

ABSTRACT
Activating mutations in the RAS family of proto-oncogenes represent some of the leading causes of cancer. Unmitigated proliferation of cells harboring oncogenic RAS mutations is accompanied by a massive increase in cellular bioenergetic demands, which offers unique opportunities for therapeutic intervention. To withstand the steep requirements for metabolic intermediates, RAS-driven cancer cells enhance endolysosome and autophagosome biogenesis. By degrading cellular macromolecules into metabolites that can be used by biosynthetic pathways, endolysosomes permit continued proliferation and survival in otherwise detrimental conditions. We recently showed that human cancers with activating mutations in HRAS elevate the expression of MCOLN1, which encodes an endolysosomal cation channel called TRPML1. Increased TRPML1 activity in HRAS-driven cancer cells is needed for the restoration of plasma membrane cholesterol that gets collaterally internalized during endocytosis. Inhibition of TRPML1 or knockdown of MCOLN1 leads to mislocalization of cholesterol from the plasma membrane to endolysosomes, loss of oncogenic HRAS from the cell surface, and attenuation of downstream signaling. Here, we discuss the implications of our findings and suggest strategies to leverage pathways that impinge upon TRPML1 as novel anti-cancer treatments.
KEYWORDS: Endolysosomes, TRPML1, lysosomes, mucolipins, HRAS, RAS, cancer, cholesterol, TFEB
Introduction
KRAS, HRAS, and NRAS are small GTPases encoded by an evolutionarily conserved family of proto-oncogenes [1,2]. These fascinating proteins operate at the nexus of growth factor receptors and mitogen-activated protein kinase (MAPK) cascades, and are responsible for the faithful transmission of signals between the two [1–3]. Over 25% of human tumors harbor “oncogenic” (i.e. activating) mutations in RAS genes [4–9], which makes their protein products some of the most important therapeutic targets in cancer [10–16]. In healthy cells, cell surface receptors are functionally coupled to RAS proteins, which incite phosphorylation cascades involving RAF–MEK–ERK or phosphatidylinositol-3-kinase (PI3K) [1–3]. Phosphorylated ERK, the activated form of a terminal MAPK, translocates to the nucleus where it induces the expression of growth-related genes [1–3,14]. Oncogenic mutations usually abolish the intrinsic GTPase activity of RAS and lock the proteins in GTP-bound constitutively active state. As a result, ERK phosphorylation and attendant cell proliferation are dramatically higher in cells harboring these mutations [4–6,11].
Despite more than three decades of concerted effort, effective anti-RAS therapies have remained elusive. The paucity of success has even prompted the notion that RAS proteins might be “undruggable” [12,13,15]. Although this idea is now being challenged by new classes of drugs [17], a traditional approach to sidestep the need to target RAS directly focused on inhibition of downstream kinases [11,13,16,18,19]. Unfortunately, these strategies were ultimately ineffective due to intractable feedback loops and the propensity for acquired resistance [18,20]. A good example is the pharmacological inhibition of BRAF, which induces paradoxical activation of RAS–ERK signaling and undesirable potentiation of cell proliferation [18,20]. Another cogent strategy to tune-down the proliferative effects of mutant RAS relies on the identification of orthogonal cellular pathways that make hyperactive RAS–ERK signaling possible. Genomic, proteomic, and other modern analytical techniques have led to the identification of pathways that are potentiated in cancers. In this arena, lysosomal proteins are emerging as an attractive group that can be targeted to mitigate tumorigenesis [21–30]. Owing to their roles in cellular metabolism, intracellular trafficking, and macromolecule recycling, lysosomes sustain hallmarks of cancer – abnormal proliferation, drug resistance, metastasis, and angiogenesis [28–34]. Based on this understanding, it has been asserted that the disruption of endolysosomal function can retard the growth of certain malignancies.
Dysregulated lysosomal biogenesis in cancer cells
Cancer cell proliferation, which requires sustained biosynthesis of a variety of macromolecules, imposes a massive requirement for nutrients. To cope with steep metabolic demands, cancer cells generate copious numbers of autophagosomes and endolysosomes, which break down cellular macromolecules to intermediates that are shunted toward growth [21,23]. Leveraging a transcriptional pathway necessary for autophagy and endolysosomal biogenesis [35–38], cancer cells upregulate several endolysosomal proteins and enzymes en masse [21,23]. The bHLH transcription factors – transcription factor EB (TFEB), transcription factor binding to IGHM enhancer 3 (TFE3), and melanocyte inducing transcription factor (MiTF) – drive the expression of many genes that encode endolysosomal proteins [38–42]. Not surprisingly, the activities of these transcription factors are coupled to nutrient availability. For instance, under nutrient-rich conditions, TFEB is phosphorylated by the mechanistic target of rapamycin complex 1 (mTORC1) and sequestered in the cytoplasm by 14-3-3 protein [39,42,43] (Figure 1). When nutrient levels drop, mTORC1 activity declines [44]. Consequently, TFEB is dephosphorylated and translocates to the nucleus, where it activates the CLEAR (coordinated lysosomal expression and regulation) gene network leading to enhanced lysosomal biogenesis [35,39,40,42] (Figure 1). Along with the emergence of the understanding that lysosomal biogenesis is a potential therapeutic avenue, elevated TFEB, MiTF, and TFE3 activities have been described in various malignancies [23,27,29,41,45,46]. In KRAS-driven pancreatic ductal adenocarcinoma (PDAC), TFEB is decoupled from mTORC1, and constitutively translocates to nuclei where it compels endolysosomal biogenesis [23,47]. In other instances, cancer-related mutations affect the expression of these transcription factors. For instance, genomic translocations in renal cell carcinoma and soft tissue sarcoma lead to TFEB and TFE3 overexpression, whereas MiTF expression is increased in melanoma and hepatocellular carcinomas [41,48–52].
Figure 1.
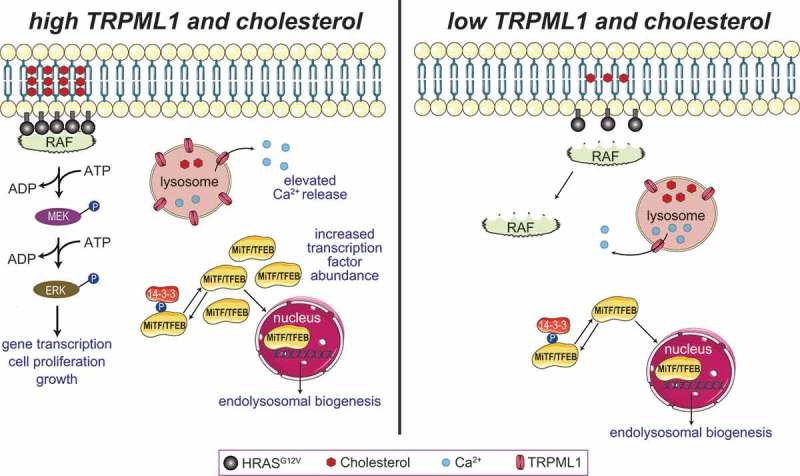
Role of TRPML1 in the regulation of ERK signaling in cells harboring HRASG12V mutations. HRAS–ERK signaling involves the assembly of HRAS monomers into nanodomains that can recruit RAF, and promote MEK and ERK phosphorylation (left). HRASG12V nanoclusters formation and plasma membrane localization are predicated upon the availability of cholesterol. When plasma membrane cholesterol is depleted or lowered (right), HRASG12V monomers cannot recruit RAF, leading to diminished ERK phosphorylation. Cancer cells harboring oncogenic HRAS showed an increased level of MiTF/TFE transcriptional factors and activation of the CLEAR gene network that leads to the lysosomal biogenesis (left). The elevated level of TRPML1 results in increased lysosomal Ca2+ release, HRASG12V nanoclusters in the plasma membrane, activation of MAPK pathway, and cell proliferation.
When thinking about translating these insights into feasible therapies, we are faced with numerous challenges. In addition to mediating endolysosomal biogenesis, TFEB, MiTF, and TFE3 are also involved in biological processes that are (at least ostensibly) unrelated to endolysosomal function. For example, MiTF is required for the specification of melanocytes [52,53]. Furthermore, some cancer cells simultaneously upregulate two or more of these proteins [29]. In this situation, inhibition of one transcription factor might not be sufficient for adequate attenuation of endolysosomal biogenesis. To make matters worse, many genes that encode non-lysosomal proteins also possess the CLEAR motif and are likely regulated by TFEB, TFE3, and MiTF. This makes the onset of unintended consequences a distinct possibility in cells treated with putative inhibitors of these transcription factors. The CLEAR motif that is bound by TFEB, TFE3, and MiTF is identical to the canonical E-box that is the target of many different bHLH transcription factors including Myc and Max [36,54,55]. Thus, Myc/Max and other bHLH transcription factors likely influence the effects of TFEB, MiTF and TFE3 to drive endolysosomal biogenesis in certain cancers. Supporting this idea, a recent study showed that Myc competes with TFEB and TFE3 for binding to CLEAR elements and thus negatively influences lysosomal biogenesis [56]. To overcome these limitations, we reasoned that targeting endolysosomal proteins that play tumor-specific roles might be a better strategy to attenuate tumor growth. The question we are faced with now is – which endolysosomal proteins should we go after?
Involvement of a lysosomal Ca2+ channel, TRPML1, in the proliferation of cells harboring oncogenic HRAS mutations
We sought to identify endolysosomal proteins that could be targeted to mitigate the growth of cancer cells. Using a genomic approach and oncogenic HRAS-driven cancer cells as models, we identified the endolysosomal Ca2+ permeable channel, TRPML1, as a potential target in cells harboring oncogenic HRAS [29]. Using datasets available at the cancer genome atlas (TCGA), we found that the gene encoding the endolysosomal cation channel, TRPML1 (MCOLN1), was upregulated in oncogenic HRAS-expressing tumors due to the combined actions of MiTF and TFEB. Accompanying the increase in MCOLN1 expression, TRPML1-mediated endolysosomal Ca2+ release was dramatically higher in HRAS-driven cancer cells compared to controls (Figure 1). Importantly, elevated expression of MCOLN1 in patients with HRAS-positive tumors correlated with poorer prognosis [29]. We went on to show that genetic or pharmacological inhibition of TRPML1 reduced the proliferation of many different oncogenic HRAS-expressing cancer cell lines via attenuation of the MEK–ERK pathway (Figure 1). TRPML1 activity depends on the vesicular phosphoinositide, PI(3,5)P2, which is synthesized by the PIK-FYVE (FYVE-containing phosphoinositide kinase) lipid kinase complex containing Fab1 and Vac14 [57,58]. Pharmacological inhibition of PIK-FYVE or knockdown of VAC14 also selectively inhibited the proliferation of mutant HRAS-driven cancer cells [29]. Interestingly, MCOLN1 knockdown or TRPML1 inhibition did not affect ERK phosphorylation and cell proliferation in cancer cells expressing wild-type HRAS, or in cells in which oncogenic HRAS was stably knocked down [29]. These data indicate that inhibition of TRPML1 imparts selective vulnerability to cells expressing oncogenic HRAS while leaving cells with normal HRAS unaffected. Thus, TRPML1 appears generally dispensable for regulating the gain of MAPK signaling, except in the context of HRAS-driven cancer cells that are marked by the profound upregulation of ERK phosphorylation. If these phenotypes were to be reproduced in the clinic, one could speculate selective attenuation of cancer cell proliferation with diminished side effects associated with perturbation of otherwise healthy cells.
The utility of blocking TRPML1 was also observed in vertebrate xenograft models and Drosophila lacking the TRPML1 homolog [29,59]. These findings paint a picture of an evolutionarily conserved pathway of endolysosomal Ca2+ release that is required for elevated RAS activity. Attenuation of cell proliferation and RAS signaling in TRPML-deficient Drosophila expressing Drosophila RasG12V or human HRASG12V also demonstrate a fundamental requirement for the vesicular channels that extends beyond the genetic “ecosystem” of cancer cells [29]. This was important from a conceptual standpoint since cancer cells carry hundreds of mutations that synergistically drive proliferation and survival [60].
How does an endolysosomal cation channel regulate HRAS–ERK signaling in cancer cells? Localization and clustering of HRAS at the plasma membrane, which is dependent on cholesterol levels, is required for the engagement of downstream effectors (Figure 1) [2,3,61–64]. Given the involvement of TRPML1 in vesicle exocytosis (Figure 2), MCOLN1 knockdown or TRPML1 inhibition diminished the movement of cholesterol from endolysosomal vesicles to the plasma membrane [29,65–69]. Accompanying these defects in cholesterol recycling, inhibition of TRPML1 also prevented the de-esterification of endocytosed cholesterol esters (Figure 2) [29]. Consequently, levels of plasma membrane cholesterol fell to an extent that was sufficient to disrupt HRASG12V nanocluster formation and plasma membrane abundance, and thereby, attenuate ERK phosphorylation and cell proliferation (Figure 1). Nanoclusters of wild-type HRAS were also perturbed by TRPML1 inhibition, but the abundance of this variant in the plasma membrane was not altered. These data explain the heightened sensitivity of HRASG12V to TRPML1 inhibition. Further supporting the involvement of cholesterol, lowering cholesterol levels by application of statins phenocopied TRPML1 inhibition, whereas supplementation of cholesterol prevented the effects of TRPML1 inhibition [29].
Figure 2.
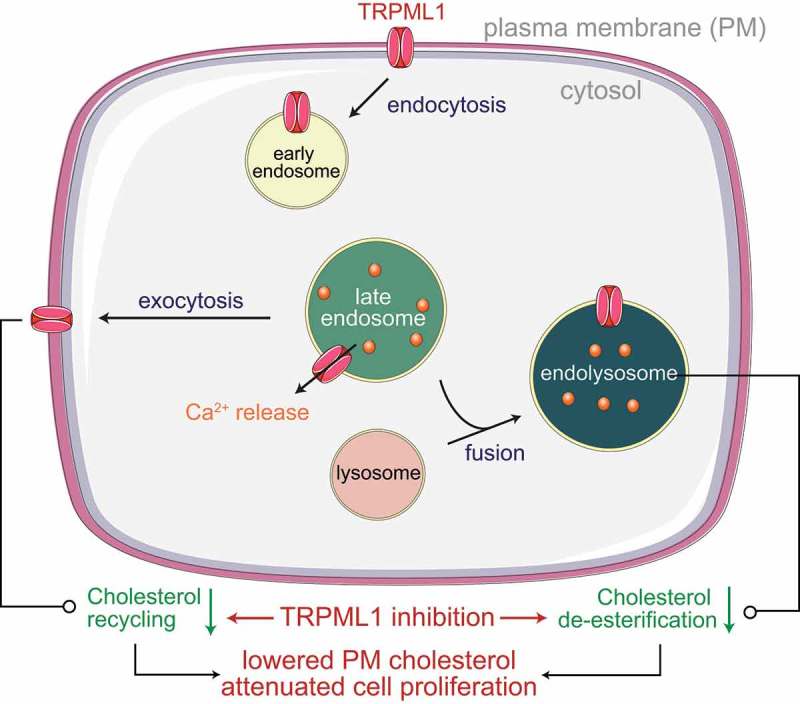
TRPML1 is necessary for vesicular trafficking and fusion of endolysosomes with lysosomes or the plasma membrane (PM). By mediating endolysosomal exocytosis, TRPML1 coordinates the restoration of PM lipids such as cholesterol that get collaterally internalized during endocytosis. Delivery of endocytosed material to lysosomes is needed for de-esterification of cholesterol esters. Defects in de-esterification and recycling lead to lowered PM cholesterol, which explains the attenuation of HRASG12V-driven ERK signaling.
Conclusions and future perspectives
Our studies raise the intriguing possibility that targeting TRPML1 function might be a viable therapeutic option for HRAS-driven cancers. The notion of inhibiting TRPML1 to attenuate tumor growth agrees with findings of MCOLN1 upregulation and a requirement for TRPML1 in proliferation and metastases of triple-negative breast cancer cells, and the survival/proliferation of melanoma cells [28,30]. Taken together, these three studies highlight some of the complexities of the relationship between TRPML1 and cancer. Similar to our findings in HRAS-driven cancer cells, Xu et al. demonstrate that TRPML1 promotes the development of triple-negative breast cancer via lysosomal exocytosis and the attendant release of ATP [25]. TRPML1 also positively regulates mTORC1 activity, and this function of the channel further promotes the breast cancer cell proliferation [25]. In a departure from this theme, TRPML1 negatively regulates both ERK phosphorylation and mTORC1 activity in melanoma [28]. Rather, the utility of blocking TRPML1 in melanoma stems from a role for the channel in potentiation of micropinocytosis [28]. As suggested by Kasitinon et al. [28], differences in the purported mechanisms by which TRPML1 promotes tumorigenesis likely reflects the presence of distinct driver mutations in the different types of cancer. If so, the qualitative similarities of the effects of TRPML1 inhibition in all three cancers are even more remarkable. The idea that endolysosomal Ca2+ release promotes tumorigenesis likely extends to other modes of cation release from these organelles [70,71]. For instance, Ca2+ permeable, nonselective endolysosomal two-pore cation channels were found to play an important role in cancer cell migration via β1-integrin trafficking and recycling [72]. Additionally, the TRPML1 paralog, TRPML2, has been shown to be involved in the onset of glioma, and inhibitors of PI(3,5)P2 biosynthesis are effective in the attenuation of cell proliferation in hepatic and hematological malignancies [73–75]. Together, these findings promise an era of cancer therapeutics that revolve around endolysosomal ionic homeostasis.
Despite our excitement, additional studies are going to be needed before we head to the clinic. Loss-of-function mutations in MCOLN1 are responsible for a severe childhood-onset lysosomal storage disease called mucolipidosis type IV (MLIV) [76–78]. Thus, the onset of potentially severe neurological dysfunction could deter the administration of TRPML1 inhibitors to humans. That being said, neurological deficits are unlikely to be a major barrier if the drug being administered is unable to inhibit the channel in the nervous system. One could consider using antisense oligonucleotides (ASOs) to diminish MCOLN1 expression in cancers. Given that ASOs are inherently unable to cross the blood–brain barrier [79], these drugs bear the potential to attenuate endolysosomal Ca2+ release in non-neuronal tumors while leaving MCOLN1 expression unchanged in the CNS. These and other possibilities ensure our ongoing enthusiasm regarding the development of new anti-cancer strategies that leverage the function of endolysosomal ion channels.
Funding Statement
Research in the K.V. laboratory is funded by NIH grants, R01NS08130 and R21AG061646. J.J. is supported by a grant from the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the ministry of education (NRF-2016R1D1A1B03932051).
Acknowledgments
We are grateful to the RASopathies network for prior support of our research.
Disclosure statement
No potential conflict of interest was reported by the authors.
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
References
- [1].Macaluso M, Russo G, Cinti C, et al. Ras family genes: an interesting link between cell cycle and cancer. J Cell Physiol. 2002;192:125–130. [DOI] [PubMed] [Google Scholar]
- [2].Hancock JF. Ras proteins: different signals from different locations. Nat Rev Mol Cell Biol. 2003;4:373–385. [DOI] [PubMed] [Google Scholar]
- [3].Plowman SJ, Muncke C, Parton RG, et al. H-ras, K-ras, and inner plasma membrane raft proteins operate in nanoclusters with differential dependence on the actin cytoskeleton. Proc Natl Acad Sci. 2005;102:15500–15505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Solus JF, Kraft S. Ras, Raf, and MAP kinase in melanoma. Adv Anat Pathol. 2013;20:217–226. [DOI] [PubMed] [Google Scholar]
- [5].Simanshu DK, Nissley DV, McCormick F. RAS proteins and their regulators in human disease. Cell. 2017;170:17–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].OXFORD G, THEODORESCU D. Review article: the role of Ras superfamily proteins in bladder cancer progression. J Urol. 2003;170:1987–1993. [DOI] [PubMed] [Google Scholar]
- [7].Satoshi S, Yoshino H, Miyamoto K, et al. Targeting HRAS as a potential therapeutic target through RAS inhibitor salirasib in bladder cancer. Eur Urol Suppl. 2018;17:e655. [DOI] [PubMed] [Google Scholar]
- [8].Theodorescu D, Cornil I, Fernandez BJ, et al. Overexpression of normal and mutated forms of HRAS induces orthotopic bladder invasion in a human transitional cell carcinoma. Proc Natl Acad Sci U S A. 1990;87:9047–9051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Prior IA, Lewis PD, Mattos C. A comprehensive survey of ras mutations in cancer. Cancer Res. 2012;72:2457–2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Lasithiotakis KG, Sinnberg TW, Schittek B, et al. Combined Inhibition of MAPK and mTOR signaling inhibits growth, induces cell death, and abrogates invasive growth of melanoma cells. J Invest Dermatol. 2008;128:2013–2023. [DOI] [PubMed] [Google Scholar]
- [11].Wang A-X, Qi X-Y. Targeting RAS/RAF/MEK/ERK signaling in metastatic melanoma. IUBMB Life. 2013;65:748–758. [DOI] [PubMed] [Google Scholar]
- [12].Cox AD, Fesik SW, Kimmelman AC, et al. Drugging the undruggable RAS: mission possible? Nat Rev Drug Discov. 2014;13:828–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Scott AJ, Lieu CH, Messersmith WA. Therapeutic approaches to RAS mutation. Cancer J (United States). 2016;22:165–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].McCubrey JA, Steelman LS, Chappell WH, et al. Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR Cascade inhibitors: how mutations can result in therapy resistance and how to overcome resistance. Oncotarget. 2012;3:1068–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Papke B, Der CJ. Drugging RAS: know the enemy. Science. 2017;355:1158–1163. [DOI] [PubMed] [Google Scholar]
- [16].Ryan MB, Der CJ, Wang-Gillam A, et al. Targeting RAS -mutant cancers: is ERK the key? Trends Cancer. 2015;1:183–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Ostrem JML, Shokat KM. Direct small-molecule inhibitors of KRAS: from structural insights to mechanism-based design. Nat Rev Drug Discov. 2016;15:771–785. [DOI] [PubMed] [Google Scholar]
- [18].Cho K, Kasai RS, Park J-H, et al. Raf inhibitors target Ras spatiotemporal dynamics. Curr Biol. 2012;22:945–955. [DOI] [PubMed] [Google Scholar]
- [19].Kidger AM, Sipthorp J, Cook SJ. ERK1/2 inhibitors: new weapons to inhibit the RAS-regulated RAF-MEK1/2-ERK1/2 pathway. Pharmacol Ther. 2018;187:45–60. [DOI] [PubMed] [Google Scholar]
- [20].Flaherty KT, Puzanov I, Kim KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363:809–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Commisso C, Davidson SM, Soydaner-Azeloglu RG, et al. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature. 2013;497:633–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Cho K, van der Hoeven D, Zhou Y, et al. Inhibition of acid sphingomyelinase depletes cellular phosphatidylserine and mislocalizes K-Ras from the plasma membrane. Mol Cell Biol. 2015;36:MCB.00719–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Perera RM, Stoykova S, Nicolay BN, et al. Transcriptional control of autophagy-lysosome function drives pancreatic cancer metabolism. Nature. 2015;524:361–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Petersen NHT, Olsen OD, Groth-Pedersen L, et al. Transformation-associated changes in sphingolipid metabolism sensitize cells to lysosomal cell death induced by inhibitors of acid sphingomyelinase. Cancer Cell. 2013;24:379–393. [DOI] [PubMed] [Google Scholar]
- [25].Mancias JD, Kimmelman AC. Targeting autophagy addiction in cancer. Oncotarget. 2011;2:1302–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Morgan MJ, Gamez G, Menke C, et al. Regulation of autophagy and chloroquine sensitivity by oncogenic RAS in vitro is context-dependent. Autophagy. 2014;10:1814–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Urbanelli L, Magini A, Ercolani L, et al. Oncogenic H-Ras up-regulates acid β-hexosaminidase by a mechanism dependent on the autophagy regulator TFEB. PLoS One. 2014;9:e89485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Kasitinon SY, Eskiocak U, Martin M, et al. TRPML1 promotes protein homeostasis in melanoma cells by negatively regulating MAPK and mTORC1 signaling. Cell Rep. 2019;28:2293–2305.e9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Jung J, Cho K, Naji AK, et al. HRAS‐driven cancer cells are vulnerable to TRPML1 inhibition. EMBO Rep. 2019;20:e46685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Xu M, Almasi S, Yang Y, et al. The lysosomal TRPML1 channel regulates triple negative breast cancer development by promoting mTORC1 and purinergic signaling pathways. Cell Calcium. 2019;79:80–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Machado E, White-Gilbertson S, van de Vlekkert D, et al. Regulated lysosomal exocytosis mediates cancer progression. Sci Adv. 2015;1:e1500603–e1500603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Zhitomirsky B, Assaraf YG. Lysosomes as mediators of drug resistance in cancer. Drug Resist Updat. 2016;24:23–33. [DOI] [PubMed] [Google Scholar]
- [33].Davidson SM, Vander Heiden MG. Critical functions of the lysosome in cancer biology. Annu Rev Pharmacol Toxicol. 2016;57:481–507. [DOI] [PubMed] [Google Scholar]
- [34].Piao S, Amaravadi RK. Targeting the lysosome in cancer. Ann N Y Acad Sci. 2016;1371:45–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Palmieri M, Impey S, Kang H, et al. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum Mol Genet. 2011;20:3852–3866. [DOI] [PubMed] [Google Scholar]
- [36].Sardiello M, Palmieri M, Di Ronza A, et al. A gene network regulating lysosomal biogenesis and function. Science. 2009;325:473–477. [DOI] [PubMed] [Google Scholar]
- [37].Settembre C, Fraldi A, Medina DL, et al. Signals from the lysosome: a control centre for cellular clearance and energy metabolism. Nat Rev Mol Cell Biol. 2013;14:283–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Settembre C, Di Malta C, Polito VA, et al. TFEB links autophagy to lysosomal biogenesis. Science. 2011;332:1429–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Martina JA, Chen Y, Gucek M, et al. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy. 2012;8:903–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Roczniak-Ferguson A, Petit CS, Froehlich F, et al. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci Signal. 2012;5:ra42–ra42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Ploper D, Taelman VF, Robert L, et al. MITF drives endolysosomal biogenesis and potentiates Wnt signaling in melanoma cells. Proc Natl Acad Sci. 2015;112:E420–E429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Martina JA, Diab HI, Lishu L, et al. The nutrient-responsive transcription factor TFE3 promotes autophagy, lysosomal biogenesis, and clearance of cellular debris. Sci Signal. 2014;7:ra9–ra9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Settembre C, De Cegli R, Mansueto G, et al. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat Cell Biol. 2013;15:647–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Zoncu R, Bar-Peled L, Efeyan A, et al. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H+-ATPase. Science. 2011;334:678–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Carreira S, Goodall J, Denat L, et al. Mitf regulation of Dia1 controls melanoma proliferation and invasiveness. Genes Dev. 2006;20:3426–3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Blessing AM, Rajapakshe K, Reddy Bollu L, et al. Transcriptional regulation of core autophagy and lysosomal genes by the androgen receptor promotes prostate cancer progression. Autophagy. 2017;13:506–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Perera RM, Bardeesy N. Pancreatic cancer metabolism : breaking it down to build it back up. Cancer Discov. 2016;118:6072–6078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Nooron N, Ohba K, Takeda K, et al. Dysregulated expression of MITF in subsets of hepatocellular carcinoma and cholangiocarcinoma. Tohoku J Exp Med. 2017;242:291–302. [DOI] [PubMed] [Google Scholar]
- [49].Kauffman EC, Ricketts CJ, Rais-Bahrami S, et al. Molecular genetics and cellular features of TFE3 and TFEB fusion kidney cancers. Nat Rev Urol. 2014;11:465–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Ladanyi M, Lui MY, Antonescu CR, et al. The der(17)t(X;17)(p11;q25) of human alveolar soft part sarcoma fuses the TFE3 transcription factor gene to ASPL, a novel gene at 17q25. Oncogene. 2001;20:48–57. [DOI] [PubMed] [Google Scholar]
- [51].Calcagnì A, Kors L, Verschuren E, et al. Modelling TFE renal cell carcinoma in mice reveals a critical role of WNT signaling. Elife. 2016;5:1–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Garraway LA, Widlund HR, Rubin MA, et al. Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature. 2005;436:117–122. [DOI] [PubMed] [Google Scholar]
- [53].Hemesath TJ, Steingrímsson E, McGill G, et al. microphthalmia, a critical factor in melanocyte development, defines a discrete transcription factor family. Genes Dev. 1994;8:2770–2780. [DOI] [PubMed] [Google Scholar]
- [54].Zeller KI, Zhao X, Lee CWH, et al. Global mapping of c-Myc binding sites and target gene networks in human B cells. Proc Natl Acad Sci U S A. 2006;103:17834–17839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Desbarats L, Gaubatz S, Eilers M. Discrimination between different E-box-binding proteins at an endogenous target gene of c-myc. Genes Dev. 1996;10:447–460. [DOI] [PubMed] [Google Scholar]
- [56].Annunziata I, van de Vlekkert D, Wolf E, et al. MYC competes with MiT/TFE in regulating lysosomal biogenesis and autophagy through an epigenetic rheostat. Nat Commun. 2019;10:3623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Zolov SN, Bridges D, Zhang Y, et al. In vivo, Pikfyve generates PI(3,5)P2, which serves as both a signaling lipid and the major precursor for PI5P. Proc Natl Acad Sci U S A. 2012;109:17472–17477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Feng X, Huang Y, Lu Y, et al. Drosophila TRPML forms PI(3,5)P 2 -activated cation channels in both endolysosomes and plasma membrane. J Biol Chem. 2014;289:4262–4272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Venkatachalam K, Long AA, Elsaesser R, et al. Motor deficit in a drosophila model of mucolipidosis type IV due to defective clearance of apoptotic cells. Cell. 2008;135:838–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Chandran UR, Ma C, Dhir R, et al. Gene expression profiles of prostate cancer reveal involvement of multiple molecular pathways in the metastatic process. BMC Cancer. 2007;7:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Sbrissa D, Ikonomov OC, Filios C, et al. Functional dissociation between PIKfyve-synthesized PtdIns5P and PtdIns(3,5)P 2 by means of the PIKfyve inhibitor YM201636. Am J Physiol Physiol. 2012;303:C436–C446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Prior IA, Muncke C, Parton RG, et al. Direct visualization of Ras proteins in spatially distinct cell surface microdomains. J Cell Biol. 2003;160:165–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Prior IA, Harding A, Yan J, et al. GTP-dependent segregation of H-ras from lipid rafts is required for biological activity. Nat Cell Biol. 2001;3:368–375. [DOI] [PubMed] [Google Scholar]
- [64].Hancock JF, Roy S, Luetterforst R, et al. Dominant-negative caveolin inhibits H-Ras function by disrupting cholesterol-rich plasma membrane domains. Nat Cell Biol. 1999;1:98–105. [DOI] [PubMed] [Google Scholar]
- [65].Ravi S, Peña KA, Chu CT, et al. Biphasic regulation of lysosomal exocytosis by oxidative stress. Cell Calcium. 2016;60:356–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Samie M, Wang X, Zhang X, et al. A TRP channel in the lysosome regulates large particle phagocytosis via focal exocytosis. Dev Cell. 2013;26:511–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Dong X, Wang X, Shen D, et al. Activating mutations of the TRPML1 channel revealed by proline-scanning mutagenesis. J Biol Chem. 2009;284:32040–32052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Sahoo N, Gu M, Zhang X, et al. Gastric acid secretion from parietal cells is mediated by a Ca 2+ efflux channel in the tubulovesicle. Dev Cell. 2017;41:262–273.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Cheng X, Zhang X, Gao Q, et al. The intracellular Ca2+ channel MCOLN1 is required for sarcolemma repair to prevent muscular dystrophy. Nat Med. 2014;20:1187–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Grimm C, Bartel K, Vollmar A, et al. Endolysosomal cation channels and cancer—a link with great potential. Pharmaceuticals. 2018;11:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Faris P, Shekha M, Montagna D, et al. Endolysosomal Ca 2+ signalling and cancer hallmarks: two-pore channels on the move, TRPML1 lags behind! Cancers (Basel). 2019;11:1–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Nguyen ONP, Grimm C, Schneider LS, et al. Two-pore channel function is crucial for the migration of invasive cancer cells. Cancer Res. 2017;77:1427–1438. [DOI] [PubMed] [Google Scholar]
- [73].Morelli MB, Nabissi M, Amantini C, et al. Overexpression of transient receptor potential mucolipin-2 ion channels in gliomas: role in tumor growth and progression. Oncotarget. 2016;7:43654–43668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Hou J, Xi Z, Niu J, et al. Inhibition of PIKfyve using YM201636 suppresses the growth of liver cancer via the induction of autophagy. Oncol Rep. 2018;41:1971–1979. [DOI] [PubMed] [Google Scholar]
- [75].Gayle S, Landrette S, Beeharry N, et al. Identification of apilimod as a first-in-class PIKfyve kinase inhibitor for treatment of B-cell non-Hodgkin lymphoma. Blood. 2017;129:1768–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Bassi MT, Manzoni M, Monti E, et al. Cloning of the gene encoding a novel integral membrane protein, mucolipidin-and identification of the two major founder mutations causing mucolipidosis type IV. Am J Hum Genet. 2000;67:1110–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Sun M, Goldin E, Stahl S, et al. Mucolipidosis type IV is caused by mutations in a gene encoding a novel transient receptor potential channel. Hum Mol Genet. 2000;9:2471–2478. [DOI] [PubMed] [Google Scholar]
- [78].Bargal R, Avidan N, Ben-Asher E, et al. Identification of the gene causing mucolipidosis type IV. Nat Genet. 2000;26:118–121. [DOI] [PubMed] [Google Scholar]
- [79].Sareen D, O’Rourke JG, Meera P, et al. Targeting RNA foci in iPSC-derived motor neurons from ALS patients with a C9ORF72 Repeat Expansion. Sci Transl Med. 2013;5:208ra149–208ra149. [DOI] [PMC free article] [PubMed] [Google Scholar]