

Abstract
Natural polymer hydrogels are one of the best biomaterials for soft tissue repair because of their excellent biocompatibility, biodegradability and low immune rejection. However, they lack mechanical strength matching that of natural tissue and desired functionality (e.g. self-healing and 3D-printability). To solve this problem, we developed a host-guest supramolecule (HGSM) with three arms covalently crosslinked with a natural polymer to construct a novel hydrogel with non-covalent bonds integrated in a covalently crosslinked network. The unique structure enabled the hydrogel to bear improved mechanical properties and show both self-healing and 3D printing capabilities. The three-armed HGSM was first prepared via the efficient non-covalent host-guest inclusion interactions between isocyanatoethyl acrylate-modified β-cyclodextrin (β-CD-AOI2) and acryloylated tetra-ethylene glycol-modified adamantane (A-TEG-Ad). Subsequently, a host-guest supramolecular hydrogel (HGGelMA) was obtained through copolymerization between the arms of HGSM and gelatin methacryloyl (GelMA) to form a covalently crosslinked network. The HGGelMA was robust, fatigue resistant, reproducible and rapidly self-healing. In HGGelMA, the covalent crosslinking maintained its overall shape whereas the weak reversible non-covalent host-guest interactions reinforced its mechanical properties and enabled it to rapidly self-heal upon fracturing. The reversible non-covalent interactions could be re-established upon breaking, so as to heal the hydrogel and dissipate energy to prevent catastrophic fracture propagation. Furthermore, the precursors of the HGGelMA were sufficiently viscous and could be rapidly photocrosslinked to produce a robust scaffold with an exquisite internal structure through 3D printing. The 3D-printed HGGelMA hydrogel scaffold was biocompatible, promoted cell adhesion and proliferation, and supported tissue in-growth. Our strategy of integrating non-covalently linked HGSM in a covalently linked hydrogel network represents a new approach to the development of natural polymers into biocompatible hydrogels with improved strength as well as desired self-healing and 3D-printability.
Graphical Abstract
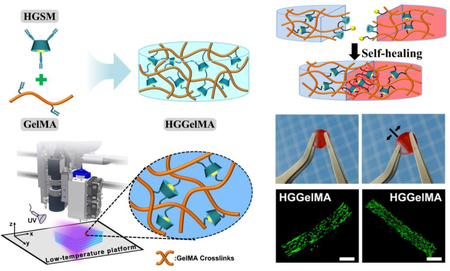
Novel 3D-printable hydrogels with host-guest non-covalent interactions and covalent crosslinking networks show robust mechanical strength, self-healing performance and excellent biocompatibility.
INTRODUCTIONS
Soft tissues are the dominant component of human organs, and the emergence of many diseases may be correlated with soft tissue injuries or defects, such as skin burns, muscular death, refractory wounds and tendon rupture.1 Traditional treatments, including the surgical debridement and grafting of autologous or allogeneic tissues, demand long treatment times and potentially lead to immunological rejection,2,3 causing secondary damage to patients. Recently, significant studies have been made to seek methods for reconstructing soft tissues.4–6 Hydrogels have attracted considerable attention because they have similar water content, biological structure and physical properties as soft tissues.7,8 However, several issues still remain to be solved. For example, since the sizes and internal structures of defected tissues vary greatly, tissue replacements should be able to conform to desired models and porous structures according to the different size scales of the cell activity.9 In addition, conventional polymer hydrogels cannot self-heal because they do not reform covalent bonds and repair the damage cracks. Furthermore, the poor mechanical strength of natural hydrogels does not match that of the surrounding tissues, disabling the practical applications of hydrogels.10 Thus, balancing good processability, great mechanical strength, and biocompatibility is still a daunting challenge for developing natural hydrogels for many biomedical applications.
3D printing is a promising manufacturing technology that can be used to fabricate personalized external or internal constructs at various length scales using computer control systems and computer-aided manufacturing (CAD/CAM) tools.11,12 3D printing provides an attractive strategy for mimicking the microarchitectures of native tissues due to its reproducibility and ability to create personalized hydrogel designs at a high resolution and accurate internal structures.13–16 In addition, the 3D porous structures of scaffolds have been known to enhance the proliferation and interactions of the cells, and increase cell densities because they enhance the exchange of oxygen and nutrients and promote the elimination of metabolites.17 Hydrogels have been widely applied for biological engineering using 3D printing to develop in vitro skin,18 cardiac,19 adipose,20 cartilage and bone tissues21,22 and hybrid tissue constructs with vascular-like networks.23 In contrast to synthetic hydrogels, natural hydrogels have significant advantages due to their good biocompatibility or bioactivity as 3D-constructed tissue regeneration materials.24,25 Unfortunately, many precursors of natural hydrogels cannot be directly extruded as a continuous fiber or rapidly stabilized to maintain a printed structure due to insufficient viscosity and solids content.26,27 These drawbacks have hindered the biomedical applications of hydrogels. To overcome these issues, gelatin methacryloyl (GelMA) has been employed as a precursor for developing 3D printed hydrogels due to its bioactivity, rapid photocrosslinking ability and temperature-adjustable viscosity.28
Despite the excellent biocompatibility and 3D printability of GelMA hydrogels, their poor mechanical properties have hampered their practical applications.29,30 For instance, soft tissues have a stiffness ranging from 200 to 500 kPa,31 while GelMA has a stiffness of only 25–100 kPa.32 Moreover, GelMA hydrogels do not have desired self-healing behaviors. Efforts have been made to improve the mechanical properties of natural hydrogels and to give them some self-healing properties.33,34 Chemical copolymerization is a common approach to increase the crosslinking density to improve mechanical strength.35–37 Specifically, the method is to add organic molecules such as acrylamide38–40 and acrylic acid41,42 to natural hydrogels to achieve copolymerization. However, these organic polymers either are non-degradable or lead to cytotoxic degradation products.43 Recently, supramolecular hydrogels have been considered as a promising reinforcement approach for overcoming the flaws of natural hydrogels.44,45 Several recent studies have shown that supramolecular hydrogels have an autonomously self-healing capacity as they are non-covalent systems where the macromolecular chains or units are crosslinked by host-guest interactions.46,47 Host-guest interactions have received attention because they can contribute to special functions, such as stimuli responsiveness,48 self-healing32,38,49,50 and distinctive mechanical properties.51,52 Many hydrogels derived from the host-guest inclusions between cyclodextrin (CD) and others have been synthesized to enhance their mechanical functions for tissue repair,53 drug carriers,54–56 and genetic engineering materials.57 These reports provided inspiration for using host-guest interactions to obtain robust and self-healing hydrogel materials. While most host-guest supramolecular hydrogels have been prepared by mixing grafted CDs with guest-molecule polymers,58–60 they have encountered problems because the host molecules cannot efficiently interact with the guest molecules due to steric effects. Moreover, a complete non-covalent crosslinking is responsible for hydrogel’s poor mechanical properties. Such impairments did not allow great improvement of the hydrogel mechanical properties and self-healing ability.
To address this challenge, we propose to fabricate a natural hydrogel with reinforced mechanical properties adequate for 3D printing applications. Specifically, we developed a novel host-guest supramolecule (HGSM) based on inclusion reaction between CD and adamantane (Ad). To this end, we mixed A-TEG-Ad, the Ad modified with acryloylated (A) tetra-ethylene glycol (TEG) (Figure 1A) and β-CD-AOI2, the β-CD conjugated with two “2-Isocyanatoethyl acrylate” (AOI2) molecules (Figure 1B), in aqueous solutions, prior to polymer grafting, to obtain host-guest supramolecules (HGSMs) through stronger host-guest interactions and to avoid problem of steric effects (Figure 1C). These HGSMs were then used as multifunctional crosslinkers to crosslink GelMA (chosen due to its excellent bioactivity61,62) during hydrogel polymerization because they had multiple active points for chemical reactions (Figure 2B). We analyzed the effects of the host-guest interactions, integrated within the polymeric network, on the mechanical properties, the reproducibility and self-healing characteristics of the resultant hydrogel (termed HGGelMA, Figure 2B). GelMA could ensure that the HGGelMA can be deposited in a layer-by-layer manner through 3D printing. We adopted a two-step curing method to improve the stabilization and accuracy of the 3D printed structure. Moreover, we expected that the 3D-printed HGGelMA scaffolds would support cell attachment and growth in vitro and are histocompatible in vivo because GelMA in the hydrogel bears RGD motifs and other motifs that can be degraded by matrix metalloproteinase.61 The HGGelMA will open up a new avenue to develop hydrogels that exhibit both reinforced mechanical properties and self-healing/3D-printability functionalities.
Figure 1.

Schematic of the HGSM synthesis. (A-B) Process for the synthesis of A-TEG-Ad (A, the guest) and β-CD-AOI2 (B, the host). (C) Preparation of HGSM. (D) Photographs showing the HGSM preparation process.
Figure 2.

Schematic illustrations of the chemical structures, photographs, SEM images. (A) The design structure of the HGGelMA before and after crosslinking. (B) photographs of the HGGelMA hydrogels under squeezing. (C) The GelMA and HGGelMA hydrogels held with common tweezers. (D) The cylindrical HGGelMA hydrogels in different concentrations. (E) Photo showing the reproducible property of HGGelMAs. Hydrogel shapes were maintained after drying-swelling cycles. (F) SEM images of various concentrations of HGGelMAs after lyophilization. The insets showed high magnification views.
RESULTS AND DISSCUSSION
Characterization of the HGSMs:
A-TEG-Ad was synthesized after the modification of 1-bromoadamantane (Figure 1A), as confirmed by the signal integral area ratio of 1H NMR spectra (Figure S1A) showing the presence of the double bond signal (m, 6H, 5.8~6.5 ppm). β-CD-AOI2 was obtained through a nucleophilic addition reaction between β-CD and AOI (Figure 1B). The Fourier transform infrared (FT-IR) spectroscopy and 1H NMR confirmed the successful grafting of AOI on β-CD (Figure S1B and C). The MALDI-TOF MS spectrum confirmed the successful preparation of β-CD-AOI2 (Figure S1D) although the final product contained a small proportion of other by-products such as β-CD-AOI1 and β-CD-AOI3. Because the interior cavity of β-CD is hydrophobic, the interior cavity of β-CD-AOI2 could hold hydrophobic molecules (e.g., Ad) to form HGSM (Figure 1C). Thus the hydrophobic and van der Waals interactions drove the formation of HGSM. In Figure 1D, we can clearly observe A-TEG-Ad (yellow oil) layers floating on the β-CD-AOI2 aqueous solution initially. Interestingly, after being stirred vigorously for 24 h at room temperature, the upper oil layer disapeared, leaving transparent solution. This change indicated that Ad of the A-TEG-Ad was successfully accommodated in the CD of the β-CD-AOI2 to form an inclusion complex (HGSM). The 2D ROESY NMR spectrum (Figure S1E) showed that the nuclear Overhauser effect (marked in red and green background) can obviously be observed between the inner protons of β-CD-AOI2 (C2–6-H) and the α, β, and γ-H protons of A-TEG-Ad. These data confirmed the inclusion of A-TEG-Ad (the guest) into the cavity of the β-CD-AOI2 (the host) and the successful preparation of HGSM. XRD patterns (Figure S2) demonstrated that the HGSM solid system exhibited an amorphous state at 2θ = 17.6° and 11.6° after physical mixing of A-TEG-Ad and β-CD-AOI2, further suggesting that Ad of A-TEG-Ad was included as a guest in the cavities of β-CD-AOI2.
Construction of the HGGelMAs:
Analysis of 1H-NMR spectrum (Figure S3A) revealed that the degree of methacryloyl modification in GelMA was ~83%. To fabricate HGGelMAs, GelMA and HGSM at different concentrations were first dissolved in the same PBS solution at 37°C. It should be noted that this temperature is important for preventing prevent the release of the HGSMs. Subsequently, the mixture was subject to the UV-initiated free radical copolymerization, resulting in the formation of robust HGGelMA supramolecular hydrogels. As shown in Figure 2A, the HGGelMA network consisted of non-covalent crosslinking and covalent crosslinking. The polymerization reaction between the double bonds of GelMA and HGSM provided the covalent crosslinks, while the host-guest interactions between Ad and CD provided the non-covalent crosslinks. The strong, permanent, covalent crosslinks maintained the overall shape of the hydrogel, whereas the weak host-guest interactions simultaneously provided several mechanical functions (e.g., fatigue resistance, robustness, and self-healing). Figure 2B and C displayed the GelMA and HGGelMA hydrogels that were prepared into cylinder and cylindrical shapes. HGGelMA exhibited an opaque character compared with GelMA (Figure 2D), indicating that the presence of HGSM increased the crystallization area of the hydrogel. The cylindrical HGGelMA hydrogel did not appear any breaches even by squeezing, which suggesting its good flexibility (Figure 2B). In contrast, GelMA hydrogel was easily destroyed during real applications due to its poor mechanical strength.
Thus, HGGelMA hydrogels exhibited good mechanical properties and could be conveniently manipulated with sharp tweezers during experimentation without cracking (Figure 2C).
The easy processability of the HGGelMA hydrogels enables their formation into multifarious shapes that can better match damaged tissue environment and minimize the mechanical stimulation of surrounding tissues. As seen in Figure 2E, the HGGelMAs could maintain their initial shapes and water content after drying-swelling cycles. This can be attributed to the reinforced crosslink networks of the hydrogels due to the crosslinking between GelMA and HGSM and the reversibility of the host-guest interactions within the HGSM. In contrast, although GelMA gels could absorb water and recover their initial size, their surfaces were damaged by repeated drying-swelling cycles, resulting in uneven and unsmooth surfaces (Figure S4). Furthermore, these reproducible properties indicated that HGGelMAs hydrogels could be conveniently stored and easily manipulated.
SEM images (Figure 2F) showed the presence of porous structures in the HGGelMAs. In contrast with pure GelMA hydrogel, the pore size decreased and the pore density increased as the concentration of HGSM increased in the HGGelMAs. These observations indicated that the HGSMs were successfully crosslinked with GelMA, and that the crosslinking density increased with the increase in the HGSM content in the HGGelMA. This effect of HGSMs on the hydrogel crosslinking density was also reflected in the swelling kinetics curves of the hydrogels (Figure S5). Thus, the swelling speed and swelling ratio decreased with the increase of the crosslinking density, indicating that increasing the HGSM content promoted the densification of the hydrogel. Next, we performed rheological tests on HGGelMAs (Figures S6 and S7). The gelation time of HGGelMA hydrogel was ≈10 s at 25°C and reached a stable modulus in ≈30 s (Figure S6). Then, G’ and G” were investigated in response to the angular frequency (ω) at a constant strain (1%) when the concentration of the HGSM was increased from 3% (w/v) to 15% (w/v). Both GelMA hydrogels and HGGelMAs had G’ values that were higher than the G” values by an order of magnitude, and their G’ values were not dependent on the frequency at 0.1–100 rad s−1 (Figure S7). These results indicate that the synthetic hydrogels had stable gel properties owing to the incorporation of both covalent and non-covalent crosslinks. Specifically, in contrast to the G’ values of hydrogels at 40 rad s−1, the G’ of GelMA and HGGelMA hydrogels increased from 23.72 kPa to 40.68 kPa due to the increase of crosslinking density.
Mechanical properties:
Most natural biopolymer hydrogels have poor mechanical properties (e.g. unstable and fragile) due to their high water content, especially for non-covalent crosslinked hydrogels. However, HGGelMAs showed greatly improved mechanical properties, even with ~400% water content. As seen in the compressive stress-strain curve (Figure 3A), HGGelMAs exhibited great compression performance compared to pure GelMA hydrogels. As the concentration of HGSM increased, the mechanical strength and compression modulus of the HGGelMA hydrogels notably improved (Figure 3B and D). Thus, as the HGSM concentration increased to 15% (w/v), the compression modulus of HGGelMAs reached 0.63 MPa, which was 5.25-fold higher than that of the pure GelMA hydrogel (0.11 MPa). To observe the mechanical strength of the hydrogel intuitively, three hydrogels were used to support a 1 kg weight (Figure 3F). The test showed that none of the HGGelMA hydrogels were destroyed and they still maintained their original state. However, the GelMA group was completely destroyed even under the pressure of a 500 g weight (Figure 3C). Thus, HGGelMA hydrogels exhibited great stiffness and strength. Furthermore, the compression modulus of HGGelMAs was as high as that of natural soft tissues (0.3–0.5 MPa).31 Similarly, the inclusion of HGSM in a polymeric network also provided versatile mechanical strength reinforcement in other natural polymer hydrogels (e.g. Hyaluronan (HA) methacrylamide hyaluronic acid (HAMe), Figure S3B and S8).
Figure 3.

Mechanical properties characterization, and schematic mechanism illustrations of HGGelMAs under loading. (A-B) Stress-strain curves of the hydrogels subject to compression (A) and tensile testing (B). (C) Digital images of GelMA hydrogels that suffered the pressure of a 500 g weight. (D) Compression modulus of the hydrogels. (E) Cyclic tensile test curves of HGGelMAs. The inset showed the overall shape of the samples after undergoing the test. (F) Digital images of HG9GelMA hydrogel that supported the pressure of a 1 kg weight. (G-I) Schematic of the possible mechanism involved in the robust and fatigue-resistant mechanical behavior of HGGelMAs.
In addition, the elongations and fracture energy at the break of the HGGelMAs were all higher than those of pure GelMA hydrogels (Figure 3B and Figure S9), which indicated that HGGelMAs presented both high compressive strength and good stretching ability. Moreover, the HGGelMAs could resist fatigue when tensile cycles were repeated. This can be judged by the result that the loading-unloading stress-strain curves remained nearly unchanged after ten cycles of tensile measurement (Figure 3E). Furthermore, HGGelMA hydrogels did not crack after ten cycles of stretching, whereas the pristine GelMA hydrogel was completely broken after the sixth stretching cycle (Figure S10).
Self-healing ability:
The HGGelMA also has the capability of rapidly self-healing because the host-guest interactions are reversible. To observe these characteristics, an HGGelMA at the swelling equilibrium was equally cut into two halves (Figure 4A). One half was dyed red, and another half did not undergo any treatment. These two halves were found to be rapidly interconnected and self-heal to form an integrated hydrogel after being in close contact for 1 h. In order to confirm the self-healing of the hydrogel, we used two tweezers to hold the sides of the healing incision. We discovered that the healed incision of HGGelMA hydrogel did not break easily even though suffering a large tensile force (Video S1, Figure 4A). The 3D rotational stereoscopic microscope images showed the gradual self-healing process, leading to the fusion of the incision and the disappearance of the crevices (Figure 4B). We proposed a possible self-healing mechanism (Figure 4C), where the “free” Ad molecules were accommodated by the “free” β-CD at the incision surface of the HGGelMA, leading to the formation of a new noncovalently bonded HGSM again and thus the rapid self-healing of the hydrogel.
Figure 4.

The self-healing ability of HGGelMA hydrogels. (A) Photographs demonstrating that the cylindrical HGGelMA was self-healed. The left photo shows the self-healed hydrogel before stretching, while the right photo shows the healed HGGelMA during the stretching process. (B) Three-dimensional rotational microscopic images showing the change of the incision during the self-healing of the hydrogel. The arrows indicated the position of the original incision. (C) Illustration of the possible mechanism for the self-healing of HGGelMA. (D) Measurements of the continuous step strain (0.5% strain→1000% strain→0.5% strain) at 37°C showing the rapid self-healing of HGGelMA. (E) Stress-strain plots of HGGelMAs in different concentrations. Solid and ashed lines represented original and self-healed hydrogels, respectively. (F) The self-healing efficiency of hydrogels with different concentrations based on rapture energy (n=3).
The dynamic properties of HGGelMA hydrogel were also examined by a continuous cyclic deformation of oscillatory strain between 1000% and 0.5% at a constant frequency of 10 rad s−1 (Figure 4D). At a higher shear strain of 1000% (the area with blue background in Figure 4D), G” of the hydrogels was greater than G’, suggesting a liquid-like behaviour and destruction of networks. When the hydrogel was subsequently subject to a lower strain of 0.5% for 140 s, the G’ increased instantly with an almost full recovery of the initial moduli value in such a short recovery time span.
These results indicated that whenever HGGelMA hydrogels were destroyed under a high shear strain, it is capable of self-healing nearly immediately through the re-establishment of the host-guest interactions.
A tensile testing was performed to evaluate the self-healing efficiency. Figure 4E showed that the stress-strain profiles were similar between the self-healed and original HGGelMAs. In addition, as the concentration of HGSM went up, the self-healing efficiency increased to 80% (Figure 4F).
The aforementioned data showed that the HGGelMAs were stiff, elastic, fatigue resistant and self-healing due to the introduction of the HGSM and the concomitant incorporation of physical noncovalent crosslinks in a network of chemical covalent crosslinks. The HGSM was homogeneously dispersed in the GelMA hydrogel network, forming dynamic supramolecular crosslinking points during random copolymerization, which resulted in a relatively high crosslinking density in the polymer network. Compared to previous studies on supramolecular hydrogels,63 we developed extraordinarily tough HGGelMA hydrogels with a network comprising of both strong and weaker crosslinks. The internal fracturing of the host-guest crosslinks effectively dissipated energy and prevented catastrophic break propagation during loading. The covalent bonds formed from the reactions of the double bonds of GelMA and HGSM served as strong crosslinks that maintained the overall shape of the HGGelMAs, while the host-guest inclusion interactions formed weak crosslinks that reinforced the mechanical properties of the HGGelMA. The experiments showed that the weaker crosslinks increased the strength of the hydrogels due to the preferential breaking of the non-covalent host-guest interactions, whereas the enhancements of the fatigue resistance and self-healing capability resulted from the reversibility of the HGSMs formation. As a result, the HGSMs enabled the formation of a special topological structure that effectively enhanced the strength and functions of natural hydrogel materials, even with high water content. As shown in Figure 3G, when the hydrogels were subject to a force, the entangled gelatin polymer chains were stretched, leading to the preferential fracturing of the noncovalent bonds between host and guest molecules (Figure 3H). With further deformation, the covalent bonds of hydrogels were eventually broken (Figure 3I). Consequently, the introduction of HGSMs in the polymeric hydrogel network forms a host-guest supramolecular topological structure, where the host-guest interactions function as a sacrificial bond in robust, elastic, and fatigue-resistant hydrogels.
3D printing of HGGelMA:
Due to the ability of 3D printing to fabricate biomaterials with features at a micrometer precision to precisely match damaged tissues on demand, 3D printing technology has been widely studied. To demonstrate that our hydrogel exhibited an excellent 3D printability (Figure 5A), a rheological study was conducted to evaluate the precursor solution (also called the printing “ink”) to comprehensively understand the 3D plotting behavior. The precursors of HGGelMAs showed a shear thinning behavior (Figures 5E), and as expected, the ink viscosity increased with the decreasing temperature (Figure 5F). Likewise, the ink underwent a “sol-gel” transformation following the change in temperature (Figure 5D). These properties indicate that the precursors of HGGelMAs have a great printability. During the 3D printing process (Figure 5A), the temperature of the printing ink was decreased to 24 °C using the LTV-Dispense Head so that it had sufficient viscosity to become a thin filament (Figure S11A, B). Then, the printed 3D scaffolds were constructed through layer-by-layer deposition (with an alternating angle of 45° or 90° between adjacent layers and the resultant scaffolds were termed HGGelMA-45 or HGGelMA-90) and rapidly cured on the low-temperature platform of the 3D-Bioplotter (step 1, Figure S11C, D and Video S2). Subsequently, the HGGelMA scaffolds were irradiated by UV light (10 mW cm−2) to initiate further crosslinking, leading to the formation of the 3D printed hydrogel scaffolds (step 2, Figures S11E).
Figure 5.

3D bioprinting of HGGelMA into scaffolds by using HGGelMA precursors as a printing ink. (A) Schematic of the 3D printing of HGGelMA. (B-C) 3D rotational microscopic images showing the swelling equilibrium scaffolds under the swelling equilibrium (B, HGGelMA-45; C, and HGGelMA-90). (D) Dependence of G’ and G” on the temperature between the gel and sol state. (E) Shear thinning curves of HGGelMA ink via rotating measurements repeated for three cycles. (F) Curves of viscosity of the ink as a function of temperature. (G) Histograms of the compression modulus of the GelMA and HGGelMAs.
To evaluate the structures and pores of the HGGelMA scaffolds, 3D rotational microscopy was carried out to visualize the water-bearing hydrogel scaffolds (Figure 5B, C). The fiber diameters were found to be approximately 500 μm, slightly larger than the setting size (400 μm) due to water absorption by the hydrogels. The fibers were uniform and quite regular in the 3D-printed scaffolds. The entire structure of the hydrogel scaffolds was regular and contained uniform pores, arising from the increased solid content due to the introduction of HGSM to the hydrogel scaffolds and the use of a two-step continuous curing procedure. In addition, the strength of the 3D-printed hydrogel scaffolds was increased due to the introduction of HGSMs (Figure 5G). These results greatly increase the potential of using 3D printed hydrogel scaffolds in biomedical applications.
Cell compatibility of the 3D printed HGGelMAs:
Cell compatibility is an important evaluation criterion for modified biomaterials. A CCK-8 test was performed to evaluate the cell densities after the cells were cultured on the 3D-printed HGGelMA scaffolds for different days (Figure 6A). The mBMSCs seeded on the HGGelMAs scaffolds exhibited a similar proliferation rate as those on the pure GelMA scaffolds. For a more intuitive observation of cell viability, mBMSCs were embedded into HGGelMA 3D printed fibers. A live/dead staining was done on mBMSCs cultured on the fibers for 12 days. The live/dead staining assay showed that the vast majority of the cells were alive (and thus dyed green) in the HGGelMA fibers (Figure 6B). Moreover, Compared with PEGDA, HGGelMA hydrogels showed the clearly visible cell filaments and pseudopodia (Figure S12), which indicate good cytocompatibility and cell proliferative capabilities of the HGGelMA hydrogels. Furthermore, GelMA displayed a higher cell proliferation rate than HGGelMA hydrogels, probably due to the higher crosslinking density in HGGelMA than in GelMA. However, although the introduction of HGSMs in GelMA enhanced the strength of GelMA hydrogels, it did not influence the cell compatibility of the hydrogels.
Figure 6.

High cell viability on the 3D-printed HGGelMA and GelMA hydrogel scaffolds. (A) mBMSCs densities of after cultured on the HGGelMA and GelMA scaffolds for different days. (B) Live/dead stained images of the encapsulated mBMSCs on the GelMA and HGGelMA printed fibers after cultured for 12 days. (C-E) Fluorescence images showing the morphology of mBMSCs after 24 h of culture on GelMA, HGGelMA and PEGDA hydrogels, respectively. F-actin, Vinculin and DAPI were stained green, red and blue, respectively.
To further visualize the cell adhesion behavior of the hydrogel materials, mBMSCs were seeded on 3D-printed HGGelMA and GelMA scaffolds as well as a control scaffold (3D-printed PEGDA hydrogel scaffold). Adhesion related proteins (Vinculin, FAK, Integrin β1) were marked with immunofluorescence staining (Figure 6C–E, S13 and S14) to confirm that mBMSCs displayed significant spreading of cytoplasma and bundling of F-actin when cultured on the HGGelMA and GelMA scaffolds, indicating active interactions between the hydrogels and cells. Moreover, the surroundings of the cells clearly developed vinculins, FAK that provided dynamic anchoring proteins for connecting the cell cytoskeleton with the extracellular matrix (ECM). Also, HGGelMA can promote cells to attach on the scaffolds via integrin receptors. Integrin β1 combined with the cell’s cytoskeleton will promote the activity of fibronectin and affect cell function by transmitting intercellular information. These characteristics indicated healthy adhesion of mBMSCs on the 3D-printed HGGelMA and GelMA hydrogel scaffolds.
However, on control PEGDA hydrogel scaffolds, mBMSCs did not present desired F-actin bundling, integrin β1 protein as well as significant vinculin secretion (Figure 6E, S14).
Formation of focal adhesion was confirmed on the HGGelMA as well as on pure GelMA hydrogels by the immunofluorescence imaging of vinculin and F-actin. These results suggested that HGGelMAs have good biocompatibility and provide favorable microenvironments for cell encapsulation, adhesion and proliferation.
Histocompatibility of the 3D-printed HGGelMAs:
To further assess the biocompatibility and histocompatibility of 3D-printed HGGelMA scaffolds, the porous HGGelMA and GelMA scaffolds were subcutaneously implanted into the pockets that were on the backs of nude mice. 40 days post-implantation, HGGelMA scaffolds were completely integrated with autogenous tissue of nude mice, and new subcutaneous muscle tissues and blood vessels were formed in their pores, but no immunological rejection occurred (Figure 7A). Histological staining results (Figure 7B) indicated that new tissue and blood vessels began to grow in the scaffold pores on the 20th day. After implantation for 40 days, more soft tissues and blood vessels were formed in the scaffold pores. At the same time, the process of ingrowth of new tissues is accompanied by partial degradation of scaffolds. Both GelMA and HGGelMA hydrogels showed good histocompatibility as well as the increasing of capillaries density in the new tissue over time. The microvessel density of HGGelMA scaffolds (marked with black arrows in the diagram) showed no significant difference with GelMA, suggesting that the introduction of HGSM has no obvious influence on the histocompatibility of GelMA. Moreover, the expression level of new tissue and blood vessel related genes and proteins (collagen I, SMA, PDGF and CD31) in the HGGelMA hydrogel scaffolds was found to be higher than that in the GelMA hydrogels via real-time PCR tests and immunehistology staining (Figure 7C and Figure S15), probably because HGGelMA hydrogel scaffolds matched mechanical microenvironment with natural tissue better than the pristine GelMA hydrogel scaffolds. Therefore, in vitro and in vivo evaluation collectively demonstrated that the novel HGGelMAs have favourable bioactivity and histocompatibility, making them useful in biomedical applications.
Figure 7.

High histocompatibility of 3D-printed HGGelMA and GelMA hydrogel scaffolds. (A) Animal experimental process. (B) The images of H&E staining at 20th days and 40th days after implantion. Black arrows indicated the new blood vessels in the pores of hydrogels. (C) The relative expression level of collagen I, SMA, PDGF and CD31 genes in implanted scaffolds by qPCR analysis.
Conclusions
We demonstrated a new strategy for developing biocompatible 3D-printable hydrogels with enhanced mechanical strength and self-healing by integrating both host-guest non-covalent crosslinking and covalent crosslinking in one hydrogel network. Specifically, HGSMs, established through the host-guest non-covalent interaction between host (β-CD) and guest (Ad) molecules (modified so that both terminated with free double bonds), were covalently linked with GelMA to form HGGelMA hydrogels throught polymerization reaction between the double bonds on HGSM and GelMA. Unlike conventional hydrogels, the developed HGGelMA hydrogels consisted of host-guest weak non-covalent bonds integrated into a network of strong covalent bonds. The covalent bonds maintained the overall shape of the hydrogels, whereas the non-covalent bonds reinforced the mechanical properties of the hydrogels and enabled the hydrogels to rapidly self-heal upon fracturing. As a result, HGGelMAs were robust, elastic, fatigue resistant, reproducible and self-healing, resulting from the reversible host-guest non-covalent interactions. The compression modulus of HGGelMA hydrogels increased by 525% compared to pure GelMA hydrogels and thus reached the level of most human soft tissues. Moreover, HGGelMA hydrogels exhibited an excellent printability. The 3D-printed HGGelMA hydrogel scaffolds showed exquisite and homogeneous porous structures, and presented good biocompatibility and histocompatibility. Overall, the HGGelMA hydrogels are a promising 3D-printable biomaterial with potential biomedical applications. Our strategy represents a new avenue to addressing the challenges of traditional hydrogels.
Supplementary Material
Conceptual insights.
The disadvantages of weak mechanical strength, low stability, non-self-healing and poor 3D-printability have hampered the development of natural polymer hydrogels as promising biomaterials. To address this issue, various methods have been reported including blending reinforcement and covalently crosslinking. However, it is still challenging to reach a balance between reasonable mechanical properties, excellent biocompatibility and desired functions (e.g. 3D-printability and self-healing). Herein, a novel three-armed host-guest supramolecule (HGSM) was developed through non-covalent host-guest recognition and allowed to form covalent bonding with biocompatible natural matrix (e.g., gelatin methacryloyl). The resultant hydrogel has noncovalent bonds embedded in a covalently linked network. The weaker noncovalent bonds can be rapidly re-established through host-guest recognition once breaking while the stronger covalent bonds maintain the network. Thus the natural polymer hydrogel not only shows improved mechanical strength, remains biocompatible, but also becomes self-healing and 3D-printable. The feature of our strategy is to allow the three-armed HGSM to covalently link the natural polymers to form hydrogels, namely, to embed noncovalent host-guest interactions in a covalently bonded network. This strategy can be applied to develop novel hydrogels that are mechanically strong, rapidly self-healing and 3D-printable. It represents a new avenue to addressing the challenges of traditional natural hydrogels.
Acknowledgements
The authors gratefully acknowledge the funding from National Nature Science Foundation of China (51502095 & 81401191), Guangdong Natural Science Funds for Distinguished Young Scholar (2016A030306018), Guangdong Young Pearl River Scholars and Science and Technology Research Foundation of Shenzhen Bureau of Science and Technology & Information (JCYJ201703061417–16014). Y.Z. and C.M. would like to acknowledge the support from National Institutes of Health (EB021339).
Footnotes
Experimental
Experimental methods are available in the ESI.
Electronic Supplementary Information (ESI) available: Experimental methods and statistical analysis including plots and figures.
Conflicts of interest
There are no conflicts to declare
References
- 1.Tie L, An Y, Han J, Xiao Y, Xiaokaiti Y, Fan S, Liu S, Chen AF and Li X, J. Nutr. Biochem, 2013, 24, 88–96. [DOI] [PubMed] [Google Scholar]
- 2.Zakrzewski JL, Van Den Brink MRM and Hubbell JA, Nat. Biotechnol, 2014, 32, 786–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yuan T, Zhang L, Li K, Fan H, Fan Y, Liang J and Zhang X, J. Biomed. Mater. Res. Part B Appl. Biomater, 2014, 102, 337–344. [DOI] [PubMed] [Google Scholar]
- 4.Badylak SF, Semin. Cell Dev. Biol, 2002, 13, 377–383. [DOI] [PubMed] [Google Scholar]
- 5.Rodriguez MJ, Brown J, Giordano J, Lin SJ, Omenetto FG and Kaplan DL, Biomaterials, 2017, 117, 105–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Staruch RMT, Glass GE, Rickard R, Hettiaratchy SP and Butler PEM, Tissue Eng. Part B Rev, 2017, 23, 183–198. [DOI] [PubMed] [Google Scholar]
- 7.Hasan A, Khattab A, Islam MA, Hweij KA, Zeitouny J, Waters R, Sayegh M, Hossain MM and Paul A, Adv. Sci, 2015, 2, 1500122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guvendiren M, Lu HD and Burdick JA, Soft Matter, 2012, 8, 260–272. [Google Scholar]
- 9.Shin SR, Zihlmann C, Akbari M, Assawes P, Cheung L, Zhang K, Manoharan V, Zhang YS, Yüksekkaya M, Wan KT, Nikkhah M, Dokmeci MR, Tang XS and Khademhosseini A, Small, 2016, 12, 3677–3689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lin YD, Ko MC, Wu ST, Li SF, Hu JF, Lai YJ, Harn HIC, Laio IC, Yeh ML, Yeh HI, Tang MJ, Chang KC, Su FC, Wei EIH, Lee ST, Chen JH, Hoffman AS, Wu WT and Hsieh PCH, Biomater. Sci, 2014, 2, 567–580. [DOI] [PubMed] [Google Scholar]
- 11.Pereira RF and Bártolo PJ, J. Appl. Polym. Sci, 2015, 4, 035005. [Google Scholar]
- 12.Hockaday LA, Kang KH, Colangelo NW, Cheung PYC, Duan B, Malone E, Wu J, Girardi LN, Bonassar LJ, Lipson H, Chu CC and Butcher JT, Biofabrication, 2012, 132, 42458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ouyang L, Highley CB, Sun W and Burdick JA, Adv. Mater, 2017, 29, 1604983. [DOI] [PubMed] [Google Scholar]
- 14.Mandrycky C, Wang Z, Kim K and Kim DH, Biotechnol. Adv, 2016, 34, 422–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chan HN, Shu Y, Tian Q, Chen Y, Chen Y and Wu H, Mater. Horiz, 2016, 3, 309–313. [Google Scholar]
- 16.Mu X, Bertron T, Dunn C, Qiao H, Wu J, Zhao Z, Saldana C and Qi HJ, Mater. Horiz, 2017, 4, 442–449. [Google Scholar]
- 17.Melchels FPW, Domingos MAN, Klein TJ, Malda J, Bartolo PJ and Hutmacher DW, Prog. Polym. Sci, 2012, 37, 1079–1104. [Google Scholar]
- 18.Lei Z, Wang Q and Wu P, Mater. Horiz, 2017, 4, 694–700. [Google Scholar]
- 19.Gaetani R, Feyen DAM, Verhage V, Slaats R, Messina E, Christman KL, Giacomello A, Doevendans PAFM and Sluijter JPG, Biomaterials, 2015, 61, 339–348. [DOI] [PubMed] [Google Scholar]
- 20.Gruene M, Pflaum M, Deiwick A, Koch L, Schlie S, Unger C, Wilhelmi M, Haverich A and Chichkov BN, Biofabrication, 2011, 3, 015005. [DOI] [PubMed] [Google Scholar]
- 21.Markstedt K, Mantas A, Tournier I, Martínez Ávila H, Hägg D and Gatenholm P, Biomacromolecules, 2015, 16, 1489–1496. [DOI] [PubMed] [Google Scholar]
- 22.Bose S, Vahabzadeh S and Bandyopadhyay A, Mater. Today, 2013, 16, 496–504. [Google Scholar]
- 23.Kolesky DB, Truby RL, Gladman AS, Busbee TA, Homan KA and Lewis JA, Adv. Mater, 2014, 26, 3124–3130. [DOI] [PubMed] [Google Scholar]
- 24.Shim JH, Kim JY, Park M, Park J and Cho DW, Biofabrication, 2011, 3, 034102. [DOI] [PubMed] [Google Scholar]
- 25.Fedorovich NE, Alblas J, de Wijn JR, Hennink WE, Verbout AJ and Dhert WJA, Tissue Eng, 2007, 13, 1905–1925. [DOI] [PubMed] [Google Scholar]
- 26.Cheng Y, Zheng F, Lu J, Shang L, Xie Z, Zhao Y, Chen Y and Gu Z, Adv. Mater, 2014, 26, 5184–5190. [DOI] [PubMed] [Google Scholar]
- 27.Jakus AE, Secor EB, Rutz AL, Jordan SW, Hersam MC and Shah RN, ACS Nano, 2015, 9, 4636–4648. [DOI] [PubMed] [Google Scholar]
- 28.Neto AI, Levkin PA and Mano JF, Mater. Horiz, 2018, 5, 379–393. [Google Scholar]
- 29.Calvert P, Adv. Mater, 2010, 21, 743–756. [Google Scholar]
- 30.Tsitsilianis C, Soft Matter, 2010, 6, 2372–2388. [Google Scholar]
- 31.Ledoux WR and Blevins JJ, J. Biomech, 2007, 40, 2975–2981. [DOI] [PubMed] [Google Scholar]
- 32.Van Den Bulcke AI, Bogdanov B, De Rooze N, Schacht EH, Cornelissen M and Berghmans H, Biomacromolecules, 2000, 1, 31–38. [DOI] [PubMed] [Google Scholar]
- 33.Shin SR, Aghaei-Ghareh-Bolagh B, Dang TT, Topkaya SN, Gao X, Yang SY, Jung SM, Oh JH, Dokmeci MR, Tang X and Khademhosseini A, Adv. Mater, 2013, 25, 6385–6391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang YS and Khademhosseini A, Science, 2017, 356, 3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Costa AMS and Mano JF, Chem. Commun, 2015, 51, 15673–15676. [DOI] [PubMed] [Google Scholar]
- 36.Jung H, Park JS, Yeom J, Selvapalam N, Park KM, Oh K, Yang JA, Park KH, Hahn SK and Kim K, Biomacromolecules, 2014, 15, 707–714. [DOI] [PubMed] [Google Scholar]
- 37.Peppas NA, Huang Y, Torres-Lugo M, Ward JH and Zhang J, Annu. Rev. Biomed. Eng, 2000, 2, 9–29. [DOI] [PubMed] [Google Scholar]
- 38.Chaudhuri O, Biomater. Sci, 2017, 5, 1480–1490. [DOI] [PubMed] [Google Scholar]
- 39.Guvendiren M, Yang S and Burdick JA, Adv. Funct. Mater, 2009, 19, 3038–3045. [Google Scholar]
- 40.Tan M, Cui Y, Zhu A, Han H, Guo M and Jiang M, Polym. Chem, 2015, 6, 7543–7549. [Google Scholar]
- 41.Gulyuz U and Okay O, Macromolecules, 2014, 47, 6889–6899. [Google Scholar]
- 42.Kim SY and Lee YM, J. Appl. Polym. Sci, 1999, 74, 1752–1761. [Google Scholar]
- 43.Zelikin AN, Breheney K, Robert R, Tjipto E and Wark K, Biomacromolecules, 2010, 11, 2123–2129. [DOI] [PubMed] [Google Scholar]
- 44.Chen G, Li J, Song M, Wu Z, Zhang W, Wang Z, Gao J, Yang Z and Ou C, Adv. Funct. Mater, 2017, 27, 1701798. [Google Scholar]
- 45.Dou XQ and Feng CL, Adv. Mater, 2017, 29, 1604062. [DOI] [PubMed] [Google Scholar]
- 46.Loh XJ, Mater. Horiz, 2014, 1, 185–195. [Google Scholar]
- 47.Wang Z, Ren Y, Zhu Y, Hao L, Chen Y, An G, Wu H, Shi X and Mao C, Angew. Chem. Int. Ed, 2018, 57, 9008–9012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tan S, Ladewig K, Fu Q, Blencowe A and Qiao GG, Macromol. Rapid Commun, 2014, 35, 1166–1184. [DOI] [PubMed] [Google Scholar]
- 49.Zeng F, Han Y, Yan ZC, Liu CY and Chen CF, Polymer, 2013, 54, 6929–6935. [Google Scholar]
- 50.Li Y, Li J, Zhao X, Yan Q, Gao Y, Hao J, Hu J and Ju Y, Chem. - A Eur. J, 2016, 22, 18435–18441. [DOI] [PubMed] [Google Scholar]
- 51.Chen JX, Cao LJ, Shi Y, Wang P and Chen JH, J. Biomed. Mater. Res. Part A, 2016, 104, 2263–2270. [DOI] [PubMed] [Google Scholar]
- 52.Guan Y, Zhao HB, Yu LX, Chen SC and Wang YZ, RSC Adv, 2014, 4, 4955–4959. [Google Scholar]
- 53.Cui H, Cui L, Zhang P, Huang Y, Wei Y and Chen X, Macromol. Biosci, 2014, 14, 440–450. [DOI] [PubMed] [Google Scholar]
- 54.Zhang J and Ma PX, Adv. Drug Deliv. Rev, 2013, 65, 1215–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kevadiya BD, Joshi GV, Mody HM and Bajaj HC, Appl. Clay Sci, 2011, 52, 364–367. [Google Scholar]
- 56.Da Hu Q, Tang GP and Chu PK, Acc. Chem. Res, 2014, 47, 2017–2025. [DOI] [PubMed] [Google Scholar]
- 57.Liu J, Hennink WE, Van Steenbergen MJ, Zhuo R and Jiang X, Bioconjug. Chem, 2016, 27, 1143–1152. [DOI] [PubMed] [Google Scholar]
- 58.Rodell CB, Kaminski AL and Burdick JA, Biomacromolecules, 2013, 14, 4125–4134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Highley CB, Rodell CB and Burdick JA, Adv. Mater, 2015, 27, 5075–5079. [DOI] [PubMed] [Google Scholar]
- 60.Lin N and Dufresne A, Biomacromolecules, 2013, 14, 871–880. [DOI] [PubMed] [Google Scholar]
- 61.Yue K, Trujillo-de Santiago G, Alvarez MM, Tamayol A, Annabi N and Khademhosseini A, Biomaterials, 2015, 73, 254–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Colosi C, Shin SR, Manoharan V, Massa S, Costantini M, Barbetta A, Dokmeci MR, Dentini M and Khademhosseini A, Adv. Mater, 2016, 28, 677–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhu F, Lin XY, Wu ZL, Cheng L, Yin J, Song Y, Qian J and Zheng Q, Polymer, 2016, 95, 9–17. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.