

Abstract
Cellular RNA levels are the result of a juggling act between RNA transcription, processing, and degradation. By tuning one or more of these parameters, cells can rapidly alter the available pool of transcripts in response to stimuli. While RNA sequencing (RNA-seq) is a vital method to quantify RNA levels genome-wide, it is unable to capture the dynamics of different RNA populations at steady-state or distinguish between different mechanisms that induce changes to the steady-state (i.e. altered rate of transcription versus degradation). The dynamics of different RNA populations can be studied by targeted incorporation of non-canonical nucleosides. 4-thiouridine (s4U) is a commonly used and versatile RNA metabolic label that allows the study of many properties of RNA metabolism from synthesis to degradation. Numerous experimental strategies have been developed that leverage the power of s4U to label newly transcribed RNA in whole cells, followed by enrichment with activated disulfides or chemistry to induce C mutations at sites of s4U during sequencing. This review presents existing methods to study RNA population dynamics genome-wide using s4U metabolic labeling, as well as a discussion of considerations and challenges when designing s4U metabolic labeling experiments.
Graphical/Visual Abstract and Caption
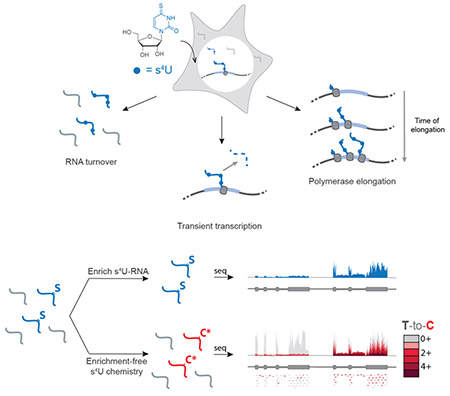
Newly transcribed RNAs can be metabolically labeled with 4-thiouridine (s4U) to study many aspects of RNA metabolism genome-wide, including RNA turnover, transient transcription, and polymerase elongation. New RNAs can be detected via biochemical enrichment (top) or nucleoside recoding to induce U to C mutations in s4U-RNA in high-throughput sequencing.
INTRODUCTION
RNA levels are determined by the tight regulation of RNA synthesis and degradation. Eukaryotes can modulate RNA levels by altering transcription, processing or decay. This dynamic regulation can lead to the same RNA steady-state levels via multiple pathways. For example, a cell can increase RNA levels by increasing transcription or decreasing degradation. Therefore, while RNA-seq can be used to detect changes in RNA levels upon stimulation, one cannot distinguish what mechanism led to a given RNA profile. Detailed studies of the kinetics of RNA transcription and degradation have been performed in response to a range of stimuli such as lipopolysaccharide (LPS) in mouse dendritic cells (Rabani et al., 2011), interleukin 7 (IL7) in mouse naïve T-cells (Li et al., 2017), and 4-hydroxytamoxifen (OHT) in mouse fibroblasts (de Pretis et al., 2017). These studies demonstrate that cells regulate RNA levels by multiple mechanisms in a transcript-specific manner. RNA turnover can be tissue-specific as well as stimulus-specific, highlighting the need to understand the contribution of RNA population dynamics in a variety of metabolic and cellular contexts.
Standard biochemical analyses have been widely used to probe specific aspects of RNA regulation. These include RNA polymerase II (RNAPII) ChIP and nuclear run-on to study transcription, as well as transcriptional shutoff using small molecules to study RNA degradation. However, metabolic labeling with non-canonical nucleosides provides a handle for analysis of transcripts throughout the lifetime of the RNA, creating a versatile platform to probe many facets of RNA metabolism in a single experimental workflow. This versatility not only streamlines experimental optimization, but also enables clearer interpretation of cellular responses where changes in RNA synthesis and degradation simultaneously occur.
In order to study the dynamics of different populations of RNA, classic studies used incorporation of radiolabeled orthophosphate or nucleotides that facilitated analysis of bulk RNA population dynamics (for early examples, see (Hokin & Hokin, 1954; Logan, Heagy, & Rossiter, 1955; Muramatsu & Busch, 1964). However, the application of non-canonical nucleosides opened the door for later genome-wide analyses by enabling the biochemical separation of new RNAs from pre-existing RNAs (reviewed by (Tani & Akimitsu, 2012). The three most common non-canonical nucleosides used for metabolic labeling of RNA are 5-bromouridine (BrU), 5-ethynyluridine (EU) and 4-thiouridine (s4U) (Box 1). BrU triphosphate can be added to isolated nuclei in global nuclear run-on sequencing (GRO-seq,(Core, Waterfall, & Lis, 2008) to study transcription, or BrU nucleoside can be incorporated into whole cells to study RNA stability (BRIC-seq,(Tani et al., 2012). Both techniques enrich BrU in newly transcribed RNAs by immunoprecipitation with anti-BrdU antibodies in order to separate new RNAs from old. Alternatively, 5-ethynyluridine (5-EU) can be incorporated into newly transcribed RNAs in whole cells and enriched using click-chemistry (Jao & Salic, 2008), a strategy which was later adapted to a high-throughput sequencing platform (Meryet-Figuiere et al., 2014). Click-compatible nucleosides hold the advantage that they are orthogonal to cellular functional groups and therefore can be used for a wide variety of applications including RNA imaging (Jao & Salic, 2008) with reduced cross reactivity with other macromolecules including proteins. The use of BrUTP in nuclear run-on experiments has been replaced with the use of biotinylated nucleotide triphosphates for streptavidin purification (PRO-seq, Kwak, Fuda, Core, & Lis, 2013). These PRO-seq experiments and analogue-free enrichment strategies based on the immunopurification of RNAPII (NET-seq, (Churchman & Weissman, 2011) can reveal the 3’-most nucleotide in a nascent transcript, providing high resolution views of transcription. Finally, s4U is rapidly incorporated into newly transcribed RNA in living cells without the need for nuclear isolation. As we will discuss in detail below, s4U provides a range of chemical opportunities. s4U-RNA can be enriched using activated disulfides conjugated to biotin, which hold the advantage that this chemistry is covalent (unlike BrU) and the disulfide bond is reversible (unlike EU), allowing for efficient elution of s4U-RNA from streptavidin beads and analysis. As an alternative to biochemical enrichment, the presence of s4U in an RNA can be identified by chemically recoding the hydrogen bonding pattern of s4U to induce apparent T-to-C mutations in genome-wide RNA-seq experiments (Herzog et al., 2017; Schofield, Duffy, Kiefer, Sullivan, & Simon, 2018). These opportunities provide a common platform to study the kinetics and regulation of the entire RNA life-cycle from synthesis to degradation.
Box 1: Metabolic labeling reagents for the capture of newly transcribed RNA.
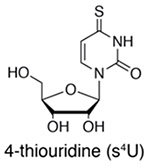 |
S4U (or 4-thiouracil in cells that express UPRT) is rapidly uptaken by cells, and s4U-RNA can be enriched with activated disulfide chemistry (e.g. HPDP-biotin [Cleary 2005], MTS-biotin [Duffy 2015], or MTS-resin [Duffy 2018]) or identified by enrichment-free conversion chemistry (Herzog 2017, Schofield 2018). Advantages: activated disulfide chemistry is both efficient and reversible, and enrichment-free chemistry offers single-nucleotide resolution. |
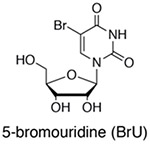 |
BrUTP is added to isolated nuclei for rapid uptake and short RNA labeling (Core 2008), while BrU is uptaken by cells at a slower rate and is applicable for pulse-chase experiments (Tani 2012). BrU-RNA is enriched using anti-BrdU antibodies. Advantages: BrU metabolic labeling methods are widely used and well validated. BrU is also compatible with immunostaining (Wansink 1993). |
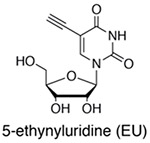 |
EU is rapidly uptaken into cells, and EU-RNA can be enriched with click chemistry (azide-conjugated biotin) or used for live-cell nascent RNA labeling (Jao 2008). Advantages: 5-EU enrichment chemistry is covalent (although irreversible) and compatible with live-cell imaging. |
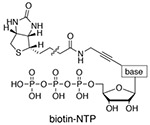 |
Biotin-NTPs are added in four separate reactions to isolated nuclei for rapid uptake and short RNA labeling, although all four biotin-NTPs can be added to the same reaction (Kwak 2013). Biotinylated RNA is enriched with streptavidin-coated magnetic beads. Advantages: Single base-pair resolution is achieved for the precise localization of RNA polymerase. |
Historically, metabolically labeled s4U-RNAs were isolated using organomercurial affinity matrices (Melvin, Milne, Slater, Allen, & Keir, 1978) based on the high affinity of thiols for mercury. While this purification strategy remains a powerful way to enrich s4U-RNA (for example, see (Kenzelmann et al., 2007; Ussuf, Anikumar, & Nair, 1995; Woodford, Schlegel, & Pardee, 1988), the practicality of s4U metabolic labeling was expanded with the development of activated disulfide purification; the endogenous uracil phosphoribosyltransferase (UPRT) activity in T. gondii was harnessed to metabolically label cells with 4-thiouracil and enrich RNAs using HPDP-biotin, a 2-pyridylthio-activated disulfide of biotin, followed by enrichment using a streptavidin matrix (Cleary, Meiering, Jan, Guymon, & Boothroyd, 2005). Later, s4U nucleosides were found to be incorporated into mammalian cells without the need for UPRT expression (Dölken et al., 2008), thereby streamlining the process of metabolic labeling and expanding the utility of this method to many different cell types.
Following the application of s4U to metabolically label newly transcribed RNAs in mammalian cells, this strategy revealed relationships between mRNA half-lives and protein stability (Friedel, Dolken, Ruzsics, Koszinowski, & Zimmer, 2009; Schwanhäusser et al., 2011). In parallel, important s4U metabolic labeling experiments were achieved in S. cerevisiae through the expression of exogenous transporters (hENT1 (C. Miller et al., 2011) or Fui1 (Swiatkowska et al., 2012) that are necessary for efficient s4U uptake and RNA labeling (unlike S. pombe that express UPRT or mammalian cells that express uridine transporters). Following these single time point analyses, multiple time point experiments were developed in yeast, including a pulse-chase approach to reveal RNA stability and a time course of s4U labeling to study RNA processing and/or stability (Munchel, Shultzaberger, Takizawa, & Weis, 2011; Neymotin, Athanasiadou, & Gresham, 2014; Windhanger et al., 2012). Transient RNAs, including introns and unstable non-coding RNAs, were enriched in yeast following 1.5 to 5 min of s4U labeling (Barrass et al., 2015), and in human cells following 5 min of s4U labeling (Schwalb et al., 2016; Windhanger et al., 2012). These techniques illustrate the diverse array of experimental strategies that leverage s4U metabolic labeling and allow for the study of RNA metabolism in many different cell types and biological contexts.
In addition to improvements in experimental design, methodologies for detecting s4U-RNAs have also advanced. While HPDP-biotin expanded the use of s4U metabolic labeling, it was later shown to be inefficient in its reactivity with s4U, leading to low yields and biases in enrichment (Duffy et al., 2015). A more efficient activated disulfide, methane thiosulfonate conjugated to biotin (MTS-biotin), was demonstrated to improve s4U metabolic labeling by increasing yields and alleviating length bias. This advance enabled the first study of s4U labeled microRNA turnover in proliferating cells, which revealed a population of mature miRNAs with rapid turnover in 293T cells (Duffy et al., 2015). Recently, methane thiosulfonate was directly coupled to magnetic sepharose, which reduces the number of handling steps through a one-step capture of s4U-RNAs (Duffy, Canzio, Maniatis, & Simon, 2018). This enables s4U-RNA enrichment from small numbers of cells and extending the utility of s4U metabolic labeling from yeast and cell culture models to primary cells including neurons. Enrichment-free techniques have also been developed that either alkylate the s4U (SLAM-seq,(Herzog et al., 2017) or recode the hydrogen bonding pattern of s4U (TimeLapse-seq, (Schofield et al., 2018) in order to induce T-to-C mutations in genome-wide RNA-seq experiments. These techniques offer an additional layer to mRNA 3’end sequencing (Herzog et al., 2017) and total RNA-seq (Schofield et al., 2018) by identifying new RNAs. Importantly nucleoside recoding approaches have been shown to be compatible with traditional enrichment-based techniques (Schofield et al., 2018), thereby combining the ability to enrich rare transcripts with stringent filtering of new reads over nonspecific background.
Here we review recent advances in s4U metabolic labeling techniques that have been used for global analysis of RNA population dynamics. We have limited our discussion to methods that have already been applied to genome-wide studies; while many other methods in principle can be extended to genome-wide, it is difficult to predict how much optimization is required in practice to establish compatibility with next-generation sequencing platforms. In addition, we present readers with important considerations and existing challenges when designing genome-wide s4U metabolic labeling experiments.
STEADY-STATE RNA POPULATION DYNAMICS
When a population of cells are at homeostasis, RNA-seq provides important information about the relative abundance of each RNA, which offers a transcriptome-wide snapshot of the steady-state. However, this freeze-frame view of the transcriptome does not capture the rich dynamics of RNA turnover that are required for cells to remain at homeostasis. In addition, mechanisms for maintaining the steady-state contribute to our understanding of how cells dynamically change their transcriptome in response to changes in their environment. For instance, transcription factors and histone mRNAs tend to be rapidly transcribed and degraded, whereas transcripts involved in cellular metabolism tend to exhibit slower turnover (Yang et al., 2003). Standard RNA-seq experiments are unable to decipher mechanisms of RNA processing and regulation, particularly when cells are at the steady-state. In order to measure transcription and degradation genome-wide, s4U can be used to label newly transcribed RNAs. Importantly, only transcripts that are actively being transcribed will incorporate s4U during the feed, which has implications for low abundance transcripts or transcripts that are tissue and/or cell cycle dependent. As there are many different ways to design a metabolic labeling experiment, each with unique acronyms, we instead provide modules for each step of the experiment in Figure 1. We also discuss considerations and challenges when designing and interpreting s4U metabolic labeling experiments for cells at the steady-state.
Figure 1:

(A) Overview of s4U metabolic labeling experiments. Different options are indicated for each module, along with references for representative examples. In the case where two references are listed, the first applies to yeast and the second to mammalian cells, as the time of s4U labeling can vary widely between organisms. (B) Representative examples of two s4U metabolic labeling experiments that leverage different module options: 4sUDRB-seq to measure RNAPII elongation and TT-TimeLapse-seq to detect unstable RNAs.
Cellular incorporation of s4U
To study RNA turnover, s4U can be added to cell culture media at a range of times and concentrations depending on the desired application (Russo, Heck, Wilusz, & Wilusz, 2017). The nucleobase 4-thiouracil can be used as a metabolic labeling reagent in any cell line that expresses UPRT, including S. cerevisiae and some archaea (Knüppel, Kuttenberger, & Ferreira-Cerca, 2017), while the nucleoside 4-thiouridine is readily uptaken by mammalian cells and converted into s4UTP by the endogenous nucleotide salvage pathway. Exogenous nucleoside transporters can be expressed in order to facilitate more efficient uptake of s4U (C. Miller et al., 2011; Swiatkowska et al., 2012). Finally, while s4UTP does not readily cross cell membranes due to its charge, s4UTP has been directly injected into zebrafish embryos for RNA metabolic labeling (Heyn et al., 2014).
While metabolic labeling experiments have been successful across a range of conditions, labeling time and s4U concentration are critical parameters to consider when designing an experiment in addition to sequencing depth and variability in enrichment (see below). RNA metabolism can be studied via approach-to-equilibrium, where cells are harvested at different times following the addition of s4U until labeling approaches the steady-state (Figure 1C), or single time point analyses, where the relative populations of s4U-RNA and unlabelled RNA allow the calculation of RNA synthesis and decay rates for each transcript. Ideally, the length of s4U metabolic labeling should be tuned in these cases such that the desired population of transcripts contain 15-90% new RNA (that has been labeled with s4U) at the end of the labeling period (Russo et al., 2017). Short labeling times decrease the amount of s4U-RNA available for analysis. This complicates handling and increasing concerns about contamination from non-s4U-labeled RNAs, although short labeling times also provide the greatest differences for certain RNAs from the steady-state. Long labeling times will eventually lead to labeling of the entire RNA population and reflect the steady-state levels. This will obscure the different kinetics in regulated RNA turnover that these experiments are designed to study, although more s4U-RNA generally improves the enrichment step. For analysis of individual transcripts, maximum sensitivity is achieved using a labeling time of approximately one half-life (Russo et al., 2017).
Alternatively, a pulse-chase approach can be used to study RNA stability, where cells are labeled with s4U for several hours (pulse), followed by the addition of a molar excess of uridine (chase) in order to inhibit further incorporation of s4U into new transcripts (Figure 1B). Cells are harvested at different times following the addition of uridine, and the relative proportion of s4U-RNA remaining can be fit to a single exponential decay function that reveals the half-life of the RNA. While ideally the length of the pulse should not affect the calculation of RNA half-life because s4U incorporation is assumed to be a homogenous process, in practice different sub-populations of RNA may have different s4U labeling and in this case a shorter pulse can be problematic. Therefore, we recommend that the length of s4U pulse be tuned such that most transcripts are at the steady-state of s4U incorporation.
The accuracy achieved when determining a half-life for an RNA also depends on the abundance of the RNA and sequencing depth. Different RNA species have different half-lives and no one labeling time is optimal for all RNAs in a genome-wide experiment. Therefore, the time of labeling should be adjusted for the RNA species of interest (Figure 1A). For example, very transient RNAs like introns, enhancer RNAs and antisense transcripts are ideally captured following 5 min of s4U (Schwalb et al., 2016), whereas most mRNAs can be sufficiently labeled following minutes to hours of s4U treatment (Russo et al., 2017; Schofield et al., 2018). For very stable transcripts (including most mature miRNAs), a much longer s4U treatment (several hours to days) may be helpful (Duffy et al., 2015). In an ideal experiment, the absolute RNA half-life should not change with the labeling time, and half-lives of RNAs between different experiments with different labeling times should be comparable. However, in practice different types of experimental bias (see below) can influence RNA half-life measurements, and therefore comparing the relative half-lives of RNAs within a single experimental condition can be more reliable than comparing the absolute half-lives of RNAs between different experiments. In addition, s4U incorporation has been demonstrated to vary widely by cell type (Russo et al., 2017), so the desired s4U incorporation for a given cell type should be determined empirically, either by qPCR of a short-lived transcript or by directly quantifying the incorporation of s4U by dot blot or mass spectrometry (Dölken et al., 2008; Hafner et al., 2010).
s4U-RNA isolation and handling
RNA handling is critical to analysis of s4U RNA, and while many of the considerations are the same as for handling unlabeled RNA (Madabusi, Latham, & Andruss, 2006), there are additional considerations when working with s4U-RNA. In some cases, the presence of s4U can lead to selective loss of these subpopulations while handling, resulting in loss of new transcripts. The thiol of s4U has the potential to form disulfides and/or oxidation products during RNA isolation and handling, and several protocols recommend the addition of reducing agents such as dithiothreitol (DTT) or β-mercaptoethanol (β-me) during RNA isolation (Duffy & Simon, 2016; Schofield et al., 2018; Spitzer et al., 2014). Care should be taken to effectively remove these added reducing reagents before downstream enrichment and/or nucleoside recoding to prevent interfering with the s4U-specific thiol chemistry.
Potential s4U toxicity
While s4U has generally been found to cause minimal toxicity in the cells, some reports have revealed that s4U can be toxic to certain cell types at high concentrations. For example, s4U has been reported to cause nucleolar stress in human U2OS cells treated with as little as 50 μM s4U (Burger et al., 2013). However, this cellular toxicity has not been widely observed in different cell types, even at higher s4U concentrations (Duffy et al., 2015; Gregersen et al., 2014; Herzog et al., 2017; Russo et al., 2017; Schofield et al., 2018). In addition, cells treated with s4U for longer labeling times are more likely to exhibit cellular toxicity. Therefore, lower concentrations of s4U are desirable for longer labeling, whereas higher concentrations of s4U may not cause adverse effects if the labeling time is very short (i.e. minutes). These concerns can be addressed by conducting s4U toxicity studies (for examples see (Burger et al., 2013; Schofield et al., 2018) when performing s4U metabolic labeling in a new cell type or at a higher concentration than previously reported. A summary of tested s4U concentrations in different cell types has been previously reported (Russo et al., 2017) which we have updated and expanded in Table 1. While the source of cell type-specific differences in cellular toxicity in the presence of s4U and other metabolic labeling reagents (e.g. BrU, EU) remain unknown, potential sources of variability include reagent cellular permeability and active transport across the cell membrane, biochemical incorporation into the available UTP pool, the rate of total RNA metabolism, potential off-target effects of the reagent and any metabolic derivatives, and possible toxicity of metabolic labeling reagent within RNA, especially ribosomal RNA.
Table 1:
Examples of studies using s4U labeling to assess mRNA stability in cell lines of different organisms.
| Human | |||
|---|---|---|---|
| Cell type | Concentration | Time of feed | Citation |
| BL41 (lymphoma) | 100 µM s4U | 1h | Dölken 2008, Friedel 2009 |
| HeLa (cervical carcinoma) | 100 µM s4U 250 µM s4U |
1h 1h |
Dölken 2008 Borowski 2014 |
| Jurkat (T tymphocyte) | 100 µM s4U 200 µM s4U |
1h 1h |
Dölken 2008 Blackinton 2016 |
| DG75 (B lymphocyte) | 100 µM s4U 500 µM s4U |
1h 5–60 min |
Dölken 2008 Windhanger 2012 |
| HEK293T (embryonic kidney) | 250 µM s4U 100 µM s4U 700 µM s4U |
1h 20 min-22 d 1h |
Borowski 2014 Duffy 2015 Gregersen 2014, Duffy 2015 |
| 293A (embryonic kidney | 2.5 µM s4U | 2h | Stubbs 2015 |
| iPS (stem cell | 400 µM s4U | 2h | Russo 2017 |
| LCL (lymphoblast) | 200 µM s4U | 2h | Duan 2013 |
| MEWO (melanoma) | 500 µM s4U | 1h | Azarkh 2011 |
| Primary T-cells | 200 µM s4U | 1-6h | Payne 2014 |
| RAJI (lymphoblast) | 300 µM s4U | 30 min | Donato 2016 |
| RCC (renal carcinoma) | 2 µM s4U | 2h | Bresson 2015 |
| SH-SY5Y (neuroblastoma) | 500 µM s4U | 30 min | Schwarzl 2015 |
| 143B (osteosarcoma | 250 µM s4U | 1h | Borowski 2014 |
| K562 | 500 µM s4U 100 µM s4U 1 mM s4U |
5 min 4h 5 min |
Schwalb 2016 Schofield 2018 Duffy 2018 |
| MDA-MB-231 (breast cancer) | 200 µM s4U | 20 min | Cho 2018 |
| MCF7 (breast cancer) | 400 µM s4U | 2h | Schwanhausser 2011 |
| Mouse | |||
| NIH-3T3 (fibroblasts) | 200 µM s4U 500 µM s4U 400 µM s4U |
1h <30 min 2h |
Dölken 2008 Dölken 2008 Schwanhausser 2011 |
| RAW 264.7 (macrophages) | 100 µM s4U | 1h | Dölken 2008 |
| SVEC 4–10 (endothelial) | 100 µM s4U | 1h | Dölken 2008 |
| MEF | 1 mM s4U 200 µM s4U |
1h 2h |
Schofield 2018 Kenzelmann 2007 |
| Primary T-cells | 250 µM s4U | 15 min | Li 2017 |
| Dendritic cells | 150 µM s4U | 45 min | Rabani 2011 |
| Yeast | |||
| S. cerevisiae + UPRT | 200 µM 4-thiouracil | 3h | Muchel 2011 |
| S. cerevisiae + hENT1 | 500 µM s4U | 3-24 min | Miller 2011 |
|
S. cerevisiae S. pombe |
5 mM 4-thiouracil 500 µM s4U |
6 min 6 min |
Sun 2012 Sun 2012 |
| S. cerevisiae + FUI1 | 100 µM s4U | 2-7 min | Swaitkowska 2012 |
| S. cerevisiae | 500 µM 4-thiouracil | 3-100 min | Neymotin 2014 |
| S. cerevisiae + FUI1 | 500 µM 4-thiouracil | 1.5-5 min | Barrass 2015 |
| S. cerevisiae | 50 µM 4-thiouracil | 16h | Miller 2018 |
| Other | |||
| H. volcanii | 300 µM 4-thiouracil | 45 min | Knüppel 2017 |
| S. acidocaldarius | 135 µM 4-thiouracil | 45 min | Knüppel 2017 |
| D. melanogaster kc167 | 500 µM s4U | 5-20 min | Pai 2017 |
| D. melanogaster kc167 | 300 µM s4U | 30-450 min | Chen and van Steensel 2017 |
| D. melanogaster S2 | 200 µM s4U | 2-6h | Khong 2017 |
| D. rerio (zebrafish) embryo | 1 nL 50 mM s4UTP | 0-9 cell divisions |
Heyn 2014 |
Biochemical enrichment of s4U-RNA
Choice of activated disulfide reagent
Biochemical enrichment with activated disulfides remains the most common method to identify s4U-RNAs for genome-wide studies. A key step in s4U biochemical enrichment is the biotinylation of thiols with activated disulfides. While historically organomercurial matrices were used to purify s4U-RNA, Cleary et al. used HPDP-biotin to enrich metabolically labeled RNA, causing a shift to this reagent for the purification of s4U-RNAs. Although this activated disulfide has been applied in numerous metabolic labeling experiments, s4U-RNA enrichment with HPDP-biotin was shown to lead to an enrichment bias (i.e. a skew toward the purification of longer RNAs with more U’s). This bias can be corrected for bioinformatically (See (C. Miller et al., 2011; Rabani et al., 2011; Rabani et al., 2014), or through shearing the RNA prior to enrichment (Schwalb et al., 2016). Labeling bias was shown to be one result of inefficient disulfide formation between HPDP-biotin and s4U, while the more efficient activated disulfide MTS-biotin alleviates this bias and increases yields in s4U-RNA enrichment (Figure 2).
Figure 2:

(A) Estimated range of RNA half-lives in mammalian cells based on data from (Schofield et al., 2018; Schwalb et al., 2016; Yang et al., 2003). (B) Simulation of RNA decay rates for a s4U pulse-chase experiment for three different RNA half-lives, represented as a fraction of the total amount of s4U-labeled RNA at the start of the chase. (C) Simulation of s4U approach-to-equilibrium kinetics for three different RNA half-lives. (D) Kinetics of s4U incorporation during 5 min of s4U labeling for a transient RNA (t1/2 = 5 min) and two mRNAs (t1/2 = 0.5h and 4h).
While there are now approaches to mitigate bias in enrichment, given the number of reports using HPDP-biotin without pre-shearing of the RNA, it is worth considering how HPDP-biotin significantly biases RNA capture toward longer RNAs. Unspliced pre-mRNAs can be significantly longer than spliced mRNAs, and an over-estimation of introns in newly transcribed RNA compromises accurate estimation of processing rates. In addition, lower enrichment yields complicate the calculation of RNA half-lives in methods where rates of transcription, processing and degradation are modeled based on the relative proportion of new RNA (enriched fraction) to pre-existing RNA (not captured fraction), although relative rates between different transcripts are still accurate. Therefore, although issues from labeling bias can be mitigated by bioinformatic modeling or biochemical shearing, an efficient biochemical purification of RNA or enrichment-free chemistry is preferable.
Nonspecific RNA background
All biochemical purifications seek to maximize the amount of real signal (in this case s4U-RNA) and minimize background contamination (non-s4U-RNA). Non-specific contamination has been estimated in some s4U-RNA enrichment protocols as high as 30% (Rabani et al., 2011), which can complicate quantification of new RNAs, particularly splicing rates as highly abundant mRNAs are most likely to contribute to background contamination. Many groups have found different ways to overcome challenges associated with nonspecific background in s4U-RNA enrichment (summarized in Box 2), including pre-blocking beads with polyvinylpyrrolidone (Pai et al., 2017) or glycogen (Swiatkowska et al., 2012), adding more stringent rinses to the streptavidin beads after incubation with biotinylated RNA (Li et al., 2017), and pre-shearing RNA before biochemical enrichment (Duffy & Simon, 2016). Notably, while RNA pre-shearing is not necessary for all metabolic labeling experiments, this fragmentation step is particularly important following short s4U labeling (5 min or less) in order to capture only the portion of the transcript that was newly made during the labeling period. For these very short experiments, failure to fragment RNA before enrichment leads to a 5’ bias in RNA coverage (Schwalb et al., 2016).
Box 2: Potential sources of nonspecific RNA background and methods to filter this contamination experimentally and bioinformatically.
| Source of background | Experimental strategy |
|---|---|
| Nonspecific RNA binding to surfaces | • Use low retention tubes • Transfer streptavidin beads or MTS resin to new tube before elution step • Perform nucleoside conversion chemistry after enrichment to identify s4U-containing reads during bioinformatic analysis |
| Nonspecific RNA binding to beads | • Fragment total RNA before enrichment • Pre-block beads with polyvinylpyrrolidone or glycogen • Add high salt rinses to RNA on beads • Compare s4U-RNA to a non-s4U RNA sample • Perform nucleoside conversion chemistry after enrichment to identify s4U-containing reads during bioinformatic analysis |
| Nonspecific RNA base-pairing with s4U-RNA on beads | • Add low salt and/or denaturing washes to RNA on beads • Perform nucleoside conversion chemistry after enrichment to identify s4U-containing reads during bioinformatic analysis |
| Contamination from endogenously thiolated RNAs (e.g. 2-thiouridine-containing tRNA) | • Compare s4U-RNA to a non-s4U RNA sample |
Others have opted to compare s4U-RNA signal to RNA enriched from cells that were not treated with s4U, as nonspecific background can be bioinformatically calculated from s4U-RNA samples (Barrass et al., 2015). However, this strategy only accounts for nonspecific background on the beads and does not account for unlabeled RNA that is retrieved through base pairing with s4U-RNA. Therefore, background contamination may be underestimated by this approach.
A strategy to avoid the multiple steps required for biotinylation, purification and capture has been developed where MTS activated disulfide was directly coupled to magnetic sepharose, allowing for more stringent rinses and higher fold enrichment of s4U, which is particularly advantageous for small-scale RNA samples including primary tissues and microdissections (Duffy et al., 2018). When using this MTS-resin for s4U-RNA enrichment, we have observed variable amounts of length bias which is not observed with MTS-biotin (Duffy et al., 2015), although these concerns can be addressed by shearing the RNA prior to enrichment.
In addition, nucleoside recoding can be used to filter non-specific background in biochemically enriched s4U-RNA during sequencing (Schofield et al., 2018). In a 5 min s4U metabolic labeling experiment, where the relative proportion of newly transcribed RNA to total RNA is low and enrichment is desirable, nucleoside recoding chemistry was applied to enriched RNA to identify bona fide metabolically labeled RNAs via T-to-C mutations in sequencing. These analyses revealed that, with MTS-biotin enrichment and RNA shearing, nonspecific RNA contamination still comprised 18% of the total signal, but with mutational information these two populations could be analysed separately and new RNAs could be validated in two independent ways (enrichment and T-to-C mutation content). Therefore, we recommend the addition of nucleoside recoding chemistry as a gold-standard for reducing nonspecific RNA background in s4U-RNA enrichment experiments.
Bioinformatic analysis of enriched s4U-RNAs
Bioinformatic analysis of s4U-RNA is largely similar to traditional RNA-seq, the best practices for which have been reviewed elsewhere (Madabusi et al., 2006). Reads are aligned to a reference genome and transcript annotation, and expression levels are quantified. However, several parameters are unique to s4U enrichment and genome-wide sequencing. As discussed above, the incorporation rate of s4U into transcripts of interest and little variability in biochemical enrichment is important for robust bioinformatic quantification. In addition, the accuracy of quantification depends on many factors including the depth of sequencing (i.e. the number of reads that map to the transcripts of interest), normalization between samples (particularly those with different s4U content), and in some cases the ability to identify transcripts from transiently expressed RNA species. These bioinformatic considerations, in conjunction with experimental design and biochemical enrichment, are critical for the interpretation and reproducibility of s4U metabolic labeling experiments.
Unstable transcript identification
Metabolic labeling experiments with short s4U labeling (< 1h), enriches for unstable RNAs including enhancer RNAs (eRNAs), antisense transcription at promoters (uaRNAs), and introns from nascent transcripts, which allows the study of transcription output in cells. These unstable RNA species are difficult to detect in the steady-state population because they are rapidly degraded after transcription and/or processing, and their steady-state levels are very low. Therefore, these transcriptional features are typically not included in gene annotations, so alternative methods are required to accurately quantify unstable transcripts in short s4U labeling experiments. Prior knowledge of the genomic context can aid in the quantification of unstable transcripts; for instance, the presence of CREB-binding protein (CBP) and H3K4me1 was used to identify thousands of activity-dependent enhancers in neurons (Kim et al., 2010). However, for this analysis only regions 1.5 kb from the CBP binding site are considered, so the length of the RNA is not determined using this analysis. In principle, unspliced RNA species including eRNAs, uaRNAs and pre-mRNAs can be identified in regions of continuous transcription. In practice, however, insufficient read depth or repetitive elements within a transcript can affect read mappability and lead to an under-estimation of unstable transcripts when requiring continuous transcription for quantification. Alternatively, several pipelines have been developed that use hidden Markov modeling (HMM) to identify transcriptional units independent of a known transcript annotation or prior knowledge of the genomic context of the transcript (for examples, see (Chae, Danko, & Kraus, 2015; Heinz et al., 2010; Spitzer et al., 2014; Zacher et al., 2017). Bioinformatic methods that were developed to analyze nuclear run-on sequencing data (GRO-seq and PRO-seq) have also been applied successfully to s4U sequencing data, such as identifying intergenic pri-miRNA transcripts (Duffy et al., 2018), suggesting that the general principles of HMM transcript identification are applicable to multiple metabolic labeling strategies, although depth of sequencing and mappability of repeat elements should be considered for these analyses as well.
Normalization
RNA-seq libraries can be normalized based on reads per kilobase per million reads (RPKM), but s4U-RNA samples cannot be directly compared to RNA-seq or even other s4U-RNA samples when different lengths of s4U labeling time are used (Figure 3A). Therefore, an exogenous spike-in must be used to normalize different s4U-RNA-seq samples in order to bioinformatically compare them. In order to control for variability in handling during s4U-RNA enrichment, an exogenous s4U-labeled RNA spike-in can be added before biotinylation, although in theory these spike-ins can be added during cell lysis to control for variability in total RNA purification as well. For the study of mammalian or S. cerevisiae RNA dynamics, S. pombe cells can be labeled with s4U and the same amount of s4U-labeled S. pombe RNA is added to each experimental sample of 4-thiouracil-treated S. cerevisiae cells (Sun et al., 2012). Following enrichment, the samples can be analyzed by a custom microarray for both S. pombe and S. cerevisiae, where S. cerevisiae s4U-RNA levels are normalized by setting the median intensity of S. pombe s4U-RNA for all samples equal to 1. A similar strategy was be used for a high-throughput sequencing format ; 4-thiouracil-labeled S. cerevisiae RNA was added to HEK293T s4U-RNA samples, and the resulting RNA was sequenced and normalized based on the upper quantile of expression for the S. cerevisiae s4U-RNA spike-in (Gregersen et al., 2014). In vitro transcribed spike-ins can also be synthesized with s4U by adding s4UTP to the transcription reaction (Munchel et al., 2011; Neymotin et al., 2014), followed by addition to total RNA and identification by high-throughput sequencing (Figure 3A).
Figure 3:

Significance of inefficient chemistry toward length bias in s4U metabolic labeling experiments. (A) Chemistry of activated disulfide reactivity with s4U. When HPDP-biotin is used as the activated disulfide, any biotin-s4U (bio-s4U) product results in a more activated disulfide due to the electron-poor pyrimidine ring of s4U, therefore favoring the reverse rather than the forward biotinylation reaction. In contrast, MTS-biotin reacts irreversibly with s4U to form the bio-s4U product under the reaction conditions, and the covalent disulfide bond can only be reversed under reducing conditions. (B) Schematic of s4U-RNA enrichment with HPDP- and MTS-biotin. Efficient activated disulfide reactivity of MTS-biotin results in greater yields of s4U-RNA and alleviates potential biases toward longer RNAs with more uridines.
While in principle, normalization with exogenous spike-ins, either synthetic or from S. pombe, are an effective way to normalize different s4U-RNA levels from different times of metabolic labeling, in practice the normalization has been shown to be variable (Lugowski, Nicholson, & Rissland, 2018). Specifically, small variability in enrichment from cells labeled with s4U-RNA for only a few minutes can significantly influence synthesis and degradation rates in an enrichment-independent manner. An alternative strategy uses introns to normalize between s4U-RNA samples in order to avoid potential variability from exogenous spike-ins (DRUID, Lugowski et al., 2018). Enrichment-free methods based on chemistry that recodes s4U to induce mutations (discussed below) can also alleviate these normalization issues, as information about steady-state transcripts and new RNAs are included in the same sample and traditional normalization strategies for RNA-seq libraries can be used to compare different samples.
Calculating rates of RNA metabolism
The final step of any genome-wide s4U metabolic labeling experiment is to use the quantification of s4U-RNAs in order to gain insight into RNA metabolism. Consequently, the models used to calculate these rates of transcription, processing and degradation should be considered carefully. While considerable progress has been made in developing computational algorithms to analyze metabolic labeling data, most models have relatively simple assumptions, which may not accurately reflect the kinetics of RNA metabolism. For example, many RNA decay assays, including s4U pulse-chase, assume simple first-order decay kinetics. RNA turnover assays that compare a single sample of enriched s4U-RNA to the steady-state assume that all s4U-RNA has been captured (Dölken et al., 2008), while enrichment with HPDP-biotin was later shown to be inefficient at capturing s4U-RNA, resulting in an under-estimation of the proportion of new transcription (Duffy et al., 2015). In addition, while properties of RNA metabolism can be studied using many variations of s4U metabolic labeling (see Figure 1), very few studies have directly compared the relative power of different methods side-by-side for the ability to accurately reflect absolute rates of RNA metabolism. Therefore, the same techniques reviewed here are the ones necessary to tease apart these differences.
Enrichment-free identification of s4U-RNA
Many of the challenges identifying s4U-RNAs through biochemical enrichment can be avoided by chemical techniques that allow nucleotide recoding (Herzog et al., 2017; Riml et al., 2017; Schofield et al., 2018). Chemical treatment of s4U-RNA can be used to convert s4U into a nucleotide with an altered hydrogen bonding pattern that is more likely to be read as a C during reverse transcription. Two chemical approaches that rely on different types of chemical reactivity have been developed to globally map sites of s4U incorporation across the transcriptome (Figure 4A). These approaches provide opportunities to identify s4U-RNAs that are complementary to enrichment-based approaches (Herzog et al., 2017; Schofield et al., 2018).
Figure 4:

Methods to normalize s4U-RNA data after high-throughput sequencing. (A) After cells are metabolically labeled with s4U and total RNA is extracted, exogenous s4U-RNA (s4U-labeled RNA from S. pombe, or in vitro-transcribed s4U-RNAs) are added to each sample and then enriched. Alternatively or in addition, exogenous RNA without s4U can also be added to samples after enrichment. Samples are then analyzed by high-throughput sequencing and normalized based on the number of reads that align to the spike-in sample. (B) Samples can also be enriched without RNA spike-ins and normalized based on the relative proportion of introns in each sample (Lugowski et al., 2018). First, intron coverage for each transcript is determined based on the number of reads that map to introns in the longest isoform of a given transcript without overlapping exons or any other isoform. Next, introns are filtered for coverage and time-dependent changes in expression. Reads that map to exons are finally normalized to the sum of all reads mapping to the well-behaved intron set.
Sites of s4U lead to low levels of T-to-C mutations upon reverse transcription (Hafner et al., 2010; Rabani et al., 2014) but these mutation levels are too low to identify most s4U-labeled transcripts. To increase the frequency of apparent U-to-C transitions, the s4U hydrogen bonding pattern can be chemically recoded. The tendency of s4U to chemically convert to products that produce T-to-C mutations in sequencing experiments has been used as part of photoactivatable ribonucleoside-enhanced crosslinking and immunoprecipitation (PAR-CLIP) to map protein-RNA interactions at nucleotide resolution (Ascanno, Hafner, Cekan, Gerstberger, & Tuschl, 2012; Hafner et al., 2010). Chemistry specifically developed to induce U to C mutations in global sequencing experiments has been developed based on the two chemical strategies: (1) Alkylating the thione of s4U to make a S4-alkyl-thiouridine (Herzog et al., 2017) (2) oxidize the thione of s4U to create a leaving group, which allows an amine to substitute into the ring yielding a N4-alkyl cytidine analogue (Schofield et al., 2018).
Chemistry to induce T-to-C mutations during sequencing
In the case of alkylation chemistry, the thione of s4U can act as a nucleophile. The nucleophilicity of the thione is also the basis of the disulfide chemistry described above. While other bases in RNA can be alkylated, especially N7 of guanosine (reviewed by (Gillingham, Geigle, & Anatole von Lilienfeld, 2016), early work demonstrated that s4U in E. coli tRNA could be specifically alkylated with chemical labels based on alkyl halides (Hara, Horiuchi, Saneyoshi, & Nishimura, 1970). For the purposes of nucleotide recoding, alkylation of the thione traps thiouridine in a tautomer with one of the hydrogen bonds recoded (from acceptor-donor-acceptor to acceptor-acceptor-acceptor, Figure 4B). This alkylation results in a base that is more likely to be read as C than U during reverse transcription. The use of iodoacetamide was developed for identifying sites of s4U in RNA in 3’-ends of RNA across the transcriptome (SLAM-seq, (Herzog et al., 2017), allowing analysis of s4U-RNA including the steady state RNA dynamics of poly-adenylated RNAs.
Alternatively, the hydrogen bonding of s4U can be fully recoded using oxidative nucleophilic aromatic substitution. In this case the thione of s4U is oxidized to create a leaving group which can be displaced with alkylamines to make cytidine analogues. Oxidative nucleophilic aromatic substitution chemistry was first used with s4U to specifically modify E. coli tRNA s4U bases (Ziff & Fresco, 1969), (Hayatsu & Yano, 1969). Extending this reactivity for use transcriptome-wide involved migrating from strong oxidants such as KMnO4 to more specific oxidants such as NaIO4 (Schofield et al., 2018). Similarly, while the use of ammonia provides the bone fide cytidine product (Hayatsu & Yano, 1969; Riml et al., 2017), amines with lower pKa values such as trifluoroethylamine can be used that remain deprotonated at neutral pH values. We refer to this reactivity as TimeLapse chemistry. The effect of TimeLapse chemistry is to convert s4U to cytosine analogues, resulting in complete recoding of s4U from the pattern of U to the pattern of C (from acceptor-donor-acceptor to donor-acceptor-acceptor). TimeLapse-seq was validated to monitor the location of s4U in transcriptome-wide RNA-seq experiments where intronic reads, non-poly-adenylated RNAs, and different transcript isoforms could be observed (Schofield et al., 2018).
Both alkylation and TimeLapse chemistry efficiently convert s4U to their respective analogue (s4U* or C*). While the mutation rate in the context of a complex RNA mixture has not been determined for either approach, both reactions convert the majority of s4U in model substrates. Reverse transcriptase enzymes have a non-zero termination rate for even natural nucleotides, so it is reasonable to wonder if the adducts induced by these transformations (S4-alkyl thiouridine for SLAM-seq, and N4-alkyl cytidine for TimeLapse-seq) induce an increase in termination in reverse transcriptase reactions. When analyzed using test substrates, neither SLAM-seq nor TimeLapse-seq led to a substantial increase in termination of reverse transcription at modified bases. The importance of full versus partial recoding of the s4U hydrogen bonding pattern will require a side-by-side analysis of both methods, which has not yet been published.
Bioinformatic analysis of T-to-C mutations
After conversion of s4U to s4U* or C*, the reverse transcriptase records the site of s4U as a G in the cDNA, and these locations are identifiable as T-to-C mutations in sequencing data. Ideally all sequencing reads could be classified as either resulting from metabolically labeled s4U-RNA or as unlabeled RNA. The combination of s4U treatment conditions and chemistries that have been developed generally lead to 1-10% T-to-C mutation rates. These rates are low enough to avoid substantial problems during read alignment using standard pipelines. Under conditions where 2% of uridines are substituted with s4U and result is chemically induced T-to-C mutations, many reads from metabolically labeled transcripts will have zero mutations (as predicted from a binomial distribution). For example, in a sequencing experiment with a single-end 50 nt reads, with a 2% mutation rate less than a quarter of the reads (22%) are expected to contain s4U-induced mutations. The number of reads with mutations can be increased by increasing s4U incorporation rates (e.g., higher [s4U] treatments) or by using a longer sequencing format. In the example here, switching from a single-end 50 nt format to a paired-end 150 nt format substantially increases the fraction of s4U-RNA reads expected to contain mutations (from 22% to 78%). The 1-10% T-to-C mutation rates observed in these experiments is well above background rates which vary experiment to experiment but tend to be less than 0.2% using standard RNA-seq workflows. It is worth noting that even these low background rates of T-to-C mutations can become problematic when there are very high numbers of unlabeled transcripts. For example, if only 1% of a transcript is labeled with s4U, the background mutations from unlabeled transcripts will frequently outnumber the s4U induced mutations.
There are two approaches that have been used to analyze steady state RNA dynamics from nucleotide recoding experiments with s4U: (1) using averaged mutation rates (Herzog et al., 2017); or (2) single-molecule analysis of sequencing reads using statistical modeling (Schofield et al., 2018). The first approach (using averaged mutation rates) was applied to a chase-style experiment where cells were treated for a long time with s4U (100 µM for 12h) prior to a chase with uridine. The change in background-subtracted average mutation rates over time was used for curve fitting to determine the RNA half-lives.
Alternatively, statistical modeling can be used to take advantage of single-molecule information that is inherent to techniques such as TimeLapse-seq (Schofield et al., 2018). This information is lost when T-to-C mutation rates are averaged. Each sequencing read reports on a single RNA transcript that was either s4U-labeled or unlabeled. The distribution of the number of T-to-C mutations in each read can be used to infer the likelihood that the read came from an RNA was labeled (Schofield et al., 2018). The distribution of mutations can be modeled as a mixture of two populations: labeled and unlabeled RNA molecules. These single-molecule distributions can be modeled using either a Poisson (using the rate of mutations per read) or a binomial model (which accounts for the number of uridines in each read). The background distributions can be determined from control experiments, and the mutation rates of s4U-labeled RNAs can be inferred from the data. Using likelihood maximization or bayesian hierarchical modeling, the fraction of each transcript (or other feature such as exon) that was labeled with s4U can be inferred with high reproducibility. The advantage of using single molecule information from the sequencing reads is particularly apparent in cases where only a small fraction of RNA is labeled. In this case, the average T-to-C mutation rate is very close to background levels, yet the increase in mutations from the labeled RNAs is apparent from the ratios of different numbers of mutations, including high numbers of mutations (which are very rare in unlabeled reads; in the example in Box 3 less than 0.002% of reads derived from pre-existing transcripts have three or greater mutations). By treating the data as single-molecule data, the frequency of mutations can be used to reproducibly infer even small fractions of labeled RNA.
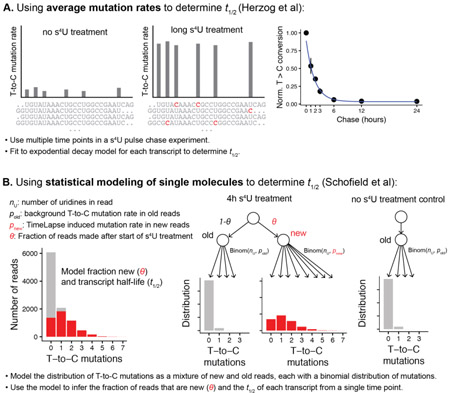
Box 3: Overveiew of two different approaches to analyse data from nucleotide recoding experiments. A. Simulated data demonstrating how average T-to-C mutation rates can be used to fit transcript half-lives. B. Simulated data demonstrating how statistically modelling of the mutations in single molecules of RNA of 100 nt reads with a 0.2% background rate of T-to-C mutations at each uridine (pold) and a 5% mutation rate of T to C mutations at each uridine in labelled reads (pnew). In this simulated example, half of the reads are from new, labelled transcripts and half are from unlabelled transcripts (θ = 0.5), representing a transcript with a 4h half-life.
Nucleotide recoding as a method to identify s4U-RNA offers several advantages over enrichment-based techniques. Nucleotide recoding requires fewer steps than biochemical approaches and many samples can be processed in parallel. Because there is no need for biochemical purification, these approaches can be performed on very little input RNA---orders of magnitude less RNA than is generally required for biochemical enrichment. One particularly useful advantage is that all the reads are sequenced in the same experiment; the data are internally normalized, avoiding the challenges described above. For nucleotide recoding approaches to be effective, the total amount of s4U-labeled material that is sequenced needs to be a significant fraction of a sequencing lane. Very short s4U treatments (such as the 5 min transient transcriptome sequencing) produce very few reads that have T-to-C mutations, and therefore biochemical enrichment is still advantageous. Even in cases that require biochemical enrichment, however, including nucleotide recoding after enrichment is useful to distinguish real signal from contaminating non-s4U-labeled background.
EXTENSIONS OF TRANSCRIPTIONAL ANALYSIS
Induced changes in RNA metabolism
While s4U metabolic labeling does not require the induction of genes or the use of reporters, many of the methods described above can be used to measure changes upon cellular perturbation as well as steady-state RNA population dynamics. For instance, s4U metabolic labeling has been used with s4U treatment at multiple time points following interferon (INF) stimulation in mouse embryonic fibroblasts (Dölken et al., 2008), lipopolysaccharide (LPS) stimulation in dendritic cells (Rabani et al., 2011), and interleukin-7 stimulation in naïve T-cells (Li et al., 2017) in order to assess the relative contribution of transcription and processing to the changes in RNA steady-state levels that occur upon stimulation. Analogous methods have been applied in S. cerevisiae to measure transcriptional changes upon osmotic stress (C. Miller et al., 2011) and Ccr4-Not knockout (Sun et al., 2012). In addition, enrichment-free nucleoside conversion has been used to observe transcriptional changes after 1 hour of heat shock in MEF cells, which revealed induction of heat shock transcripts that were not detectable by RNA-seq alone at this short time scale (Schofield et al., 2018).
An important component of these protocols is using bioinformatic analyses to assess the relative contribution of transcription, processing, and decay to changes in total RNA levels. To model synthesis, processing, and degradation rates, Rabani et al. developed a computational approach that infers degradation rates based on steady-state RNA levels from total RNA and s4U-RNA populations (Rabani et al., 2014). The simplest model (“constant degradation”) assumes that no degradation of s4U-RNA occurs during the metabolic labeling period, whereas the “varying degradation” model assumes that degradation occurs during labeling. Transcripts that significantly reject the constant degradation model are highly transient, so the degradation rate significantly contributes to changes in steady-state levels. Processing rates can be estimated by comparing the ratio of intronic reads to spliced reads that contain exon-exon junctions. These methods were later refined and formalized into publicly available computational pipelines available in different programming languages (DRiLL (Rabani et al., 2014) is available in MATLAB, and INSPeCT (de Pretis et al., 2015) in R). Therefore, small changes in experimental design and bioinformatic analysis can easily extend our understanding of steady-state RNA dynamics to mechanisms of inducible changes in RNA metabolism that allow cells to respond to the surrounding environment.
RNAPII elongation
Recent genome-wide experiments have revealed that RNAPII pausing, as well as the rate of elongation following release from pausing, are tightly coupled to co-transcriptional processes such as splicing, termination, and RNA stability (reviewed by (Jonkers & Lis, 2015). Measuring RNAPII elongation rates genome-wide requires quantifying the distance that the polymerase travels as a function of elapsed time. This can be achieved by treatment with 5,6-dichloro-1-β-D-ribofuranosylbenzimidazole (DRB), a small molecule that inhibits pTEFb phosphorylation and therefore prevents RNAPII from releasing from the pause site into productive elongation. Even if s4U is added during the DRB treatment, a paused polymerase will not incorporate s4U. Upon washout of DRB, new transcripts are labeled with s4U, and cells are harvested at multiple times following washout. Because RNAPII becomes effectively synchronized at the pause site during DRB treatment, the distance RNAPII travels directly measures the elongation rate, and rates can be measured genome-wide (Fuchs et al., 2014). RNAPII elongation rates calculated by this method are unable to detect variations in elongation within genes, and only relatively long genes can be used. During the same metabolic labeling period, RNA produced from small genes would be synthesized and exported to the cytoplasm faster than larger genes, and therefore the rates of RNAPII elongation at these short genes requires temporal resolution that is not currently feasible. Regardless, genome-wide DRB-seq methods offer improvements over previous methods that use RT-PCR (Singh & Padgett, 2009) or fluorescent labeling (Darzacq et al., 2007) to measure elongation rates of only a handful of genes at a time. In addition, DRB inhibition combined with s4U metabolic labeling was recently used to demonstrate that genome-wide variation in RNAPII elongation rate is consistent between cell culture models and mouse primary neurons (Duffy et al., 2018).
Tissue-specific transcription
In addition to capturing new RNAs with temporal resolution, s4U metabolic labeling can also be leveraged for spatial resolution of new transcripts in a whole organism (TU-tagging, (M. R. Miller, Robinson, Cleary, & Doe, 2009). The UPRT enzyme, which converts 4-thiouracil into 4-thio-UTP, can be expressed in the cell type of interest via targeted transgene expression, followed by injection of the nucleobase 4-thiouracil (4TU). Only cells that express the UPRT transgene are able to incorporate 4TU into newly transcribed RNA, thus allowing cell type-specific RNA enrichment from whole tissues. While in principle TU tagging can also give temporal resolution of transcripts via the methods described above, in practice the kinetics of 4TU incorporation into new transcripts in a whole organism are not well characterized, which may complicate analyses of RNA stability or transcription rate. Regardless, this approach is particularly advantageous over methods that isolate cell type specific translating ribosomes (TRAP,(Heiman et al., 2008) because TU tagging captures non-coding RNAs in addition to protein coding transcripts, or methods that isolate cell-specific nuclei (INTACT,(Deal & Henikoff, 2010) because only nuclear RNAs are detected.
Conclusion
Methods that enable the study of RNA population dynamics give a rich picture of how steady-state RNA levels are regulated and how cells dynamically respond to environmental stimuli. 4-thiouridine has proven to be a versatile metabolic label for understanding RNA transcription, processing and degradation in a variety of organisms. For example, short pulses of s4U have been used to measure differences in transcriptional efficiency of TATA-less versus TATA-containing promoters in yeast (Donczew & Hahn, 2018) as well as the influence of the RNA helicase DDX54 on pre-mRNA splicing efficiency in response to DNA damage (Milek et al., 2018). RNA half-life measurements have demonstrated the contribution of RNA helicase MOV10 RNA stability (Gregersen et al., 2014) as well as the importance of m6A to regulate RNA stability during interleukin-7-dependent T-cell maturation (Li et al., 2017). Approach-to-equilibrium experiments have helped to identify cellular factors that facilitate rapid mRNA degradation in response to changes in environmental nitrogen in S. cerevisiae (D. Miller, Brandt, & Gresham, 2018) and revealed a subset of mature miRNAs with rapid turnover in proliferating HEK293T cells (Duffy et al., 2015). In addition, transcript isoforms like ASXL1 in human cells have been shown to have different stabilities, which suggests that differential isoform stability may be a more widespread consequence of alternative splicing (Schofield et al., 2018).
These methods can also be modified and combined to add additional power to s4U-RNA identification, as demonstrated in Figure 1. For example, tissue-specific expression of the UPRT enzyme enables the spatial and temporal analysis of RNA dynamics in living organisms (TU-tagging). In general, these strategies are also compatible with other RNA-seq workflows. For example, s4U-RNAs enriched after a short pulse of s4U can be treated with nucleoside recoding chemistry in order to bioinformatically filter out nonspecific RNA background using the presence of T-to-C mutations in enriched reads (TT-TimeLapse-seq, Schofield et al. 2018). Enrichment is required in these cases in order to detect the small proportion of s4U-RNAs that would not be abundant enough to easily detect with nucleoside recoding strategies alone, and the addition of T-to-C mutations in the RNA-seq data allows the precise calculation of RNA contamination levels and removal of non-s4U reads from downstream analysis. Given that all of the information from biochemical enrichment experiments is retained when T-to-C mutations are added, there is good reason to apply recoding chemistry when performing biochemical enrichment experiments.
We anticipate that recent improvements to s4U metabolic labeling techniques, including more efficient chemistry, single-step capture of s4U, improved normalization strategies and enrichment-free nucleoside conversion chemistry, will provide a robust toolkit of experimental strategies to understand RNA population dynamics in many biological contexts. Recent advances in single-cell RNA sequencing also present the opportunity to apply s4U metabolic labeling to understand cellular heterogeneity of RNA population dynamics. In addition, improvements to sample normalization, as well as the addition of nucleoside conversion chemistry to filter nonspecific RNA contamination, should enable more robust measurements of RNA transcription and processing events genome-wide. With proper considerations for experimental design, RNA handling and bioinformatic analysis, s4U metabolic labeling holds the power to help reveal the numerous strategies that organisms have evolved in order to regulate RNA population dynamics.
Figure 5:

(A) Hydrogen bonding pattern of modified or recoded s4U nucleosides following enrichment-free chemistry. (B) Example TimeLapse-seq tracks adapted from Schofield et al. 2018 for faster turnover (Ier3) and slower turnover (Srsf3) transcripts in MEF cells treated with 1 mM s4U for 1h.
Acknowledgments
We thank members of the Simon lab for helpful discussions and comments on the manuscript. Our apologies to authors whose work could not be cited or discussed here because of space limitations. This work was supported by National Science Foundation [DGE1122492 to E.E.D.]; the National Institute of Health [T32GM007223 to E.E.D., 1DP2HD083992 to M.D.S.]; and the Searle Scholars Program [to M.D.S.].
Contributor Information
Erin E. Duffy, Department of Molecular Biophysics & Biochemistry, Yale University, New Haven, CT 06511, USA, Chemical Biology Institute, Yale University, West Haven, CT 06516, USA, Erin.Duffy@yale.edu.
Jeremy A. Schofield, Department of Molecular Biophysics & Biochemistry, Yale University, New Haven, CT 06511, USA, Chemical Biology Institute, Yale University, West Haven, CT 06516, USA, Jeremy.Schofield@yale.edu.
Matthew D. Simon, Department of Molecular Biophysics & Biochemistry, Yale University, New Haven, CT 06511, USA, Chemical Biology Institute, Yale University, West Haven, CT 06516, USA, Matthew.Simon@yale.edu.
References
- Ascanno M, Hafner M, Cekan P, Gerstberger S, & Tuschl T (2012). Identification of RNA-protein interaction networks using PAR-CLIP. Wiley Interdisciplinary Reviews: RNA, 3(2), 159–177. doi: 10.1002/wrna.1103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrass JD, Reid JEA, Huang Y, Hector RD, Sanguinetti G, Beggs JD, & Granneman S (2015). Transcriptome-wide RNA processing kinetics revealed using extremely short 4tU labeling. Genome Biology, 16, 282. doi: 10.1186/s13059-015-0848-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burger K, Mühl B, Kellner M, Rohrmoser M, Gruber-Eber A, Windhager L, … Eick D (2013). 4-thiouridine inhibits rRNA synthesis and causes a nucleolar stress response. RNA biology, 10(10), 1623–1630. doi: 10.4161/rna.26214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chae M, Danko CG, & Kraus WL (2015). groHMM: a computational tool for identifying unannotated and cell type-specific transcription units from global run-on sequencing data. BMC Bioinformatics, 16, 222. doi: 10.1186/s12859-015-0656-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchman LS, & Weissman JS (2011). Nascent trascript sequencing visualizes trascription at nucleotide resolution. Nature, 469(7330), 368–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleary M, Meiering C, Jan E, Guymon R, & Boothroyd J (2005). Biosynthetic labeling of RNA with uracil phosphoribosyltransferase allows cell-specific microarray analysis of mRNA synthesis and decay. Nature Biotechnology, 23(2), 232–237. doi: 10.1038/nbt1061 [DOI] [PubMed] [Google Scholar]
- Core LJ, Waterfall JJ, & Lis JT (2008). Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science, 322(5909), 1845–1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darzacq X, Shav-Tal Y, de Turris V, Brody Y, Shenoy SM, Phair RD, & Singer RH (2007). In vivo dynamics of RNA polymerase II transcription. Nature Structural & Molecular Biology, 14, 796–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Pretis S, Kress T, Morelli MJ, Melloni GEM, Riva L, Amati B, & Pelizzola M (2015). INSPEcT: a computational tool to infer mRNA synthesis, processing, and degradation dynamics from RNA- and 4sU-seq time course experiments. Bioinformatics, 31(17), 2829–2835. [DOI] [PubMed] [Google Scholar]
- de Pretis S, Kress T, Morelli MJ, Sabo A, Locarno C, Verrecchia A, … Pelizzola M (2017). Integrative analysis of RNA polymerase II and transcripitonal dynamics upon MYC activation. Genome Research, 27, 1658–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deal RB, & Henikoff S (2010). A simple method for gene expression and chromatin profiling of individual cell types within a tissue. Developmental Cell, 18(6), 1030–1040. doi: 10.1016/j.devcel.2010.05.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dölken L, Ruzsics Z, Rädle B, Friedel C, Zimmer R, Mages J, … Koszinowski U (2008). High-resolution gene expression profiling for simultaneous kinetic parameter analysis of RNA synthesis and decay. RNA (New York, N.Y.), 14(9), 1959–1972. doi: 10.1261/rna.1136108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donczew R, & Hahn S (2018). Mechanistic differences in transcription initiation at TATA-less and TATA-containing promoters. Molecular and Cellular Biology, 38(1), e00448–00417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy EE, Canzio D, Maniatis T, & Simon MD (2018). Solid phase chemistry to covalently and reversibly capture thiolated RNA. Nucleic Acids Res, gky556. doi: 10.1093/nar/gky556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy EE, Rutenberg-Schoenberg M, Stark CD, Kitchen RR, Gerstein MB, & Simon MD (2015). Tracking Distinct RNA Populations Using Efficient and Reversible Covalent Chemistry. Molecular Cell, 59(5), 858–866. doi: 10.1016/j.molcel.2015.07.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy EE, & Simon MD (2016). Enriching s4U-RNA Using Methane Thiosulfonate (MTS) Chemistry. Current Protocols in Chemical Biology, 8(4), 234–250. doi: 10.1002/cpch.12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedel CC, Dolken L, Ruzsics Z, Koszinowski U, & Zimmer R (2009). Conserved principles of mammalian transcription regulation revealed by RNA half-life. Nucleic Acids Research, 37(17), e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs G, Voichek Y, Benjamin S, Gilad S, Amit I, & Oren M (2014). 4sUDRB-seq: measuring genomewide transcriptional elongation rates and initiation frequencies within cells. Genome Biology, 15(5), R69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillingham D, Geigle S, & Anatole von Lilienfeld O (2016). Properties and reactivity of nucleic acids relevant to epigenomics, transcriptomics, and therapeutics. Chem Soc Rev, 45(9), 2637–2655. [DOI] [PubMed] [Google Scholar]
- Gregersen LH, Schueler M, Munschauer M, Mastrobuoni G, Chen W, Kempa S, … Landthaler M (2014). Mov10 is a 5’ to 3’ RNA helicase contributing to UPF1 mRNA target degradation by translocation along 3’ UTRs. Mol Cell, 54(4), 573–585. doi: 10.1016/j.molcel.2014.03.017 [DOI] [PubMed] [Google Scholar]
- Hafner M, Landthaler M, Burger L, Khorshid M, Hausser J, Berninger P, … Tuschl T (2010). Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell, 141(1), 129–141. doi: 10.1016/j.cell.2010.03.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara H, Horiuchi T, Saneyoshi M, & Nishimura S (1970). 4-Thiouridine-specific spin-labeling of E. coli transfer RNA. Biochem Biophys Res Commun, 38(2), 305–311. [DOI] [PubMed] [Google Scholar]
- Hayatsu H, & Yano M (1969). Permanganate oxidation of 4-thiouracil derivatives. Isolation and properties of 1-substituted 2-pyrimidone 4-sulfonates. Tetrahedron Letters, 10, 755–758. [DOI] [PubMed] [Google Scholar]
- Heiman M, Schaefer A, Gong S, Peterson JD, Day M, Ramsey KE, … Heintz N (2008). A translational profiling approach for the molecular characterization of CNS cell types. Cell, 135(4), 738–748. doi: 10.1016/j.cell.2008.10.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, … Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell, 38(4), 576–589. doi: 10.1016/j.molcel.2010.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzog VA, Reichholf B, Neumann T, Rescheneder P, Bhat P, Burkard TR, … Ameres SL(2017). Thiol-linked alkylation of RNA to assess expression dynamics. Nature Methods, 14(12), 1198–1204. doi: 10.1038/nmeth.4435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyn P, Kircher M, Dahl A, Kelso J, Tomancak P, Kalinka AT, & Neugebauer KM (2014). The earliest transcribed zygotic genes are short, newly evolved, and different across species. Cell Reports, 6(2), 285–292. doi: 10.1016/j.celrep.2013.12.030 [DOI] [PubMed] [Google Scholar]
- Hokin LE, & Hokin MR (1954). The incorporation of 32P into the nucleotides of ribonucleic acid in pancreas slices during enzyme synthesis and secretion. Biochemica et Biophysica Acta, 13, 401–412. [DOI] [PubMed] [Google Scholar]
- Jao C, & Salic A (2008). Exploring RNA transcription and turnover in vivo by using click chemistry. Proceedings of the National Academy of Sciences of the United States of America, 105(41), 15779–15784. doi: 10.1073/pnas.0808480105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonkers I, & Lis JT (2015). Getting up to speed with transcription elongation by RNA polymerase II. Nature Reviews Molecular Cell Biology, 16, 167–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenzelmann M, Maertens S, Haergenhahn M, Kueffer S, Hotz-Wagenblatt A, Li L, … Schütz G (2007). Microarray analysis of newly synthesized RNA in cells and animals. Proc Natl Acad Sci U S A, 104(15), 164–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim T-K, Hemberg M, Gray JM, Costa AM, Bear DM, Wu J, … Greenberg ME (2010). Widespread transcription at neuronal activity-regulated enhancers. Nature, 465(7295), 182–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knüppel R, Kuttenberger C, & Ferreira-Cerca S (2017). Toward time-resolved analysis of RNA metabolism in archaea using 4-thiouracil. Frontiers in Microbiology, 8, 286. doi: 10.3389/fmicb.2017.00286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak H, Fuda NJ, Core LJ, & Lis JT (2013). Precise maps of RNA polymerase reveal how promoters direct initiation and pausing. Science, 339(6122), 950–953. doi: 10.1126/science.1229386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li HB, Tong J, Zhu S, Batista PJ, Duffy EE, Zhao J, … Flavell RA. (2017). m6A mRNA methylation controls T cell homeostasis by targeting the IL-7/STAT5/SOCS pathways. Nature, 548(7667), 338–342. doi: 10.1038/nature23450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logan JE, Heagy FC, & Rossiter RJ (1955). Phosphorous metabolism of the liver: effect of hypophysectomy, adrenalectomy, and administration of ACTH on the incorporation of radioactive phosphate into RNA nucleotides. Canadian Journal of Biochemistry and Physiology, 33(1), 54–61. [PubMed] [Google Scholar]
- Lugowski A, Nicholson B, & Rissland OS (2018). DRUID: a pipeline for transcriptome-wide measurements of mRNA stability. RNA, 24(5), 623–632. doi: 10.1261/rna.062877.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madabusi LV, Latham GJ, & Andruss BF (2006). RNA extraction for arrays. Methods Enzymol, 411, 1–14. doi: 10.1016/S0076-6879(06)11001-0 [DOI] [PubMed] [Google Scholar]
- Melvin WT, Milne HB, Slater AA, Allen HJ, & Keir HM (1978). Incorporation of 6-thioguanosine and 4-thiouridine into RNA. Application to isolation of newly synthesised RNA by affinity chromatography. European Journal of Biochemistry, 92(2), 373–379. [DOI] [PubMed] [Google Scholar]
- Meryet-Figuiere M, Alaei-Mahabadi B, Ali MM, Mitra S, Subhash S, Pandey GK, … C. K (2014). Temporal separation of replication and transcription during S-phase progression. Cell Cycle, 13(20), 3241–3248. doi: 10.4161/15384101.2014.953876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milek M, Imami K, Mukherjee N, De Bortoli F, Zinnall U, Hazapis O, … Landthaler M (2018). DDX54 regulates transcriptome dynamics during DNA damage response. Genome Research, 27, 1344–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller C, Schwalb B, Maier K, Schulz D, Dümcke S, Zacher B, … Cramer P (2011). Dynamic transcriptome analysis measures rates of mRNA synthesis and decay in yeast. Molecular systems biology, 4(7), 458. doi: 10.1038/msb.2010.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller D, Brandt N, & Gresham D (2018). Systematic identification of factors mediating accelerated mRNA degradation in response to changes in enviornmental nitrogen. PLoS Genetics, 14(5), p1–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MR, Robinson KJ, Cleary MD, & Doe CQ (2009). TU-tagging: cell type-specific RNA isolation from intact complex tissues. Nature Methods, 6, 439–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munchel SE, Shultzaberger RK, Takizawa N, & Weis K (2011). Dynamic profiling of mRNA turnover reveals gene-specific and system-wide regulation of mRNA decay. Molecular Biology of the Cell, 22(15), 2787–2795. doi: 10.1091/mbc.E11-01-0028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muramatsu M, & Busch H (1964). Studies on nucleolar RNA of the Walker 25 carcinoma and the liver of the rat. Cancer Research, 24, 1028–1034. [PubMed] [Google Scholar]
- Neymotin B, Athanasiadou R, & Gresham D (2014). Determination of in vivo RNA kinetics using RATE-seq. RNA, 20, 1645–1652. doi: 10.1261/rna.045104.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pai AA, Henriques T, McCue K, Burkholder A, Adelman K, & Burge CB (2017). The kinetics of pre-mRNA splicing in the Drosophila genome and the influence of gene architecture. Elife, 6, e23537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabani M, Levin JZ, Fan L, Adiconis X, Raychowdhury R, Garber M, … Regev A (2011). Metabolic labeling of RNA uncovers principles of RNA production and degradation dynamics in mammalian cells. Nature Biotechnology, 29(5), 436–442. doi: 10.1038/nbt.1861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabani M, Raychowdhury R, Jovanovic M, Rooney M, Stumpo DJ, Pauli A, … Regev A (2014). High-resolution sequencing and modeling identifies distinct dynamic RNA regulatory strategies. Cell, 159(7), 1698–1710. doi: 10.1016/j.cell.2014.11.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riml C, Amort T, Rieder D, Gasser C, Lusser A, & Micura R (2017). Osmium-Mediated Transformation of 4-Thiouridine to Cytidine as Key To Study RNA Dynamics by Sequencing. Angew Chem Int Ed Engl, 56(43), 13479–13483. doi: 10.1002/anie.201707465 [DOI] [PubMed] [Google Scholar]
- Russo J, Heck AM, Wilusz J, & Wilusz CJ (2017). Metabolic labeling and recovery of nascent RNA to accurately quantify mRNA stability. Methods, 120, 39–48. doi: 10.1016/j.ymeth.2017.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schofield JA, Duffy EE, Kiefer L, Sullivan MC, & Simon MD (2018). TimeLapse-seq: adding a temporal dimension to RNA sequencing through nucleoside recoding. Nature Methods. doi: doi: 10.1038/nmeth.4582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwalb B, Michel M, Zacher B, Frühauf K, Demel C, Tresch A, … Cramer P (2016). TT-seq maps the human transient transcriptome. Science, 352(6290), 1225–1228. doi: 10.1126/science.aad9841 [DOI] [PubMed] [Google Scholar]
- Schwanhäusser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, … Selbach M (2011). Global quantification of mammalian gene expression control. Nature, 473, 337–342. [DOI] [PubMed] [Google Scholar]
- Singh J, & Padgett RA (2009). Rates of in situ trascription and splicing in large human genes. Nature Structural & Molecular Biology, 16, 1128–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitzer J, Hafner M, Landthaler M, Ascano M, Farazi T, Wardle G, … Tuschl T (2014). PAR-CLIP (Photoactivatable Ribonucleoside-Enhanced Crosslinking and Immunoprecipitation): a Step-By-Step Protocol to the Transcriptome-Wide Identification of Binding Sites of RNA-Binding Proteins In Lorsch J (Ed.), Laboratory Methods in Enzymology: Protein Part B (Vol. 539, pp. 113–161). Methods in Enzymology: Elsevier. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun M, Schwalb B, Schulz D, Pirkl N, Etzold S, Larivire L, … Cramer P (2012). Comparative dynamic transcriptome analysis (cDTA) reveals mutual feedback between mRNA synthesis and degradation. Genome Research, 22, 1350–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swiatkowska A, Wlotzka W, Tuck A, Barrass JD, Beggs JD, & Tollervey D (2012). Kinetic analysis of pre-ribosome structure in vivo. RNA, 18, 2187–2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tani H, & Akimitsu N (2012). Genome-wide technology for determining RNA stability in mammalian cells. RNA biology, 9(10), 1233–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tani H, Mizutani R, Salam KA, Tano K, Ijiri K, Wakamatsu A, … Akimitsu N (2012). Genome-wide determination of RNA stability reveals hundreds of short-lived noncoding transcripts in mammals. Genome Research, 22(5), 947–956. doi: 10.1101/gr.130559.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ussuf KK, Anikumar G, & Nair PM (1995). Newly synthesized mRNA as a probe for identification of wound responsive genes from potatoes. Indian J Biochem Biophys, 32(2), 78–83. [PubMed] [Google Scholar]
- Windhanger L, Bonfert T, Burger K, Ruzsics Z, Krebs S, Kauffman S, … Dolken L (2012). Ultrashort and progressive 4sU-tagging reveals key characteristics of RNA processing at nucleotide resolution. Genome Research, 22(10), 2031–2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodford TA, Schlegel R, & Pardee AB (1988). Selective isolation of newly synthesized mammalian mRNA after in vivo labeling with 4-thiouridine or 6-thioguanosine. Anal Biochem, 171(1), 166–172. [DOI] [PubMed] [Google Scholar]
- Yang E, van Nimwegen E, Zavolan M, Rajewsky N, Schroeder M, Magnasco M, & Darnell JEJ (2003). Decay rates of human mRNAs: correlation with functional characteristics and sequence attributes. Genome Research, 13(8), 1863–1872. doi: 10.1101/gr.1272403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zacher B, Michel M, Schwalb B, Cramer P, Tresch A, & Gagneur J (2017). Accurate Promoter and Enhancer Identification in 127 ENCODE and Roadmap Epigenomics Cell Types and Tissues by GenoSTAN. PLoS One, 12(1), e0169249. doi: 10.1371/journal.pone.0169249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziff EB, & Fresco JR (1969). A method for locating 4-thiuridylate in the primary structure of transfer ribonucleic acids. Biochemistry, 8(8), 3242–3248. [DOI] [PubMed] [Google Scholar]