

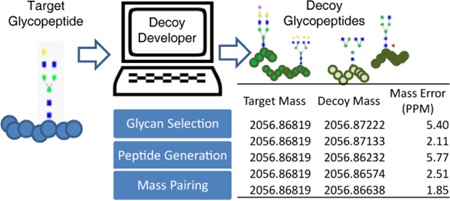
Abstract
Glycopeptide analysis is a growing field that is struggling to adopt effective, automated tools. Many creative workflows and software apps have emerged recently that offer promising capabilities for assigning glycopeptides to MS data in an automated fashion. The effectiveness of these tools is best measured and improved by determining how often they would select a glycopeptide decoy as a spectral match, instead of its correct assignment, yet generating the appropriate number and type of glycopeptide decoys can be challenging. To address this need, we have designed DecoyDeveloper, an on-demand decoy glycopeptide generator that can produce a high volume of decoys with low mass differences. DecoyDeveloper has a simple user interface and is capable of producing large sets of decoys containing complete, biologically relevant glycan and peptide sequences. We demonstrate the tool’s efficiency by applying it to a set of 80 glycopeptide targets. This tool is freely available and can be found at http://glycopro.chem.ku.edu/J1.php
Keywords: N-linked glycans, glycosylation, glycopeptides, bioinformatics, decoys, false discovery rate, tandem mass spectrometry
Graphical abstract
INTRODUCTION
Protein glycosylation is the most common post translational modification (PTM) and a vital biological process that impacts processes such as protein folding1 and immune response.2,3 Aberrant protein glycosylation can be indicative of disease, and glycoproteins serve as biomarkers for a number of cancers.4,5
Mass spectrometry is a fast and sensitive method that provides information en masse about the glycoproteome; glycopeptide analysis following a proteolytic digest is particularly useful as it provides information down to the level of site-specific glycan occupancy.6 However, due to the heterogeneity of protein glycosylation, data generated from MS/MS experiments is complex, and the analysis of the data is laborious. The need for a tool that can aid researchers by reducing the number of hours spent on time-consuming, manual analysis has led to the development of software that assigns glycopeptide spectra from MS/MS data.
At least 30 different algorithms for assigning glycopeptides have been described in the last 12 years,7–36 but little has been done to compare their efficacy, and validation in some cases is insufficient. For example, when SpectraST-MassAnalyzer was described, validation data on a single protein with a total of two glycosylation sites was included.11 The validation protocol for gFinder was not described; instead, the authors declared that the software output “seemed accurate.”21 By contrast, some groups10,17 manually assign hundreds of spectra for software validation, increasing confidence that their algorithms work as designed. This laborious manual validation approach requires significant effort and expertise, which is likely why it is not broadly adopted.
A better way forward, for assessing existing and emerging glycopeptide software tools’ output, is to perform automated testing by challenging the tools’ assignments with large panels of decoy glycopeptides and calculating accurate false discovery rates (FDRs).37,38 The FDR is found by analyzing a known set of targets mixed together with a set of decoys; it is calculated by dividing the number of false positives (incorrect assignments) by total assignments, giving the rate that a scoring algorithm incorrectly assigns spectra. In proteomics studies a 1:1 ratio of targets and decoys is commonly used; however, the same target-decoy approach is not well suited to evaluate the quality of automated glycopeptide tools. When a 1:1 ratio of decoys is used with existing and emerging glycoproteomics tools, and assignments are manually evaluated, the assignment accuracy is significantly worse than what is predicted by the FDR.39,40 Other studies have shown that using 1:1 target-decoy based approaches can fail to identify deficiencies in scoring algorithms that are revealed using a higher decoy:target ratio.40 One solution has been to increase the ratio of decoy glycopeptides in the data above the typical 1:1 ratio, as findings from Zhu et al indicate that the FDR is more accurately reflected with a higher decoy:target ratio.37 Identifying deficiencies in automated software using these methods can guide researchers to specific areas of scoring algorithms that need improvement.41
Constructing decoy sequences can be done through simple processes such as the reversal or shuffling of peptide sequences.15,18,24,29,36 The number of decoys that can be generated per target using these approaches is limited, and this approach does not address the glycan component of the glycopeptide, since scrambling the peptide does not allow for any change to the glycan mass. Constructing whole glycopeptide sequences de novo would allow analysts to create as many decoys as needed. Unfortunately, doing so is computationally challenging because of the need to balance the mass changes between the glycan and peptide components. Approaches have been used where either a glycan or peptide sequence is computationally generated or selected from a large database while the other component is treated as a static mass.37,40 Using a static mass is a computationally efficient method to balance decoy glycopeptide masses with their respective targets; however, this strategy limits decoy sets to fragmentation techniques where only one portion of the glycopeptide is expected to be fragmented in MS/MS spectra. As hybrid fragmentation techniques such as AI-ETD,42 EThcD,43 and stepped HCD44 continue to emerge, which generate information about peptide and glycan components in a single spectrum, automated software tools adapted for these techniques need to be evaluated and improved. A tool that generates multiple decoy glycopeptide sequences with single digit PPM mass-error, comparable to the mass accuracy achieved by high resolution mass spectrometers, does not currently exist, yet it will be vital in improving automated glycopeptide analysis. Herein, we provide this tool with DecoyDeveloper, an online software tool that generates decoy glycopeptides consisting of full glycan and peptide sequences. We use DecoyDeveloper to generate a large number of decoys for two sets of 80 N-linked glycopeptides, showing the capability of this powerful new aid in the analysis of glycoproteomics software. In addition to its valuable use as a stand-alone product, DecoyDeveloper is open-source and may be incorporated into new or existing tools that require glycopeptide decoys to properly assess the FDR of glycopeptide assignments.
EXPERIMENTAL
The software uses a database of 245 biologically relevant glycans for the glycan portion of the glycopeptide decoy; these are provided in the excel file found in the Supporting Information on Sheet S2. Decoy peptide sequences were composed of two parts: PEP1, a variable length peptide sequence generated from amino acid distributions containing differing relative abundances of amino acids, and PEP2, a tetrapeptide selected from a list of 4,252 tetrapeptides. The distributions used to generate PEP1 (Sheet S3) and the list of tetrapeptides (Sheet S4) can also be found in Supporting Information. Also found in Supporting Information is a detailed algorithm that describes the logic used to build the software. The tool is freely available at: http://glycopro.chem.ku.edu/J1.php
To test the software, 80 glycopeptides were selected as targets. These targets are provided in Supplemental Information on Sheet S5. Two sets of 1,600 decoys, 20 for each target glycopeptide, were generated. In one set of decoys, PEP1 was generated using variable amino acid frequencies, in the second set, all amino acids frequencies were equal in PEP1. The two sets of decoys were compared to measure the efficiency of generating PEP1 when using the custom-tailored amino acid distributions. The settings for “Target N,” the number of times a randomized glycan is used in an attempt to generate a decoy, and “Error Threshold,” the tolerated mass error for successfully generated decoys (in PPM), during decoy generation were 2 and 10, respectively. The number of successful and rejected attempts at decoy generation were then recorded and used to calculate the efficiency of the software. To validate that the software’s output was repeatable, a second set of 80 glycopeptide targets was used to generate an additional 1,600 decoys, and the computational efficiency between the two datasets was compared.
RESULTS AND DISCUSSION
User interface overview.
DecoyDeveloper is a new tool that generates glycopeptide decoy sequences de novo, providing a simple means to create large libraries of decoys for the assessment and improvement of scoring algorithms used in automated software tools. Figure 1 shows the user interface; this tool is free to access at http://glycopro.chem.ku.edu/J1.php. To generate a decoy, the peptide and glycan sequences of a target glycopeptide are entered along with Target N and the mass error threshold (in PPM). The efficiency of the software will generally increase with a lower value for Target N and a higher error threshold.
Figure 1:
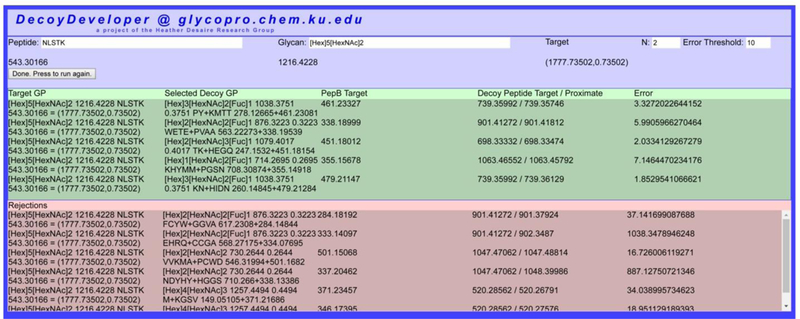
User interface for DecoyDeveloper. Peptide and glycan sequences are entered at the top of the page, and the software automatically generates decoys with the specified mass tolerance. Successfully generated decoys appear in the green portion of the output section while rejected decoys are logged in the red portion. Rejected decoys are those that the algorithm computed but do not satisfy the mass accuracy threshold that the user selected.
Once all sequences and settings are entered, the software can be run, and a decoy will be generated; the algorithm for generating decoys will automatically repeat itself until a decoy is successfully generated; the decoy will then appear in the top (green) portion of the output menu. Rejected decoys are stored at the bottom of the output menu (red). Once a decoy is generated the “run” button will read “Done. Press to run again.” The algorithm can be re-run as many times as needed to create a sufficiently large set of decoys.
Workflow for Decoy Generation.
Figure 2 shows the workflow that DecoyDeveloper uses to create decoy glycopeptides. The algorithm assembles decoys by generating three components: a glycan (DGLY) and two separate peptide components (PEP1 and PEP2). Splitting the peptide into two components, (PEP1 and PEP2) allows most of the mass of the decoy to be generated with a randomly-generated peptide; the remainder of the mass is obtained from a small database of peptides. Had the entire peptide been selected from a database, instead of mostly generated randomly, the database would need to be enormous (>1020 entries) to account for the large range of possible masses.
Figure 2:
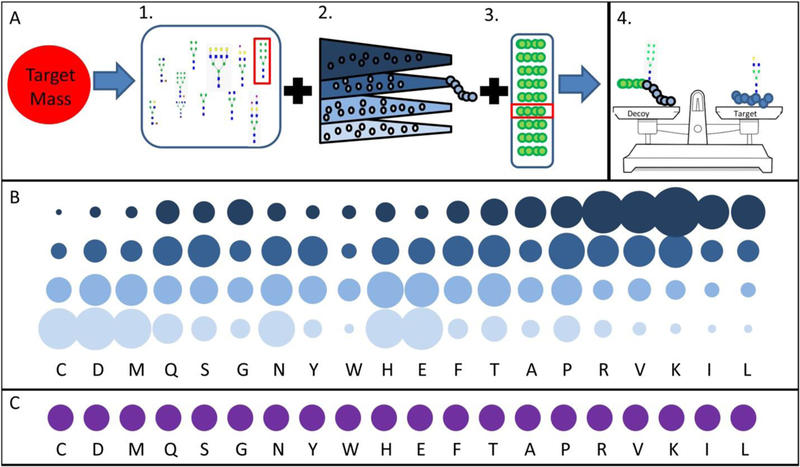
(A) Workflow for decoy generation. A decoy is created for a glycopeptide target in four steps: selection of a decoy glycan (1), generation of a variable length peptide sequence (2), selection of a tetrapeptide closest to the remaining mass (3), and checking that the mass of the decoy glycopeptide is within the specified tolerance of the target (4).
(B) Custom distributions of amino acid frequencies used to generate PEP1. Amino acids are ordered based on their ratio of mass defect to mass; the area of the circles is proportional to their respective abundances in the four distributions.
(C) Equivalent distribution of amino acid frequencies, used for testing purposes only.
The first step in decoy generation is the random selection of a glycan from a database of 245 N-linked, biologically-relevant glycans. The structures in this database contain all of the glycans that were identified in a large-scale glycopeptide analysis reported previously.45 In addition to the previously reported glycans, additional sialylated and fucosylated forms were added to further increase the glycan diversity. The glycans are distributed across a wide range of masses, and the three classes of N-linked glycan: high-mannose, complex, and hybrid, are represented. The number of N-glycan structures in the database is comparable to the number of glycans previously reported in a comprehensive glycomics study.46
After selection of the glycan from the database, the target mass of the decoy is adjusted by subtracting the glycan mass. Next a peptide sequence must be derived that matches the remainder of the mass. In order to reproducibly generate peptide sequences with the necessary low mass error, peptide generation is split into two steps: first, a randomly generated peptide sequence that varies in length decreases the remaining mass into a small, searchable space (PEP1), and then a tetrapeptide that is likely to exist within this space (PEP2) is identified.
The primary objective in generating PEP1 is to consume all the mass needed for the decoy peptide, minus the amount that could be made up by a four amino-acid peptide, which can be selected from a database of tetrapeptides. Two values, the mass and the mass defect, must be considered when generating the sequence for PEP1. The relative rate that the mass defect and mass must decrease depends on the target glycopeptide and decoy glycan already selected; if the mass defect to mass ratio is not adjusted on a case-to-case basis, there will be a high failure rate for some decoys because the mass following PEP1 generation will exist in a space where little to no tetrapeptide solutions exist. This concept is illustrated in Figure 3. To consume the target mass and mass defect at the correct proportion, the software sorts decoys into one of four amino acid distributions for PEP1 generation; for each distribution amino acid frequencies have been weighted so the generated sequences will have a ratio of mass defect to mass closely resembling the target mass. A graphical representation showing the principal behind these distributions, and their importance in PEP1 generation, can be seen in Figure 3.
Figure 3:
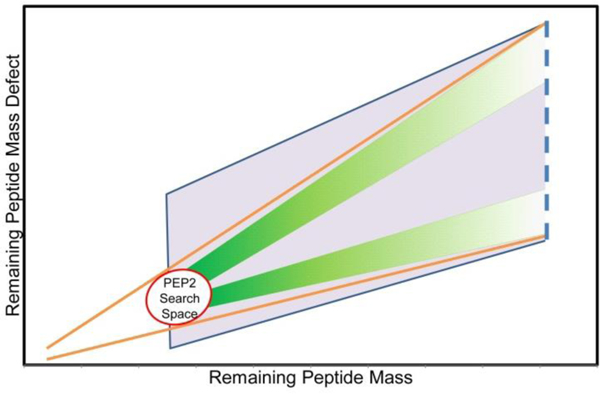
Conceptual illustration showing why the amino acid frequencies in PEP1 should vary for different target peptide masses. All peptide sequences exist within the two orange lines on the graph. Peptides larger than four amino acids appear on the right side of the circle, which contains all possible tetrapeptides. The decoy’s total peptide mass is represented by the blue dotted line. This mass will be comprised of PEP1, which is a randomly generated peptide, and PEP2, which is a tetrapeptide in the circle. By changing the amino acid frequencies used to generate PEP1 sequences based on the initial position along the dotted line, the mass-defect-to-mass ratio of PEP1 can be varied so that the remaining mass reaches the PEP2 search space. Two examples, where different amino acid frequencies are used to generate PEP1, are shown by the two green regions. If all the amino acids have an equal probability of being used to generate PEP1, then the generated PEP1 sequences will have a much wider distribution on the graph (purple region), and it is less likely that PEP1 could be paired with a tetrapeptide partner to successfully generate a decoy.
Following PEP1 generation the remaining mass is balanced with a tetrapeptide. The database of tetrapeptides contains 4,252 entries, representing all possible tetrapeptide masses. (While there are 160,000 possible tetrapeptide sequences that could be generated from 20 amino acids, many of these are isomers, and only a single entity per mass was selected for the database.) The database of 4,252 tetrapeptides represents a computationally efficient set of solutions for the final component needed to construct the decoy. In our workflow the remaining mass (after selection of glycan and PEP1) is compared to the masses in the tetrapeptide database and the last four amino acids, PEP2, are selected. The mass of PEP1, PEP2, and the DGLY are combined, and the algorithm checks to determine if the total mass is within the mass error threshold specified by the user. If the mass error is too high, the decoy is rejected; and the algorithm will recalculate another decoy, either by generating PEP1 again or re-selecting DGLY. The number of times that the algorithm attempts to use the same DGLY is specified by the “Target N” setting in the user interface.
Evaluation of Software Performance.
In preliminary testing, DecoyDeveloper was used to successfully generate decoys for a very broad range of glycopeptides; and almost all attempts were successful. After initial testing verified that the tool worked as designed, we asked the question: What advantage does using the custom amino-acid distributions for generating PEP1 provide to the algorithm? Note, we focused attention on the selection of PEP1 because DGLY and PEP2 are selected from rather small curated lists; therefore, the efficiency of the algorithm relies on PEP1 being consistently generated such that the mass remainder is within the ranges of mass and mass defect present in the tetrapeptide masses used for PEP2.
To test the efficiency of the algorithm, 80 glycopeptides were used as targets for decoy generation. For each target, 20 decoys were generated using: 1) the optimized algorithm, which contains the four sets of amino acid frequencies for PEP1 shown in Figure 2B, which are tailored to match different mass defect/mass ratios; (henceforth referred to as the custom distributions) and 2) a distribution of amino acid frequencies for PEP1 where all amino acids were in the same abundance, henceforth referred to as the equal distribution. By comparing the results for the custom distributions to the results for the equal distribution, the effect of varying the frequencies of amino acids used to generate PEP1 based on the ratio of mass defect to mass can be measured, showing the importance of generating different peptide sequences for different subsets of target glycopeptides.
Using custom distributions in the algorithm yielded successful decoys more frequently than using equivalent distributions, the former requiring 3.9 computations per decoy generated, and the latter, 11.9. The results are shown in Table 1. When the algorithm built peptides whose mass and mass defect placed them in distributions two and three, a marginal increase in efficiency was observed for searches using the custom distributions, requiring 2.5 and 2.7 calculations per success, respectively, compared to 2.6 and 3.4 calculations per success when using the equivalent distribution. When the algorithm built peptides whose mass and mass defect placed them in distributions one and four, a pronounced increase in efficiency was observed when the custom distributions were used. Peptides that fell into distribution one required 3.1 calculations per success for the custom distribution compared to 8.6 calculations per success for the equivalent distribution. For peptides in distribution four, the calculations per success were 8.0 compared to 66.3 for the custom and equivalent distributions, respectively. Peptides in distributions one and four encompass cases where the mass defect to mass ratio is farther from the average values of the equivalent distribution, so it is not surprising that these cases benefit the most from using custom distributions.
Table 1:
Comparison of decoy generation efficiency using custom amino acid distributions vs using an equivalent amino acid distribution for PEP1 sequences.
| Distribution set |
Distribution groupa |
MD/M ×104b | Successful Decoys |
Rejected Decoys |
Calculations per Successc |
|---|---|---|---|---|---|
| Custom Distribution | 1 | <3.97 | 262 | 540 | 3.1 |
| 2 | >3.97–4.67 | 351 | 521 | 2.5 | |
| 3 | >4.67–5.70 | 616 | 1044 | 2.7 | |
| 4 | >5.70 | 371 | 2586 | 8.0 | |
| Total | 1600 | 4691 | 3.9 | ||
| Equivalent Distribution | 1 | <3.97 | 233 | 1760 | 8.6 |
| 2 | >3.97–4.67 | 394 | 634 | 2.6 | |
| 3 | >4.67–5.70 | 771 | 1851 | 3.4 | |
| 4 | >5.70 | 202 | 13193 | 66.3 | |
| Total | 1600 | 17438 | 11.9 |
Prior to the generation of PEP1 sequences, decoys are sorted into one of four groups depending on the ratio of mass defect to mass required for the decoy peptide. For the “Custom Distribution” set, the frequencies of amino acids in PEP1 were variable depending on the distribution group, as shown in Figure 2B. For the “Equivalent Distribution” set, the frequencies of amino acids were constant and equal for each of the four distribution groups, as shown in Figure 2C.
MD/M is the range of mass defect to mass ratios sorted into a given distribution.
Calculations per success is equal to the sum of rejected and successful decoys divided by the number of successful decoys.
While 1600 decoys were generated in the above experiment, it is theoretically possible that testing a larger set of decoys would uncover inefficiencies in the algorithm. To assess this possibility, the experimental dataset was doubled. A total of 1600 new decoys were generated, starting with a completely different set of 80 glycopeptide targets, In this case, the 80 glycopeptide targets were systematically chosen from identified glycopeptides described in reference 45, and they can be found in the supporting information on Sheet S6. The number of computations per successfully generated decoy was comparable for each of the four amino acid distributions; in fact the algorithm was marginally more efficient at generating decoys for the second dataset. The validation test reiterates the point that the algorithm can efficiently generate decoys. A comparison between the algorithm’s efficiency for the first and second set of decoys can be found in the supplemental information (Table S1).
CONCLUSION
The number of fragmentation techniques and software tools available to researchers assigning glycopeptide spectra continues to grow in number and diversity. Detecting deficiencies in scoring algorithms can be done using a high ratio of decoy glycopeptides to target glycopeptides;40 however, the ability to generate the necessary number of decoys using full glycan and peptide sequences is currently lacking. DecoyDeveloper is an online software tool presented and tested herein that provides decoy glycopeptides on demand. The workflow allows for the efficient production of high decoy:target ratios, a vital tool for researchers interested in testing and improving automated software. The algorithm used to build the tool is provided in Supplemental Data, and it can be incorporated into any new or existing glycopeptide analysis tool that uses decoys in assigning an FDR.
Supplementary Material
Acknowledgements
We are grateful to NIH for providing the financial resources necessary to carry out this work. This study was supported by projects 1R35GM130354, R01GM103547, and T32-GM008359.
Footnotes
SUPPORTING INFORMATION
DecoyDeveloper_SupportingInformation.docx. S1 Cover Page. S2 Table of Contents.S3-S5 detailed outline of algorithm used to build the described software tool. S6 Table S1: Comparison of decoy generation efficiency using custom amino acid distributions for two sets of glycopeptide targets.
DecoyDeveloper_Workbook.xlsx. Excel workbook including: amino acid masses and example calculations described in DecoyDeveloper_Algorithm.docx (Sheet S1), list of 245 decoy glycans used for DGLY (Sheet S2), characteristics of amino acid distributions used to construct PEP1 (Sheet S3), list of 4,252 tetrapeptides used for PEP2 (Sheet S4), 80 target glycopeptides (Decoy Set 1) used for software testing (Sheet S5), 80 target glycopeptides (Decoy Set 2) used for software validation (Sheet S6).
References
- 1.Shental-Bechor D; Levy Y Effect of Glycosylation on Protein Folding: A Close Look at Thermodynamic Stabilization. Proc. Natl. Acad. Sci. 2008, 105(24), 8256–8261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rudd PM; Elliott T; Cresswell P; Wilson IA; Dwek RA Glycosylation and the Immune System. Science, 2001, 291, 2370–2376. [DOI] [PubMed] [Google Scholar]
- 3.Nimmerjahn F; Ravetch JV Fcγ Receptors as Regulators of Immune Responses. Nat. Rev. Immuno. 2008, 8, 34–47. [DOI] [PubMed] [Google Scholar]
- 4.Duffy MJ; Shering S; Sherry F; McDermott E; O’Higgins N CA 15–3: A Prognostic Marker in Breast Cancer. Int. J. Biol. Markers. 2000, 15(4), 330–333. [DOI] [PubMed] [Google Scholar]
- 5.Drake RR; Jones EE; Powers TW; Nyalwidhe JO Altered Glycosylation in Prostate Cancer. Adv. Cancer Res. 2015, 126, 345–382. [DOI] [PubMed] [Google Scholar]
- 6.Dalpathado D; Desaire H Glycopeptide Analysis by Mass Spectrometry. Analyst, 2008, 133, 731–738. [DOI] [PubMed] [Google Scholar]
- 7.Zhu Z; Hua D; Clark DF; Go EP; Desaire H GlycoPep Detector: A tool for assigning mass spectrometry data of N-linked glycopeptides on the basis of their electron transfer dissociation spectra. Anal. Chem. 2013, 85 (10), 5023–5032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ahn J; Gillece-Castro B; Berger S BiopharmaLynx: A new bioinformatics tool for automated LC/MS peptide mapping assignment. Corporation, W., Ed. Milford, MA, US, 2008. [Google Scholar]
- 9.Irungu J; Go EP; Dalpathado DS; Desaire H Simplification of Mass Spectral Analysis of Acidic Glycopeptides Using GlycoPep ID. Anal. Chem. 2007, 79 (8), 3065–3074. [DOI] [PubMed] [Google Scholar]
- 10.Woodin CL; Hua D; Maxon M; Rebecchi KR; Go EP; Desaire H GlycoPep Grader: A Web-Based Utility for Assigning the Composition of N-linked Glycopeptides. Anal. Chem. 2012, 84 (11), 4821–4829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pai PJ; Hu Y; Lam H Direct Glycan Structure Determination of Intact N-linked Glycopeptides by Low-Energy Collision-Induced Dissociation Tandem Mass Spectrometry and Predicted Spectral Library Searching. Analytica Chimica Acta. 2016, 934, 152–162. [DOI] [PubMed] [Google Scholar]
- 12.Ren JM; Rejtar T; Li L; Karger BL N-Glycan Structure Annotation of Glycopeptides Using a Linearized Glycan Structure Database (GlyDB). J. Proteome Res. 2007, 6(8), 3162–3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu Y; Mechref Y; Klouckova I; Mayampurath A; Novotny MV; Tang H Mapping Site-Specific Protein N-glycosylations through Liquid Chromatography/Mass Spectrometry and Targeted Tandem Mass Spectrometry. Rapid Comm. Mass Spectrom. 2010, 24(7), 965–972. [DOI] [PubMed] [Google Scholar]
- 14.He L; Xin L; Shan B; Lajoie GA; Ma B GlycoMaster DB: Software to Assist the Automated Identification of N-linked Glycopeptides by Tandem Mass Spectrometry. J. Proteome Res. 2014, 13 (9), 3881–3895. [DOI] [PubMed] [Google Scholar]
- 15.Mayampurath A; Yu CY; Song E; Balan J; Mechref Y; Tang H Computational Framework for Identification of Intact Glycopeptides in Complex Samples. Anal. Chem. 2014, 86(1), 453–463. [DOI] [PubMed] [Google Scholar]
- 16.Eshghi ST; Shah P; Yang W; Li X; Zhang H GPQuest: A Spectral Library Matching Algorithm for Site-Specific Assignment of Tandem Mass Spectra to Intact N-glycopeptides. Anal. Chem. 2015, 87(10), 5181–5188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yu CY; Mayampurath A; Zhu R; Zacharias L; Song E; Wang L; Mechref Y; Tang H Automated Glycan Sequencing from Tandem Mass Spectra of N-Linked Glycopeptides. Anal. Chem. 2016, 88(11), 5725–5732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Park GW; Kim JY; Hwang H; Lee JY; Ahn YH; Lee HK; Ji ES; Kim KH; Jeong HK; Yun KN; Kim YS; Ko JH; An HJ; Kim JH; Paik YK; Yoo JS Integrated GlycoProteome Analyzer (I-GPA) for Automated Identification and Quantitation of Site-Specific N-Glycosylation. Sci. Rep., 2016, 6, 21175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zeng WF; Liu MQ; Zhang Y; Wu JQ; Fang P; Peng C; Nie A; Yan G; Cao W; Liu C; Chi H; Sun RX; Wong CCL; He SM; Yang P pGlyco: A Pipeline for the Identification of Intact N-Glycopeptides by Using HCD- and CID-MS/MS and MS3. Sci. Rep., 2016, 6, 25102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nasir W; Toledo AG; Noborn F; Nilsson J; Wang M; Bandeira N; Larson G SweetNET: A Bioinformatics Workflow for Glycopeptide MS/MS Spectral Analysis. J. Proteome Res. 2016, 15(8), 2826–2840. [DOI] [PubMed] [Google Scholar]
- 21.Kim JW; Hwang H; Lim JS; Lee HJ; Jeong SK; Yoo JS; Paik YK gFinder: A Web-Based Bioinformatics Tool for the Analysis of N-Glycopeptides. J. Proteome Res, 2016. 15, 4116–4125. [DOI] [PubMed] [Google Scholar]
- 22.Sun W; Liu Y; Lajoie G; Ma B; Zhang K An Improved Approach for N-linked Glycan Structure Identification from HCD MS/MS Spectra. IEEE/ACM Trans. Comput. Biol. Bioinform, 2017, 99, 1–1. [DOI] [PubMed] [Google Scholar]
- 23.Apte A; Meitei NS; Bioinformatics in glycomics: Glycan Characterization with Mass Spectrometric Data Using SimGlycan™ Functional Glycomics: Methods and Protocols, J Li., Ed. Humana Press: Totowa, NJ, 2010, 269–281. [DOI] [PubMed] [Google Scholar]
- 24.Wu SW; Liang SY; Pu TH; Chang FY; Khoo KH Sweet-Heart — An Integrated Suite of Enabling Computational Tools for Automated MS2/MS3 Sequencing and Identification of Glycopeptides. J. Proteomics. 2013, 84, 1–16. [DOI] [PubMed] [Google Scholar]
- 25.Goldberg D; Bern M; Parry S; Sutton-Smith M; Panico M; Morris HR; Dell A Automated N-Glycopeptide Identification Using a Combination of Single- and Tandem-MS. J. Proteome Res, 2007, 6(10), 3995–4005. [DOI] [PubMed] [Google Scholar]
- 26.Ozohanics O; Krenyacz J; Ludányi K; Pollreisz F; Vékey K; Drahos L GlycoMiner: A New Software Tool to Elucidate Glycopeptide Composition. Rapid Commun. Mass Spectrom, 2008, 22(20), 3245–3254. [DOI] [PubMed] [Google Scholar]
- 27.Pompach P; Chandler KB; Lan R; Edwards N; Goldman R Semi-Automated Identification of N-Glycopeptides by Hydrophilic Interaction Chromatography, Nano-Reverse-Phase LC–MS/MS, and Glycan Database Search. J. Proteome Res. 2012, 11(3), 1728–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Serang O; Froehlich JW; Muntel J; McDowell G; Steen H; Lee RS; Steen JA SweetSEQer, Simple de novo Filtering and Annotation of Glycoconjugate Mass Spectra. Mol. Cell. Proteomics, 2013, 12(6), 1735–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Strum JS; Nwosu CC; Hua S; Kronewitter SR; Seipert RR; Bachelor RJ; An HJ,; Lebrilla CB Automated Assignments of N- and O-site Specific Glycosylation with Extensive Glycan Heterogeneity of Glycoprotein Mixtures. Anal. Chem, 2013, 85 (12), 5666–5675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lynn KS; Chen CC; Lih TM; Cheng CW; Su WC; Chang CH; Cheng CY; Hsu WL; Chen YJ; Sung TY MAGIC: An Automated N-Linked Glycoprotein Identification Tool Using a Y1-Ion Pattern Matching Algorithm and in Silico MS2 Approach. Anal. Chem, 2015, 87(4), 2466–2473. [DOI] [PubMed] [Google Scholar]
- 31.Bern M, Kil YJ, Becker C. Byonic: Advanced Peptide and Protein Identification Software. Curr. Protoc. Bioinformatics, / editoral board, Andreas D. Baxevanis … [et al.] 2012, Chapter 13, Unit13.20-Unit13.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhu Z; Su X; Clark DF; Go EP; Desaire H Characterizing O-linked Glycopeptides by Electron Transfer Dissociation: Fragmentation Rules and applications in Data Analysis. Anal Chem. 2013, 85, 8403–8411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Go EP; Rebecchi KR; Dalphathado DS; Bandu ML; Zhang Y; Desaire H GlycoPep DB: A Tool for Glycopeptide Analysis Using a “Smart Search. Anal. Chem. 2007, 79(4), 1708–1713. [DOI] [PubMed] [Google Scholar]
- 34.Cheng K; Chen R; Seebun D; Ye M; Figeys D; Zou H Large-scale Characterization of Intact N-glycopeptides Using an Automated Glycoproteomic Method. J. Proteom. 2014, 110, 145–154. [DOI] [PubMed] [Google Scholar]
- 35.Pioch M; Hoffmann M; Pralow A; Reichl U; Rapp E glyXtoolMS: An Open-Source Pipeline for Semiautomated Analysis of Glycopeptide Mass Spectrometry Data. Anal. Chem. 2018, 90(20), 11908–11916. [DOI] [PubMed] [Google Scholar]
- 36.Sun W; Liu Y; Zhang K An Approach for N-Linked Glycan Identification from MS/MS Spectra by Target-Decoy Strategy. Comput. Biol. Chem, 2018, 74, 391–398. [DOI] [PubMed] [Google Scholar]
- 37.Zhu Z; Su X; Go EP; Desaire H New Glycoproteomics Software, GlycoPep Evaluator, Generates Decoy Glycopeptides de Novo and Enables Accurate False Discovery Rate Analysis for Small Data Sets. Anal. Chem. 2014, 86, 9212–9219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hu H; Khatri K; Zaia J Algorithms and Design Strategies Towards Automated Glycoproteomics Analysis. Mass Spectrom. Rev. 2017, 36, 475–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu S-W; Pu T-H; Viner R; Khoo K-H Novel LC-MS2 Product Dependent Parallel Data Acquisition Function and Data Analysis for Sequencing and Identification of Intact Glycopeptides. Anal. Chem, 2014, 86(11), 5478–5486. [DOI] [PubMed] [Google Scholar]
- 40.Lakbub JC; Su X; Zhu Z; Patabandige MW; Hua D; Go EP; Desaire H Two New Tools for Glycopeptide Analysis Researchers: A Glycopeptide Decoy Generator and a Large Data Set of Assigned CID Spectra of Glycopeptides. J. Proteome Res. 2017, 16, 3002–3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lakbub JC; Su X; Hua D; Go EP; Desaire H Dissecting the Dissociation Patterns of Fucosylated Glycopeptides Undergoing CID: A Case Study in Improving Automated Glycopeptide Analysis Scoring Algorithms. Anal. Methods. 2018, 10, 256–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ledvina AR; Beauchene NA; McAlister GC; Syka JEP; Schwartz JC; Griep-Raming J; Westphall MS; Coon JJ Activated-Ion ETD (AI-ETD) Improves the Ability of ETD to Identify Peptides in a Complex Mixture. Anal. Chem. 2010, 82(24), 10068–10074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yu Q; Wang B; Chen Z; Urabe G; Glover MS; Shi X; Gua L; Kent KC; Li L Electron-Transfer/Higher-Energy Collision Dissociation (EThcD)-enabled Intact Glycopeptide/Glycoproteome Characterization. J. Am. Soc. Mass Spectrom. 2017, 28(9), 1751–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bollineni RC; Koehler CJ; Gislefoss RE; Anonsen JH; Thiede B Large-Scale Intact Glycopeptide Identification by Mascot Database Search. Sci. Rep. 2018, 8, 2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Go EP; Hua D; Desaire H Glycosylation and Disulfide Bond Analysis of Transiently and Stably Expressed Clade C HIV 1 gp140 Trimers in 293T Cells Identifies Disulfide Heterogeneity Present in Both Proteins and Differences in O Linked Glycosylation. J. Proteome Res. 2014, 13, 4012–4027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hua S; Hyun JA; Ozcan S; Ro GS; Soares S; DeVere-White R; Lebrilla CB Comprehensive Native Glycan Profiling with Isomer Separation and Quantitation for the Discovery of Cancer Biomarkers. Analyst. 2011, 136, 3663–3671. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.