

Abstract
The influenza A M2 protein is an acid-activated proton channel responsible for the acidification of the inside of the virus, a critical step in the viral life cycle. This channel has four central histidine residues that form an acid-activated gate, binding protons from the outside until an activated state allows proton transport to the inside. While previous work has focused on proton transport through the channel, the structural and dynamic changes that accompany proton flux and enable activation have yet to be resolved. In this study, extensive Multiscale Reactive Molecular Dynamics simulations with explicit Grotthuss-shuttling hydrated excess protons are used to explore detailed molecular-level interactions that accompany proton transport in the +0, +1, and +2 histidine charge states. The results demonstrate how the hydrated excess proton strongly influences both the protein and water hydrogen-bonding network throughout the channel, providing further insight into the channel’s acid-activation mechanism and rectification behavior. We find that the excess proton dynamically, as a function of location, shifts the protein structure away from its equilibrium distributions uniquely for different pH conditions consistent with acid-activation. The proton distribution in the xy-plane is also shown to be asymmetric about the channel’s main axis, which has potentially important implications for the mechanism of proton conduction and future drug design efforts.
Graphical Abstract
Authors are required to submit a graphic entry for the Table of Contents (TOC) that, in conjunction with the manuscript title, should give the reader a representative idea of one of the following: A key structure, reaction, equation, concept, or theorem, etc., that is discussed in the manuscript. Consult the journal’s Instructions for Authors for TOC graphic specifications.
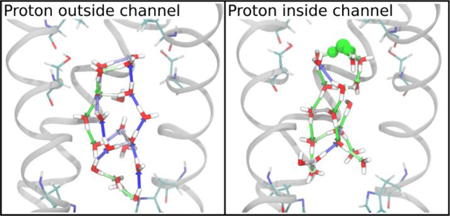
INTRODUCTION
Proton transport (PT) is a ubiquitous process in bio-molecular systems and a functional component of many channels and transporters.1–4 However, accurately modeling an excess proton in large, complex biomolecular systems is difficult due to the quantum-chemical nature of the solvated proton – it does not exist as a hydronium ion, but rather in a complex hydrogen bonded network that constantly rearranges as bonds break and form dynamically,5 allowing the protons involved in the structure to move according to the Grotthuss shuttling mechanism.6–7 (Note that the phrase “excess proton” in this work refers to the hydrated proton structure, which at its core is H3O+ but generally is a more complicated structure including additional water molecules.8 It is helpful to think of the solvated proton not as a fixed particle that hops from water to water, but rather as a positive charge spread across a water complex that dynamically alters its structure and dynamic properties in response.) Moreover, the PT process can be coupled to other complex processes such as hydration changes, electron transfer, and protein conformational changes. One critical implication of these potential couplings is that the mechanism of PT cannot be easily inferred from standard molecular dynamics (MD) simulations without an explicit excess proton actually being in the system, even if “water-wires” are otherwise present (or not). In fact, such inferences may be wrong. These couplings of PT to other processes also present further complications for computer modeling, due to the difference in timescales at play: proton shuttling between any two water molecules can occur on the femto- to picosecond timescales, whereas other processes influencing the overall PT may take nano- to milliseconds or longer. To overcome these challenges, Multiscale Reactive Molecular Dynamics (MS-RMD)8–11 was developed (and Multi-State Empirical Bond, MS-EVB, before it; see, e.g., Ref. 12) to efficiently and accurately capture the dynamic solvation and delocalization of the excess proton in complex aqueous phase and biomolecular systems. Since MS-RMD evolves the system according to deterministic Newtonian dynamics, it is capable of accessing the longer timescales needed to realize couplings with slower degrees of freedom and to describe the rare event PT processes that are often biologically relevant. This method (and MS-EVB before it) has successfully been applied to several protein systems to predict and explain mechanisms of PT.13–22
The M2 channel is a homo-tetrameric protein in influenza A viral capsids that is responsible for the acidification of the viral interior, a critical step in the viral life cycle.23–25 It is the target of the antiviral drugs amantadine and rimantadine,26 but the prevalence of drug-resistant mutants has necessitated a continued effort to understand the PT mechanism and channel properties in order to better inform drug-design efforts. A critical histidine residue (His37) located at the center of the transmembrane portion of each helix acts as a “pH-sensor” and is responsible for enabling activation as the pH is lowered.27 Each His37 can bind one additional proton, such that the tetrad can have a total charge ranging from +0 to +4, referred to as the His37 tetrad charge state. As the charge increases and approaches the activated state, electrostatic repulsion between these residues causes the channel to widen, opening the Trp41 gate located just below the His37 tetrad28–32 and enabling proton flux to the viral interior.13, 16, 33–37 It is hypothesized that the channel becomes activated in the +2 state, such that conduction primarily occurs as a cycle through the +2 and +3 states,38 based on experimentally determined pKa’s, pH-dependent conduction, and computer simulations.13, 16, 39–42 Another important characteristic of the channel is its rectification behavior—channel activation and inward proton flux occurs when the exterior pH is lowered, but there is no outward proton flux observed when the interior pH is lowered.43–44
Recently, we carried out multiscale simulations to investigate the PT mechanism and pH-dependent activation behavior in the influenza A M2 proton channel.13, 16 In contrast to other prior simulation studies, our work employed multiscale simulations (including MS-RMD and quantum mechanics/molecular mechanics) with explicit, reactive excess protons to calculate the free energy profiles of PT through the full transmembrane M2 channel in the +0, +1, and +2 protonation states, thereby providing definitive insight into both the acid activation mechanism of M2 and the critical role of the His37 tetrad. This effort characterized the physical basis of PT through extensive sampling with an explicit proton and explained the rectification behavior of the channel through the asymmetry of the PT free energy profiles and estimated rate calculations. Importantly, the calculated rates of deprotonation and conductance quantitatively agreed with proton exchange rates measured by NMR45 and experimentally determined conductance values.33, 44, 46 While this previous study revealed some molecular understanding of the acid activation and rectification mechanisms, the detailed interactions between the excess proton and the channel that contribute to these behaviors were not explored. In this work, our previously published simulations are further analyzed to resolve the channel’s dynamic response as an excess charge moves through the channel and how these dynamic changes enable acid activation.
Water-mediated PT in proteins occurs through three dominant regimes: 1) water is excluded from the lowest energy structures, but fluctuations allow short water net-works to transiently form; 2) water forms a single-file hydrogen bonded path in narrow regions; and 3) water exists in clusters in larger pore regions. The M2 channel exhibits both cases 1 and 3, and is thus an excellent system for studying the dynamic collaboration between the protein, porelining residues, and water that enables PT.
We note that a recent computational study from Chen et al. used constant pH MD (CpHMD) to describe the conformational activation of the M2 channel.47 By simulating the channel at several different pH’s, rather than in fixed protonation states, they elucidate the pH-dependence of equilibrium conformational changes that occur as part of the activation mechanism. Our work here, in contrast, is focused on the changes coupled to an explicit proton’s movement through the channel in order to determine the molecular-level details of the PT mechanism, which should be viewed in tandem with CpHMD results. While they sample the configuration space at a given pH, we sample the configuration space of the dominant protonation state in several pH ranges as a function of the excess proton’s transport through the channel.
To determine the channel’s dynamic response during explicit PT, we focus on the water structure in different regions of the channel and how the number and orientation of the water molecules fluctuate based on where the excess proton is in the channel. Importantly, we find that the channel structure is transiently affected in the presence of the proton, especially for the +0 state, which exhibits distinct conformations when the excess proton is in different regions of the channel. Additionally, the three-dimensional spatial distribution of the excess proton is explored for the first time, revealing an asymmetric path due to asymmetric water distributions and interactions with pore-lining residues. This analysis further emphasizes that having an explicit, reactive excess proton MD description is crucial for understanding such a PT mechanism. It is difficult, if not impossible, to predict or observe the changes and fluctuations that are coupled with PT from non-reactive empirical MD simulations that do not explicitly include a dynamically evolving excess proton in them.
METHODS
The analysis performed here was done using previously published simulations; readers are referred to ref16 for more details on the simulation set-up and potential of mean force (PMF) free energy profile calculations. Briefly, the simulations were initiated from a crystal structure of the transmembrane portion of the M2 channel (this construct is referred to as M2TM) resolved at room-temperature and high pH (PDB: 4QKL48) embedded in a 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) bilayer solvated with water. We have shown that the presence of amphipathic helices, included in the full-length M2 protein, do not significantly influence the PT mechanism,16 and experiments have indicated proton conduction is similar for M2TM with or without the amphipathic helices.49 Following classical equilibration of the +0, +1, and +2 His37 tetrad charge states, MS-RMD simulations were run using the replica-exchange umbrella sampling method.50 The collective variable (CV) for umbrella sampling was defined as the z-coordinate difference between the excess proton center of excess charge (CEC) and the center of mass of the 4 Gly34 alpha carbons, referred to as CECZ. The excess proton CEC was defined as:51
where is the center-of-charge of the ith diabatic state, and is the EVB amplitude of that state. The sum is over all N states. The CV was restrained over 75–80 windows every 0.5 Å for 1–2 ns each after equilibration of the system, including any hydration changes. Frames were saved every 10 ps. The PMF calculations were performed with both MS-RMD and quantum mechanics/molecular mechanics (QM/MM) simulations. While the MS-RMD methodology allows for reactive amino acids, there is currently no reliable procedure for fitting the MS-RMD forcefield for four highly coupled residues in a complex environment (i.e., the His37 tetrad), so QM/MM was used for this region of the channel (CECZ = 1.5 to 11.5 Å). Due to the significantly improved sampling enabled by the efficiency of MS-RMD simulations, only the MS-RMD trajectories are used in the following analysis. Thus, proton-dependent properties are only calculated for CECZ = [−22.0, 1.0] and CECZ = [12.0, 22.0] Å. Simulations were run until the PMF was converged. Simulations were additionally extended, following the same protocol, as needed for increased sampling in this study.
The analyses in this paper were performed using all of the MS-RMD umbrella sampling trajectories to delineate changes as the proton moves through the channel. Since we did not track unbiased dynamics, our conclusions are based on the assumption that PT through each part of the channel is slower than the observed hydration and conformational changes. As partial, though not conclusive, support of this assumption, we note that the reported changes occur and equilibrate on the sub-nanosecond timescale, while the rate of proton flux is multiple orders of magnitude slower (100–1000 sec−1 at pH 6).46 In the water and radial distribution analyses, channel properties are calculated with respect to the protein’s principal axis (PA) rather than the z-axis, which we label Z’ (see Figure S1 for visualization of this difference). The z-coordinate of the CEC in this new frame of reference is defined as CECZ’. This is the z-component of the CEC position along the protein’s PA, rather than the z-axis, with respect to the center of mass of the 4 Gly34 alpha carbons such that CECZ’ ≈ CECZ. This was done due to the channel’s slightly tilted axis (see Results and Discussion). Thus, Z’ refers to the channel’s principal axis, and CECZ’ refers to the CEC position along that axis. Detailed descriptions of all analysis calculations are in the supporting information (SI).
RESULTS AND DISCUSSION
Acid-Activation Mechanism.
In our previous work, multiscale simulation methods were used to calculate PMFs for the movement of an excess proton through the channel in the +0, +1, and +2 His37 protonation states to gain insight into the acid-activation mechanism, shown in Figure 1. These PMF profiles show deep local free energy minima for the excess proton to bind to the His37 tetrad in the +0 and + 1 states, with large barriers for deprotonation towards the viral interior. The deprotonation barrier in the +2 state, however, is lower and the calculated conductance matches the experimental value, indicating how the channel reaches an activated state at low external pH.
Figure 1.
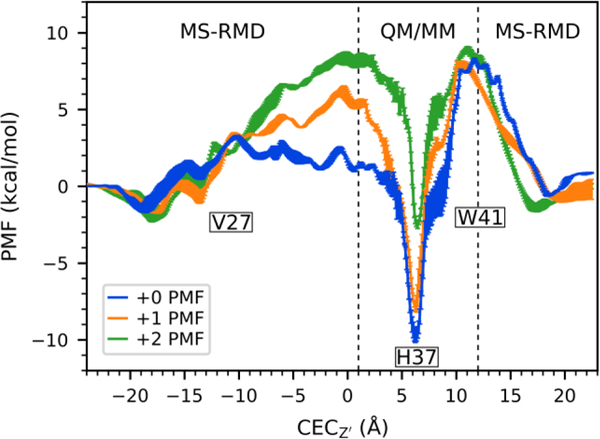
Previously calculated and published PMFs for proton transport through M2 in the +0, +1, and +2 states. Reproduced from ref 11.
Water Structure and Hydrogen Bonding Networks are Sensitive to Proton Position.
It has previously been shown that a hydrated excess proton can change the water dynamics in confined areas, including increasing the level of hydration and rearranging hydrogen bonds.12, 52–53 Here, we evaluate and show the extent to which an excess proton affects the water structure in different regions of the M2 channel. To quantify these effects, the direction of each water-water hydrogen bond was calculated as the cosine of the angle between the hydrogen bond vector and the z-axis, taking on values from −1.0 to 1.0, as defined in Figure 2b. This value was calculated for all hydrogen bonds and averaged over all hydrogen bonds in each region to determine the average direction of water hydrogen bonding in a given configuration. This was done for all simulations and proton positions and then plotted as a function of the proton position to quantify the change as the proton moves through the channel. See the Supporting Information (SI) for more details.
Figure 2.
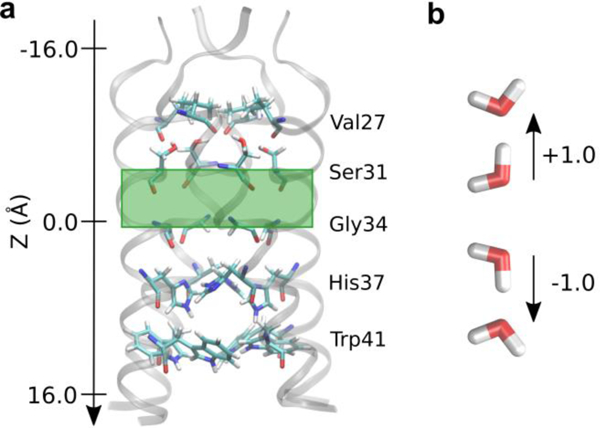
(a) The M2 channel with pore-lining residues shown as sticks. The green box highlights the region of water analyzed in Figure 3a. The direction of the z-axis indicates the direction of proton flux from the exterior to the viral interior. (b) Definition of hydrogen bond directions, with respect to the channel as shown, used throughout.
Figure 3 shows how a cluster of water in the middle of the channel (comprised of all water contained in the region indicated by a green box in Figure 2a, from roughly Z’ = −5.0 A to Z’ = 0.0 A) interacts with and responds to the hydrated excess proton charge for three His37 charge states as a function of the CEC position along the Z’ axis. The average direction of the hydrogen bonds in this middle region is positive before the proton enters and declining as it moves down the channel. The difference is maximized when the excess proton CEC is directly above this middle region (CECZ’ = −6.0 Å), at which point the waters are completely reoriented. However, this effect initiates when the excess proton is several layers of water away as the rearrangement “trickles down” from the excess charge. The water follows this trend in all sections of the channel – the average hydrogen bond direction reaches its minimum value when the proton is situated just above that region, and its maximum value when the proton is just below that region. As the excess proton moves through the region, there is a transition as the hydrogen bonds change polarity due to the excess charge passing through.
Figure 3.
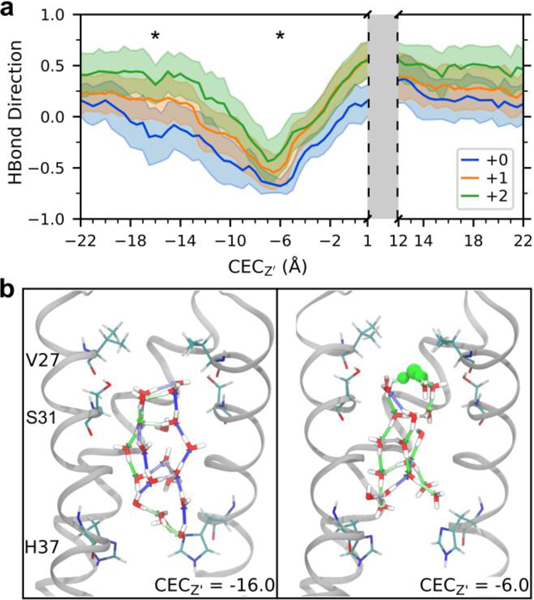
Hydrogen bond rearrangement as proton moves through the channel. (a) Average and standard deviation of the direction of water-water hydrogen bonds in the central region of M2 (see green box in Figure 2a) as a function of CEC position. (b) Example water structures in the middle region of the +1 channel for CECZ’ = −16.0 (the proton is near the entrance of the channel, not in view) and −6.0 Å (above this central region – see stars in 3a). Hydrogen bonds are shown as arrows, colored as a gradient based on direction where blue is +1.0 and green is −1.0. The most hydronium-like water is shown in green. Front helix and residues on back helix are removed for clarity.
The backbone and sidechain atoms also play an important, but dynamic, role in stabilizing water structures through hydrogen bonding. As the hydrated excess proton moves through the channel, these interactions transiently shift both in strength and orientation. The detailed results and discussion of Ser31 hydrogen bonding dependency on the excess proton position are in the SI (see Figure S3 and corresponding text).
Altogether, these changes in hydrogen bonding clearly demonstrate that PT in M2 is a highly dynamic and variable process, with a range of water reorganization trends in response to both the presence of an excess proton charge and the protein structural changes associated with the PT. This analysis also provides insight into water interactions that enable PT through water clusters (regime 3). Considering a hydrated proton approaching from the virus exterior, the hydrogen bonds of connecting water generally have a negative orientation (as defined in Figure 2b) for facile PT in accordance with the Grotthuss shuttling mechanism. The re-arrangement of water as shown above indicates how the proton can, in a sense, “pave its own path.” Before the proton enters the M2 channel, the water is not arranged optimally for PT, but the water structure is flexible and rearranges in response to the passage of the excess proton. Additionally, the changes in water-protein hydrogen bonding shows the flexibility of such interactions – the presence of the excess proton can result in both increased and decreased frequencies of these interactions, as well as shift the positions of involved water molecules. These types of proton-induced changes are expected to play a critical role in many biomolecular (and other) systems involving PT.
The Water Structure in +2 State is “primed” for PT.
The water and hydrogen bonding analysis can be used to understand aspects of the acid activation mechanism in terms of the free energy of PT through the channel. The barrier for the proton to deprotonate the His37 tetrad and move past the Trp41 tetrad is large (~18, 16 kcal/mol) in the +0 and +1 states, but only ~10 kcal/mol in the +2 state (Figure 1). Comparison with the relatively lower barriers of deprotonation towards the viral exterior indicates that these high inward-deprotonation barriers, particularly in the +0 state, are not just due to the favorable His-binding. To better understand the role of the water structure in PT from the His37 tetrad to the viral interior, and how it may contribute to the high deprotonation barrier, we calculated the number of waters and the average direction of hydrogen bonding (as in the previous section) of water in the region just below the His37 tetrad and above the Trp41 tetrad, which approximately corresponds to the range 6 < Z’ < 11 in Figure 2a. At CECZ’ = 12.0 Å, the excess proton is sufficiently beyond the His37 tetrad such that no His37-bound states significantly contribute to its charge delocalization, as previously determined.13, 16 This is also the region where PMF calculations from MS-RMD, which does not account for His37 contributions to delocalization, and QM/MM, which does, overlap. Thus, the water structure below the His37 tetrad, when the proton is at that position, represents the average water structure needed to facilitate proton unbinding from the His37 tetrad towards the interior. As shown in Figure 4a, the number of waters in this region increases by 3 to 5 water molecules in the +0 and +1 states as the proton passes through (comparing CECZ’ = 1.0 with CECZ’ = 12.0 Å). However, the number of waters is relatively constant in the +2 state. Additionally, the average direction of hydrogen bonds between waters in this region also shows tetrad-charge-dependent changes (Figure 4b). When the proton is out of the channel (CECZ’ = −22.0 or 22.0 Å) the +0 state has significantly positive (pointing upward) hydrogen bonds in this region, averaging ~0.4, which need to reorient to achieve a negative direction for PT to the His37 tetrad. The +1 and +2 states, however, start negative and do not need to change as much.
Figure 4.
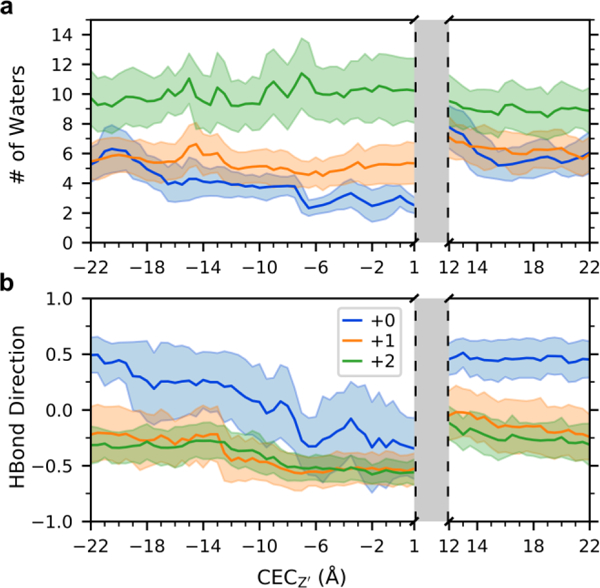
Analysis of water in the region below the His37 tetrad and above the Trp41 tetrad. See SI for specific definition. (a) The average and fluctuation (standard deviation) of the number of waters in this region as the proton moves through the channel. (b) The average and standard deviation of the direction of water-water hydrogen bonds in this region.
The same analysis was also done for the excess proton approaching the His37 tetrad from the viral exterior, focusing on the water in the region directly above the tetrad, below the Gly34 residues in Figure S2. The PMF deprotonation barriers as the proton moves from the His37 tetrad towards the viral exterior are relatively similar for the three charge states: ~12, 14, 11 kcal/mol in the +0, +1, and +2 states, respectively. We can consider how the water in this region responds to the proton approaching the tetrad by comparing the water when CECZ’ = 1.0 Å, which corresponds to the point at which the proton is completely past the His37 tetrad (by same logic as above). The number of waters in this region at CECZ’ = 1.0 is nearly the same as when the proton is out of the channel (ie, CECZ’= −22.0). In the +1 and +2 states, the number of waters above the His37 tetrad is relatively constant as the proton moves through the channel, but it varies in the +0 state by 3–4 waters. The average direction of hydrogen bonding in this region is also similar for all three charge-states. Before the excess proton enters the channel, all three have a slightly positive value corresponding to hydrogen bonds pointing away from the His37 tetrad, and all three show a similar response to the proton as it moves through the channel. The +0 state noticeably becomes more negative more quickly, due to the lack of positive charge on the His37 tetrad to strongly direct hydrogen bonding. The small changes in the number of waters and the similarity in the direction of hydrogen bonding across the three charge states suggest that, in contrast to below the His37 tetrad, the water structure does not significantly contribute to free energy differences of PT above the His37 tetrad in the different states.
Together, these results indicate how the +2 state is “primed” for PT. Physiologically, the +0, +1, and +2 states represent the dominate charge state in pH ranges of 8–9, 7–8, and 6–7 (according to recent constant pH simulations47); these results thus also imply the acid-activation mechanism of PT. For a proton to be released inward from the His37 tetrad, both the +0 and +1 states require more water to move into the region to solvate the excess charge. The water in the +0 state additionally must reorient to a greater degree than the water in the +1 and +2 states. The +2 state, however, does not need more water to enter this region, nor does the water need to completely reorient. This stability of the water as the proton passes through corroborates with the lower barrier in the +2 PMF, corresponding to proton conduction at low pH.
Water Structure Implications for Rectification Behavior.
This water analysis can also be considered with regards to the fascinating rectification behavior (i.e., how the channel activates to allow for inward proton flux when the exterior pH is lowered, but does not activate to allow outward proton flux if the internal pH is lowered). Our previous work provides a qualitative explanation for rectification based on the asymmetry of the +0 PMF and the different relative free energy barriers between protonation and deprotonation in both the inward and outward directions. Essentially, the calculated rate for His37 protonation from the viral exterior is faster than that of His37 deprotonation towards the viral interior, such that the channel can move from the +0 to the +1 state, and similarly from the +1 to the activated +2 state. The relative rates of the reverse process, however, are flipped—the rate for His37 protonation from the viral interior is slower than that of His37 deprotonation towards the viral exterior. Thus, the rate of deprotonation (koff) to the exterior outcompetes the rate of protonation from the interior (kon) and the channel remains stuck in the +0 state instead of progressing to the +1 and the activated +2 states when the internal pH is lowered.
The water structure analysis provides further molecular insight into this rectification behavior. In the +0 state, proton movement from the interior to the His37 tetrad must be accompanied by several additional waters moving into the channel, requiring the channel to dilate around Trp41 (the radius also increases ~1Å, see below). Additionally, once the channel reaches the +1 state, the water hydrogen bond network surrounding the His37 tetrad is directed away from the tetrad, suggesting it would be easier for that proton to move off the tetrad (His37 to exterior) than an additional proton to move in. By comparing the average number of water molecules in the regions above and below the His37 tetrad when the tetrad is protonated (estimated from +1 simulations when the excess proton is out of the channel) versus when the excess proton is just above or below the tetrad (CECZ’= 1.0 or 12.0 Å respectively), we estimate that an additional ~1 waters must move into the region above the His37 tetrad for deprotonation towards the exterior, and ~2 below for interior deprotonation. From this analysis, one excess proton near the tetrad may more easily be solvated above the tetrad, as the change in the number of waters above the tetrad is smaller than that below the tetrad, requiring less rearrangement. This is consistent with our previous analysis of the rectification mechanism, showing that when the pH is lowered in the interior, the channel remains stuck in the +0 state because the koff from the His37 tetrad to exterior bulk outcompetes the kon from the interior to the His37 tetrad. This is not true in the opposite direction, so the channel jumps from the +0 to the +1 state only when the external pH is lowered. Here, we see the explanation of this asymmetry in the +0 PMF is due to the challenge of hydrating the region below the His tetrad and ease of hydrating the region above it.
The Proton Transiently Affects the Protein Structure.
The radii profiles of the channel were calculated using the program HOLE54 for each excess proton position to determine correlations between the proton position and the protein structure. First, the radius of the channel was calculated for each frame and then averaged over each proton position. Figure 5 shows these averaged radii profiles for all proton positions, for each charge state, where each line is the average for a proton position along the channel. Clearly, the +2 state is more open below the Trp41 tetrad and in the His37 tetrad region. The narrowest part of the channel in the +0 and +1 states occurs at the His37 tetrad (~Z’ = 5 Å), and a secondary closure at the Val27 tetrad is seen near Z’ = −10 Å. To more clearly distinguish how the channel radii profile changes are dependent on the proton position, the profiles are plotted separately for each state in Figure S4. Most striking is the variation seen in the +0 state that is not seen in the + 1 and +2 states.
Figure 5.
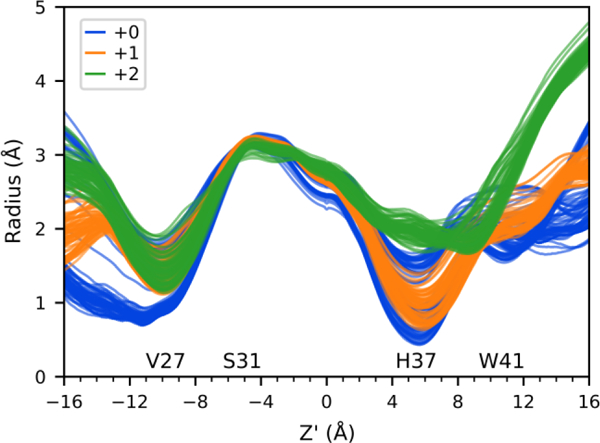
Radii profiles for 3 charge states. Each line is the average for one proton position binned every 0.5 Å, shown for CECZ’ = −22.0 to 1.0 and 10.5 to 22.0. The position of pore lining residues is indicated. See Figure S4 for profiles plotted separately for each state.
The +0 state can be divided into three main conformers based on clustering the radii profiles, shown in Figure 6. Following the excess proton’s path from the top of the channel: when the proton is out of the channel and just starting to enter at the top, the channel is narrowest at the top near Val27, and slightly more open at the bottom of the channel (at this point the radius near the His37 tetrad is still less than that in the +2 state). As the excess proton approaches and passes through the Val27 gate, the top of the channel opens up and the bottom of the channel closes. As the proton moves through the middle of the channel, before reaching the His37 tetrad, both ends of the channel are closed. Finally, as the proton moves past the Trp41 gate, the channel returns to its initial conformation. The larger radius near the Trp41 residues when the proton passes by (seen in the variation near Z’ = 12.0 Å in panel 3 of Figure 6b) indicates how the channel must open to accommodate additional water molecules moving into that region in tandem with His37 tetrad deprotonation. Figure S5 shows a difference plot of these profiles.
Figure 6.
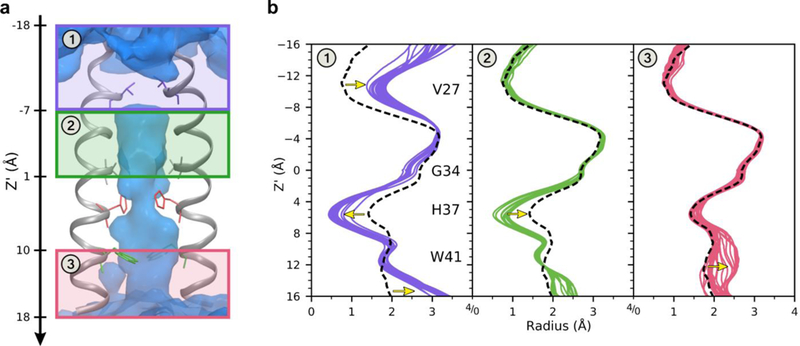
Distinct conformations observed in the +0 state. The channel can be divided into 3 main conformers when the excess proton is in different regions, labeled 1–3, based on radii profiles. (a) The three regions of the channel, indicated by purple, green, and pink boxes. The backbone of two helices are shown in gray, with pore lining residues as sticks. The average water oxygen density isosur-face is shown in blue. (b) The radii profiles of the channel for all proton positions every 0.5 Å, separated based on which region the proton is located. Each colored line is an average radius profile for one proton position in that region. The black dashed line is the average radius profile when the proton is out of the channel. Yellow arrows highlight the largest deviations from the average.
The +1 and +2 states also respond to the excess proton position, but to a much smaller degree. In the +1 state, the top of the channel opens slightly to accommodate the proton moving through the Val27 gate while the bottom of the pore becomes slightly narrower; the bottom of the channel opens slightly as the excess proton passes through that portion. The +2 state exhibits less dependence on the proton position. The different extent of response in each state is consistent with ssNMR results that indicate one conformational state with greater conformational fluctuations at low pH, but the presence of distinct states with small fluctuations at high pH in thick bilayers.29 This suggests that the charged states (+1, +2) are more flexible and may have increased natural fluctuations that allow the proton to pass through, whereas the +0 state is more rigid and requires minor, but distinct, transitions to accommodate the proton.
Channel Asymmetry Distorts Proton Path.
The three-dimensional (3D) path of the excess proton has not previously been studied, nor has the effect of the channel’s asymmetry on the proton path. The M2 channel is not perfectly aligned with the z-axis, but instead is tilted as seen in Figure S6. In these simulations, the +2 state is most tilted and the +1 state is least titled, on average. The protein (initial structure from PDB: 4QKL) was initially aligned with the z-axis in the set-up for these simulations and became tilted during classical equilibration in fixed protonation states. It should be noted that on longer timescales the protein will sample tilts around the z-axis, such that the average principal axis would align with the z-axis. Additionally, we note that only the transmembrane portion of M2 is used in these simulations, which may affect the protein’s degree of tilt in the membrane.
To investigate the 3D path of the proton through the channel, system configurations were first binned according to CECZ’, and the two-dimensional (2D) histogram of CEC values (every 100 fs) was then calculated in the xy-plane for each bin and smoothed using Gaussian interpolation. Since CEC values were saved much more frequently than the co-ordinates of the entire system, the CEC coordinate was projected onto the Z’ axis of the frame nearest in time. Figure 7 depicts the 3D path of the excess proton based on most probable locations, built from 2D histograms such as those shown in Figure 8. It is clear that the proton’s path is increasingly asymmetric in the +1 and +2 charge states. In combination with the average radius and center of the channel, as well as average positions of protein atoms involved in hydrogen bonding in each plane, we are able to identify the distribution of excess proton CEC values with respect to key pore-lining residues (see Figure 8 and SI Movie 1). This again shows that the proton does not follow a symmetric path about the z-axis as it moves through the channel, but tends to cluster near important residues. This is not an artifact of insufficient sampling, but instead indicates a real preference for the proton to be near the wall of the channel in certain xy-planes. Interestingly, this preference also corresponds to the off-centered position of the ammonium group of amantadine and rimantadine,55 which will be further explored in future work. The uneven distribution along the x- and y-axes is due to the asymmetric internal environment and density of water in the channel, which is evident in disparate hydrogen-bonding interactions between water and the channel (see Table S1). The slightly skewed orientation of each helix relative to the others as a result of the channel’s tilt within the membrane may contribute to this asymmetry. The protonation of the His residues in the +1 and +2 also contributes to this asymmetry. A more detailed description of the excess proton’s path is included in the SI.
Figure 7.
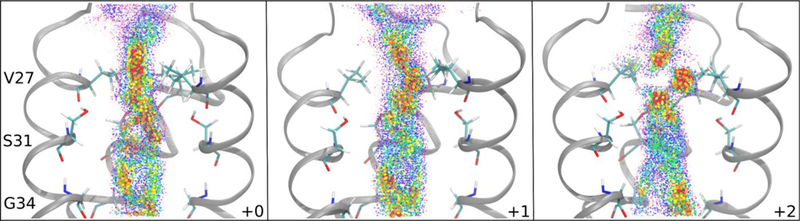
Histogram values of CEC density for each charge state. Each colored dot represents one 2D histogram bin value, colored and sized according to value. Values from high to low (indicating high to low probability of the CEC being found in that bin) are large to small, and red to purple. Bin densities less than 0.01 not shown. The structure is taken from a trajectory with the excess proton outside the channel and does not show changes associated with proton position. The front helix is removed for clarity. See movies 2–4 in SI for a better 3D perspective.
Figure 8.
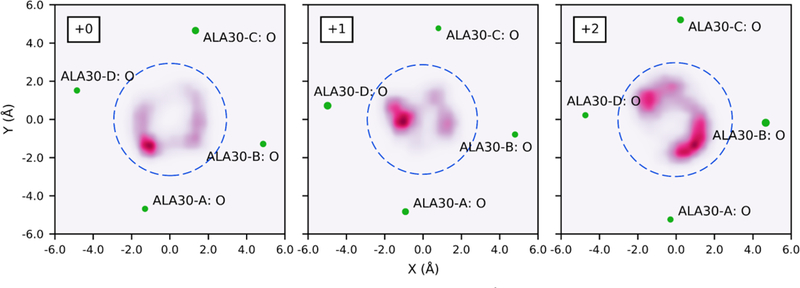
Density of CEC positions in the xy-plane for bin centered at CECZ’ = −1.6 Å. Blue dashed circles represent the average radius of the channel at Z’ = −1.6 Å for this proton position, centered at the average center of the channel at Z’ = −1.6 Å based on the center of mass of backbone atoms. Relevant nearby atoms shown in green, with size indicating proximity (larger atoms are closer to this plane). Atoms are labeled by residue and chain. See movie 1 in SI for equivalent slices moving through the channel.
Furthermore, due to the protein’s tilt, this analysis shows that the cross-section of the pore is not well-defined as a circle all through the channel, but is more elliptical on the nanosecond timescale – the distributions at CECZ’ = −10.0 Å, for example, appear to be outside the channel but in fact the Val27 residues transiently shift to open more space along the edge of the pore. The method used here to calculate the pore radius does not account for this asymmetry.
Understanding this asymmetric proton path could be helpful for structure-based drug design efforts, as it is can be used to identify particular “sticky” spots for the hydrated excess proton structure. This result also points to the dynamic shape of the pore, which could influence new drug shapes and sizes. It is critical as well to realize that the channel structure and helix tilts are dependent on lipid composition,29, 56–58 and the precise asymmetries may vary depending on the composition.
CONCLUSIONS
In this work, extensive MS-RMD simulations were used to understand in molecular-level detail the specific interactions and structural perturbations that occur during PT in the M2 channel in the +0, +1, and +2 charge states of the His37 tetrad. Our results reveal how the proton causes subtle shifts in the protein structure to increase the pore volume and water density in its vicinity, as well as larger conformational changes in the +0 state. Asymmetry in the channel (persistent on the timescale of these simulations) is shown to influence the proton pathway, causing sharp deviations from a purely symmetric path. Additionally, water molecules move in and out of the channel and hydrogenbond networks rearrange in response to the positive charge defect as the hydrated proton “paves its own path” through the channel. Specifically, the observed water rearrangements below the His37 tetrad provide further insight into the acid-activation mechanism, showing how, unlike the +2 state, the +0 and +1 states do not have enough optimally oriented water below the tetrad to facilitate proton conduction. In contrast, above the His37 tetrad, the hydration required for PT is reasonably facile in all charge states. This helps to explain the rectification mechanism: when the pH is lowered in the interior of the virus, the system remains stuck in the +0 state (it is not activated) because the barrier for proton uptake from the inside to the His37 tetrad is large and the barrier for release from the His37 tetrad is low. Thus, the rate of deprotonation of the His37 tetrad to the outside outcompetes protonation from the inside, all due to the asymmetry in the ease of hydration above and below the His37 tetrad.
Altogether, these results provide a more detailed look at the PT mechanism and give helpful insight for drug-design efforts to exploit channel asymmetry and hydration changes associated with acid activation. For example, designing molecules that destabilize hydration above the His37 tetrad could effectively block PT by switching the dominance of koff versus kon. Also, designing molecules that do not just block the channel, but increase the barrier for PT past the Val27 region by binding above the channel and stabilizing a tightly closed structure, could be an effective strategy. Finally, we emphasize how this study again illustrates that PT is a highly dynamic process in which hydration and structural changes are induced during PT. Simulations that include an explicit, reactive excess proton are necessary to capture these correlated changes. Thus, looking only for “water wires” in experimental structures or empirical non-reactive MD simulations that omit an explicit excess proton may sometimes suggest misleading conclusions.
Supplementary Material
ACKNOWLEDGMENT
R.L., J.M.J.S., and G.A.V. were supported by the National Institute of General Medical Sciences (NIGMS) of the National Institutes of Health (NIH Grant R01-GM053148). W.F.D. was supported by NIH Grant GM122603. LCW received funding from Department of Energy (DOE) Computational Science Graduate Fellowship under grant DE-FG02-97ER25308. The authors acknowledge the University of Chicago Research Computing Center and the U.S. Department of Defense High Performance Computing Modernization Program for providing computing resources.
Footnotes
ASSOCIATED CONTENT
Supporting Information.
Illustration of Z’ axis.
Analysis of number of waters and hydrogen bond direction in region above His37.
Illustration of water hydrogen bonds with Ser31 and calculation of average hydrogen bond direction.
Radii profiles displayed separately for each charge state.
Water hydrogen bond prevalence with Gly34.
Contour plot of protein principal axis projection onto xy-plane.
Movies of structures in Figure 7 rotating.
Movie of all 2D-histograms for all CECZ’ positions through the top of the channel as in Figure 8.
This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
REFERENCES
- 1.Decoursey TE, Voltage-gated proton channels and other proton transfer pathways. Physiol. Rev. 2003, 83 (2), 475–579. [DOI] [PubMed] [Google Scholar]
- 2.Mitchell P, Coupling of Phosphorylation to Electron and Hydrogen Transfer by a Chemi-Osmotic Type of Mechanism. Nature 1961, 191, 144–8. [DOI] [PubMed] [Google Scholar]
- 3.Stryer L, Chapters 17 and 22 In Biochemistry, W. H. Freeman & Co: New York, 1988. [Google Scholar]
- 4.Wraight CA, Chance and design-Proton transfer in water, channels and bioenergetic proteins. Bba-Bioenergetics 2006, 1757 (8), 886–912. [DOI] [PubMed] [Google Scholar]
- 5.Knight C; Voth GA, The Curious Case of the Hydrated Proton. Acc. Chem. Res 2012, 45 (1), 101–109. [DOI] [PubMed] [Google Scholar]
- 6.de Grotthuss CJT, Sur la décomposition de l’eau et des corps qu’elle tient en dissolution á l’aide de l’électricité galvanique. Ann. Chim 1806, LVIII, 54–74. [Google Scholar]
- 7.Agmon N, The Grotthuss Mechanism. Chem. Phys. Lett 1995, 244, 456–462. [Google Scholar]
- 8.Knight C; Lindberg GE; Voth GA, Multiscale reactive molecular dynamics. J Chem Phys 2012, 137 (22), 22A525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yamashita T; Peng Y; Knight C; Voth GA, Computationally Efficient Multiconfigurational Reactive Molecular Dynamics. J Chem Theory Comput 2012, 8 (12), 4863–4875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nelson JG; Peng Y; Silverstein DW; Swanson JM, Multiscale Reactive Molecular Dynamics for Absolute pKa Predictions and Amino Acid Deprotonation. J Chem Theory Comput 2014, 10 (7), 2729–2737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee S; Liang R; Voth GA; Swanson JM, Computationally Efficient Multiscale Reactive Molecular Dynamics to Describe Amino Acid Deprotonation in Proteins. J Chem Theory Comput 2016, 12 (2), 879–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Swanson JM; Maupin CM; Chen H; Petersen MK; Xu J; Wu Y; Voth GA, Proton solvation and transport in aqueous and biomolecular systems: insights from computer simulations. J Phys Chem B 2007, 111 (17), 4300–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liang R; Li H; Swanson JM; Voth GA, Multiscale simulation reveals a multifaceted mechanism of proton permeation through the influenza A M2 proton channel. Proc Natl AcadSci USA 2014, 111 (26), 9396–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Taraphder S; Maupin CM; Swanson JM; Voth GA, Coupling Protein Dynamics with Proton Transport in Human Carbonic Anhydrase II. J Phys Chem B 2016, 120 (33), 8389–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee S; Swanson JM; Voth GA, Multiscale Simulations Reveal Key Aspects of the Proton Transport Mechanism in the ClC-ec1 Antiporter. Biophys J 2016, 110 (6), 1334–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liang R; Swanson JM; Madsen JJ; Hong M; DeGrado WF; Voth GA, Acid activation mechanism of the influenza A M2 proton channel. Proc Natl Acad Sci U S A 2016, 201615471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liang R; Swanson JM; Peng Y; Wikstrom M; Voth GA, Multiscale simulations reveal key features of the proton-pumping mechanism in cytochrome c oxidase. Proc Natl Acad Sci U SA 2016, 113 (27), 7420–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee S; Mayes HB; Swanson JM; Voth GA, The Origin of Coupled Chloride and Proton Transport in a Cl(−)/H(+) Antiporter. J Am Chem Soc 2016, 138 (45), 14923–14930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liang R; Swanson JMJ; Wikstrom M; Voth GA, Understanding the essential proton-pumping kinetic gates and decoupling mutations in cytochrome c oxidase. Proc Natl Acad Sci USA 2017, 114 (23), 5924–5929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Parker JL; Li C; Brinth A; Wang Z; Vogeley L; Solcan N; Ledderboge-Vucinic G; Swanson JMJ; Caffrey M; Voth GA; Newstead S, Proton movement and coupling in the POT family of peptide transporters. Proc Natl Acad Sci US A 2017, 114 (50), 13182–13187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang Z; Swanson JMJ; Voth GA, Modulating the Chemical Transport Properties of a Transmembrane Antiporter via Alternative Anion Flux. J Am Chem Soc 2018, 140 (48), 16535–16543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mayes HB; Lee S; White AD; Voth GA; Swanson JMJ, Multiscale Kinetic Modeling Reveals an Ensemble of Cl(–)/H(+) Exchange Pathways in ClC-ec1 Antiporter. J Am Chem Soc 2018, 140 (5), 1793–1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pinto LH; Lamb RA, The M2 proton channels of influenza A and B viruses. J Biol Chem 2006, 281 (14), 8997–9000. [DOI] [PubMed] [Google Scholar]
- 24.Pinto LH; Holsinger LJ; Lamb RA, Influenza virus M2 protein has ion channel activity. Cell 1992, 69 (3), 517–28. [DOI] [PubMed] [Google Scholar]
- 25.Chizhmakov IV; Geraghty FM; Ogden DC; Hayhurst A; Antoniou M; Hay AJ, Selective proton permeability and pH regulation of the influenza virus M2 channel expressed in mouse erythroleukaemia cells. J Physiol 1996, 494 (Pt 2), 329–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang C; Takeuchi K; Pinto LH; Lamb RA, Ion channel activity of influenza A virus M2 protein: characterization of the amantadine block. J Virol 1993, 67 (9), 5585–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang C; Lamb RA; Pinto LH, Activation of the M2 ion channel of influenza virus: a role for the transmembrane domain histidine residue. Biophys J 1995, 69 (4), 1363–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tang Y; Zaitseva F; Lamb RA; Pinto LH, The gate of the influenza virus M2 proton channel is formed by a single tryptophan residue. J Biol Chem 2002, 277 (42), 39880–6. [DOI] [PubMed] [Google Scholar]
- 29.Hu F; Luo W; Cady SD; Hong M, Conformational plasticity of the influenza A M2 transmembrane helix in lipid bilayers under varying pH, drug binding, and membrane thickness. Biochim Biophys Acta 2011, 1808 (1), 415–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hu F; Luo W; Hong M, Mechanisms of Proton Conduction and Gating in Influenza M2 Proton Channels from Solid-State NMR. Science 2010, 330 (6003), 505–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li C; Qin H; Gao FP; Cross TA, Solid-state NMR characterization of conformational plasticity within the transmembrane domain of the influenza A M2 proton channel. Biochim Biophys Acta 2007, 1768 (12), 3162–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schnell JR; Chou JJ, Structure and mechanism of the M2 proton channel of influenza A virus. Nature 2008, 451 (7178), 591–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sharma M; Yi M; Dong H; Qin H; Peterson E; Busath DD; Zhou HX; Cross TA, Insight into the mechanism of the influenza A proton channel from a structure in a lipid bilayer. Science 2010, 330 (6003), 509–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Polishchuk AL; Lear JD; Ma C; Lamb RA; Pinto LH; Degrado WF, A pH-dependent conformational ensemble mediates proton transport through the influenza A/M2 protein. Biochemistry 2010, 49 (47), 10061–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Acharya R; Carnevale V; Fiorin G; Levine BG; Polishchuk AL; Balannik V; Samish I; Lamb RA; Pinto LH; DeGrado WF; Klein ML, Structure and mechanism of proton transport through the transmembrane tetrameric M2 protein bundle of the influenza A virus. Proc Natl Acad Sci U S A 2010, 107 (34), 15075–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wei C; Pohorille A, Activation and proton transport mechanism in influenza A M2 channel. Biophys J 2013, 105 (9), 2036–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Williams JK; Zhang Y; Schmidt-Rohr K; Hong M, pH-dependent conformation, dynamics, and aromatic interaction of the gating tryptophan residue of the influenza M2 proton channel from solid-state NMR. Biophys J 2013, 104 (8), 1698–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hong M; DeGrado WF, Structural basis for proton conduction and inhibition by the influenza M2 protein. Protein Sci 2012, 21 (11), 1620–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miao Y; Fu R; Zhou HX; Cross TA, Dynamic Short Hydrogen Bonds in Histidine Tetrad of Full-Length M2 Proton Channel Reveal Tetrameric Structural Heterogeneity and Functional Mechanism. Structure 2015, 23 (12), 2300–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liao SY; Yang Y; Tietze D; Hong M, The influenza m2 cytoplasmic tail changes the proton-exchange equilibria and the backbone conformation of the transmembrane histidine residue to facilitate proton conduction. J Am Chem Soc 2015, 137 (18), 6067–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Colvin MT; Andreas LB; Chou JJ; Griffin RG, Proton association constants of His 37 in the Influenza-A M218–60 dimer-of-dimers. Biochemistry 2014, 53 (38), 5987–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hu J; Fu R; Nishimura K; Zhang L; Zhou HX; Busath DD; Vijayvergiya V; Cross TA, Histidines, heart of the hydrogen ion channel from influenza A virus: toward an understanding of conductance and proton selectivity. Proc Natl Acad Sci U S A 2006, 103 (18), 6865–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chizhmakov IV; Ogden DC; Geraghty FM; Hayhurst A; Skinner A; Betakova T; Hay AJ, Differences in conductance of M2 proton channels of two influenza viruses at low and high pH. Journal of Physiology 2003, 546, 427–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mould JA; Li HC; Dudlak CS; Lear JD; Pekosz A; Lamb RA; Pinto LH, Mechanism for proton conduction of the M(2) ion channel of influenza A virus. J Biol Chem 2000, 275 (12), 8592–9. [DOI] [PubMed] [Google Scholar]
- 45.Hu F; Schmidt-Rohr K; Hong M, NMR detection of pH-dependent histidine-water proton exchange reveals the conduction mechanism of a transmembrane proton channel. J Am Chem Soc 2012, 134 (8), 3703–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Leiding T; Wang J; Martinsson J; DeGrado WF; Arskold SP, Proton and cation transport activity of the M2 proton channel from influenza A virus. Proc Natl Acad Sci US A 2010, 107 (35), 15409–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen W; Huang Y; Shen J, Conformational Activation of a Transmembrane Proton Channel from Constant pH Molecular Dynamics. J Phys Chem Lett 2016, 7 (19), 3961–3966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Thomaston JL; Alfonso-Prieto M; Woldeyes RA; Fraser JS; Klein ML; Fiorin G; DeGrado WF, High-resolution structures of the M2 channel from influenza A virus reveal dynamic pathways for proton stabilization and transduction. Proc Natl Acad Sci USA 2015, 112 (46), 14260–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ma C; Polishchuk AL; Ohigashi Y; Stouffer AL; Schon A; Magavern E; Jing X; Lear JD; Freire E; Lamb RA; DeGrado WF; Pinto LH, Identification of the functional core of the influenza A virus A/M2 proton-selective ion channel. Proc Natl Acad Sci US A 2009, 106 (30), 12283–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sugita Y; Kitao A; Okamoto Y, Multidimensional replica-exchange method for free-energy calculations. Journal of Chemical Physics 2000, 113 (15), 6042–6051. [Google Scholar]
- 51.Day TJF; Soudackov AV; Čuma M; Schmitt UW; Voth GA, A second generation multistate empirical valence bond model for proton transport in aqueous systems. The Journal of Chemical Physics 2002, 117 (12), 5839–5849. [Google Scholar]
- 52.Peng Y; Swanson JM; Kang SG; Zhou R; Voth GA, Hydrated Excess Protons Can Create Their Own Water Wires. J Phys Chem B 2015, 119 (29), 9212–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li H; Chen H; Zeuthen T; Conrad C; Wu B; Beitz E; Voth GA, Enhancement of Proton Conductance by Mutations of the Selectivity Filter of Aquaporin-1. J. Mol. Biol 2011, 407, 607–620. [DOI] [PubMed] [Google Scholar]
- 54.Smart OS; Neduvelil JG; Wang X; Wallace BA; Sansom MSP, HOLE: A program for the analysis of the pore dimensions of ion channel structural models. Journal of Molecular Graphics 1996, 14 (6), 354–360. [DOI] [PubMed] [Google Scholar]
- 55.Thomaston JL; Polizzi NF; Konstantinidi A; Wang J; Kolocouris A; DeGrado WF, Inhibitors of the M2 Proton Channel Engage and Disrupt Transmembrane Networks of Hydrogen-Bonded Waters. J Am Chem Soc 2018, 140 (45), 15219–15226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.zhou H-X; Cross TA, Modeling the membrane environment has implications for membrane protein structure and function: Influenza A M2 protein. Protein Science 2013, 22 (4), 381–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Luo W; Cady SD; Hong M, Immobilization of the influenza A M2 transmembrane peptide in virus envelope-mimetic lipid membranes: a solid-state NMR investigation. Biochemistry 2009, 48 (27), 6361–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lin CW; Mensa B; Barniol-Xicota M; DeGrado WF; Gai F, Activation pH and Gating Dynamics of Influenza A M2 Proton Channel Revealed by Single-Molecule Spectroscopy. Angew Chem Int Ed Engl 2017, 56 (19), 5283–5287. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.