

Abstract
Chronic kidney disease (CKD) is a heterogeneous range of disorders affecting up to 11% of the world’s population. The majority of patients with CKD die of cardiovascular disease (CVD) before progressing to end-stage renal disease. CKD patients have an increased risk of atherosclerotic disease as well as a unique cardiovascular phenotype. There remains no clear aetiology for these issues and a better understanding of the pathophysiology of CKD-associated CVD is urgently needed. Although nonanimal studies can provide insights into the nature of disease, the whole-organism nature of CKD-associated CVD means that high-quality animal models, at least for the immediate future, are likely to remain a key tool in improving our understanding in this area. We will discuss the methods used to induce renal impairment in rodents and the methods available to assess cardiovascular phenotype and in each case describe the applicability to humans.
Keywords: animal models, cardiovascular disease, chronic kidney disease, translational models
Introduction
Chronic kidney disease (CKD) is the term given to a heterogeneous range of disorders affecting the structure and function of the kidney. It is a global health problem, affecting up to anywhere between 9 and 11% of the world’s population 1. It is driven by a broad range of aetiologies and can progress to end-stage renal disease, a condition that can only be treated by dialysis or renal transplantation. However, populations with CKD pose a major healthcare burden aside from provision of renal replacement therapy, mainly attributable to cardiovascular (CV) morbidity and mortality. Indeed, the majority of patients with CKD will die from cardiovascular disease (CVD) before progressing to end-stage renal disease 2.
Compared with the general population, patients with CKD have a higher burden of traditional CVD risk factors. These in turn behave in a more accelerated fashion, for example, CKD patients have more severe coronary artery atherosclerotic plaque formation compared with the general population 3,4 and the risk of myocardial infarction among CKD patients is twice that of patients without CKD 5. Similarly, peripheral artery disease 6 and stroke 7 all show increased risk as the estimated glomerular filtration rate begins to fall below 60 ml/min/1.73m2.
Alongside increased risks of atherosclerotic disease, CKD also leads to a distinctive CV phenotype characterized by prominent endothelial dysfunction, arterial stiffening and calcification along with left ventricular hypertrophy, fibrosis and cardiac dysrhythmia. CKD-associated CVD typically leads to clinical outcomes such as admission for heart failure and sudden cardiac death. It is these clinical events that constitute the leading causes of morbidity and mortality in patients receiving dialysis 8. Although a number of underlying risk factors and mechanisms for this clinical syndrome have been described including proteinuria 9, anaemia 10, salt retention 11, retention of uraemic toxins 12, inflammation 13 and oxidative stress 14, there remains no clear aetiological framework for these problems. A better understanding of the pathophysiology of CKD-associated CVD is therefore urgently required both to better understand the nature of the risk and to develop novel therapies aimed at reducing the burden of CVD in this population. Although epidemiological, clinical translational and in-vitro studies can provide insights into the nature of disease, the whole-organism nature of CVD-associated CVD means that high-quality animal models, at least for the immediate future, are likely to remain a key tool towards improving our understanding in this area.
Most scientists aspire to a future where the use of cell-based systems and cross-platform bioinformatics approaches makes animal experimentation redundant, but currently rodent disease models provide a unique insight into whole-organism physiology and pathophysiology. However, although animal models of disease remain necessary, all studies should be designed to minimize suffering and in accordance with the ‘three R’s’, that is, ‘Replacement’ of animals with other methods if possible, ‘Refinement’ of animal models to maximize data gained per animal and ‘Reduction’ of animals used where possible 15.
There is a long history of modelling chronic renal injury in rodents 16. These models can be used to (i) simulate specific diseases that cause CKD, for example diabetes 17 or systemic lupus erythematous 18; (ii) examine the final common fibrosis pathways of progressive CKD; or (iii) to investigate CKD complications such as CKD-associated CVD. It is this third aim that will be the focus of this review.
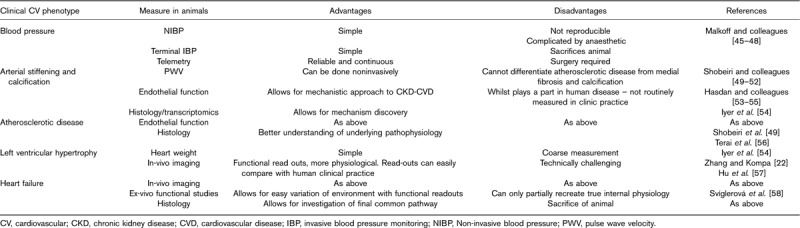
Although atherosclerotic disease and the consequences of athero-occlusive events have been modelled in animals in the context of renal injury (e.g. ApoE KO 19 or coronary artery occlusion in rat models 20), we focus on models aimed at investigating the distinctive CV phenotype of CKD-associated CVD in this review. We will discuss the methods used to induce renal impairment in rodents (Table 1) and the methods available to assess CV phenotype (Table 2) and in each case describe the applicability to humans.
Table 1.
Chronic kidney disease phenotyping in animal models

Table 2.
Cardiovascular phenotyping in animal models
Rodent models of chronic kidney disease
Unilateral ureteric obstruction
The unilateral ureteric obstruction model has been used in rats and mice. One ureter is tied off, causing increased urinary tract pressure proximal to the lesion, followed by an interstitial inflammatory response with subsequent cellular invasion and eventual tubulointerstitial fibrosis and atrophy. The degree of fibrosis is proportional to the length of time that the ureter is tied off 42. In general, the contralateral kidney in-situ provides a control ‘normal’ kidney to be analysed histologically alongside the obstructed one. The advantage of this model is that it is technically less challenging than nephrectomy, it is easily reproducible and works in most strains of both mice and rats. The renal damage occurs rapidly, reaching a peak within 7 days. However, although human CKD can be caused by urinary obstruction 43, overall, it is not a major cause of CKD in the adult population, and even when it is, partial rather than complete obstruction is typical 59. Furthermore, in rodent models, the remaining functioning kidney also goes onto compensate for loss of function of the obstructed kidney. Thus, biomarkers of renal failure, such as serum urea and creatinine or proteinuria, are often not clearly elevated using this model. Efforts to better reflect human disease, for example partial or reversible obstruction, have been explored 44, but are not in widespread use because of their surgical difficulty, therefore limiting the utility of this type of model.
Surgical nephron reduction
Subtotal nephrectomy mimics the consequences of reducing functional renal mass. It is most often used in rats and encompasses two different methodologies. The first is a ligation model, where one kidney is removed, with consequent ligation of polar branches of the renal artery of the contralateral kidney. In the other method, the rat undergoes a nephrectomy and then roughly 50% of the contralateral kidney is excised 1–2 weeks later. The latter model does not tend to manifest with hypertension, whereas the former does 21. In both models, the rats develop progressive renal failure by week 2, with renal histology showing progressive glomerular and interstitial fibrosis with renal dysfunction and proteinuria, which is similar to that found in human disease 22. Conversely, mice are typically resistant to induction of chronic renal injury by nephron reduction 23, although there is considerable interstrain variability 60. Furthermore, the challenges of carrying out surgery in small animals should not be underestimated 61.
Uninephrectomy deoxycorticosterone acetate/salt models
The mineralocorticoid deoxycorticosterone acetate (DOCA), when administered with a high-salt diet and unilateral nephrectomy, induces renal injury, low renin levels and hypertension in both rat and mouse models. It consists of a 2–6-week period of high salt and mineralocorticoid exposure. The DOCA itself is administered by either subcutaneous pellet insertion or oral supplementation. Animals then develop hypertension, followed by proteinuria, glomerular sclerosis, tubulointerstitial inflammation and fibrosis in keeping with progressive CKD 35, although the severity of the phenotype varies between different rodent strains. For example, 129/SV mice have markedly higher DOCA-induced blood pressure (BP), glomerulosclerosis, interstitial fibrosis and albuminuria compared with C57BL/6 mice 36.
This model has been used to investigate the relationship between the renin–angiotensin–aldosterone system activation and CKD 37 as well as the therapeutic effect of angiotensin-converting enzyme inhibitors 38. The CV phenotype probably reflects only one of the potential mechanisms (i.e. sodium retention±oxidative stress) through which kidney injury impacts on the CV system. Therefore, although this approach has been used extensively to study hypertension and the final common pathway of CKD, it may not provide a comprehensive model for investigating the fundamental mechanisms of CKD-associated CVD.
Nephrotoxic models of chronic kidney disease
Nephrotoxic models of CKD are attractive as they typically cause less suffering and are technically less challenging than surgical models. First proposed as a viable CKD model in 1982 24, adenine, when administered in high quantities, saturates the adenine phosphoribosyl transferase pathway, causing it to be oxidized into 2,8-dihydroxyadenine by xanthine oxidase. 2,8-dihydroxyadenine is renally excreted and precipitates within the tubule, leading to tubulointerstitial inflammation and fibrosis 25. The adenine model leads to severe CKD, with marked biochemical renal failure and associated vascular calcification 49. By adjusting the concentration of adenine in rat chow, Shobeiri et al. 49 could produce stable CKD at 5, 8 and 11 weeks. Sex plays a key role in this model, with female rats requiring higher adenine concentrations in their diet to achieve the same severity of kidney injury 26. The model has also been validated in mice 27. Hyperphosphataemia, secondary hyperparathyroidism, renal osteodystrophy and vascular calcification are prominent in this model 28, a phenotype consistent with the consequences of the CKD-mineral bone disorder (CKD-MB) observed in patients.
A single dose of 250 mg/kg of folic acid causes an acute kidney injury in mice. Animals show the development of crystals within the tubular lumen, causing acute tubular damage, followed by tubulointerstitial fibrosis over 2 weeks 32. By alkalinizing the urine, using sodium bicarbonate, folic acid crystals can be reduced, but tubular damage still occurs, suggesting that a direct nephrotoxic effect also contributes in this model 33. Acute damage occurs within 2 weeks, with the chronic fibrosis becoming apparent between 4 and 6 weeks. Biochemical markers of renal impairment occur in parallel with renal fibrosis 34, making this a particularly useful model for studying the consequences of CKD of different severities. This model is increasingly being used; for example Rattanasinganchan 62 used this approach to investigate urinary biomarkers of tubulointerstitial fibrosis.
Aristolochia, when administered to animals (rats at a dose of 10 mg/kg daily subcutaneous injections for 35 days, for mice 3 mg/kg, intraperitoneal, every 3 days for 6 weeks), induces proteinuria, elevated serum creatinine with associated tubular necrosis, atrophy and interstitial fibrosis by day 35 29,30. These findings closely resemble those found in Chinese-herb nephropathy in humans, a disease in which aristolochia has been shown to be the causative agent 63. As such, this is a model of human tubulointerstitial nephritis and has predominantly been used to study the molecular basis of kidney fibrosis rather than to investigate CKD-associated complications. However, given that albuminuria is a feature of this model, some studies have suggested that aristolochia may also lead to glomerular damage, with podocyte effacement noted on electron microscopy 31. This model has predominantly been investigated in mice (NMRI, FVB, C76BL/6 and C3H/He strains have all been studied), and there is some evidence that there may be a degree of renal recovery after 9–15 weeks 30.
Immunological models
Of the immunological models, the anti-Thy1 model in rats has been most studied. Thy1 is an antigen found on glomerular mesangial cells. When rats are injected with a single dose of either antithymocyte serum (containing anti-Thy1 antibodies) or a mouse anti-Thy1 monoclonal antibody, animals develop a glomerulonephritis 39. Histologically, there is initially mesangiolysis with an inflammatory cell infiltrate, followed by mesangial matrix expansion with the occasional extracellular crescent 40. This process takes roughly 1 week, with subsequent repair from 3 weeks onwards. The renal phenotype of proteinuria, haematuria and renal impairment reflects the abnormalities observed in human glomerulonephritis. Although in humans there is hypertension (as opposed to normotension in animals) and immune complex deposits histologically, these are not observed in the animal model. This model has also been combined with uninephrectomy to produce a glomerulonephritis model more typical of a glomerulonephritis-associated CKD 41. Note that this model is not viable in mice.
Other models
We have only outlined the most commonly used approaches to induce renal injury in rodents. Many others are described, particularly those aimed at modelling specific diseases; these include spontaneous models of glomerular and interstitial injury such as NZB/W lupus nephritis model 64 or the Sprague-Dawley rat ageing model 65, spontaneously hypertensive rats; 66 genetically engineered models such as the mouse Alports model lacking collagen α 3 (IV); 67 acquired immunological models such as models of antiglomerular basement disease; 68 and acquired nonimmunological models such as radiation nephropathy 69 or cyclosporine nephropathy 70.These approaches have been reviewed extensively elsewhere 16.
Cardiovascular phenotyping
BP is the simplest CV phenotype and is typically elevated in CKD. Noninvasive methods involve the use of a tail-cuff. This can be performed on awake, restrained rodents. However, restraint induces agitation in mice and rats unless they have been suitably conditioned and tail-cuff readings generally correlate poorly with invasive measures 45. Terminal invasive methods require the use of anaesthetic agents that may interfere with BP readings 46. Conversely, telemetry devices enable BP measurement during normal activity 47 and represent the gold-standard approach; however, this requires additional animal procedures, the surgery is technically challenging and the equipment is expensive. BP measurements have typically been performed in all the models described above. Hypertension is typically observed in the nephron reduction, DOCA-salt, folic acid and the adenine models; however, there are marked differences between species and strains 48.
Pulse wave velocity (PWV) using ultrasound noninvasively or invasively directly relates to the burden of atherosclerotic disease in both animals 50 and humans 51. However, it is also a key measure of arterial stiffness in the absence of atherosclerosis and therefore an important characteristic of CKD-associated CVD. PWV is quantified in animals by measuring two pressures a known distant apart with high-fidelity transducers and then determining the delay between waveforms. The adenine model shows increased PWV; this occurs early within 5 days of model induction 52 and continues as the animals vasculature becomes more calcified 71.
Abnormal endothelial function has long been linked to the pathophysiology of CKD-associated CVD with a number of implicated mechanisms including alterations in nitric oxide (NO), asymmetric dimethylarginine and advanced glycation end products 72. Hypertension is known to cause endothelial dysfunction and in nephrectomy rat models 53 and DOCA-salt models, aberrant levels of endothelial mediators are found 54. However, the folic acid 73, adenine 74,75 and unilateral ureteric obstruction 76 models have all been used to investigate the mechanisms and potential therapeutics of endothelial dysfunction in CKD. Most simply, these methods are ex-vivo transcriptomic assays; however, there have been some attempts to model in-vivo endothelial function using high-resolution ultrasound to measure flow-mediated vasodilation in both rats and mice in response to different endothelial activators or inhibitors 55,77.
Arterial histology in CKD-associated CVD typically shows medial thickening or fibrosis along with calcification in the smooth muscle layer and this phenotype has been demonstrated in animal models. Initially, the adenine model involved feeding the animals a high-phosphate diet that markedly reduced the animals’ weight and confounded results; however, an updated model involving a high initial adenine dose combined with a normal diet has resolved this issue 56. However, in nonadenine models, combining a high-phosphate diet with the existing model can heighten the calcific phenotype 49. Histopathology can also indicate accelerated plaque formation when examining models of atherosclerotic disease in the context of chronic renal injury.
Functional cardiac imaging is also possible in small animals. Patients with CKD-associated CVD typically have left ventricular hypertrophy, ventricular dysfunction and cardiac fibrosis. Teams such as Zhang et al. 22 have utilized echocardiography to analyse systolic and diastolic functioning of the hearts in animal models of renal injury, observing impairments in both. Cardiac MRI has also been used to illustrate cardiac hypertrophy and fibrosis in rodent models of CKD 57.
Others have shown ex-vivo functional changes for example, decreased contractility in a subtotal nephrectomy model in rats 58. Cardiac histology (and weight) can also be used to demonstrate increased heart size and fibrosis. For example, the DOCA-salt model (rats and mice) also shows a cardiac phenotype in keeping with hypertension with increased heart size and cardiac fibrosis 54.
To what degree the above animal models of chronic kidney injury recapitulate the CV phenotype, other than hypertension and left ventricular hypertrophy, observed in patients has been explored inadequately in all except a few cases. However, some of the above models do show vascular features suggestive of some aspects of arterial disease observed in CKD and are worth additional comment.
For example, the adenine model, alongside hypertension, mimics the CKD-MBD with evidence of arterial calcification and hyperphosphataemia and secondary hyperparathyroidism. Evidence from this model suggests that vascular smooth muscle cells transform into an osteochondrogenic-like phenotype 78–80.
Reduced NO bioavailability has long been suggested to underlie vascular pathology in CKD. Rats that have undergone subtotal nephrectomy develop a worsening systolic function and cardiac fibrosis (both features of CVD-associated CKD) when treated with a low dose of a NO synthase inhibitor, providing support to this theory 81. Similarly, a nephrectomy rat model has been used to highlight the role of the uraemic toxin indoxylsulphate in cardiac fibrosis, as well as test out monoclonal antibodies against cardiotonic steroids implicated in uraemic cardiomyopathy 82,83.
Therefore, the evidence for the mechanisms underlying the kidney–vascular link remains sparse and certainly there is no comprehensive understanding of the pathological pathways leading to the development of CKD-associated CVD. This will not only require induction of a range of different models of CKD but the systematic characterization of the CV phenotype using whole-organism physiological measures, imaging and interrogation of isolated tissues across multiple time points.
General considerations when designing experiments on the basis of animal models of chronic kidney disease-associated cardiovascular disease
A number of further considerations arise when planning experiments on the basis of animal models of CKD-associated CVD, both general and specific to the research question. The generalizability of the model, given the age, sex, species and strain of the animals included in the experiment, is fundamental to the conclusions being drawn. For example although experiments are typically conducted in mice at age 8–12 weeks of age, older animals 84 are likely to better reflect the pathophysiology observed in human CKD, a disease of middle and old age 85. Similarly, the sex of animals has been shown to influence the phenotype of models across a wide range of organ systems 86.
Species and strain are also important. Although mouse models are becoming more popular than rats because of the ability for genetic manipulation 16 and the lower cost, many of the CKD models rely on surgical procedures and CV measurements that are technically less challenging in larger animals such as rats. Furthermore, several strains of mice seem uniquely resistant to developing disease phenotype in several of the models described above (Table 1). In addition, rats used in experiments are typically outbred; thus, standardized nomenclature describing each strain, for example, ‘Wistar’, may be misleading as there can be a great deal of genetic heterogeneity within these broad strain descriptions 87.
Focusing on models of CKD-associated CVD, it is important to ensure that key CV findings are not related to the method of inducing kidney injury in the animal. Hence, any proposed pathway or intervention should be examined in at least two differing models of CKD, for example, one surgical and nephrotoxic model, and ideally across both rats and mice. Similarly, the heterogeneity of human CKD must be recognized, again implying any important findings are replicated across surgical, toxic and immunologically induced models of renal injury. Finally, the complexity of some of the procedures, particularly those related to CV phenotyping, means that only teams with extensive experience of performing this type of work can achieve high levels of reproducibility and minimize adverse events for experimental animals. To ensure compliance with the ‘three R’s’, investigators without appropriate skills should ensure that they collaborate with those who do.
Conclusion
Although advances in technology may mean that this approach becomes redundant, animal models will likely continue to be a useful method to describe pathophysiology and test potential therapies in CKD-related CVD for decades to come. Thoughtful design of experiments, replication using different models and in-depth phenotyping using a wide range of techniques mean that we can maximize the benefits from these studies and, over the longer term, produce real benefits for patients.
Acknowledgements
Omid Sadeghi-Alavijeh is a National Institute for Health Research (NIHR)-funded academic clinical fellow.
Conflicts of interest
There are no conflicts of interest.
References
- 1.Hallan SI, Coresh J, Astor BC, Asberg A, Powe NR, Romundstad S, et al. International comparison of the relationship of chronic kidney disease prevalence and ESRD risk. J Am Soc Nephrol 2006; 17:2275–2284. [DOI] [PubMed] [Google Scholar]
- 2.Berl T, Henrich W. Kidney-heart interactions: epidemiology, pathogenesis, and treatment. Clin J Am Soc Nephrol 2006; 1:8–18. [DOI] [PubMed] [Google Scholar]
- 3.Baber U, Stone GW, Weisz G, Moreno P, Dangas G, Maehara A, et al. Coronary plaque composition, morphology, and outcomes in patients with and without chronic kidney disease presenting with acute coronary syndromes. JACC Cardiovasc Imaging 2012; 5:S53–S61. [DOI] [PubMed] [Google Scholar]
- 4.Muntner P, He J, Astor BC, Folsom AR, Coresh J. Traditional and nontraditional risk factors predict coronary heart disease in chronic kidney disease: results from the atherosclerosis risk in communities study. J Am Soc Nephrol 2005; 16:529–538. [DOI] [PubMed] [Google Scholar]
- 5.Collins AJ, Foley RN, Gilbertson DT, Chen S-C. United States renal data system public health surveillance of chronic kidney disease and end-stage renal disease. Kidney Int Suppl (2011) 2015; 5:2–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wattanakit K, Folsom AR, Selvin E, Coresh J, Hirsch AT, Weatherley BD. Kidney function and risk of peripheral arterial disease: results from the atherosclerosis risk in communities (ARIC) study. J Am Soc Nephrol 2007; 18:629–636. [DOI] [PubMed] [Google Scholar]
- 7.Abramson JL, Jurkovitz CT, Vaccarino V, Weintraub WS, Mcclellan W. Chronic kidney disease, anemia, and incident stroke in a middle-aged, community-based population: the ARIC study. Kidney Int 2003; 64:610–615. [DOI] [PubMed] [Google Scholar]
- 8.Collins AJ, Foley R, Herzog C, Chavers B, Gilbertson D, Ishani A, et al. United States Renal Data System 2007 Annual Data Report Abstract. Am J Kidney Dis 2008; 51:A6–A7. [DOI] [PubMed] [Google Scholar]
- 9.Mykkänen L, Zaccaro DJ, O’Leary DH, Howard G, Robbins DC, Haffner SM. Microalbuminuria and carotid artery intima-media thickness in nondiabetic and NIDDM subjects. Stroke 1997; 28:1710–1716. [DOI] [PubMed] [Google Scholar]
- 10.Besarab A, Bolton WK, Browne JK, Egrie JC, Nissenson AR, Okamoto DM, et al. The effects of normal as compared with low hematocrit values in patients with cardiac disease who are receiving hemodialysis and epoetin. N Engl J Med 1998; 339:584–590. [DOI] [PubMed] [Google Scholar]
- 11.Sarnak MJ, Levey AS, Schoolwerth AC, Coresh J, Culleton B, Hamm LL, et al. Kidney disease as a risk factor for development of cardiovascular disease. Circulation 2003; 108:2154–2169. [DOI] [PubMed] [Google Scholar]
- 12.Caplin B, Nitsch D, Gill H, Hoefield R, Blackwell S, MacKenzie D, et al. Circulating methylarginine levels and the decline in renal function in patients with chronic kidney disease are modulated by DDAH1 polymorphisms. Kidney Int 2010; 77:459–467. [DOI] [PubMed] [Google Scholar]
- 13.Yeun JY, Levine RA, Mantadilok V, Kaysen GA. C-Reactive protein predicts all-cause and cardiovascular mortality in hemodialysis patients. Am J Kidney Dis 2000; 35:469–476. [DOI] [PubMed] [Google Scholar]
- 14.Himmelfarb J, Joannidis M, Molitoris B, Schietz M, Okusa MD, Warnock D, et al. Evaluation and initial management of acute kidney injury. Clin J Am Soc Nephrol 2008; 3:962–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fenwick N, Griffin G, Gauthier C. The welfare of animals used in science: how the ‘Three Rs’ ethic guides improvements. Can Vet J 2009; 50:523. [PMC free article] [PubMed] [Google Scholar]
- 16.Hewitson TD, Ono T, Becker GJ. Small animal models of kidney disease: a review. Methods Mol Biol 2009; 466:41–57. [DOI] [PubMed] [Google Scholar]
- 17.Betz B, Conway BR. An update on the use of animal models in diabetic nephropathy research. Curr Diab Rep 2016; 16:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cohen PL, Maldonado MA. Animal models for SLE. Curr Protoc Immunol 2003. Chapter 15:Unit 15.20. [DOI] [PubMed] [Google Scholar]
- 19.Bro S, Bentzon JF, Falk E, Andersen CB, Olgaard K, Nielsen LB. Chronic renal failure accelerates atherogenesis in apolipoprotein E-deficient mice. J Am Soc Nephrol 2003; 14:2466–2474. [DOI] [PubMed] [Google Scholar]
- 20.Cho E, Kim M, Ko YS, Lee HY, Song M, Kim MG, et al. Role of inflammation in the pathogenesis of cardiorenal syndrome in a rat myocardial infarction model. Nephrol Dial Transplant 2013; 28:2766–2778. [DOI] [PubMed] [Google Scholar]
- 21.Chow KM, Liu ZC, Chang TMS. Animal remnant kidney model of chronic renal failure revisited. Hong Kong J Nephrol 2003; 5:57–64. [Google Scholar]
- 22.Zhang Y, Kompa AR. A practical guide to subtotal nephrectomy in the rat with subsequent methodology for assessing renal and cardiac function. Nephrology 2014; 19:552–561. [DOI] [PubMed] [Google Scholar]
- 23.Kren S, Hostetter TH. The course of the remnant kidney model in mice. Kidney Int 1999; 56:333–337. [DOI] [PubMed] [Google Scholar]
- 24.Yokozawa T, Oura H, Okada T. Metabolic effects of dietary purine in rats. J Nutr Sci Vitaminol (Tokyo) 1982; 28:519–526. [DOI] [PubMed] [Google Scholar]
- 25.Koeda T, Wakaki K, Koizumi F, Yokozawa T, Oura H. Early changes of proximal tubules in the kidney of adenine-ingesting rats, with special reference to biochemical and electron microscopic studies. Nihon Jinzo Gakkai Shi 1988; 30:239–246. [PubMed] [Google Scholar]
- 26.Ogirima T, Tano K, Kanehara M, Gao M, Wang X, Guo Y, et al. Sex difference of adenine effects in rats: renal function, bone mineral density and sex steroidogenesis. Endocr J 2006; 53:407–413. [DOI] [PubMed] [Google Scholar]
- 27.Jia T, Olauson H, Lindberg K, Amin R, Edvardsson K, Lindholm B, et al. A novel model of adenine-induced tubulointerstitial nephropathy in mice. BMC Nephrol 2013; 14:116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Katsumata K, Kusano K, Hirata M, Tsunemi K, Nagano N, Burke SK, Fukushima N. Sevelamer hydrochloride prevents ectopic calcification and renal osteodystrophy in chronic renal failure rats. Kidney Int 2003; 64:441–450. [DOI] [PubMed] [Google Scholar]
- 29.Debelle FD, Nortier JL, De Prez EG, Garbar CH, Vienne AR, Salmon IJ, et al. Aristolochic acids induce chronic renal failure with interstitial fibrosis in salt-depleted rats. J Am Soc Nephrol 2002; 13:431–436. [DOI] [PubMed] [Google Scholar]
- 30.Huang L, Scarpellini A, Funck M, Verderio EAM, Johnson TS. Development of a chronic kidney disease model in C57BL/6 mice with relevance to human pathology. Nephron Extra 2013; 3:12–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou Y, Bian X, Fang L, He W, Dai C, Yang J. Aristolochic acid causes albuminuria by promoting mitochondrial DNA damage and dysfunction in podocyte. PLoS One 2013; 8:e83408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Doi K, Okamoto K, Negishi K, Suzuki Y, Nakao A, Fujita T, et al. Attenuation of folic acid-induced renal inflammatory injury in platelet-activating factor receptor-deficient mice. Am J Pathol 2006; 168:1413–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yuan HT, Li XZ, Pitera JE, Long DA, Woolf AS. Peritubular capillary loss after mouse acute nephrotoxicity correlates with down-regulation of vascular endothelial growth factor-A and hypoxia-inducible factor-1 alpha. Am J Pathol 2003; 163:2289–2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Eddy AA, López-Guisa JM, Okamura DM, Yamaguchi I. Investigating mechanisms of chronic kidney disease in mouse models. Pediatr Nephrol 2012; 27:1233–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mohammed-Ali Z, Cruz GL, Lu C, Carlisle RE, Werner KE, Ask K, et al. Development of a model of chronic kidney disease in the C57BL/6 mouse with properties of progressive human CKD. Biomed Res Int 2015; 2015:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hartner A, Cordasic N, Klanke B, Veelken R, Hilgers KF. Strain differences in the development of hypertension and glomerular lesions induced by deoxycorticosterone acetate salt in mice. Nephrol Dial Transplant 2003; 18:1999–2004. [DOI] [PubMed] [Google Scholar]
- 37.Kirchhoff F, Krebs C, Abdulhag UN, Meyer-Schwesinger C, Maas R, Helmchen U, et al. Rapid development of severe end-organ damage in C57BL/6 mice by combining DOCA salt and angiotensin II. Kidney Int 2008; 73:643–650. [DOI] [PubMed] [Google Scholar]
- 38.Hatta T, Nakata T, Harada S, Kiyama M, Moriguchi J, Morimoto S, et al. Lowering of blood pressure improves endothelial dysfunction by increase of nitric oxide production in hypertensive rats. Hypertens Res 2002; 25:455–460. [DOI] [PubMed] [Google Scholar]
- 39.Ishizaki M, Masuda Y, Fukuda Y, Sugisaki Y, Yamanaka N, Masugi Y. Experimental mesangioproliferative glomerulonephritis in rats induced by intravenous administration of anti-thymocyte serum. Pathol Int 2008; 36:1191–1203. [DOI] [PubMed] [Google Scholar]
- 40.Liu N, Makino T, Nogaki F, Kusano H, Suyama K, Muso E, et al. Coagulation in the mesangial area promotes ECM accumulation through factor V expression in MsPGN in rats. AJP. Ren Physiol 2004; 287:F612–F620. [DOI] [PubMed] [Google Scholar]
- 41.Wada Y, Morioka T, Oyanagi-Tanaka Y, Yao J, Suzuki Y, Gejyo F, et al. Impairment of vascular regeneration precedes progressive glomerulosclerosis in anti-Thy 1 glomerulonephritis. Kidney Int 2002; 61:432–443. [DOI] [PubMed] [Google Scholar]
- 42.Klahr S, Morrissey J. Obstructive nephropathy and renal fibrosis. Am J Physiol Ren Physiol 2002; 283:F861–F85. [DOI] [PubMed] [Google Scholar]
- 43.Roth KS, Koo HP, Spottswood SE, Chan JC. Obstructive uropathy: an important cause of chronic renal failure in children. Clin Pediatr (Phila) 2002; 41:309–314. [DOI] [PubMed] [Google Scholar]
- 44.Josephson S, Robertson B, Claesson G, Wikstad I. Experimental obstructive hydronephrosis in newborn rats. I. Surgical technique and long-term morphologic effects. Invest Urol 1980; 17:478–483. [PubMed] [Google Scholar]
- 45.Malkoff J. Non-invasive blood pressure for mice and rats. Animal Lab News 2005; 1:1–2. [Google Scholar]
- 46.Parasuraman S, Raveendran R. Measurement of invasive blood pressure in rats. J Pharmacol Pharmacother 2012; 3:172–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Huetteman DA, Bogie H. Direct blood pressure monitoring in laboratory rodents via implantable radio telemetry. Methods Mol Biol 2009; 573:57–73. [DOI] [PubMed] [Google Scholar]
- 48.Kurtz TW, Griffin KA, Bidani AK, Davisson RL, Hall JE. Recommendations for blood pressure measurement in humans and experimental animals. Hypertension 2005; 45:299–310. [DOI] [PubMed] [Google Scholar]
- 49.Shobeiri N, Adams MA, Holden RM. Vascular calcification in animal models of CKD: a review. Am J Nephrol 2010; 31:471–481. [DOI] [PubMed] [Google Scholar]
- 50.Di Lascio N, Stea F, Kusmic C, Sicari R, Faita F. Non-invasive assessment of pulse wave velocity in mice by means of ultrasound images. Atherosclerosis 2014; 237:31–37. [DOI] [PubMed] [Google Scholar]
- 51.Blacher J, Asmar R, Djane S, London GM, Safar ME. Aortic pulse wave velocity as a marker of cardiovascular risk in hypertensive patients. Hypertension 1999; 33:1111–1117. [DOI] [PubMed] [Google Scholar]
- 52.Nguy L, Johansson ME, Grimberg E, Lundgren J, Teerlink T, Carlström M, et al. Rats with adenine-induced chronic renal failure develop low-renin, salt-sensitive hypertension and increased aortic stiffness. Am J Physiol Regul Integr Comp Physiol 2013; 304:R744–R752. [DOI] [PubMed] [Google Scholar]
- 53.Hasdan G, Benchetrit S, Rashid G, Green J, Bernheim J, Rathaus M. Endothelial dysfunction and hypertension in 5/6 nephrectomized rats are mediated by vascular superoxide. Kidney Int 2002; 61:586–590. [DOI] [PubMed] [Google Scholar]
- 54.Iyer A, Chan V, Brown L. The DOCA-salt hypertensive rat as a model of cardiovascular oxidative and inflammatory stress. Curr Cardiol Rev 2010; 6:291–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schuler D, Sansone R, Freudenberger T, Mateos AR, Weber G, Momma T, et al. Measurement of endothelium-dependent vasodilation in mice. Arterioscler Thromb Vasc Biol 2014; 34:2651–2657. [DOI] [PubMed] [Google Scholar]
- 56.Terai K, Nara H, Takakura K, Mizukami K, Sanagi M, Fukushima S, et al. Vascular calcification and secondary hyperparathyroidism of severe chronic kidney disease and its relation to serum phosphate and calcium levels. Br J Pharmacol 2009; 156:1267–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hu MC, Shi M, Cho HJ, Adams-Huet B, Paek J, Hill K, et al. Klotho and phosphate are modulators of pathologic uremic cardiac remodeling. J Am Soc Nephrol 2015; 26:1290–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Svíglerová J, Kuncová J, Nalos L, Tonar Z, Rajdl D, Stengl M. Cardiovascular parameters in rat model of chronic renal failure induced by subtotal nephrectomy. Physiol Res 2010; 59 (Suppl 1): S81–S88. [DOI] [PubMed] [Google Scholar]
- 59.Levey AS, Eckardt KU, Tsukamoto Y, Levin A, Coresh J, Rossert J, et al. Definition and classification of chronic kidney disease: a position statement from Kidney Disease: Improving Global Outcomes (KDIGO). Kidney Int 2005; 67:2089–2100. [DOI] [PubMed] [Google Scholar]
- 60.Ma LJ, Fogo AB. Model of robust induction of glomerulosclerosis in mice: importance of genetic background. Kidney Int 2003; 64:350–355. [DOI] [PubMed] [Google Scholar]
- 61.Pritchett-Corning KR, Luo Y, Mulder GB, White WJ. Principles of rodent surgery for the new surgeon. J Vis Exp 2011; 47:2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rattanasinganchan P. A folic acid-induced rat model of renal injury to identify biomarkers of tubulointerstitial fibrosis from urinary exosomes. Asian Biomed 2016; 0:491. [Google Scholar]
- 63.Vanherweghem JL, Tielemans C, Abramowicz D, Depierreux M, Vanhaelen-Fastre R. Vanhaelen M, et al. Rapidly progressive interstitial renal fibrosis in young women: association with slimming regimen including Chinese herbs. Lancet 1993; 341:387–391. [DOI] [PubMed] [Google Scholar]
- 64.Morel L, Wakeland EK. Susceptibility to lupus nephritis in the NZB/W model system. Curr Opin Immunol 1998; 10:718–725. [DOI] [PubMed] [Google Scholar]
- 65.Goldstein RS, Tarloff JB, Hook JB. Age-related nephropathy in laboratory rats. FASEB J 1988; 2:2241–2251. [DOI] [PubMed] [Google Scholar]
- 66.Ofstad J, Iversen BM. Glomerular and tubular damage in normotensive and hypertensive rats. Am J Physiol Renal Physiol 2005; 288:F665–F672. [DOI] [PubMed] [Google Scholar]
- 67.Miner JH, Sanes JR. Molecular and functional defects in kidneys of mice lacking collagen alpha 3(IV): implications for Alport syndrome. J Cell Biol 1996; 135:1403–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Reynolds J, Mavromatidis K, Cashman SJ, Evans DJ, Pusey CD. Experimental autoimmune glomerulonephritis (EAG) induced by homologous and heterologous glomerular basement membrane in two substrains of Wistar-Kyoto rat. Nephrol Dial Transplant 1998; 13:44–52. [DOI] [PubMed] [Google Scholar]
- 69.Stevens G, Joiner M, Joiner B, Johns H, Denekamp J. Early detection of damage following bilateral renal irradiation in the mouse. Radiother Oncol 1991; 20:124–131. [DOI] [PubMed] [Google Scholar]
- 70.Young BA, Burdmann EA, Johnson RJ, Andoh T, Bennett WM, Couser WG, Alpers CE. Cyclosporine A induced arteriolopathy in a rat model of chronic cyclosporine nephropathy. Kidney Int 1995; 48:431–438. [DOI] [PubMed] [Google Scholar]
- 71.Shobeiri N, Pang J, Adams MA, Holden RM. Cardiovascular disease in an adenine-induced model of chronic kidney disease. J Hypertens 2013; 31:160–168. [DOI] [PubMed] [Google Scholar]
- 72.Endemann DH, Schiffrin EL. Endothelial dysfunction. J Am Soc Nephrol 2004; 15:1983–1992. [DOI] [PubMed] [Google Scholar]
- 73.Tomlinson JA, Caplin B, Boruc O, Bruce-Cobbold C, Cutillas P, Dormann D, et al. Reduced renal methylarginine metabolism protects against progressive kidney damage. J Am Soc Nephrol 2015; 26:3045–3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Van TV, Watari E, Taketani Y, Kitamura T, Shiota A, Tanaka T, et al. Dietary phosphate restriction ameliorates endothelial dysfunction in adenine-induced kidney disease rats. J Clin Biochem Nutr 2012; 51:27–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Inami Y, Hamada C, Seto T, Hotta Y, Aruga S, Inuma J, et al. Effect of AST-120 on endothelial dysfunction in adenine-induced uremic rats. Int J Nephrol 2014; 2014:164125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Xavier S, Vasko R, Matsumoto K, Zullo JA, Chen R, Maizel J, et al. Curtailing endothelial TGF-β signaling is sufficient to reduce endothelial-mesenchymal transition and fibrosis in CKD. J Am Soc Nephrol 2015; 26:817–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Heiss C, Sievers RE, Amabile N, Momma TY, Chen Q, Natarajan S, et al. In vivo measurement of flow-mediated vasodilation in living rats using high-resolution ultrasound. Am J Physiol Heart Circ Physiol 2008; 294:H1086–H1093. [DOI] [PubMed] [Google Scholar]
- 78.Proudfoot D, Skepper JN, Hegyi L, Bennett MR, Shanahan CM, Weissberg PL. Apoptosis regulates human vascular calcification in vitro: evidence for initiation of vascular calcification by apoptotic bodies. Circ Res 2000; 87:1055–1062. [DOI] [PubMed] [Google Scholar]
- 79.London GM, Marchais SJ, Guérin AP, Métivier F. Arteriosclerosis, vascular calcifications and cardiovascular disease in uremia. Curr Opin Nephrol Hypertens 2005; 14:525–531. [DOI] [PubMed] [Google Scholar]
- 80.Bucay N, Sarosi I, Dunstan CR, Morony S, Tarpley J, Capparelli C, et al. Osteoprotegerin-deficient mice develop early onset osteoporosis and arterial calcification. Genes Dev 1998; 12:1260–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bongartz LG, Braam B, Verhaar MC, Cramer MJ, Goldschmeding R, Gaillard CA, et al. Transient nitric oxide reduction induces permanent cardiac systolic dysfunction and worsens kidney damage in rats with chronic kidney disease. Am J Physiol Regul Integr Comp Physiol 2010; 298:R815–R823. [DOI] [PubMed] [Google Scholar]
- 82.Lekawanvijit S, Kompa AR, Manabe M, Wang BH, Langham RG, Nishijima F, et al. Chronic kidney disease-induced cardiac fibrosis is ameliorated by reducing circulating levels of a non-dialysable uremic toxin, indoxyl sulfate. PLoS One 2012; 7:e41281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Haller ST, Kennedy DJ, Shidyak A, Budny GV, Malhotra D, Fedorova OV, et al. Monoclonal antibody against marinobufagenin reverses cardiac fibrosis in rats with chronic renal failure. Am J Hypertens 2012; 25:690–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rivers JR, Ashton JC. Age matching animal models to humans-theoretical considerations. Curr Drug Discov Technol 2013; 10:177–181. [DOI] [PubMed] [Google Scholar]
- 85.Levey AS, De Jong PE, Coresh J, Nahas ME, Astor BC, Matsushita K, et al. The definition, classification, and prognosis of chronic kidney disease: a KDIGO Controversies Conference report. Kidney Int 2011; 80:17–28. [DOI] [PubMed] [Google Scholar]
- 86.Si H, Banga RS, Kapitsinou P, Ramaiah M, Lawrence J, Kambhampati G, et al. Human and murine kidneys show gender- and species-specific gene expression differences in response to injury. PLoS One 2009; 4:e4802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Festing MF. Inbred strains should replace outbred stocks in toxicology, safety testing, and drug development. Toxicol Pathol 2010; 38:681–690. [DOI] [PubMed] [Google Scholar]