HMGB1 release induced by liver ischemia involves Toll-like receptor 4 dependent reactive oxygen species production and calcium-mediated signaling. J Exp Med. 2007;204:2913–2923. doi: 10.1084/jem.20070247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim DI, Park MJ, Choi JH, Kim IS, Han HJ, Yoon KC, Park SW, Lee MY, Oh KS, Park SH. PRMT1 and PRMT4 Regulate Oxidative Stress-Induced Retinal Pigment Epithelial Cell Damage in SIRT1-Dependent and SIRT1-Independent Manners. Oxid Med Cell Longev. 2015;2015:617919. doi: 10.1155/2015/617919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lou PH, Lucchinetti E, Scott KY, Huang Y, Gandhi M, Hersberger M, Clanachan AS, Lemieux H, Zaugg M. Alterations in fatty acid metabolism and sirtuin signaling characterize early type-2 diabetic hearts of fructose-fed rats. Physiol Rep. 2017;5:pii: e13388. doi: 10.14814/phy2.13388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Warner DR, Liu H, Miller ME, Ramsden CE, Gao B, Feldstein AE, Schuster S, McClain CJ, Kirpich IA. Dietary Linoleic Acid and Its Oxidized Metabolites Exacerbate Liver Injury Caused by Ethanol via Induction of Hepatic Proinflammatory Response in Mice. Am J Pathol. 2017;187:2232–2245. doi: 10.1016/j.ajpath.2017.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Geng C, Xu H, Zhang Y, Gao Y, Li M, Liu X, Gao M, Wang X, Liu X, Fang F, Chang Y. Retinoic acid ameliorates high-fat diet-induced liver steatosis through sirt1. Sci China Life Sci. 2017;60:1234–1241. doi: 10.1007/s11427-016-9027-6. [DOI] [PubMed] [Google Scholar]
- 26.Chang B, Xu MJ, Zhou Z, Cai Y, Li M, Wang W, Feng D, Bertola A, Wang H, Kunos G, Gao B. Short- or long-term high-fat diet feeding plus acute ethanol binge synergistically induce acute liver injury in mice: An important role for CXCL1. Hepatology. 2015;62:1070–1085. doi: 10.1002/hep.27921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang M, Antoine DJ, Weemhoff JL, Jenkins RE, Farhood A, Park BK, Jaeschke H. Biomarkers distinguish apoptotic and necrotic cell death during hepatic ischemia/reperfusion injury in mice. Liver Transpl. 2014;20:1372–1382. doi: 10.1002/lt.23958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yu Y, Tang D, Kang R. Oxidative stress-mediated HMGB1 biology. Front Physiol. 2015;6:93. doi: 10.3389/fphys.2015.00093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhao P, Ye T, Yan X, Hu X, Liu P, Wang X. HMGB1 release by H2O2-induced hepatocytes is regulated through calcium overload and 58-F interference. Cell Death Discov. 2017;3:17008. doi: 10.1038/cddiscovery.2017.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ding RB, Bao J, Deng CX. Emerging roles of SIRT1 in fatty liver diseases. Int J Biol Sci. 2017;13:852–867. doi: 10.7150/ijbs.19370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pittelli M, Formentini L, Faraco G, Lapucci A, Rapizzi E, Cialdai F, Romano G, Moneti G, Moroni F, Chiarugi A. Inhibition of nicotinamide phosphoribosyltransferase: Cellular bioenergetics reveals a mitochondrial insensitive NAD pool. J Biol Chem. 2010;285:34106–34114. doi: 10.1074/jbc.M110.136739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Robu M, Shah RG, Purohit NK, Zhou P, Naegeli H, Shah GM. Poly(ADP-ribose) polymerase 1 escorts XPC to UV-induced DNA lesions during nucleotide excision repair. Proc Natl Acad Sci U S A. 2017;114:E6847–E6856. doi: 10.1073/pnas.1706981114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hanzlikova H, Kalasova I, Demin AA, Pennicott LE, Cihlarova Z, Caldecott KW. The Importance of Poly(ADP-Ribose) Polymerase as a Sensor of Unligated Okazaki Fragments during DNA Replication. Mol Cell. 2018;71:319–331.e3. doi: 10.1016/j.molcel.2018.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rajamohan SB, Pillai VB, Gupta M, Sundaresan NR, Birukov KG, Samant S, Hottiger MO, Gupta MP. SIRT1 promotes cell survival under stress by deacetylation-dependent deactivation of poly(ADP-ribose) polymerase 1. Mol Cell Biol. 2009;29:4116–4129. doi: 10.1128/MCB.00121-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cantó C, Sauve AA, Bai P. Crosstalk between poly(ADP-ribose) polymerase and sirtuin enzymes. Mol Aspects Med. 2013;34:1168–1201. doi: 10.1016/j.mam.2013.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]