

Abstract
Background:
The homologous plasma proteins prekallikrein and factor XI (FXI) circulate as complexes with high molecular weight kininogen. Although evidence supports an interaction between the prekallikrein-kininogen complexes and vascular endothelium, there is conflicting information regarding FXI binding to endothelium.
Objective:
To study the interaction between FXI and blood vessels in mice.
Methods:
C57Bl/6 wild-type or F11−/− mice in which variants of FXI were expressed by hydrodynamic tail vein injection, received intravenous infusions of saline, heparin, polyphosphates, protamine, or enzymes that digest glycosaminoglycans (GAGs). Blood was collected after infusion and plasma was analyzed by western blot for FXI.
Results and conclusions:
Plasma FXI increased 5- to 10-fold in wild-type mice after infusion of heparin, polyphosphates, protamine, or GAG-digesting enzymes, but not saline. Similar treatments resulted in a much smaller change in plasma FXI levels in rats, and infusions of large boluses of heparin did not change FXI levels appreciably in baboons or humans. The releasable FXI fraction was reconstituted in F11−/− mice by expressing murine FXI, but not human FXI. We identified a cluster of basic residues on the apple 4 domain of mouse FXI that is not present in other species. Replacing the basic residues with alanine prevented the interaction of mouse FXI with blood vessels, whereas introducing the basic residues into human FXI allowed it to bind to blood vessels. Most FXI in mice is noncovalently associated with GAGs on blood vessel endothelium and does not circulate in plasma.
Keywords: factor XI, glycosaminoglycans, heparin, mice, protamine
1 |. INTRODUCTION
The blood plasma protein factor XI (FXI) is the precursor of factor XIa (FXIa), a serine protease that contributes to thrombin generation through proteolytic activation of factor IX and other coagulation factors.1 FXI is a 160-kDa dimeric protein containing two identical 80-KDa subunits.2–6 Hydrophobic interactions between subunits are critical to dimer formation. In most species, a disulfide bond involving Cys321 also provides a covalent link between subunits. Each FXI subunit is organized into four 90 to 91 amino acid repeats called apple domains (A1 to A4 from the N-terminus) and a catalytic domain belonging to the trypsin family of serine proteases.7 FXI shares this organization with its monomeric homolog plasma prekal-likrein (PK).8–10 In humans, almost all FXI and most (75%−90%) PK circulates in plasma in non-covalent complexes with the glycoprotein high molecular weight kininogen (HK).11,12
Factor XI and PK are components of the plasma contact system, and their activation and activity are regulated and influenced by a variety of “surfaces,” many of which carry a negative charge.13 Human FXI binds polyanions such as heparins, polyphosphates, and nucleic acids through two anion binding sites (ABSs) comprising clusters of surface-exposed positively charged basic amino acids.14–20 ABS1 is located on the apple 3 (A3) domain (Arg250-Ile-Lys-Lys-Ser-Lys), whereas ABS2 is located on the 170-loop of the catalytic domain (Lys529-Arg-Tyr-Arg).19 ABS1 and ABS2 contribute to FXI activation and to regulation of FXIa by protease inhibitors. They are required for proper FXI function on most polyanions/surfaces. In contrast, PK, which lacks a site comparable to ABS1 in FXI, appears to rely on HK to facilitate proper binding to negatively charged surfaces.20
A serendipitous observation laid the foundation for the work presented in this manuscript. We observed that intravenous infusion of heparin into mice resulted in a marked increase in the plasma level of FXI. We conclude that most FXI in mice is bound to glycosaminoglycans (GAGs) on blood vessels through interactions requiring ABSs.
2 |. METHODS
2.1 |. Materials
The following materials were used: pooled normal plasma; George King (Overland Park, KS); human FXI; Haematologic Technologies (Burlington, VT); S-2366 (L-pyro-Glu-L-Pro-L-Arg-p-nitroanilide); DiaPharma; heparinase I/III, hyaluronidase, and chondroitinase ABC, Sigma-Aldrich (St Louis, MO)’ heparin (1000 USP units/mL), Hospira, Inc. (Lake Forest, IL); and protamine (10 mg/mL); Fresenius Kabi (Lake Zurich, IL). Polyphosphate (poly-P, 60 to 100 phosphate units) was a gift from Dr. Thomas Renne (U. Hamburg-Eppendorf).
2.2 |. Antibodies
Goat horseradish peroxidase (HRP)-anti-human FXII, sheep HRP-anti-human PK, and goat HRP-anti-human C1-INH immunoglobulin G (IgG); Enzyme Research Laboratories (South Bend, IN). Monoclonal IgG to mouse FXI (14E11).21 Rabbit anti-mouse HK IgG (anti-mHK) was raised against a peptide representing residues 607–638 on the mouse Kng1 gene product.22
2.3 |. Rodents
Procedures with mice and rats were approved by the Vanderbilt University Animal Care and Use Committee. C57Bl/6 mice deficient in FXI (F11−/−),23 FXII (F12−/−),24 PK (Klkb1−/−),25 or HK (Kng1−/−)26 were back-crossed through at least 10 generations against wild-type (WT) C57BI/6 mice from Jackson Laboratory (Bar Harbor, ME). Male and female mice at least 8 weeks of age were used in experiments. Sprague-Dawley male rats were from Charles River Laboratories (Wilmington, MA).
2.4 |. Recombinant FXI
cDNAs for human FXI (huFXI),5 mouse FXI (muFXI),27 and full-length messages for mouse HK (muHK) encoded by the Kng1 (NM_001102411.1) and Kng2 (NM_201375.2) genes26 were modified to add sequence encoding an eight amino acid hemagglutinin tag to the C-termini. cDNAs were inserted into vector pJVCMC and expressed in HEK293 cells as described previously.27 Tagged recombinant proteins were purified from conditioned media on a Pierce anti-HA IgG Agarose affinity column (ThermoFisher, USA), and stored in 25 mmol/L Tris-HCl pH 7.4 with 100 mmol/L NaCl at −80°C. Untagged muFXI was expressed in a similar manner.27
2.5 |. Western blots
One microliter samples of mouse plasma mixed with non-reducing SDS-sample buffer were size fractionated on 7.5% acrylamide-SDS gels and transferred to nitrocellulose membranes. FXI was detected with biotinylated IgG 14E11, and HRP-conjugated streptavidin. FXII and PK were detected with HRP-conjugated polyclonal IgGs to FXII or PK, respectively. HK was detected with anti-mHK, and HRP-conjugated goat anti-rabbit IgG. Detection in all cases was by chemiluminescence. Factor XI signals were quantified by densitometry of blots using Image J software.
2.6 |. Binding of FXI to Heparin-Sepharose
A 1-mL heparin-sepharose column attached to an AKTA FPLC workstation (GE Healthcare Life Sciences, Piscataway, NJ) was equilibrated with buffer A (20 mmol/L N-2-hydroxyethylpiperazine- N-2-ethane sulfonic acid, 50 mmol/L NaCl, pH 7.4). Plasma FXI, huFXI-HA, or muFXI-HA (10 μg) was loaded on the column in 0.5 mL buffer A. Protein was eluted with a 15-mL linear NaCl gradient (50–1000 mmol/L), and 0.5 mL fractions were collected. Fractions were tested for FXI by western blot.
2.7 |. Intravenous infusions
WT C57BI/6 mice received intravenous infusions (100 μL) through tail or jugular veins of saline (0.9% NaCI); unfractionated heparin (400–10 000 U/kg, 10 mice treated with 2000 U/kg); poly-P (60 mg/ Kg, 4 mice); protamine (40 mg/Kg, 5 mice), or an enzyme that digests GAGs (2000 U/kg of heparinase [4 mice], chondroitinase ABC [2 mice], or hyaluronidase [2 mice]). Blood samples were collected from the tail before infusion, and then 5 minutes postinfusion for saline, heparin, poly-P, or protamine from the inferior vena cava. For infusions of enzymes, samples were collected before infusion and 0.5, 1, and 2 hours postinfusion.
Sprague-Dawley rats (2 animals for each type of infusion) received infusions of heparin (8000 U/kg), protamine (1 mg/30 g) or heparinase (40 U/animal) into the jugular vein. Blood samples were collected from the carotid artery before and 5 minutes after heparin or protamine infusion, and at various times after heparinase. An olive baboon (Papio anubis) received a 2000 U/kg bolus of unfractionated heparin (total dose 27 000 units). Blood samples were drawn into 3.2% sodium citrate from a peripheral vein before heparin infusion, and 10 minutes after heparin infusion. The animal was euthanized after the procedure.
2.8 |. Expression of FXI variants in mice by hydrodynamic tail-vein injection
cDNAs for WT huFXI and muFXI (no hemagglutinin tags), and variants with alanine replacing basic residues in FXI ABSs (Figure S1) were introduced into an EEV600A expression vector (System Biosciences, Mountain View, CA). F11−/− mice (5 mice per construct, for each time point) anesthetized with pentobarbital (50 mg/ kg intraperitoneally) received infusions of FXI/EEV600A constructs (3.5 μg) in 2 mL of lactated Ringer’s solution through a tail vein over 30 seconds.28,29 Twenty-four hours or seven days after HTI, mice were tested with infusions, as described previously.
2.9 |. Ferric chloride arterial thrombosis model
WT mice, F11−/− mice, and F11−/− mice expressing FXI by HTI (five mice per group) were tested in a model of carotid artery occlusion induced by exposing the vessel to 3.5% FeCl3.29 Mice were anesthetized with 50 mg/kg pentobarbital intraperitoneally. The right common carotid artery was exposed and fitted with a Doppler probe (Model 0.5 VB, Transonic System, Ithaca, NY). Thrombus formation was induced by applying two 1 × 1.5 mm filter papers (GB003, Schleicher & Schuell, Keene, NH) saturated with a 3.5% (0.16 mol/L) solution of FeCl3 to opposite sides of the artery for 3 minutes and flow was monitored for 30 minutes.
2.10 |. Statistical analysis
For comparing FXI levels before and after interventions, the FXI bands from western blots were quantified by densitometry using ImageJ. Data are presented by bar charts showing fold-change after intervention using preintervention values as baseline. Paired Student t test was used to test the significance of the difference between results for WT mice treated with saline or 2000 U/kg heparin.
For the FeCl3 thrombosis model, data are presented as average time to occlusion (±SD). Groups were compared using one-way analysis of variance with post-hoc Tukey’s multiple comparison. P values <0.05 were considered significant.
3 |. RESULTS
3.1 |. Plasma FXI increases following heparin administration in mice
WT mice received intravenous boluses of unfractionated heparin ranging from 2 to 20 000 U/kg. An increase in plasma FXI was noted 5 minutes postheparin administration starting at a dose of 40 U/ kg (Figure 1A). The effect was not enhanced by increasing time between heparin administration and blood collection (not shown). A 2000 U/kg heparin dose was used in subsequent experiments. Using control curves constructed with known concentrations of muFXI (Figure S2), the average baseline plasma FXI concentration in mice (~1.3 μg/mL or 8 nmol/L), is ~3- to 4-fold lower than in humans (~5 μg/mL or 30 nmol/L). The data shown in Figure 1B are consistent with the impression that the FXI concentration is higher in humans than in mice. In WT mice, plasma FXI concentrations increased to ~13 μg/mL (80 nmol/L) after heparin infusion (Figure 1C), an ~10- fold increase over baseline, and consistent with data in Figure 1A. In contrast, there was little change in the plasma concentrations of other contact factors (FXII, PK, and HK; Figure 1D).
FIGURE 1.
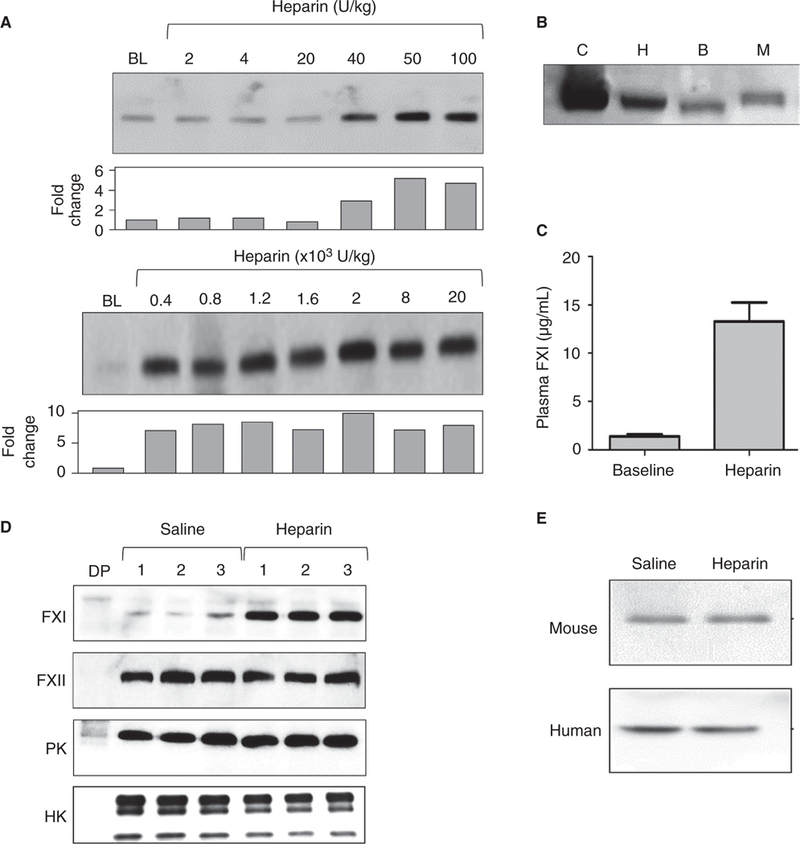
A heparin-releasable pool of FXI in mice. (A) Shown are representative western blots for muFXI. C57BI/6 WT mice were infused with doses of heparin from 2 to 20 000 U/kg. The bar graphs below each blot show results of a densitometric analysis of FXI bands showing fold-changes compared with baseline (no heparin). (B) Western blot showing FXI in 1-μL samples of normal human, baboon, and mouse plasma. The control is a 100-ng sample of pure muFXI. (C) Plasma FXI antigen concentrations in WT mouse plasma before (baseline) and after infusion of unfractionated heparin (2000 U/kg). Blots underwent densitometric analysis and results were compared with a control curve constructed with pure muFXI (Figure S2). (D) Representative western blots for FXI, FXII, PK, and HK. WT mice were infused with saline and blood was collected. The same mice then received infusions of heparin (2000 U/kg) and blood was collected 5 minutes later. (E) Representative western blot for FXI in plasma from a WT mouse and a healthy human volunteer. Blood was collected into sodium citrate, then saline or heparin (final concentration 50 U/mL) was added. Abbreviations: B, baboon; BL, baseline; C, control; DP, mouse plasma deficient in the respective coagulation factor the blot was probed for; H, human; M, mouse. All experiments were run in triplicates
Because FXI increases rapidly after heparin infusion, we postulated that the source was protein bound to the blood vessel or to cellular components of the blood. Adding heparin to a final concentration of 50 U/mL (estimated to be the equivalent of the peak plasma concentration after giving a mouse a heparin bolus of 2000 U/kg) to whole blood collected from WT mice or human volunteers did not increase plasma FXI (Figure 1E), indicating blood cells are not the source of heparin-releasable FXI.
3.2 |. A non-circulating pool of FXI forms through non-covalent interactions with GAGs
Based on the ability of heparin, a highly sulfated GAG, to displace FXI into the plasma, we postulated that FXI binds GAGs on blood vessels through charge-based interactions. Consistent with this, WT mice receiving intravenous chondroitinase ABC, hyaluronidase, or a mixture of heparinases I and III to digest cell-surface GAGs showed 3- to 7-fold increases in plasma FXI 30 minutes posttreatment (Figure 2A).
FIGURE 2.
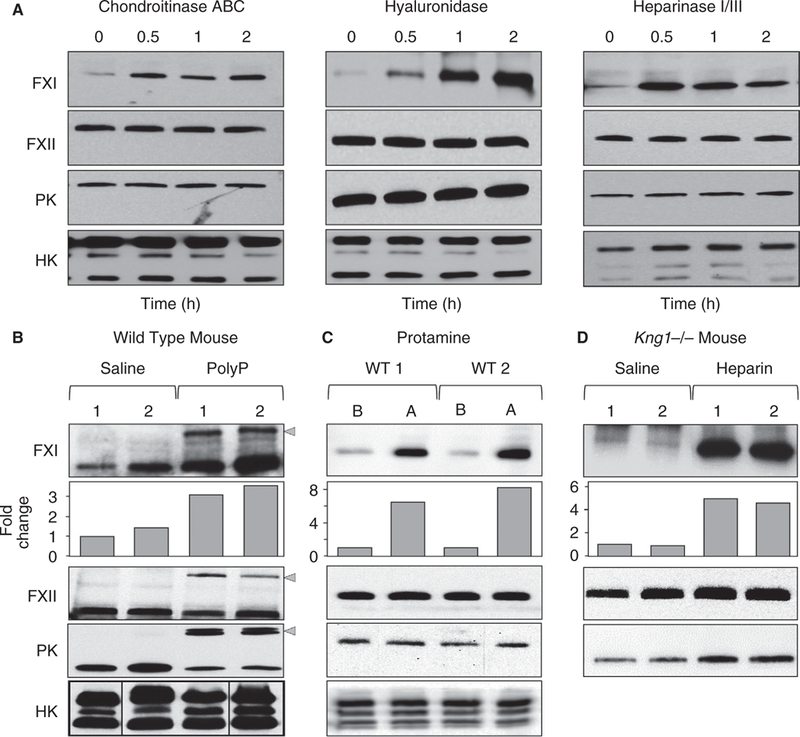
The FXI pool in mice is associated with GAGs. (A) Shown are representative western blots for FXI, FXII, PK, and HK in WT mice at various times after infusion of chondroitinase ABC (left), hyaluronidase (middle), or heparinase I/III (right). (B,C) WT mice were infused with normal saline and blood was collected after 5 minutes. The same animals then received infusions of (B) poly-P (60 mg/kg) or (C) protamine (40 mg/kg). Blood was collected 5 minutes after treatment. (D) Kng1−/−- mice received infusions of normal saline and blood was collected after 5 min. The same mice received heparin infusions (2000 U/kg) and blood was collected 5 minutes after. (B) Gray arrowheads indicate potential complexes of activated proteases with serpin inhibitors. Below FXI western blots in panels B-D are bar charts showing results of densitometry analysis for FXI bands (fold-change from baseline). (C) Abbreviations B and A indicate before and after protamine infusion. Panels B-D show representative western blots
Species of poly-P similar to those released from platelet granules (60–100 phosphate units) bind FXI30 and enhance its activation in a manner that depends on FXI ABSs.19 Infusing poly-P into WT mice resulted in an ~3-fold increase in plasma FXI, whereas levels of FXII and PK appeared to decrease modestly (Figure 2B). Poly-P induces contact activation in plasma, and we suspect the higher molecular weight species (indicated by gray arrows in Figure 2B) in blots for FXI, FXII, and PK represent activated forms of these proteins in complex with plasma serine protease inhibitors. The decrease in PK and FXII is consistent with consumption during contact activation.31 FXI is also consumed in contact activation, so the observation that its plasma level actually increases after poly-P infusion emphasizes the significant amount of FXI released into the plasma by the infusion. Protamine is a polycation isolated from fish sperm that is used in clinical practice to neutralize the anticoagulant activity of heparin by displacing antithrombin and coagulation proteases from heparin.32,33 Protamine infusion into WT mice increased the plasma FXI level ~7-fold (Figure 2C), consistent with the importance of charge interactions in establishing the non-circulating FXI pool.
3.3 |. HK is not required to establish the non-circulating FXI pool
FXI circulates in plasma as a complex with HK, a protein that serves as a cofactor that facilitates FXI binding to certain types of surfaces.34 Mice have two closely linked kininogen genes (Kng1 and Kng2)26 that encode similar (but not identical) forms of HK. Kng1 appears to encode most of the HK expressed in liver and adrenal tissue, whereas the Kng2 product is expressed mostly in kidney.26 Using an anti-HK antibody that recognizes both gene products (Figure S3A), we observed that Kng1−/− mice lack detectable HK protein in plasma at baseline (Figure S3A), and after infusion of heparinase (Figure S3B), indicating that the source of HK in the circulation of mice is the Kng1 gene product.
mKng1−/− mice have modestly reduced FXI levels (~60% of normal plasma, data not shown), perhaps resulting from loss of a stabilizing effect from HK. Infusing heparin into mKng1−/− mice results in a 4- to 5-fold increase in plasma FXI (Figure 2D). These results indicate HK is not required to establish the non-circulating FXI pool, although it does not rule out the possibility that HK binds to vessel-bound FXI in the same manner as it binds to FXI in blood.
3.4 |. Reconstituting the heparin-releasable FXI pool in FXI-deficient mice
F11−/− mice were transfected with an EEV600A-1 expression plasmid containing the muFXI cDNA by hydrodynamic tail vein injection (HTI). This results in substantial amounts of FXI in the plasma within 24 hours of injection (Figure 3A). Previously, we showed that a similar construct for huFXI supported expression for at least 6 weeks in F11−/− mice.35 Heparin and protamine releasable pools of muFXI were established 1 week post-HTI (~5- to 6-fold increase with heparin and ~3.5-fold increase with protamine) (Figure 3B). Although plasma FXI levels reach a relatively stable level 24–48 hours post-HTI, the heparin releasable pool was not consistently evident at these early time points, suggesting it requires several days to become established. Interestingly, when huFXI was expressed in FXI−/− mice, a heparin/protamine releasable pool was not established (Figure 3C), suggesting muFXI and huFXI bind GAGs with different affinities. Although we have demonstrated here that HK is not necessary for establishing the non-circulating pool of murine FXI, we assessed the possibility that results with huFXI could be due to poor binding to muHK. Using native agarose gels,36 we were able to demonstrate that huFXI and muHK form a complex (Figure S4), consistent with our observation that huFXI can reconstitutes the aPTT of F11−/− mouse plasma and restores the WT phenotype in F11−/− mice in thrombosis models.19
FIGURE 3.
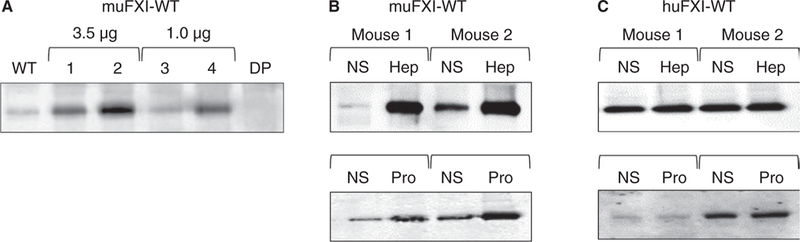
muFXI reconstitutes the non-circulating FXI pool in F11−/− mice. (A) Expression of muFXI in F11−/− mice by HTI. F11−/− mice received either 3.5 μg (mouse 1 and 2) or 1.0 μg (mouse 3 and 4) muFXI/EEV600A construct. Plasma samples were collected 24 hours later and analyzed by western blot. Samples from untreated WT and F11−/− (DP) mice are shown for comparison. F11−/− mice expressing (B) muFXI or (C) huFXI (by HTI) received infusions of normal saline (NS) and blood was collected 5 minutes later. The same animals then received infusions of heparin (Hep, 2000 U/kg) or protamine (Pro, 40 mg/kg), and blood was again collected 5 minutes later. Plasma samples were tested for FXI expression by western blot. In panels A-C, representative western blots are shown
3.5 |. ABSs are required to establish the non-circulating FXI pool
Previously, we identified two ABSs (ABS1 and ABS2) on each subunit of the huFXI dimer that mediate interactions with heparin and polyphosphate (Figure 4A).15–19 These sites are present on muFXI (Figure 4A), although ABS1 is less developed than in huFXI (Figure 4B). We created muFXI cDNAs in which basic residues in ABS1, ABS2, or both ABS1 and ABS2 are replaced with alanine (Figure S1). Variants lacking ABS2 failed to reconstitute the non-circulating pool, while loss of ABS1 appeared to reduce the pool (Figure 5A). Although the ABS2 sites are similar in muFXI and huFXI, the ABS1 sites differ somewhat (Figure 4B). Changing the huFXI ABS1 site to the mouse sequence (Lys252 to Thr and Lys255 to His) did not confer the ability to reconstitute the releasable pool on huFXI (Figure 5B). These results show that ABS1 and ABS2 are required, but are not sufficient, to establish the non-circulating FXI pool.
FIGURE 4.
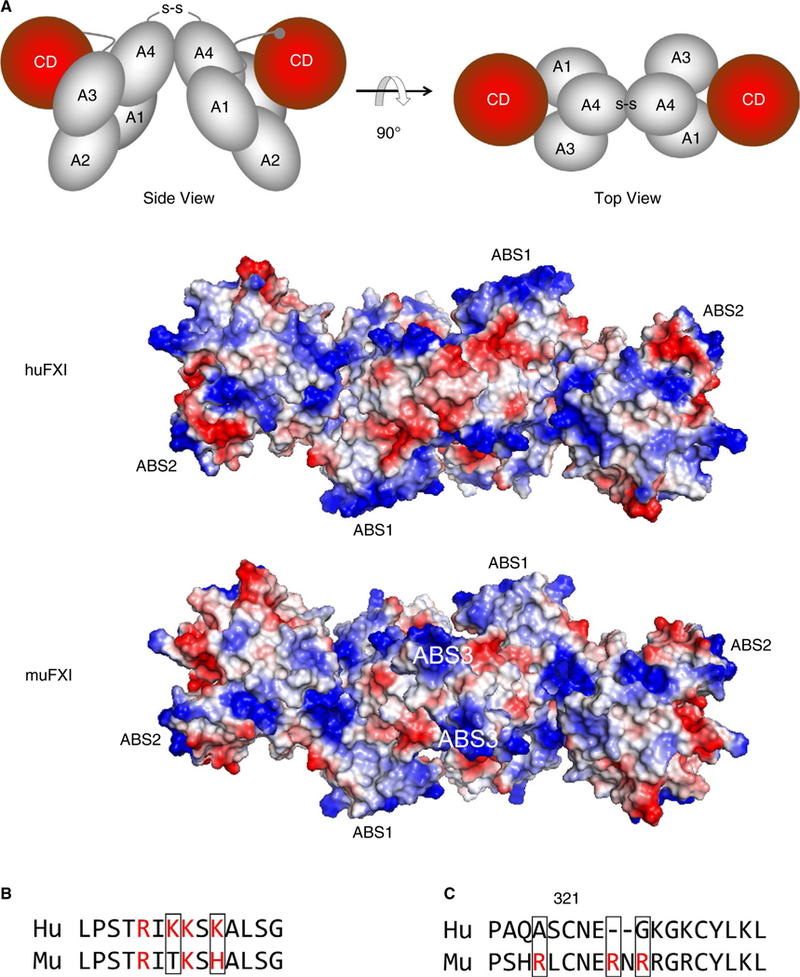
FXI anion binding sites (ABSs). (A) The images at the top are schematic representations of the human FXI dimer based on the crystal structure reported by Papagrigoriou et al7 in “side view” (left) and rotated 90 degrees for a “top view (right). The four apple domains (A1-A4) and catalytic domains (CD [red]) are shown. The disulfide bond (S-S) between Cys321 on each A4 domain indicates the position of the dimer interface. The RGB structural images below the schematic diagrams depict top views of huFXI and muFXI FXI dimers, with positively charged regions indicated in blue and negatively charged regions in red. Positions of ABS1 and ABS2 for both species, and ABS3 in muFXI are indicated. The three ABSs are positioned in a roughly linear arrangement, crossing the dimer interface created by the A4 domains. (B) A comparison of the amino acid sequences in ABS1 on the Apple 3 domain in human (Hu) and mouse (Mu) FXI. Basic residues are indicated in red. (C) Amino acid sequences of human (Hu) and murine (Mu) FXI in the Apple 4 domain in the vicinity of Cys321. Basic residues are indicated in red
FIGURE 5.
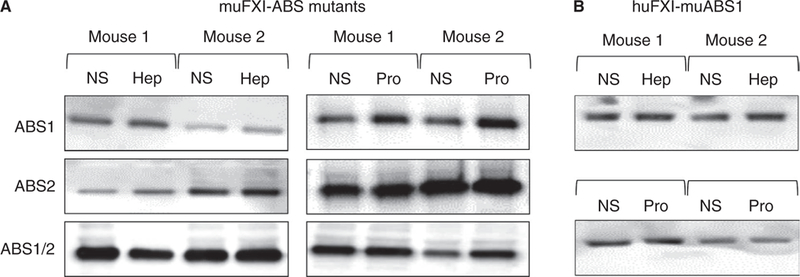
ABS1 and ABS2 are required to form the non-circulating FXI pool. (A) Representative western blots of muFXI variants in the blood of F11−/− mice. muFXI in which alanine residues replace basic residues in ABS1, ABS2, or both ABS1 and ABS2 (ABS1/2) were expressed in F11−/− mice by HTI. Seven days post-HTI, mice received infusions of normal saline (NS) and blood was collected 5 minutes later. The same animals then received infusions of heparin (Hep, 2000 U/kg) or protamine (Pro, 40 mg/kg), and blood was collected 5 minutes later. (B) Representative western blots of huFXI in which the sequence for ABS1 was changed to that of muFXI (see Figure 4B; basic residues are indicated in red, and those that were changed are within the black squares) was expressed in F11−/− mice by HTI. Seven days post-HTI, mice received infusions of NS and blood was collected 5 minutes later. The same animals then received infusions of heparin (Hep, 2000 U/ kg) or protamine (Pro, 40 mg/kg), and blood was again collected 5 minutes later. All experiments were run in triplicate
3.6 |. ABS3 on muFXI
Aligning amino acid sequences for muFXI and huFXI (Figure 4C) revealed a cluster of basic amino acids (Arg319, Arg324, and Arg326) on the A4 domain of muFXI in proximity to Cys321 that are not present in huFXI. The space-filling model in Figure 4A indicates these charged groups (designated ABS3) are located along lines of basic residues extending from ABS1 on one subunit to ABS2 on the opposite subunit. ABS3 would increase the positive charge in the vicinity of the dimer interface in muFXI.
Replacing Arg319, Arg324, and Arg326 on muFXI with alanines results in a protein (muFXI-Ala319,324,326) that does not reconstitute the heparin/protamine-releasable FXI pool in mice (Figure 6A). Furthermore, replacing Ala319, Gly324, and Gln326 in huFXI with arginines conferred the ability to form the non-circulating pool on huFXI (~6-fold increase with heparin and ~3.5-fold with protamine) (Figure 6B). These data indicate that ABS3, in conjunction with ABS1 and ABS2, permit muFXI to bind more tightly to GAGs than does huFXI. This was confirmed by studying binding of huFXI and muFXI to heparin-sepharose (Figure 6C). When the column was eluted with a sodium chloride gradient, human plasma FXI and huFXI eluted similarly (peaks 490–500 nmol/L NaCl), whereas muFXI eluted at a higher salt concentration (peak 580–590 nmol/L NaCl).
FIGURE 6.
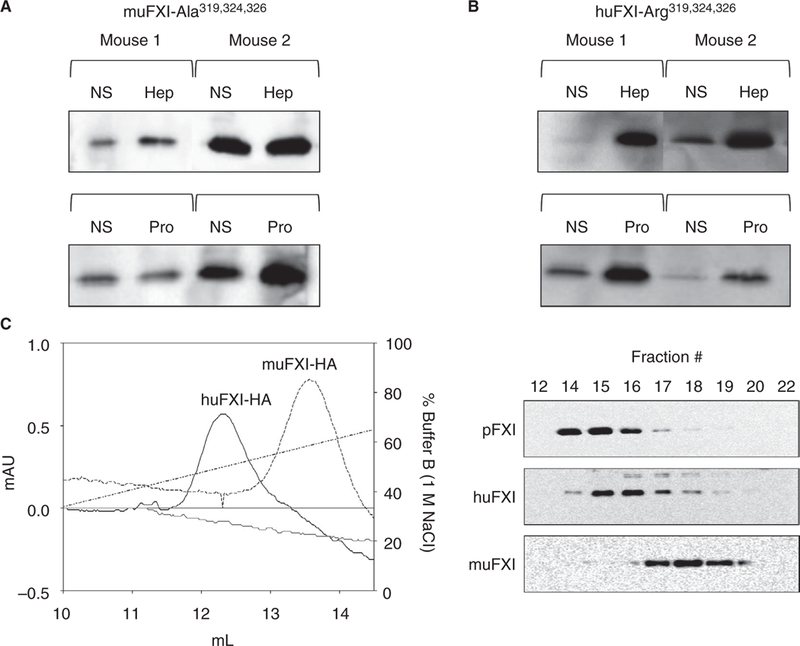
ABS3 is required to form the non-circulating FXI pool. Representative western blots of (A) muFXI in which alanine residues replace basic residues Arg319, Arg324, and Arg326 or (B) huFXI in which residues 319, 324, and 326 were replaced with arginine (as in muFXI) were expressed in F11−/− mice by HTI. Seven days post-HTI, mice received infusions of normal saline (NS) and blood was collected 5 minutes later. The same animals then received infusions of heparin (Hep, 2000 U/Kg) or protamine (Pro, 40 mg/Kg), and blood was again collected 5 minutes later. (C) muFXI-HA and huFXI-HA were applied to a heparin-sepharose column and eluted with a linear sodium chloride gradient. Plasma-derived human FXI eluted at a position identical to huFXI-HA (data not shown). (Right) Western blots of elution fractions from the heparin-sepharose column, probed with an anti-FXI antibody. Note that muFXI elutes at a higher salt concentration than huFXI. Shown are representative results of studies run in triplicate
3.7 |. MuFXI lacking ABS 3 supports carotid artery occlusion in F11−/− mice
FXI−/− mice are resistant to thrombosis induced by applying FeCl3 to the exterior of a blood vessel.37 Previously, we showed that infusing human plasma FXI, or expressing huFXI by HTI in F11−/− mice restored the WT phenotype in response to FeCl3 (Figure 7A).29 Because huFXI does not restore the non-circulating FXI pool in FXI−/− mice, this suggests non-circulating FXI is not required for FeCl3-induced thrombosis. Alternatively, huFXI may have properties distinct from those of muFXI that allow it to function without interacting with blood vessel GAGs. We expressed muFXI or the murine variant lacking ABS3 (muFXI-Ala319,324,326) in F11−/− mice. Twenty-four hours post-HTI, mice were studied in the FeCl3-induced carotid artery thrombosis model. Both proteins restored thrombus formation in response to 3.5% FeCl3 (Figure 7A, P > 0.5 compared with huFXI or WT mice), indicating neither ABS3 nor the FXI interaction with GAGs on the vessel surface are required in this model.
FIGURE 7.
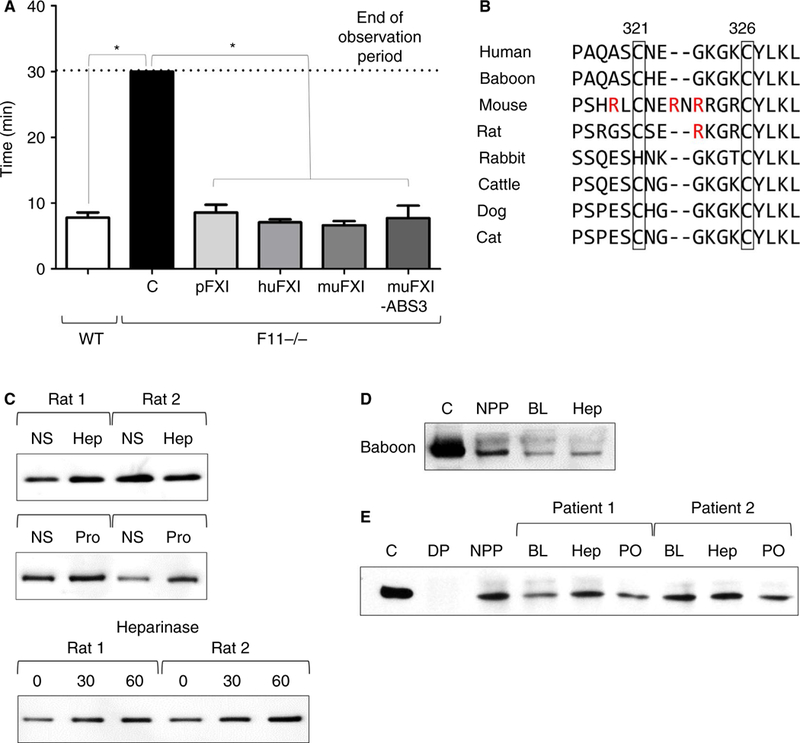
FXI ABS3 is not required for thrombosis in mice, and appears to be species specific. (A) Bar graph comparing the times to carotid artery occlusion induced by FeCl3 in WT, (C) F11−/− mice, F11−/− mice that received an infusion of human plasma FXI (10 μg), or F11−/− mice expressing huFXI, muFXI, or muFXI-Ala319,324,326 (muFXI-ABS3) (n = 5 per group). For mice subject to HTI, thrombosis studies were conducted 7 days after HTI. (B) FXI amino acid sequences in the apple 4 domain in the vicinity of Cys321 and Cys326. Residues in red indicate basic residues of ABS3 in the mouse. Note that with the exception of one basic residue in the rat, Arg319, Arg324, and Arg326 are not conserved across species. (C) Western blots of plasma FXI. Rats received infusions of normal saline (NS) and blood was collected 5 minutes later. The same animals then received infusions of heparin (Hep, 2000 U/kg) or protamine (Pro, 40 mg/kg), and blood was collected 5 minutes later. (Lower) Rats were treated with heparinase I/III and blood was collected at various times (in minutes) after the infusion. (D) A western blot for FXI in plasma from a single baboon at baseline (BL) or 10 minutes after infusion of 2000 U/kg heparin. (E) Western blots from two patients at baseline (BL), 5 minutes after a priming does of heparin (400 U/kg) before cardiopulmonary bypass (Hep), and postoperatively (PO) after protamine reversal. Abbreviations: C, control of 100 ng purified human FXI; DP, FXI-deficient plasma; NPP, pooled normal human plasma
3.8 |. Evidence for a heparin-releasable FXI pool in other species
Aligning FXI amino acid sequences from mammalian species revealed ABS3 is most prominent in mice (Figure 7B). Rat FXI have some basic residues in the vicinity of Cys321. When rats were infused with heparin, protamine, or heparinase, there was perhaps a slight increase in the FXI level on western blots (Figure 7C). The FXI sequence around Cys321 in the baboon Papio anubis is similar to the human sequence. There was no obvious change in plasma FXI in a baboon receiving 2000 U/Kg unfractionated heparin (Figure 7D). The results are similar to those for two patients receiving 400 units/kg unfractionated heparin before cardiopulmonary bypass (Figure 7E) and a single patient exposed to a considerably higher heparin dose (Figure S5). Although these studies do not exclude that some FXI is bound to blood vessels in most mammalian species, mice appear to be unusual in that the majority of FXI is bound.
4 |. DISCUSSION
As components of the plasma contact system, FXI and its homolog PK interact with a variety of biological and non-biological substances that often carry a net negative charge.13,14 Such “surfaces” accelerate FXI and PK activation by factor XIIa and facilitate factor XII activation by FXIa and kallikrein. Although FXI and PK binding to inorganic earths such as kaolin or silica is facilitated by HK,33 inter-actions with polyanions such as poly-P, nucleic acids, and GAGs appear to differ.19,20 HK is required for optimal PK activation on poly-P or nucleic acids, but acceleration of FXI activation requires ABS1 and ABS2 on FXI.19,20 The ABSs also contribute to FXI/FXIa binding to heparin,15–18 and are conserved across mammalian species, implying functional importance; however, there is sufficient variation in ABS sequences between species to suggest different affinities for polyanions.
We describe a difference between mouse and human FXI. muFXI contains charged residues (ABS3) not found in huFXI that contribute to a higher affinity for heparin and, with ABS1 and ABS2, result in most muFXI binding to GAGs on blood vessels. The interaction does not require HK. HK binds to the face of the FXI apple domain disk opposite the extended negative patch created by the ABSs.1,3,7,9 Supporting the conclusion that the FXI-GAG interaction is electrostatic, heparin, protamine, and poly-P displace muFXI from its binding site. Reversible charge-based interactions with blood vessels have been described for several blood constituents including enzymes (lipoprotein lipase, hepatic triglyceride lipase, superoxide dismutase), cytokines (midkine, pleiotrophin), and coagulation proteins (platelet factor 4, tissue plasminogen activator, tissue factor pathway inhibitor [TFPI]).38–43 It is interesting to note that FXIa cleaves TFPI, neutralizing its ability to inhibit coagulation.44 It is tempting to speculate that positioning FXI in proximity to TFPI on blood vessels may be of importance in this regard, but this has not been demonstrated.
Mice have been used extensively as model organisms for studying FXI in processes relevant to human health including its contributions to thrombosis,21,37,45–48 hemostasis,23,35,49 ischemia-reperfusion injury,46,50 inflammation,46,51–58 and susceptibility to infection.52,53,55,58–60 The work is predicated on structural and enzymatic similarities between human and murine FXI that suggest comparable biology in primates and rodents.27 In support of this, FXI-deficient humans and mice are both resistant to thrombosis,45,47,61,62 and huFXI restores thrombus formation in F11−/− mice.37,45 As with F11−/− mice, baboons treated with anti-FXI antibodies are resistant to thrombosis21,63,64 and have blunted inflammatory responses when challenged with bacteria65; however, the different intravascular distributions of FXI in mice and humans raises the possibility that the protein functions differently in some situations in the two species. Although FXI binding to GAGs on blood vessels does not appear to be required for FeCl3-induced thrombus formation, it may be relevant to other processes.
A significant fraction of FXI-deficient humans have a trauma- associated bleeding disorder.66 FXI-deficient domesticated animals (cattle, dog, cat) may also bleed excessively with surgery or trauma.67–69 FXI-deficient mice, in contrast, have not consistently shown a hemostatic defect.35,45,49 During hemostasis, the major FXIa substrate is factor IX. In mice, a substantial fraction of factor IX is bound to collagen 4 in blood vessels walls, and a similar situation likely occurs in humans.70 The interaction between factor IX and blood vessels is required for normal function because mice expressing factor IX that binds weakly to collagen 4 have a hemostatic defect.71 It is tempting to speculate that muFXI bound to blood vessels is positioned in proximity to factor IX to facilitate its activation; however, the implications for hemostasis remain uncertain given the lack of a bleeding phenotype in F11−/− mice. Furthermore, FXIa activation of factor IX is required for clot formation in mouse thrombosis models,35,45 and our results indicate FXI does not need to bind to vessels to support thrombosis.
FXI deficiency blunts systemic inflammation and improves survival in mice undergoing cecal ligation and puncture52,55 or infection with Listeria monocytogenes.53 An antibody to FXI has a similar beneficial effect in baboons challenged with a lethal dose of heat-inactivated Staphylococcus aureus,72 pointing to a role for FXI in promoting inflammation. In contrast, Stroo and colleagues reported that F11−/− mice have increased leukocyte infiltration and local cytokine production compared with WT or F12−/− mice when bacteria are introduced into the lungs.60 F11−/− mice also exhibit increased pulmonary inflammation in response to allergen inhalation.57 Plasminogen-deficient mice have a propensity to deposit fibrin in multiple organs, including the lungs.73 We showed that superimposing FXI deficiency on plasminogen deficiency results in robust pulmonary leukocyte infiltration leading to early death.51 Enhanced susceptibility to lung injury has not been reported in FXI-deficient humans. A recent analysis by Salomon and coworkers of a database from a large health care provider in Israel indicated that FXI-deficient patients are not at increased risk for pneumonia and do not have a more severe clinical course if they get pneumonia, compared with patients with normal FXI levels.74 Although FXI may influence inflammatory pathways, the nature of the contribution may differ in mice and humans. It is not clear at this point if this can be attributed to the non-circulating FXI pool in mice.
In conclusion, we determined that most FXI in mice forms a non-circulating pool bound non-covalently to GAGs, most likely on the surfaces of blood vessels. Mice appear to differ from humans in this regard. This observation, in conjunction with the enhanced susceptibility of F11−/− mice to pulmonary infection, suggests that some aspects of the biology of FXI in mice may not be applicable to human physiology and pathology. It may be possible in some situations to address this by using F11−/− mice expressing human FXI. However, incompatibilities between human and murine proteins may introduce new variables that would need to be considered.
Supplementary Material
Essentials.
The majority of FXI in mice is in a non-circulating pool associated with vascular glycosaminoglycans.
FXI binds to GAGs via electrostatic interactions involving anion binding sites (ABSs).
The non-circulating FXI pool is prominent in mice, but not in rats, baboons, or humans.
A mouse-specific ABS on the FXI apple 4 domain is required for binding to blood vessel GAGs.
ACKNOWLEDGEMENT
The authors acknowledge support from awards: HL58837, HL81326, and HL140025 (D.G.); HL130081 (I.M.V.); HL128016 (AG); HL101972 (O.J.T.M. and A.G.); GM116194 (O.J.T.M.) from the National Institutes of Health; and postdoctoral fellowship award 18P0ST34030076 from American Heart Association (B.M.M.). We also acknowledge support from the Rat Metabolic Physiology Core of the Metabolic Shared Resource at Vanderbilt University Medical Center (supported by 5P30 DK020593–41).
Funding information
National Heart, Lung, and Blood Institute, Grant/Award Number: 5P30DK020593–41, GM116194, HL101972, HL128016, HL130081, HL140025, HL58837 and HL81326; American Heart Association, Grant/Award Number: 18POST34030076
Footnotes
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section at the end of the article.
CONFLICT OF INTEREST
D. Gailani is a consultant and receives consultant’s fees from several pharmaceutical companies. A. Gruber is an employee of Aronora Inc.
REFERENCES
- 1.Mohammed BM, Matafonov A, Ivanov I, Sun M-F, Cheng Q, Dickeson SK, et al. An update on factor XI structure and function. Thromb Res. 2018;161:94–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gailani D, Geng Y, Verhamme I, Sun M, Bajaj SP, Messer A, et al. The mechanism underlying activation of factor IX by factor XIa. Thromb Res. 2014;133:S48–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Emsley J, McEwan PA, Gailani D. Structure and function of factor XI. Blood. 2010;115:2569–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bouma BN, Griffin JH. Human blood coagulation factor XI. Purification, properties, and mechanism of activation by activated factor XII. J Biol Chem. 1977;252:6432–7. [PubMed] [Google Scholar]
- 5.Fujikawa K, Chung DW, Hendrickson LE, Davie EW. Amino acid sequence of human factor XI, a blood coagulation factor with four tandem repeats that are highly homologous with plasma prekallikrein. Biochemistry. 1986;25:2417–24. [DOI] [PubMed] [Google Scholar]
- 6.McMullen BA, Fujikawa K, Davie EW. Location of the disulfide bonds in human coagulation factor XI: the presence of tandem apple domains. Biochemistry. 1991;30:2056–60. [DOI] [PubMed] [Google Scholar]
- 7.Papagrigoriou E, McEwan PA, Walsh PN, Emsley J. Crystal structure of the factor XI zymogen reveals a pathway for transactivation. Nat Struct Mol Biol. 2006;13:557–8. [DOI] [PubMed] [Google Scholar]
- 8.McMullen BA, Fujikawa K, Davie EW. Location of the disulfide bonds in human plasma prekallikrein: the presence of four novel apple domains in the amino-terminal portion of the molecule. Biochemistry. 1991;30:2050–6. [DOI] [PubMed] [Google Scholar]
- 9.Hooley E, Mcewan PA, Emsley J. Molecular modeling of the prekallikrein structure provides insights into high-molecular-weight kininogen binding and zymogen activation. J Thromb Haemost. 2007;5:2461–6. [DOI] [PubMed] [Google Scholar]
- 10.Ponczek MB, Gailani D, Doolittle RF. Evolution of the contact phase of vertebrate blood coagulation. J Thromb Haemost. 2008;6:1876–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mandle RJ, Colman RW, Kaplan AP. Identification of prekallikrein and high-molecular-weight kininogen as a complex in human plasma. Proc Natl Acad Sci. 1976;73:4179–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thompson RE, Mandle R, Kaplan AP. Association of factor XI and high molecular weight kininogen in human plasma. J Clin Invest. 1977;60:1376–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schmaier AH. The contact activation and kallikrein/kinin systems: pathophysiologic and physiologic activities. J Thromb Haemost. 2016;14:28–39. [DOI] [PubMed] [Google Scholar]
- 14.Naudin C, Burillo E, Blankenberg S, Butler L, Renné T. Factor XII contact activation. Semin Thromb Hemost. 2017;43:814–26. [DOI] [PubMed] [Google Scholar]
- 15.Zhao M, Abdel-Razek T, Sun M-F, Gailani D. Characterization of a heparin binding site on the heavy chain of factor XI. J Biol Chem. 1998;273:31153–9. [DOI] [PubMed] [Google Scholar]
- 16.Badellino KO, Walsh PN. Localization of a heparin binding site in the catalytic domain of factor XIa. Biochemistry. 2001;40:7569–80. [DOI] [PubMed] [Google Scholar]
- 17.Yang L, Sun M, Gailani D, Rezaie AR. Characterization of a heparin-binding site on the catalytic domain of factor XIa: mechanism of heparin acceleration of factor XIa inhibition by the serpins anti-thrombin and C1-inhibitor. Biochemistry. 2009;48:1517–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ho DH, Badellino K, Baglia FA, Walsh PN. A binding site for heparin in the Apple 3 domain of factor XI. J Biol Chem. 1998;273:16382–90. [DOI] [PubMed] [Google Scholar]
- 19.Geng Y, Verhamme IM, Smith SA, Cheng Q, Sun M, Sheehan JP, et al. Factor XI anion-binding sites are required for productive interactions with polyphosphate. J Thromb Haemost. 2013;11:2020–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ivanov I, Shakhawat R, Sun M-F, Dickeson SK, Puy C, McCarty OJT, et al. Nucleic acids as cofactors for factor XI and prekallikrein activation: different roles for high-molecular-weight kininogen. Thromb Haemost. 2017;117:671–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cheng Q, Tucker EI, Pine MS, Sisler I, Matafonov A, Sun M-F, et al. A role for factor XIIa-mediated factor XI activation in thrombus formation in vivo. Blood. 2010;116:3981–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ivanov I, Matafonov A, Sun M-F, Mohammed BM, Cheng Q, Dickeson SK, et al. A mechanism for hereditary angioedema with normal C1 inhibitor: an inhibitory regulatory role for the factor XII heavy chain. Blood. 2019;133:1152–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gailani D, Lasky NM, Broze JG. A murine model of factor XI deficiency. Blood Coagul Fibrinolysis. 1997;8:134–44. [DOI] [PubMed] [Google Scholar]
- 24.Pauer H-U, Renné T, Hemmerlein B, Legler T, Fritzlar S, Adham I, et al. Targeted deletion of murine coagulation factor XII gene-a model for contact phase activation in vivo. Thromb Haemost. 2004;92:503–8. [DOI] [PubMed] [Google Scholar]
- 25.Liu J, Gao B-B, Clermont AC, Blair P, Chilcote TJ, Sinha S, et al. Hyperglycemia-induced cerebral hematoma expansion is mediated by plasma kallikrein. Nat Med. 2011;17:206–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Merkulov S, Zhang W-M, Komar AA, Schmaier AH, Barnes E, Zhou Y, et al. Deletion of murine kininogen gene 1 (mKng1) causes loss of plasma kininogen and delays thrombosis. Blood. 2008;111:1274–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gailani D, Sun MF, Sun Y. A comparison of murine and human factor XI. Blood. 1997;90:1055–64. [PubMed] [Google Scholar]
- 28.Liu F, Song Y, Liu D. Hydrodynamics-based transfection in animals by systemic administration of plasmid DNA. Gene Ther. 1999;6:1258–66. [DOI] [PubMed] [Google Scholar]
- 29.Geng Y, Verhamme IM, Smith SB, Sun M, Matafonov A, Cheng Q, et al. The dimeric structure of factor XI and zymogen activation. Blood. 2013;121:3962–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Choi SH, Smith SA, Morrissey JH. Polyphosphate is a cofactor for the activation of factor XI by thrombin. Blood. 2011;118:6963–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ivanov I, Matafonov A, Gailani D. Single-chain factor XII: a new form of activated factor XII. Curr Opin Hematol. 2017;24:411–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chargaff E. Studies on the chemistry of blood coagulation VII. Protamines and blood clotting. J Biol Chem. 1938;125:671–6. [Google Scholar]
- 33.Wiggins RC, Bouma BN, Cochrane CG, Griffin JH. Role of high- molecular-weight kininogen in surface-binding and activation of coagulation Factor XI and prekallikrein. Proc Natl Acad Sci. 1977;74:4636–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mahdi F, Shariat-Madar Z, Schmaier AH. The relative priority of prekallikrein and factors XI/XIa assembly on cultured endothelial cells. J Biol Chem. 2003;278:43983–90. [DOI] [PubMed] [Google Scholar]
- 35.Mohammed BM, Cheng Q, Matafonov A, Monroe DM, Meijers JCM, Gailani D. Factor XI promotes hemostasis in factor IX-deficient mice. J Thromb Haemost. 2018;16:2044–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim R. Native agarose gel electrophoresis of multiprotein complexes. Cold Spring Harb Protoc. 2011;7:884–7. [DOI] [PubMed] [Google Scholar]
- 37.Rosen ED, Gailani D, Castellino FJ. FXI is essential for thrombus formation following FeCl3-induced injury of the carotid artery in the mouse. Thromb Haemost. 2002;87:774–6. [PubMed] [Google Scholar]
- 38.Rao AK, Niewiarowski S, James P, Holt JC, Harris M, Elfenbein B, et al. Effect of heparin on the in vivo release and clearance of human platelet factor 4. Blood. 1983;61:1208–14. [PubMed] [Google Scholar]
- 39.Huber K, Resch I, Rosc D, Probst P, Kaindl F, Binder BR. Heparin induced increase of t-PA antigen plasma levels in patients with unstable angina: no evidence for clinical benefit of heparinization during the initial phase of treatment. Thromb Res. 1989;55:779–84. [DOI] [PubMed] [Google Scholar]
- 40.Novotny WF, Palmier M, Wun TC, Broze GJ, Miletich JP. Purification and properties of heparin-releasable lipoprotein-associated coagulation inhibitor. Blood. 1991;78:394–400. [PubMed] [Google Scholar]
- 41.Frost PH, Shore VG, Havel RJ. Purification of canine post-heparin hepatic lipase. Biochim Biophys Acta. 1982;712:71–8. [DOI] [PubMed] [Google Scholar]
- 42.Karlsson K, Marklund SL. Heparin-induced release of extracellular superoxide dismutase to human blood plasma. Biochem J. 1987;242:55–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vilaró S, Llobera M, Bengtsson-Olivecrona G, Olivecrona T. Lipoprotein lipase uptake by the liver: localization, turnover, and metabolic role. Am J Physiol. 1988;254:G711–22. [DOI] [PubMed] [Google Scholar]
- 44.Puy C, Tucker EI, Matafonov A, Cheng Q, Zientek KD, Gailani D, et al. Activated factor XI increases the procoagulant activity of the extrinsic pathway by inactivating tissue factor pathway inhibitor. Blood. 2015;125:1488–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang X, Cheng Q, Xu L, Feuerstein GZ, Hsu M-Y, Smith PL, et al. Effects of factor IX or factor XI deficiency on ferric chloride-induced carotid artery occlusion in mice. J Thromb Haemost. 2005;3:695–702. [DOI] [PubMed] [Google Scholar]
- 46.Kleinschnitz C, Stoll G, Bendszus M, Schuh K, Pauer H-U, Burfeind P, et al. Targeting coagulation factor XII provides protection from pathological thrombosis in cerebral ischemia without interfering with hemostasis. J Exp Med. 2006;203:513–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang X, Smith PL, Hsu M-Y, Gailani D, Schumacher WA, Ogletree ML, et al. Effects of factor XI deficiency on ferric chloride-induced vena cava thrombosis in mice. J Thromb Haemost. 2006;4:1982–8. [DOI] [PubMed] [Google Scholar]
- 48.Chan JC, Ganopolsky JG, Cornelissen I, Suckow MA, Sandoval- Cooper MJ, Brown EC, et al. The characterization of mice with a targeted combined deficiency of protein c and factor XI. Am J Pathol. 2001;158:469–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ay C, Hisada Y, Cooley BC, Mackman N. Factor XI-deficient mice exhibit increased bleeding after injury to the saphenous vein. J Thromb Haemost. 2017;15:1829–33. [DOI] [PubMed] [Google Scholar]
- 50.Lorentz CU, Verbout NG, Cao Z, Liu L, Hinds MT, McCarty OJT, et al. Factor XI contributes to myocardial ischemia-reperfusion injury in mice. Blood Adv. 2018;2:85–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cheng Q, Zhao Y, Lawson WE, Polosukhin VV, Johnson JE, Blackwell TS, et al. The effects of intrinsic pathway protease deficiencies on plasminogen-deficient mice. Blood. 2005;106:3055–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tucker EI, Verbout NG, Leung PY, Hurst S, McCarty OJT, Gailani D, et al. Inhibition of factor XI activation attenuates inflammation and coagulopathy while improving the survival of mouse polymicrobial sepsis. Blood. 2012;119:4762–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Luo D, Szaba FM, Kummer LW, Johnson LL, Tucker EI, Gruber A, et al. Factor XI-deficient mice display reduced inflammation, coagulopathy, and bacterial growth during listeriosis. Infect Immun. 2012;80:91–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shnerb Ganor R, Harats D, Schiby G, Gailani D, Levkovitz H, Avivi C, et al. Factor XI deficiency protects against atherogenesis in apolipoprotein E/factor XI double knockout mice. Arterioscler Thromb Vasc Biol. 2016;36:475–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bane CE, Ivanov I, Matafonov A, Boyd KL, Cheng Q, Sherwood ER, et al. Factor XI deficiency alters the cytokine response and activation of contact proteases during polymicrobial sepsis in mice. PLoS ONE. 2016;11:e0152968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kossmann S, Lagrange J, Jäckel S, Jurk K, Ehlken M, Schönfelder T, et al. Platelet-localized FXI promotes a vascular coagulation-inflammatory circuit in arterial hypertension. Sci Transl Med. 2017;9:pii: eaah4923. [DOI] [PubMed] [Google Scholar]
- 57.Stroo I, Yang J, de Boer JD, Roelofs JJTH, van ‘t Veer C, Castellino FJ, et al. Factor XI deficiency enhances the pulmonary allergic response to house dust mite in mice independent of factor XII. Am J Physiol Lung Cell Mol Physiol 2017;312:L163–71. [DOI] [PubMed] [Google Scholar]
- 58.Tucker EI, Gailani D, Hurst S, Cheng Q, Hanson SR, Gruber A. Survival advantage of coagulation factor XI-deficient mice during peritoneal sepsis. J Infect Dis. 2008;198:271–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Luo D, Szaba FM, Kummer LW, Plow EF, Mackman N, Gailani D, et al. Protective roles for fibrin, tissue factor, plasminogen activator inhibitor-1, and thrombin activatable fibrinolysis inhibitor, but not factor XI, during defense against the gram-negative bacterium Yersinia enterocolitica. J Immunol. 1950;2011(187):1866–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stroo I, Zeerleder S, Ding C, Luken BM, Roelofs JJTH, de Boer JD, et al. Coagulation factor XI improves host defence during murine pneumonia-derived sepsis independent of factor XII activation. Thromb Haemost. 2017;117:1601–14. [DOI] [PubMed] [Google Scholar]
- 61.Weitz JI, Fredenburgh JC. Factors XI and XII as targets for new anticoagulants. Front Med. 2017;4:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tillman BF, Gruber A, McCarty OJT, Gailani D. Plasma contact factors as therapeutic targets. Blood Rev. 2018;32:433–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tucker EI, Marzec UM, White TC, Hurst S, Rugonyi S, McCarty OJT, et al. Prevention of vascular graft occlusion and thrombus- associated thrombin generation by inhibition of factor XI. Blood. 2009;113:936–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gruber A, Hanson SR. Factor XI-dependence of surface- and tissue factor-initiated thrombus propagation in primates. Blood. 2003;102:953–5. [DOI] [PubMed] [Google Scholar]
- 65.Popescu NI, Silasi R, Keshari RS, Girton A, Burgett T, Zeerleder SS, et al. Peptidoglycan induces disseminated intravascular coagulation in baboons through activation of both coagulation pathways. Blood. 2018;132:849–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wheeler AP, Gailani D. Why factor XI deficiency is a clinical concern. Expert Rev Hematol. 2016;9:629–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gentry PA, Ross ML. Coagulation factor XI deficiency in Holstein cattle: expression and distribution of factor XI activity. Can J Vet Res. 1994;58:242–7. [PMC free article] [PubMed] [Google Scholar]
- 68.Knowler C, Giger U, Dodds WJ, Brooks M. Factor XI deficiency in Kerry Blue Terriers. J Am Vet Med Assoc. 1994;205:1557–61. [PubMed] [Google Scholar]
- 69.Troxel MT, Brooks MB, Esterline ML. Congenital factor XI deficiency in a domestic shorthair cat. J Am Anim Hosp Assoc. 2002;38:549–53. [DOI] [PubMed] [Google Scholar]
- 70.Gui T, Lin H-F, Jin D- Y, Hoffman M, Straight DL, Roberts HR, et al. Circulating and binding characteristics of wild-type factor IX and certain Gla domain mutants in vivo. Blood. 2002;100:153–8. [DOI] [PubMed] [Google Scholar]
- 71.Gui T, Reheman A, Ni H, Gross PL, Yin F, Monroe D, et al. Abnormal hemostasis in a knock-in mouse carrying a variant of factor IX with impaired binding to collagen type IV. J Thromb Haemost. 2009;7:1843–51. [DOI] [PubMed] [Google Scholar]
- 72.Silasi R, Keshari R, van Rensburg W, Regmi G, Lupu C, Lortenz C. Inhibition of factor XI activation by factor XIIa blocks coagulopathy and provides organ protection and survival benefit in a baboon model of S. aureus sepsis. Res Pract Thromb Haemost. 2017;1:115. [Google Scholar]
- 73.Bugge TH, Flick MJ, Daugherty CC, Degen JL. Plasminogen deficiency causes severe thrombosis but is compatible with development and reproduction. Genes Dev. 1995;9:794–807. [DOI] [PubMed] [Google Scholar]
- 74.Salomon O, Preis M, Abu Shtaya A, Kotler A, Stein N, Saliba W. Factor XI deficiency is not associated with an increased risk of pneumonia and pneumonia-related mortality. Haemophilia. 2018;24:634–40. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.