

Abstract
Collagens carry out critical extracellular matrix (ECM) functions by interacting with numerous cell receptors and ECM components. Single glycine substitutions in collagen III, which predominates in vascular walls, result in vascular Ehlers–Danlos syndrome (vEDS), leading to arterial, uterine, and intestinal rupture and an average life expectancy of <50 years. Collagen interactions with integrin α2β1 are vital for platelet adhesion and activation; however, how these interactions are impacted by vEDS-associated mutations and by specific amino acid substitutions is unclear. Here, we designed collagen-mimetic peptides (CMPs) with previously reported Gly → Xaa (Xaa = Ala, Arg, or Val) vEDS substitutions within a high-affinity integrin α2β1-binding motif, GROGER. We used these peptides to investigate, at atomic-level resolution, how these amino acid substitutions affect the collagen III–integrin α2β1 interaction. Using a multitiered approach combining biological adhesion assays, CD, NMR, and molecular dynamics (MD) simulations, we found that these substitutions differentially impede human mesenchymal stem cell spreading and integrin α2–inserted (α2I) domain binding to the CMPs and were associated with triple-helix destabilization. Although an Ala substitution locally destabilized hydrogen bonding and enhanced mobility, it did not significantly reduce the CMP–integrin interactions. MD simulations suggested that bulkier Gly → Xaa substitutions differentially disrupt the CMP–α2I interaction. The Gly → Arg substitution destabilized CMP–α2I side-chain interactions, and the Gly → Val change broke the essential Mg2+ coordination. The relationship between the loss of functional binding and the type of vEDS substitution provides a foundation for developing potential therapies for managing collagen disorders.
Keywords: collagen, integrin, protein-protein interaction, cell surface receptor, extracellular matrix protein, cell spreading, collagen mutations, connective tissue disorder, triple helix, vascular Ehlers-Danlos syndrome
Introduction
Vascular Ehlers–Danlos syndrome (vEDS)4 is life-threatening and results from abnormal synthesis of, or pathogenic mutations to, collagen III in blood vessel walls and distensible organ linings. This leads to arterial aneurysms; rupture to arterial, uterine, and intestinal walls; and thin, translucent skin (1, 2). vEDS is one of several debilitating genetic collagen diseases primarily caused by single Gly → Xaa mutations in the triple-helical (Gly-Xaa-Xaa′)n repeating domain of fibrillar collagens that include osteogenesis imperfecta (OI), achondrogenesis type II, spondyloepiphyseal dysplasia syndrome, and Stickler syndrome. Because glycines stabilize the interior of collagen triple helices through an intricate hydrogen-bonding network (3–5) in the canonical (GXX′)n repeating domain, mutation of conserved glycines in fibrillar collagens may cause deficiencies in collagen structure, assembly, or production (6–8).
The specific triple-helical conformation of collagen is important for recognition by its numerous binding partners in the extracellular matrix (ECM). Interactions of collagen III with cellular receptors are critical for cell homeostasis, wound healing, and platelet adhesion and activation (9–12). In this study, we focus on the collagen III interaction with the endothelial and platelet receptor integrin α2β1, which plays a pivotal role in cell–ECM adhesion and firm platelet arrest, adhering to exposed subendothelial ECM upon vascular injury (13–16). The collagen-binding integrins interact with native collagens via their inserted (I) domain located in the α subunit (αI) of the integrin headpiece (17–21). Isolated, αI retains the specificity and affinity of the parent integrins for collagen and has been used as a model of integrin in biological and structural studies (22–26). Integrins recognize a specific binding motif in the collagen sequence, GXX′GEX″ (in which X is any amino acid, X′ is usually Hyp, and X″ is frequently Arg or Asn), and the binding is metal-mediated, in which the Glu of collagen is coordinated to a divalent metal cation in the metal ion–dependent adhesion site (MIDAS) of the αI (27, 28). The collagen III sequence GROGER has been determined to be a high-affinity integrin-binding site (29) and is in a region that is especially overrepresented in reported cases of Gly → Xaa mutations (Fig. 1A). Here, we investigated how Gly → Xaa mutations within this domain impact integrin α2β1 binding and the molecular basis of any functional interference.
Figure 1.

Reported vEDS mutations and cell receptor binding. A, the number of Gly → Xaa mutant cases per residue of 611 total reported in the Ehlers–Danlos Syndrome Variant Database (https://eds.gene.le.ac.uk,5 updated May 14, 2019) (97, 98) along the collagenous domain of collagen III. Regions that contain stretches of eight or more consecutive Gly residues that have been reported to have vEDS mutations are highlighted in yellow. B, major ligand-binding sites are plotted along the collagenous domain for comparison with regions dense in vEDS mutations. Ligand-binding sites plotted in gray include Yersinia adhesin A (Yad A), Gly177–Hyp185 (56); platelet fibrinogen receptor, Pro226–Asp230 (53); heparin, Lys263–Arg266 and Lys1106–Arg1109 (57, 58); pigment epithelium–derived factor (PEDF), Met262–Gly273 and Ile1105–Gly1116 (59, 60); von Willebrand factor (VWF), Arg572–Phe580 (48); secreted protein acidic and rich in cysteine (SPARC), Gly576–Hyp581 and Gly870–Hyp875 (61); discoidin domain receptor-2 (DDR2), Gly576–Hyp581 (51); kindlin-3, Lys655–Lys662 (50); leukocyte-associated immunoglobulin-like receptor-1 (LAIR-1), Gly690–Hyp716, Gly834–Lys860, and Gly1068–Lys1094 (54, 55); glycoprotein VI (GpVI), Gly690–Hyp716 (49); osteoclast-associated receptor (OSCAR), Gly804–Hyp815 and Gly867–Arg878 (52); matrix metalloproteinases (MMPs), Gly945–Ala959 (63); and decoron, Gly1032–Glu1038 and Lys1060–Glu1066 (62). Integrin α2β1–binding sites, Gly237–Arg242, Gly288–Arg293, Gly303–Arg308, Gly678–Arg683, Gly726–Arg731, and Gly987–Arg992 (28, 29), are highlighted in orange. C, percentages of Gly → Xaa substituted amino acids of the total cases reported. Amino acids are colored based on type (negatively charged, red; positively charged, blue; hydrophobic, green; small, yellow).
Previous studies have shown that substitution of these integral Gly residues causes structural deformities to the triple helix, and the extent of triple-helix destabilization depends on the amino acid substituted (30, 31) and the surrounding sequence context (32–36). Recently, elegant studies have investigated the impact of Gly → Xaa mutations near integrin-binding sites in collagen I (31, 37) using recombinant bacterial collagen systems, which can incorporate long spans of collagen-like sequence and can probe selective mutation sites. However, these models lack hydroxyprolines that are important for triple-helix folding and are unable to model the heterotrimeric collagen I triple helix. Here, we use collagen-mimetic peptides (CMPs) that incorporate a native collagen III sequence. These are good physiological models of collagen III fragments given its homotrimeric nature, and incorporation of native hydroxyprolines allows us to more closely model effects of Gly → Xaa mutations on triple-helix structure and dynamics.
Structural consequences of Gly substitutions in collagen triple helices have been extensively studied in the context of OI mutations to collagen I (35, 38–42). OI is an autosomal dominant disorder, resulting from mutations to COL1A1 or COL1A2 genes. Because collagen I is a heterotrimer of α1(I) and α2(I) α-chains, only one or two mutant α-chains will be incorporated into the collagen I triple helix. In the homotrimeric collagen III, vEDS mutations may result in incorporation of up to three mutant α-chains. For OI, clinical phenotypes have been classified based on severity, and larger Gly substitutions more frequently result in lethal OI (43–45). No classification system has been established for vEDS. However, a clinical study found that survival of individuals with vEDS Gly → Xaa mutations was correlated with the substituting amino acid identity, with the trend Ser > Arg/Asp > Val (46). That is, those individuals with large hydrophobic Val and charged Arg or Asp substitutions exhibited shorter life spans than those with small Ser substitutions (46). This suggests that the structural impact of different amino acids on the collagen III triple helix may be relevant to disease outcome.
Three amino acid substitutions at Gly240, in the integrin-binding GROGER motif, have been associated with vEDS: Ala, Arg, and Val (https://eds.gene.le.ac.uk)5 (97, 98). In this work, we employed CMPs incorporating these naturally occurring vEDS substitutions within the GROGER α2I-binding site. We probed how the specific amino acid substitutions impact functional cell spreading and α2I adhesion and elucidated the underlying atomic-level structural and dynamic perturbations to the collagen triple helix and the CMP–α2I interaction interface. We found that although the Ala mutation locally disrupts the canonical triple-helix hydrogen bonding and enhances the backbone dynamics, it is not sufficient to significantly reduce α2I interactions, in the context of recombinant protein or on human mesenchymal stem cells (hMSCs). However, larger substitutions, Arg and Val, do impede α2I interactions through different mechanisms involving breakage of side-chain interactions or Mg2+ coordination, as suggested by MD simulations. Novel insight into this sequence–structure/dynamics–function relationship sets the foundation for new drug therapy techniques to combat collagen disorders that lead to compromised collagen interactions.
Results
Clusters of vEDS mutations coincide with several functional domains and pose a hindrance to cellular function
The Ehlers–Danlos Syndrome Variant Database reports 611 cases of Gly → Xaa mutations that result in vEDS (https://eds.gene.le.ac.uk,5 updated May 14, 2019) (97, 98). By plotting the distribution of cases within the collagenous domain of collagen III, it is apparent that mutations are concentrated in four regions of the collagenous domain: 1) the terminus, 2) Gly681–Gly744, 3) Gly924–Gly987, and 4) the C terminus (Fig. 1A). These regions contain long stretches of eight or more glycines for which vEDS mutations have been reported. Several binding domains for critical ligands have been determined along the collagen III sequence (47), including those for von Willebrand factor (48), glycoprotein VI (49), integrins α1β1 and α2β1 (28, 29), kindlin-3 (50), discoidin domain receptor-2 (51), osteoclast-associated receptor (52), platelet fibrinogen receptor (53), leukocyte-associated immunoglobulin-like receptor-1 (54, 55), Yersinia adhesin A (56), heparin (57, 58), pigment epithelium–derived factor (59, 60), secreted protein acidic and rich in cysteines (61), decoron (62), and matrix metalloproteinases (63) (Figs. 1B and S1; integrin α2β1–binding motifs are highlighted in orange). Modifications to glycines in these domains may perturb the local conformation and impede receptor binding, causing detriment to cell–ECM adhesion and cell signaling (9–11, 64). Known binding domains of integrin α2β1 (28, 29) are indicated in the schematic in Fig. 1B. These binding domains occur within or near regions where Gly → Xaa mutations are prevalent. Additionally, mutations to the C terminus may hinder global triple-helix folding, which proceeds in the C-terminal to N-terminal direction when there is a nucleation domain at the C terminus (65, 66).
Gly → Xaa mutations within the collagenous domain are not evenly distributed between amino acid types but rather are highly skewed toward charged amino acids, which make up 61% of Gly → Xaa mutations of all cases reported (Fig. 1C). To determine the impact of reported vEDS mutations of different amino acid types on triple-helix conformation and function, we pursued a sequence–structure/dynamics–function study of CMPs that contain an integrin α2β1–binding domain in the context of the collagen III sequence.
Gly → Xaa vEDS mutations reduce binding of the integrin α2I domain and hinder cell spreading
The collagen III sequence 237GROGER242 was identified to be an α2I-binding site by rotary shadowing and electron microscopy (EM) and was determined to be a high-affinity binding motif through solid-phase assays using synthetic CMPs (29). Previously, a CMP containing the native GROGER sequence was found to be biologically functional, as it supported cell adhesion to human lung fibroblast MRC-5 cells (29), which express α2β1 and α1β1 integrins (67). This binding site is in the N-terminal region of collagen III in which 27 consecutive glycines have at least one reported mutation that results in vEDS. We have designed a series of CMPs, referred to as T3-237, that are composed of 12 native residues of the collagen III sequence, including the high-affinity α2I-binding motif, 237GROGERGLOGPO248, flanked by (GPO)4 at the N terminus and (GPO)3 at the C terminus to promote triple-helix formation, GPC at the N and C termini for enhanced adhesion to microtiter plates (68), and GY at the C terminus to monitor peptide concentration by absorbance. We have incorporated the three naturally occurring mutations at Gly240: T3-237 G240A, G240V, and G240R.
We expected that mutation of this binding motif may hinder the ability of integrin α2β1 to interact with collagen III at this site. We investigated the adhesion and spreading behavior of hMSCs, which express this integrin receptor (69, 70), on three of these synthesized CMPs, T3-237 WT, G240A, and G240V, as well as a positive control (collagen III) and negative controls (BSA and a hexapeptide, GROGER, which cannot form the necessary triple helix for cell binding) conditions. Cultures were followed for 1 day, with representative images captured at 4, 7, and 24 h after cell seeding. After 4 h, hMSCs cultured on collagen III exhibited attachment and cell morphology consistent with strong adhesion (Fig. 2A). By comparison, cells attached to the negative controls were fewer in number and displayed a “pancake”-shaped morphology that is typical of weak adhesion (Fig. 2, B and C). When grown on T3-237 WT and G240A, hMSCs showed cell spreading trends comparable with collagen III (Fig. 2, D and E), whereas the cell morphology on G240V was consistent with the negative controls (Fig. 2F). Similar observations were made from images collected after 7 h in culture. Interestingly, by 24 h, cells on all conditions, including the negative controls, demonstrated attachment and morphology consistent with the collagen III condition, perhaps indicating that the hMSCs had deposited new matrix to foster cell adhesion.
Figure 2.

Cell spreading assay to CMPs. A–E, morphology of hMSCs (green) after 4 h in culture adhered to collagen III (positive control) (A), BSA (negative control) (B), GROGER peptide (negative control) (C), T3-237 WT (D), G240A (E), and G240V (F). Cell attachment and morphology on the WT and G240A match that on the positive control, whereas attachment and morphology on G240V are similar to that on the negative controls.
To probe the impact of the collagen III mutations specifically on α2I domain binding, we used in vitro solid-phase enzyme-linked immunosorbent assays (ELISAs). The α2I–collagen interaction depends upon coordination of a divalent metal cation with the Glu in the recognition domain (27, 28). Therefore, binding assays were performed in the presence and absence of Mg2+ cations to control for unspecific binding. Specificity for the GXX′GEX″ binding motif was also assessed by a negative control, (GPP)10, which does not contain an integrin-binding motif, and BSA, a globular protein with no known specific interaction for the integrin α2I domain. Both negative controls show significantly reduced adhesion relative to T3-237 WT. The relative adhesion of the α2I domain to each CMP was assessed by measuring absorbance at 450 nm. Integrin α2I has the highest adhesion to T3-237 WT and binds less to the mutants, decreasing in the order Ala > Arg > Val (Figs. 3 and S2), trending with the decrease of triple-helical stability when substituted into the Gly position (71) and consistent with the trend of the cell adhesion assay.
Figure 3.
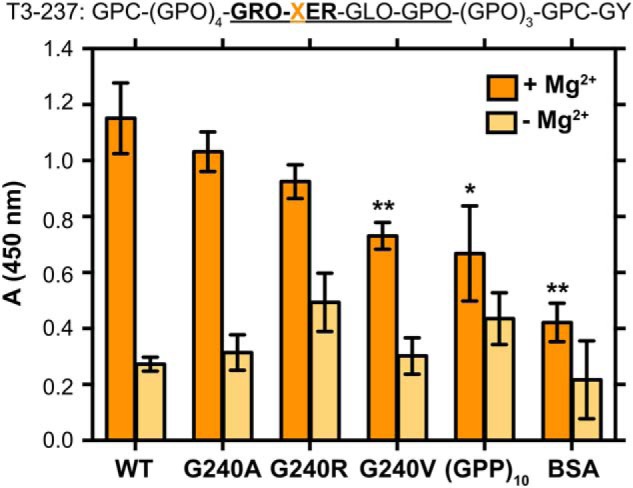
ELISA binding assay of α2I to T3-237 CMP variants. Recombinant α2I was incubated in Immunolon 2HB 96-well plates coated with the T3-237 CMP variants. Adhesion was measured in the presence of 5 mm MgCl2 (dark orange) or 5 mm EDTA (light orange). The EDTA condition is a negative control for unspecific binding independent of the divalent metal cations. BSA was used to block any uncoated surfaces and as a control for unspecific binding to the wells. (GPP)10 is used as a control for unspecific binding of α2I to collagen triple-helical segments that do not incorporate an integrin-binding motif. Experiments were performed in triplicate on one plate with 10 μg/ml indicated CMP/well and 10 μg/ml α2I/well. Error bars indicate the standard deviation between the triplicate repeats. Statistical analysis was performed with an unpaired t test relative to WT binding in GraphPad Prism. *, p ≤ 0.05; **, p ≤ 0.01. The peptide sequence is given at the top. The integrin-binding motif is in bold, and native collagen III residues are underlined.
T3-237 CMPs with naturally occurring vEDS mutations maintain the triple-helix conformation but have decreased thermal stability
We characterized the conformation and dynamics of the T3-237 CMPs to determine the molecular underpinnings of reduced α2I adhesion with bulkier mutations. The triple-helix conformation is a prerequisite for interaction with integrin α2β1 (72). At 4 °C, each of the T3-237 variants have CD wavelength profiles characteristic of triple helices, that is a minimum negative molar residue ellipticity (MRE) near 198 nm and a maximum positive MRE near 225 nm, with little deviation in intensity (Fig. 4A). This indicates that the triple-helical composition between these variants is unchanged at this low temperature. The Gly → Xaa mutations do perturb the sensitivity of these triple helices to temperature, however. We found that the melting points of G240R and G240V are decreased by ∼10 °C relative to T3-237 WT (47.8 ± 0.5–38.1 ± 0.7 °C) (Fig. 4B) by monitoring a CD melt.
Figure 4.
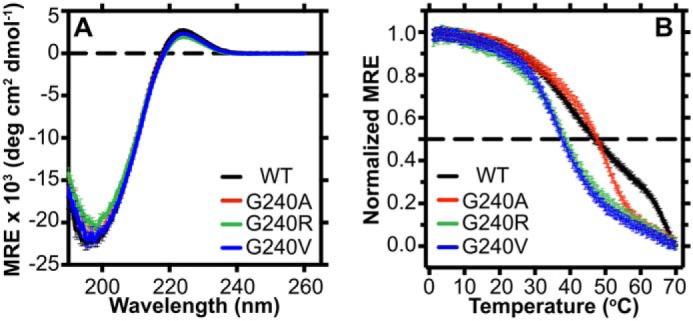
Characterization of the triple-helix global conformation and stability. A, CD wavelength scan at 4 °C of each CMP (0.5 mg/ml) in 10 mm acetic acid from 190 to 260 nm. B, temperature dependence of the CD signal at 224 nm for each CMP (1 mg/ml) in 10 mm acetic acid from 0 to 70 °C. Fraction folded was based on the assumption that the peptides are fully folded at 0 °C and fully unfolded at 70 °C. The dashed line denotes a 50% folded population. deg, degrees.
vEDS mutations perturb conformation and dynamics of the triple helix local to the mutation site
To understand how this decreased stability impacts the ability for the α2I domain to interact with its binding domain, we probed for residue-specific perturbations to conformation and dynamics by integrating NMR with MD simulations. Three CMPs, T3-237 WT, G240A, and G240V, were specifically 15N-labeled at Gly16, Xaa19 (mutation site), and Gly28 (Fig. 5A). The isotopic labels on Gly16 and Xaa19 allow us to probe perturbation of local structure and dynamics within the binding site and near the mutation. Enrichment of Gly28 allows us to probe perturbations within a GPO-rich segment that is expected to have a stable triple-helical structure distant from the mutation site. Within the triple helix, each of the three α-chains is staggered by one residue from the adjacent chain. This creates degeneracy between like residues in the α-chains, giving distinct cross-peaks for isotopically labeled residues in each of the three chains in 1H–15N heteronuclear single quantum correlation (HSQC) spectra (Fig. 5A). A population of monomer species is also present for each residue. Because no adjacent residues were isotopically labeled, we could not unequivocally assign cross-peaks to specific α-chains. Gly28 residues in all three α-chains of a GPO-rich segment have identical chemical environments and thus have a single overlapping triple-helical cross-peak (Fig. 5A). By comparison of 1H–15N HSQC spectra, Gly16 has vastly different 1H and 15N chemical shifts between the three variants (Figs. 5A and S3), indicating that mutation of the Gly19 residue perturbs the conformation and/or chemical environment of the Gly16 backbone amide three residues N-terminal.
Figure 5.

Residue-specific characterization of the vEDS mutation influence on triple-helical CMPs. A, top, schematic of staggered α-chains of the T3-237 CMP series. 15N isotopically labeled residues (Res) are highlighted in yellow. Bottom, two-dimensional 1H–15N HSQC spectra of T3-237 WT (gray), T3-237 G240A (red), and T3-237 G240V (blue). Data were collected at 25 °C. Residue assignments are indicated on the spectra (T, triple helix; M, monomer). B, amide 1H chemical shift changes of triple-helical residues with increasing temperature from 5 to 40 °C, in increments of 5 °C, for the indicated 15N-labeled residues in the T3-237 CMP variants. Data for mutated residue 19 are indicated in color (gray, Gly19; red, Ala19; blue, Val19). The dashed line at −4.5 ppb/°C is the cutoff for hydrogen-bond formation. Chemical shift changes more positive than −4.5 ppb/°C are indicative of stable hydrogen bonds (73). C, {1H}–15N heteronuclear NOEs, measured at 15 °C, of T3-237 WT, T3-237 G240A, and T3-237 G240V. The mutation site is indicated in color as in B. Error bars are propagated from the noise level of the NMR spectra. All NMR data were collected at 600-MHz 1H Larmor frequency. Peptide solutions for NMR were of 1.8–3 mm monomer concentration in 10 mm acetic acid, 5 mm tris(2-carboxyethyl)phosphine, 10% D2O.
The structural integrity of the triple helix is provided by a critical hydrogen-bond network formed between the amide proton of each Gly and the carbonyl oxygen of the Xaa residue in the adjacent chain. As shown in Fig. 5B, in T3-237 WT, Gly16, Gly19, and Gly28 all have amide chemical shift perturbations less than 4.5 ppb/°C (dashed line) upon increasing temperature, indicating that their associated hydrogen-bond network is not significantly modulated by temperature (73) up to 40 °C. Within mutant peptides T3-237 G240A and G240V, Gly16 and Gly28 remain hydrogen-bonded as shown by their only minor chemical shift perturbations with temperature. However, each of the triple-helix α-chains of the mutated residues, Ala19 and Val19, show considerable temperature-dependent chemical shift changes. This indicates that even the small mutation of Gly → Ala abolishes the local hydrogen-bond network. Regardless of the small (Ala) or larger hydrophobic (Val) mutation, however, the disruption to the conformation is only local to the mutation site, as Gly16, only three residues N-terminal to the mutation site, maintains its hydrogen-bonding capacity.
The backbone dynamics of the 15N-labeled residues within the triple helices of each CMP were probed by heteronuclear {1H}–15N NOEs. Lower NOEs indicate increased mobility on the fast, ps–ns timescale. In the WT CMP, {1H}–15N NOEs at 15 °C were similar for all triple-helical isotopically enriched glycines (Fig. 5C). Upon mutation to G240A, the small Gly → Ala mutation leads to a significantly more mobile Ala19 backbone in only one α-chain. However, a larger, hydrophobic Gly → Val mutation increases the flexibility of all three α-chains dramatically, leading to {1H}–15N NOE values near zero. In both variants, the increase in backbone flexibility is only local to the mutation site, as {1H}–15N NOE values for triple-helical Gly16 and Gly28 are not decreased. In the case of G240V, one Gly16 actually has an increased NOE value, indicating a rigidification of the α-chain backbone in this position.
Modeling α2I interactions with vEDS Gly → Xaa variants
To understand how perturbations to the triple helix impact α2I binding, we performed 500-ns all-atom MD simulations on each CMP variant as a free triple helix and in complex with α2I. The MD simulations predict ns-timescale dynamics for all residues within the CMPs and α2I domain, including those residues not probed by NMR. This provides information on the span of CMP residues around the substitution site that is impacted by dynamic perturbations due to a Gly → Xaa mutation. The diameter of the triple helix at a specific site is reflective of local unfolding. We therefore monitored how incorporation of the three mutations changed the diameter of the triple-helix backbone in the free state. Fig. 6A shows the average diameter of the triple helix around the integrin-binding site. The positions are numbered by the residue of the leading strand, and the diameters are measured from the circle encompassing three in-register Cα atoms of the three α-chains at the indicated positions (74). The in-register residues for each position are indicated in the schematic in Fig. 6A. This places the mutations in the leading, middle, and trailing strands at positions 19, 20, and 21, respectively. We found that the triple helix is expanded, relative to the WT CMP, at each position that includes a Gly → Xaa mutation for all variants, and the effect asymmetrically propagates at least three positions C-terminal of the mutation sites, impacting functional domains. This asymmetric effect of Gly → Xaa mutations was found previously by Yigit et al. (37) in that Gly → Ser substitutions interfered with hydrogen bonding up to three triplets C-terminal of the mutation. In the T3-237 CMPs, we found that the diameter expansion extends two triplets in the C-terminal direction, from Xaa19 to Hyp24 of the leading chain (Fig. 6A). Val and Arg mutations have the greatest effect. Representative snapshots of the free triple-helix MD simulations show the diameter expansions for each CMP, with G240V and G240R having the greatest triple-helix distortion (Fig. 6B). Notably, the site of greatest expansion is at the position of Glu20M, the metal-coordinating Glu for CMP–α2I binding. We would expect this distortion directly at the binding site to inhibit the α2I interaction.
Figure 6.

MD simulations of T3-237 variants. A, average triple-helix diameter at each position (Pos) within the α2I-binding domain. The schematic shows the stagger of the three α-chains with the positions numbered by the residue of the leading chain. The plotted region is underlined. The mutation site in each chain is highlighted in orange and indicated on the x axis. B, representative snapshots of each T3-237 variant during the 500-ns MD simulations. The full T3-237 WT CMP is shown on the top. The highlighted region near the integrin-binding site is shown in the box for comparison between the different amino acid substitutions. For each variant, the mutation site is indicated in color in all three α-chains.
We probed for the impact of the mutations on complex formation with α2I. We have confirmed that the proper Mg2+ coordination and the van der Waals contacts, hydrogen bonds, and salt bridges that have been reported previously (29, 75) are present and stable in the simulated complex formed with the WT CMP (Table 1). These interactions remain unchanged upon introduction of an Ala mutation in the Xaa19 position, consistent with the retention of α2I binding and hMSC spreading over the G240A CMP observed experimentally. However, in the case of an Arg mutation, several interactions become lost over the course of the simulation. The Mg2+–α2I Ser155 coordination becomes unstable within 100 ns in complex with the G240R CMP (Fig. S4). The Mg2+ coordination is fulfilled by α2I Asp151, maintaining three coordinates with α2I. However, only two direct interactions between the triple-helical CMP and α2I remain, an Arg21M backbone–His258 side-chain hydrogen bond (Fig. S5) and an Arg21M–Asp219 salt bridge (Fig. S6). Other stabilizing hydrogen bonds and salt bridges formed between CMP and α2I side chains are destabilized upon introduction of the G240R mutation, as given in Table 1 and shown in Figs. S5 and S6. Upon mutation to Val, α2I loses Mg2+ coordination at two residues, Ser153 and Ser155, which is replaced by additional coordination to the CMP Glu20M and a water molecule (Figs. 7C and S4). Although in the MD simulations of both Arg and Val substitutions, the Mg2+ coordination state is disturbed, the α2I in the G240R complex maintains three coordinates; however, the α2I in the G240V complex is able to stabilize just one Mg2+ coordinate. This extreme loss in α2I–Mg2+ coordination in the case of the T3-237 G240V complex provides an explanation for the greatest reduction in α2I adhesion to T3-237 G240V.
Table 1.
Summary of CMP–α2I interactions observed in MD simulations of complexes formed with each variant
A gain of interaction relative to WT is shaded in green; a loss of interaction relative to WT is shaded in red.
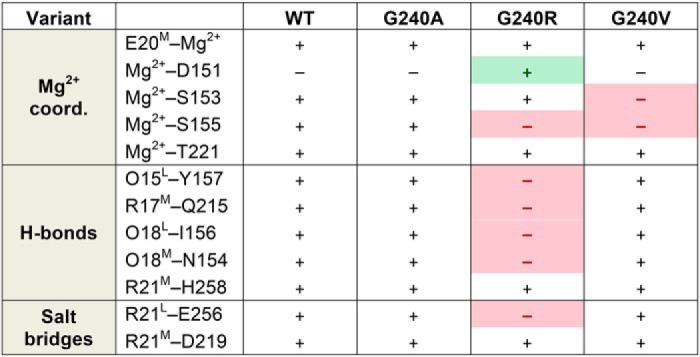
Figure 7.

Simulating T3-237 variant complex formation. A, collagen III (col III) in blood vessel walls interacts with integrin on endothelial cell and platelet membranes. B, natively, collagen III folds into a triple helix of three polypeptide chains. Glycines are involved in the critical hydrogen-bonding network that stabilizes the triple helix. Integrin αI domain interactions occur primarily via divalent metal ion coordination to a Glu C-terminal of the Gly mutated in this study. C, snapshots at 500-ns time points of CMP–α2I complex MD simulations, highlighting the Mg2+ coordination between integrin α2I (tan) and T3-237 WT (gray) or T3-237 G240V (blue). Residues that coordinate the Mg2+ in the WT complex are labeled (CMP, Glu20M; α2I, Ser153, Ser155, and Thr221).
Discussion
We have used an integrative approach, combining biological adhesion assays, CD, NMR spectroscopy, and MD simulations, to gain insight into the molecular underpinnings of the reduced α2I–CMP adhesion in the presence of Gly → Xaa mutations in collagen III. We observed that substitution of a Gly in the integrin-binding site interferes with α2I–CMP adhesion, dependent upon the identity of the Gly substitution, decreasing in the order WT > G240A > G240R > G240V, and T3-237 G240V substantially reduces hMSC spreading functionality relative to WT or G240A. Interestingly, in contrast to previous reports (31) using recombinant bacterial collagen models, a Gly → Ala mutation in the integrin-binding motif does not abolish α2I binding, which may in part be due to the presence of hydroxyproline in our CMPs that stabilizes the α2I–CMP interaction through hydrogen bonds with α2I side chains. This highlights the importance of native hydroxyprolines in collagen interactions. The results indicate that, even within this short CMP, relative to the full-length collagen III, a single Gly → Xaa mutation does not abolish triple-helix formation, but the thermal stability of the triple helix formed is disrupted, depending upon the side chain of the substituted amino acid.
In concert with the insight gained from ELISA, CD, NMR, and MD, the cell adhesion trends of hMSCs, which express α2β1 integrins on the cell surface, on CMPs could be predicted based on the substituted amino acid. The reduced cell adhesion of T3-237 G240V relative to WT and G240A can be explained by local backbone mobility and diameter expansion of the triple helix upon substitution of a bulky side chain. However, because the melting temperature of G240V is 10 °C lower compared with WT and G240A, a more significant fraction of the G240V may be unfolded. Based on the CD analysis, at 37 °C, ∼70% of the α-chains for both WT and G240A are in the triple-helical conformation, whereas only ∼50% of the α-chains for G240V have attained triple helix. This difference in the percentage of triple-helix structure presented by CMPs might present a reduced number of optimal integrin-binding sites to which the cells and recombinant α2I can adhere. Hence, it is imperative to realize whether the mutation itself leads to reduced cell and α2I adhesion or the variation in the number of triple-helical integrin-binding sites offered by the CMPs.
vEDS is one of several debilitating connective tissue disorders due to glycine mutations in fibrillar collagens (5, 76, 77). OI is another connective tissue disorder in which Gly → Xaa mutations in collagen I have been extensively studied (30–36, 40, 42, 78–84). The frequencies of substituted amino acids for OI are much different from those reported for vEDS. Over all OI mutations, Ser is the most substituted amino acid, accounting for ∼41% of Gly → Xaa mutations (44). Small-residue mutations make up nearly 63% of all OI Gly → Xaa substitutions (44). Conversely, in vEDS, small substitutions make up only ∼19% of Gly → Xaa mutations (Fig. 1C), with the most common substitutions being bulky Asp, Arg, Val, and Glu. Thus, Gly → Xaa mutations in the context of OI and vEDS cannot be treated as equal. It is not yet understood whether this is due to tissue specificity, the difference in sequence environment between the collagens, hetero- versus homotrimeric nature of the triple helices, or other factors.
When substituted into the Gly position, the charged and bulky mutations reported in vEDS are the most triple helix–destabilizing amino acids (71), and Gly → Xaa substitutions of these amino acids in collagen I most often result in the lethal form of OI (43, 44). Thus, it has been previously proposed that there is a correlation between OI phenotype and genetic mutation identity (44). In the case of vEDS, survival of a cohort of afflicted individuals was indeed found to trend with the identity of the substituted amino acid, in that those with Val, Arg, and Asp substitutions had the lowest survivability and that Ser substitutions were less severe (46). Our results show that substitution of the small amino acid Ala into a critical integrin α2β1-binding site only moderately disrupts α2I adhesion to the Gly → Ala CMP, and hMSC spreading on the Gly → Ala CMP is minimally affected. Conversely, substitution of a larger Val mutation substantially reduced α2I adhesion and hMSC spreading. This suggests that, despite the local structural and dynamic perturbations imparted on the triple helix by the Gly → Ala mutation, the CMP–α2I interaction maintains some plasticity wherein cellular functions are still able to occur. The ability for the small Gly → Ala mutation to maintain its functionality may be reflective of the underrepresentation of small mutations in vEDS, as milder phenotypes may go unreported. However, genotype–phenotype relationships will need to be further investigated for verification.
The collagen III–integrin α2β1 interaction investigated here is critical for platelet adhesion and signaling and endothelial cell adhesion in blood vessel walls (Fig. 7, A and B). Disruption of the collagen III triple helix through even a single Gly → Xaa mutation may hinder both the structural integrity of collagen III–rich tissues such as blood vessel walls and distensible organs but also inhibit vital cellular interactions with the ECM. The severity of vEDS is attributed largely to the potential for aortic aneurysms and arterial ruptures, which may be exacerbated by reduced platelet activity. These novel atomic-level insights into how the identity of vEDS Gly → Xaa mutations in an integrin-binding site impact the interaction of α2I with its recognition motif on collagen III provide a foundation for new drug therapy techniques to combat debilitating collagen disorders that compromise collagen interactions.
Experimental procedures
Preparation of CMPs
All CMP variants were purchased from LifeTein LLC (Somerset, NJ) as purified peptides. CMPs were dissolved in assay buffer from lyophilized powder and equilibrated at 4 °C overnight before use. For CD studies, CMPs were further purified using PD Midi-trap G10 desalting columns (GE Healthcare). Concentrations of peptides were determined by measuring the absorbance at 280 nm using a molar extinction coefficient of 1280 m−1 cm−1, with the exception of the GROGER hexapeptide (GenScript), which does not have a Tyr residue. The concentrations of this peptide were determined by weight, considering the net peptide content and HPLC purity as provided by the synthesis company.
Cell culture
hMSCs were continuously cultured in complete MEM-α containing 10% fetal bovine serum, 1% penicillin-streptomycin, 1% l-glutamine, and 0.001% basic fibroblast growth factor. Growth medium was changed every alternate day. The cells were detached by TrypLE and split at a ratio of 1:3 in a T175 flask upon reaching 80% confluence. The cells were incubated at 37 °C in a cell incubator with 95% air and 5% CO2.
Cell spreading assays
Cell spreading assays were performed as follows. 96-well solid white polystyrene plates (Corning, catalog number 3917) were coated with 100 μl/well multiple concentrations of CMPs in 10 mm acetic acid overnight at 4 °C. 50 μg/ml collagen III in 10 mm acetic acid was used as a positive control. 75 μg/ml GROGER hexapeptide in 10 mm acetic acid and 5% (w/v) BSA in 1× sterile-filtered PBS were used as negative controls. The GROGER hexapeptide consists of the sequence 237GROGER242 in a single chain and does not form a triple-helical structure, leading to no cell adhesion or spreading. The next day, peptide solutions were aspirated, and the wells were then blocked with 200 μl/well BSA solution (5% (w/v) BSA in 1× sterile-filtered PBS) for 2 h at room temperature with the exception of the tissue culture treated wells used as positive controls. Meanwhile, near-confluent hMSCs were labeled with 3 μm CellTracker Green 5-chloromethylfluorescein diacetate (CMFDA) dye (Thermo Fisher Scientific, catalog number C7025) in MEM-α (serum- and thiol-free) according to the manufacturer's instructions. After the blocking step, the BSA solution was aspirated, and the wells were washed three times with 200 μl/well PBS. After the washes, hMSCs were harvested from culture and reseeded onto the peptide-coated plates at a density of 5,000 cells/well in complete MEM-α and returned to the incubator. At 4, 7, and 24 h, plates were imaged using an Olympus IX81 inverted epifluorescence microscope (Olympus Scientific, Waltham, MA) with a Hamamatsu ORCA digital camera (Hamamatsu Photonics, Bridgewater, NJ).
Expression and purification of integrin α2I
The α2I domain used in these studies corresponds to residues 142–336 of the integrin α2 subunit. Integrin α2I was recombinantly expressed in Escherichia coli BL21(DE3) cells by induction with 1 mm isopropyl 1-thio-β-d-galactopyranoside overnight at 25 °C. The cells were lysed using a 20% sucrose TES buffer. The α2I domain was purified by Ni2+–nitrilotriacetic acid–agarose affinity chromatography (Qiagen) and buffer-exchanged with PD-10 desalting columns (GE Healthcare). Protein concentration was determined by measuring the absorbance at 280 nm using a molar extinction coefficient of 20,400 m−1 cm−1.
ELISA
Adhesion of recombinant α2I to T3-237 CMP variants was determined colorimetrically in solid-phase ELISAs. Immunolon 2HB 96-well plates (Thermo Fisher) were coated with 100 μl of CMP (10 μg/ml in 10 mm acetic acid) overnight at 4 °C. The wells were then blocked with 200 μl of 5% (w/v) BSA in PBS, pH 7.4, for 1 h at room temperature. From this point, for each α2I–CMP adhesion, half of the wells were treated with washing and binding buffers that consisted of PBS, pH 7.4 + 0.5% (w/v) BSA in the presence of 5 mm MgCl2 or 5 mm EDTA. After three washes with 200 μl of washing buffer, 100 μl of α2I (10 μg/ml in binding buffer) was incubated with each CMP for 1 h at room temperature. After washing three times with 200 μl of washing buffer, 100 μl of mouse anti-α2I mAb (1:2000 (v/v) dilution; Thermo Fisher, catalog number MA5-16571) in binding buffer was incubated in the wells for 45 min at room temperature. After washing three times with 200 μl of washing buffer, 100 μl of goat horseradish peroxidase–conjugated anti-mouse IgG antibody (1:5000 (v/v) dilution; GenScript, catalog number A00160, lot number 17B001197) in binding buffer was incubated in each well for 30 min at room temperature. Following a final four washes, α2I binding was detected using a 3,3′,5,5′-tetramethylbenzidine substrate kit (Pierce) as directed by the manufacturer's instructions. Absorbance was measured at 450 nm using a Tecan Infinite F50 plate reader equipped with Magellan software.
CD spectroscopy
CD wavelength profiles and temperature scans of each CMP were acquired on an AVIV Model 400 CD spectrometer (AVIV Biomedical Inc.). Wavelength scans were obtained at 4 °C from 260 to 190 nm, collecting points every 0.5 nm with a 1-nm bandwidth for 4 s, averaging three scans for each sample. Temperature scans were acquired by measuring MRE at 224 nm from 0 to 70 °C with a 10-s averaging time and 1.5-nm bandwidth. Samples were equilibrated for 2 min at each temperature before acquiring. The melting temperature was determined by first normalizing the melting curves assuming a fully folded state at 0 °C and a fully unfolded state at 70 °C and then calculating the temperature at which 50% of the population is folded based on a linear fit of the central temperature-dependent decay in the melting curves using GraphPad Prism.
NMR spectroscopy
T3-237 WT, G240A, and G240V CMPs were selectively 15N-labeled at sites Gly16, Xaa19, and Gly28. Peptides were dissolved in 10 mm acetic acid with 5 mm tris(2-carboxyethyl)phosphine and 10% D2O to monomeric concentrations of 1.8–3 mm. Triple-helix formation was equilibrated at 4 °C overnight before performing experiments.
All NMR experiments were performed on a Bruker Avance III 600-MHz spectrometer equipped with a TXI probe. Amide proton temperature gradient experiments were acquired as 1H–15N HSQC (85, 86) experiments at temperatures from 5 to 40 °C in increments of 5 °C. The samples were equilibrated for at least 1 h between temperature changes. Amide proton temperature gradients were calculated by linear fitting of the amide proton chemical shifts versus temperature. The amide proton gradient is taken as the slope of the line. Heteronuclear {1H}-15N NOE experiments were performed at 15 °C. All data were processed with NMRPipe (87) and analyzed in Sparky (88).
MD simulations
The initial model of the T3-237 WT CMP was built based on the GFOGER peptide from the crystal structure of the GFOGER–α2I complex (Protein Data Bank (PDB) code 1DZI) (75). The Phe residue in the GFOGER peptide was replaced by an Arg residue using PyMOL (Schrödinger, LLC). The extra GPO repeats, the terminal GPCs, and the C-terminal Tyr of T3-237 were built using the Triple-Helical Collagen Building Script (89). The N and C termini were capped with acetyl and NH2 groups, respectively. The initial models for the mutants were generated by replacing Gly19 of T3-237 with Ala, Val, and Arg residues using PyMOL.
The initial coordinates for the T3-237 CMP–α2I complex were taken from the X-ray structure (PDB code 1DZI) (75). The coordinates for T3-237 were generated by aligning the backbone to the X-ray structure of the GFOGER peptide. The Co2+ ion was replaced by a Mg2+ ion. All of the water molecules in the X-ray structure were retained.
The T3-237 CMP and CMP–α2I complex models were laid in a cubic box of TIP3P water molecules (90) with the box border at least 10 Å away from any atoms of the CMP or α2I. Extra Cl− ions were added to neutralize the positive charges.
The protein was treated with the ff14SB force field (91). The simulations were performed with the CUDA version of the pmemd module of the AMBER 2018 package (92). Periodic boundary conditions were used, and electrostatic interactions were calculated by the particle mesh Ewald method (93, 94), with the nonbonded cutoff set to 8 Å. The SHAKE algorithm (95) was applied to bonds involving hydrogens, and a 2-fs integration step was used. Pressure was held constant at 1 atm with a relaxation time of 2.0 ps. The temperature was held at 300 K with Langevin dynamics with a collision frequency of 2.0 ps−1.
Prior to MD simulations, the systems were subjected to energy minimizations and equilibration. The minimization started with 1000 steps of steepest descent minimization followed by 4000 steps of conjugate gradient minimization with 10 kcal mol−1 Å−2 position restraints on the CMP and α2I. The following minimization was carried out without any restraints. Then the system was heated from 0 to 300 K for 100 ps with position restraints of 10 kcal mol−1 Å−2 on the CMP and α2I. The system was first equilibrated for 1 ns at a constant temperature of 300 K and pressure of 1 atm with position restraints of 2 kcal mol−1 Å−2 on the CMP and α2I. The following equilibration was conducted without any restraints. The production runs for all of the models were 500 ns. The trajectories were analyzed using CPPTRAJ (96).
Author contributions
C. L. H., A. P. K., B. W., S. G., M. G., J. Z., D. A. C., D. I. S., and J. B. conceptualization; C. L. H., A. P. K., B. W., S. G., M. G., J. Z., and H. R. W. data curation; C. L. H., A. P. K., B. W., S. G., M. G., J. Z., H. R. W., and D. I. S. formal analysis; C. L. H., M. G., D. A. C., D. I. S., and J. B. supervision; C. L. H., M. G., H. R. W., D. I. S., and J. B. funding acquisition; C. L. H., B. W., S. G., M. G., J. Z., D. A. C., D. I. S., and J. B. validation; C. L. H., A. P. K., B. W., S. G., M. G., J. Z., H. R. W., D. I. S., and J. B. investigation; C. L. H., A. P. K., B. W., S. G., M. G., J. Z., H. R. W., D. A. C., D. I. S., and J. B. visualization; C. L. H., A. P. K., B. W., S. G., M. G., J. Z., H. R. W., D. A. C., D. I. S., and J. B. methodology; C. L. H., S. G., and J. B. writing-original draft; C. L. H., A. P. K., B. W., S. G., M. G., J. Z., H. R. W., D. A. C., D. I. S., and J. B. writing-review and editing; D. A. C. resources; D. I. S. and J. B. project administration.
Supplementary Material
Acknowledgment
This research used resources from the Rutgers Discovery Informatics Institute, which is supported by Rutgers and the State of New Jersey.
This work was supported in part by National Institutes of Health Grant GM 45302 (to J. B.) and American Heart Association Postdoctoral Fellowship 17POST33410326 (to C. L. H.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S6.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- vEDS
- vascular Ehlers–Danlos syndrome
- ECM
- extracellular matrix
- I
- inserted
- MD
- molecular dynamics
- CMP
- collagen-mimetic peptide
- OI
- osteogenesis imperfecta
- hMSC
- human mesenchymal stem cell
- MRE
- molar residue ellipticity
- HSQC
- heteronuclear single quantum correlation
- O or Hyp
- hydroxyproline
- MEM
- minimal essential medium
- TES
- 2-{[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]amino}ethanesulfonic acid.
References
- 1. Beighton P., De Paepe A., Steinmann B., Tsipouras P., and Wenstrup R. J. (1998) Ehlers-Danlos syndromes: revised nosology, Villefranche, 1997. Ehlers-Danlos National Foundation (U.S.A.) and Ehlers-Danlos Support Group (UK). Am. J. Med. Genet 77, 31–37 [DOI] [PubMed] [Google Scholar]
- 2. Pepin M., Schwarze U., Superti-Furga A., and Byers P. H. (2000) Clinical and genetic features of Ehlers–Danlos syndrome type IV, the vascular type. N. Engl. J. Med. 342, 673–680 10.1056/NEJM200003093421001 [DOI] [PubMed] [Google Scholar]
- 3. Ramachandran G. N., and Kartha G. (1955) Structure of collagen. Nature 176, 593–595 10.1038/176593a0 [DOI] [PubMed] [Google Scholar]
- 4. Rich A., and Crick F. H. (1955) The structure of collagen. Nature 176, 915–916 10.1038/176915a0 [DOI] [PubMed] [Google Scholar]
- 5. Kuivaniemi H., Tromp G., and Prockop D. J. (1997) Mutations in fibrillar collagens (types I, II, III, and XI), fibril-associated collagen (type IX), and network-forming collagen (type X) cause a spectrum of diseases of bone, cartilage, and blood vessels. Hum. Mutat. 9, 300–315 [DOI] [PubMed] [Google Scholar]
- 6. Bonadio J., and Byers P. H. (1985) Subtle structural alterations in the chains of type I procollagen produce osteogenesis imperfecta type II. Nature 316, 363–366 10.1038/316363a0 [DOI] [PubMed] [Google Scholar]
- 7. Forlino A., Kuznetsova N. V., Marini J. C., and Leikin S. (2007) Selective retention and degradation of molecules with a single mutant α1(I) chain in the Brtl IV mouse model of OI. Matrix Biol. 26, 604–614 10.1016/j.matbio.2007.06.005 [DOI] [PubMed] [Google Scholar]
- 8. Makareeva E., Aviles N. A., and Leikin S. (2011) Chaperoning osteogenesis: new protein-folding disease paradigms. Trends Cell Biol. 21, 168–176 10.1016/j.tcb.2010.11.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Heino J. (2007) The collagen family members as cell adhesion proteins. BioEssays 29, 1001–1010 10.1002/bies.20636 [DOI] [PubMed] [Google Scholar]
- 10. Kadler K. E., Baldock C., Bella J., and Boot-Handford R. P. (2007) Collagens at a glance. J. Cell Sci. 120, 1955–1958 10.1242/jcs.03453 [DOI] [PubMed] [Google Scholar]
- 11. Shoulders M. D., and Raines R. T. (2009) Collagen structure and stability. Annu. Rev. Biochem. 78, 929–958 10.1146/annurev.biochem.77.032207.120833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hynes R. O. (2009) The extracellular matrix: not just pretty fibrils. Science 326, 1216–1219 10.1126/science.1176009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Broos K., De Meyer S. F., Feys H. B., Vanhoorelbeke K., and Deckmyn H. (2012) Blood platelet biochemistry. Thromb. Res. 129, 245–249 10.1016/j.thromres.2011.11.002 [DOI] [PubMed] [Google Scholar]
- 14. Moroi M., and Jung S. M. (1998) Integrin-mediated platelet adhesion. Front. Biosci. 3, d719–d728 10.2741/A315 [DOI] [PubMed] [Google Scholar]
- 15. Savage B., Ginsberg M. H., and Ruggeri Z. M. (1999) Influence of fibrillar collagen structure on the mechanisms of platelet thrombus formation under flow. Blood 94, 2704–2715 [PubMed] [Google Scholar]
- 16. Nieswandt B., and Watson S. P. (2003) Platelet-collagen interaction: is GPVI the central receptor? Blood 102, 449–461 10.1182/blood-2002-12-3882 [DOI] [PubMed] [Google Scholar]
- 17. Tulla M., Pentikäinen O. T., Viitasalo T., Käpylä J., Impola U., Nykvist P., Nissinen L., Johnson M. S., and Heino J. (2001) Selective binding of collagen subtypes by integrin α1I, α2I, and α10I domains. J. Biol. Chem. 276, 48206–48212 10.1074/jbc.M104058200 [DOI] [PubMed] [Google Scholar]
- 18. Barczyk M., Carracedo S., and Gullberg D. (2010) Integrins. Cell Tissue Res. 339, 269–280 10.1007/s00441-009-0834-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Diamond M. S., Garcia-Aguilar J., Bickford J. K., Corbi A. L., and Springer T. A. (1993) The I domain is a major recognition site on the leukocyte integrin Mac-1 (CD11b/CD18) for four distinct adhesion ligands. J. Cell Biol. 120, 1031–1043 10.1083/jcb.120.4.1031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Springer T. A. (1997) Folding of the N-terminal, ligand-binding region of integrin α-subunits into a β-propeller domain. Proc. Natl. Acad. Sci. U.S.A. 94, 65–72 10.1073/pnas.94.1.65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hynes R. O. (2002) Integrins: bidirectional, allosteric signaling machines. Cell 110, 673–687 10.1016/S0092-8674(02)00971-6 [DOI] [PubMed] [Google Scholar]
- 22. Bahou W. F., Potter C. L., and Mirza H. (1994) The VLA-2 (α2β1) I domain functions as a ligand-specific recognition sequence for endothelial cell attachment and spreading: molecular and functional characterization. Blood 84, 3734–3741 [PubMed] [Google Scholar]
- 23. Kamata T., and Takada Y. (1994) Direct binding of collagen to the I domain of integrin α2β1 (VLA-2, CD49b/CD29) in a divalent cation-independent manner. J. Biol. Chem. 269, 26006–26010 [PubMed] [Google Scholar]
- 24. Tuckwell D., Calderwood D. A., Green L. J., and Humphries M. J. (1995) Integrin α2 I-domain is a binding site for collagens. J. Cell Sci. 108, 1629–1637 [DOI] [PubMed] [Google Scholar]
- 25. Knight C. G., Morton L. F., Onley D. J., Peachey A. R., Messent A. J., Smethurst P. A., Tuckwell D. S., Farndale R. W., and Barnes M. J. (1998) Identification in collagen type I of an integrin α2β1-binding site containing an essential GER sequence. J. Biol. Chem. 273, 33287–33294 10.1074/jbc.273.50.33287 [DOI] [PubMed] [Google Scholar]
- 26. Knight C. G., Morton L. F., Onley D. J., Peachey A. R., Ichinohe T., Okuma M., Farndale R. W., and Barnes M. J. (1999) Collagen-platelet interaction: Gly-Pro-Hyp is uniquely specific for platelet Gp VI and mediates platelet activation by collagen. Cardiovasc. Res. 41, 450–457 10.1016/S0008-6363(98)00306-X [DOI] [PubMed] [Google Scholar]
- 27. Farndale R. W., Lisman T., Bihan D., Hamaia S., Smerling C. S., Pugh N., Konitsiotis A., Leitinger B., de Groot P. G., Jarvis G. E., and Raynal N. (2008) Cell-collagen interactions: the use of peptide Toolkits to investigate collagen-receptor interactions. Biochem. Soc. Trans. 36, 241–250 10.1042/BST0360241 [DOI] [PubMed] [Google Scholar]
- 28. Siljander P. R., Hamaia S., Peachey A. R., Slatter D. A., Smethurst P. A., Ouwehand W. H., Knight C. G., and Farndale R. W. (2004) Integrin activation state determines selectivity for novel recognition sites in fibrillar collagens. J. Biol. Chem. 279, 47763–47772 10.1074/jbc.M404685200 [DOI] [PubMed] [Google Scholar]
- 29. Kim J. K., Xu Y., Xu X., Keene D. R., Gurusiddappa S., Liang X., Wary K. K., and Höök M. (2005) A novel binding site in collagen type III for integrins α1β1 and α2β1. J. Biol. Chem. 280, 32512–32520 10.1074/jbc.M502431200 [DOI] [PubMed] [Google Scholar]
- 30. Xiao J., Madhan B., Li Y., Brodsky B., and Baum J. (2011) Osteogenesis imperfecta model peptides: incorporation of residues replacing Gly within a triple helix achieved by renucleation and local flexibility. Biophys. J. 101, 449–458 10.1016/j.bpj.2011.06.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Qiu Y., Mekkat A., Yu H., Yigit S., Hamaia S., Farndale R. W., Kaplan D. L., Lin Y. S., and Brodsky B. (2018) Collagen Gly missense mutations: effect of residue identity on collagen structure and integrin binding. J. Struct. Biol. 203, 255–262 10.1016/j.jsb.2018.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bhate M., Wang X., Baum J., and Brodsky B. (2002) Folding and conformational consequences of glycine to alanine replacements at different positions in a collagen model peptide. Biochemistry 41, 6539–6547 10.1021/bi020070d [DOI] [PubMed] [Google Scholar]
- 33. Hyde T. J., Bryan M. A., Brodsky B., and Baum J. (2006) Sequence dependence of renucleation after a Gly mutation in model collagen peptides. J. Biol. Chem. 281, 36937–36943 10.1074/jbc.M605135200 [DOI] [PubMed] [Google Scholar]
- 34. Xiao J., Cheng H., Silva T., Baum J., and Brodsky B. (2011) Osteogenesis imperfecta missense mutations in collagen: structural consequences of a glycine to alanine replacement at a highly charged site. Biochemistry 50, 10771–10780 10.1021/bi201476a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bryan M. A., Cheng H., and Brodsky B. (2011) Sequence environment of mutation affects stability and folding in collagen model peptides of osteogenesis imperfecta. Biopolymers 96, 4–13 10.1002/bip.21432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chhum P., Yu H., An B., Doyon B. R., Lin Y. S., and Brodsky B. (2016) Consequences of glycine mutations in the fibronectin-binding sequence of collagen. J. Biol. Chem. 291, 27073–27086 10.1074/jbc.M116.753566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yigit S., Yu H., An B., Hamaia S., Farndale R. W., Kaplan D. L., Lin Y. S., and Brodsky B. (2016) Mapping the effect of Gly mutations in collagen on α2β1 integrin binding. J. Biol. Chem. 291, 19196–19207 10.1074/jbc.M116.726182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sun X., Liu Z., Zhao S., Xu X., Wang S., Guo C., and Xiao J. (2019) A self-assembling collagen mimetic peptide system to simultaneously characterize the effects of osteogenesis imperfecta mutations on conformation, assembly and activity. J. Mater. Chem. B 7, 3201–3209 10.1039/C9TB00086K [DOI] [Google Scholar]
- 39. Sun X., Liu S., Yu W., Wang S., and Xiao J. (2016) CD and NMR investigation of collagen peptides mimicking a pathological Gly-Ser mutation and a natural interruption in a similar highly charged sequence context. Protein Sci. 25, 383–392 10.1002/pro.2828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Xiao J., Yang Z., Sun X., Addabbo R., and Baum J. (2015) Local amino acid sequence patterns dominate the heterogeneous phenotype for the collagen connective tissue disease osteogenesis imperfecta resulting from Gly mutations. J. Struct. Biol. 192, 127–137 10.1016/j.jsb.2015.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Li Y., Brodsky B., and Baum J. (2009) NMR conformational and dynamic consequences of a Gly to Ser substitution in an osteogenesis imperfecta collagen model peptide. J. Biol. Chem. 284, 20660–20667 10.1074/jbc.M109.018077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Xu K., Nowak I., Kirchner M., and Xu Y. (2008) Recombinant collagen studies link the severe conformational changes induced by osteogenesis imperfecta mutations to the disruption of a set of interchain salt bridges. J. Biol. Chem. 283, 34337–34344 10.1074/jbc.M805485200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bodian D. L., Madhan B., Brodsky B., and Klein T. E. (2008) Predicting the clinical lethality of osteogenesis imperfecta from collagen glycine mutations. Biochemistry 47, 5424–5432 10.1021/bi800026k [DOI] [PubMed] [Google Scholar]
- 44. Persikov A. V., Pillitteri R. J., Amin P., Schwarze U., Byers P. H., and Brodsky B. (2004) Stability related bias in residues replacing glycines within the collagen triple helix (Gly-Xaa-Yaa) in inherited connective tissue disorders. Hum. Mutat. 24, 330–337 10.1002/humu.20091 [DOI] [PubMed] [Google Scholar]
- 45. Beck K., Chan V. C., Shenoy N., Kirkpatrick A., Ramshaw J. A., and Brodsky B. (2000) Destabilization of osteogenesis imperfecta collagen-like model peptides correlates with the identity of the residue replacing glycine. Proc. Natl. Acad. Sci. U.S.A. 97, 4273–4278 10.1073/pnas.070050097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pepin M. G., Schwarze U., Rice K. M., Liu M., Leistritz D., and Byers P. H. (2014) Survival is affected by mutation type and molecular mechanism in vascular Ehlers-Danlos syndrome (EDS type IV). Genet. Med. 16, 881–888 10.1038/gim.2014.72 [DOI] [PubMed] [Google Scholar]
- 47. Parkin J. D., San Antonio J. D., Persikov A. V., Dagher H., Dalgleish R., Jensen S. T., Jeunemaitre X., and Savige J. (2017) The collagen III fibril has a “flexi-rod” structure of flexible sequences interspersed with rigid bioactive domains including two with hemostatic roles. PLoS One 12, e0175582 10.1371/journal.pone.0175582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lisman T., Raynal N., Groeneveld D., Maddox B., Peachey A. R., Huizinga E. G., de Groot P. G., and Farndale R. W. (2006) A single high-affinity binding site for von Willebrand factor in collagen III, identified using synthetic triple-helical peptides. Blood 108, 3753–3756 10.1182/blood-2006-03-011965 [DOI] [PubMed] [Google Scholar]
- 49. Jarvis G. E., Raynal N., Langford J. P., Onley D. J., Andrews A., Smethurst P. A., and Farndale R. W. (2008) Identification of a major GpVI-binding locus in human type III collagen. Blood 111, 4986–4996 10.1182/blood-2007-08-108472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Maurice P., Legrand C., and Fauvel-Lafeve F. (2004) Platelet adhesion and signaling induced by the octapeptide primary binding sequence (KOGEOGPK) from type III collagen. FASEB J. 18, 1339–1347 10.1096/fj.03-1151com [DOI] [PubMed] [Google Scholar]
- 51. Konitsiotis A. D., Raynal N., Bihan D., Hohenester E., Farndale R. W., and Leitinger B. (2008) Characterization of high affinity binding motifs for the discoidin domain receptor DDR2 in collagen. J. Biol. Chem. 283, 6861–6868 10.1074/jbc.M709290200 [DOI] [PubMed] [Google Scholar]
- 52. Barrow A. D., Raynal N., Andersen T. L., Slatter D. A., Bihan D., Pugh N., Cella M., Kim T., Rho J., Negishi-Koga T., Delaisse J. M., Takayanagi H., Lorenzo J., Colonna M., Farndale R. W., et al. (2011) OSCAR is a collagen receptor that costimulates osteoclastogenesis in DAP12-deficient humans and mice. J. Clin. Investig. 121, 3505–3516 10.1172/JCI45913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hayashi Y., Katada J., Sato Y., Igarashi K., Takiguchi Y., Harada T., Muramatsu M., Yasuda E., and Uno I. (1998) Discovery and structure–activity relationship studies of a novel and specific peptide motif, Pro-X-X-X-Asp-X, as a platelet fibrinogen receptor antagonist. Bioorg. Med. Chem. 6, 355–364 10.1016/S0968-0896(97)10050-5 [DOI] [PubMed] [Google Scholar]
- 54. Lebbink R. J., de Ruiter T., Adelmeijer J., Brenkman A. B., van Helvoort J. M., Koch M., Farndale R. W., Lisman T., Sonnenberg A., Lenting P. J., and Meyaard L. (2006) Collagens are functional, high affinity ligands for the inhibitory immune receptor LAIR-1. J. Exp. Med. 203, 1419–1425 10.1084/jem.20052554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lebbink R. J., van den Berg M. C., de Ruiter T., Raynal N., van Roon J. A., Lenting P. J., Jin B., and Meyaard L. (2008) The soluble leukocyte-associated Ig-like receptor (LAIR)-2 antagonizes the collagen/LAIR-1 inhibitory immune interaction. J. Immunol. 180, 1662–1669 10.4049/jimmunol.180.3.1662 [DOI] [PubMed] [Google Scholar]
- 56. Leo J. C., Elovaara H., Bihan D., Pugh N., Kilpinen S. K., Raynal N., Skurnik M., Farndale R. W., and Goldman A. (2010) First analysis of a bacterial collagen-binding protein with collagen Toolkits: promiscuous binding of YadA to collagens may explain how YadA interferes with host processes. Infect. Immun. 78, 3226–3236 10.1128/IAI.01057-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. San Antonio J. D., Lander A. D., Karnovsky M. J., and Slayter H. S. (1994) Mapping the heparin-binding sites on type I collagen monomers and fibrils. J. Cell Biol. 125, 1179–1188 10.1083/jcb.125.5.1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sweeney S. M., Guy C. A., Fields G. B., and San Antonio J. D. (1998) Defining the domains of type I collagen involved in heparin-binding and endothelial tube formation. Proc. Natl. Acad. Sci. U.S.A. 95, 7275–7280 10.1073/pnas.95.13.7275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sekiya A., Okano-Kosugi H., Yamazaki C. M., and Koide T. (2011) Pigment epithelium-derived factor (PEDF) shares binding sites in collagen with heparin/heparan sulfate proteoglycans. J. Biol. Chem. 286, 26364–26374 10.1074/jbc.M111.252684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Meyer C., Notari L., and Becerra S. P. (2002) Mapping the type I collagen-binding site on pigment epithelium-derived factor. Implications for its antiangiogenic activity. J. Biol. Chem. 277, 45400–45407 10.1074/jbc.M208339200 [DOI] [PubMed] [Google Scholar]
- 61. Giudici C., Raynal N., Wiedemann H., Cabral W. A., Marini J. C., Timpl R., Bächinger H. P., Farndale R. W., Sasaki T., and Tenni R. (2008) Mapping of SPARC/BM-40/osteonectin-binding sites on fibrillar collagens. J. Biol. Chem. 283, 19551–19560 10.1074/jbc.M710001200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Orgel J. P., Eid A., Antipova O., Bella J., and Scott J. E. (2009) Decorin core protein (decoron) shape complements collagen fibril surface structure and mediates its binding. PLoS One 4, e7028 10.1371/journal.pone.0007028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Miller E. J., Finch J. E. Jr, Chung E., Butler W. T., and Robertson P. B. (1976) Specific cleavage of the native type III collagen molecule with trypsin. Similarity of the cleavage products to collagenase-produced fragments and primary structure at the cleavage site. Arch. Biochem. Biophys. 173, 631–637 10.1016/0003-9861(76)90300-3 [DOI] [PubMed] [Google Scholar]
- 64. Sweeney S. M., Orgel J. P., Fertala A., McAuliffe J. D., Turner K. R., Di Lullo G. A., Chen S., Antipova O., Perumal S., Ala-Kokko L., Forlino A., Cabral W. A., Barnes A. M., Marini J. C., and San Antonio J. D. (2008) Candidate cell and matrix interaction domains on the collagen fibril, the predominant protein of vertebrates. J. Biol. Chem. 283, 21187–21197 10.1074/jbc.M709319200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Bächinger H. P., Bruckner P., Timpl R., Prockop D. J., and Engel J. (1980) Folding mechanism of the triple helix in type-III collagen and type-III pN-collagen. Role of disulfide bridges and peptide bond isomerization. Eur. J. Biochem. 106, 619–632 10.1111/j.1432-1033.1980.tb04610.x [DOI] [PubMed] [Google Scholar]
- 66. Buevich A. V., Silva T., Brodsky B., and Baum J. (2004) Transformation of the mechanism of triple-helix peptide folding in the absence of a C-terminal nucleation domain and its implications for mutations in collagen disorders. J. Biol. Chem. 279, 46890–46895 10.1074/jbc.M407061200 [DOI] [PubMed] [Google Scholar]
- 67. Humtsoe J. O., Kim J. K., Xu Y., Keene D. R., Höök M., Lukomski S., and Wary K. K. (2005) A streptococcal collagen-like protein interacts with the α2β1 integrin and induces intracellular signaling. J. Biol. Chem. 280, 13848–13857 10.1074/jbc.M410605200 [DOI] [PubMed] [Google Scholar]
- 68. Slatter D. A., Bihan D. G., Jarvis G. E., Stone R., Pugh N., Giddu S., and Farndale R. W. (2012) The properties conferred upon triple-helical collagen-mimetic peptides by the presence of cysteine residues. Peptides 36, 86–93 10.1016/j.peptides.2012.04.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Semon J. A., Nagy L. H., Llamas C. B., Tucker H. A., Lee R. H., and Prockop D. J. (2010) Integrin expression and integrin-mediated adhesion in vitro of human multipotent stromal cells (MSCs) to endothelial cells from various blood vessels. Cell Tissue Res. 341, 147–158 10.1007/s00441-010-0994-4 [DOI] [PubMed] [Google Scholar]
- 70. Popov C., Radic T., Haasters F., Prall W. C., Aszodi A., Gullberg D., Schieker M., and Docheva D. (2011) Integrins α2β1 and α11β1 regulate the survival of mesenchymal stem cells on collagen I. Cell Death Dis. 2, e186 10.1038/cddis.2011.71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Persikov A. V., Ramshaw J. A., and Brodsky B. (2000) Collagen model peptides: sequence dependence of triple-helix stability. Biopolymers 55, 436–450 [DOI] [PubMed] [Google Scholar]
- 72. Morton L. F., Peachey A. R., Zijenah L. S., Goodall A. H., Humphries M. J., and Barnes M. J. (1994) Conformation-dependent platelet adhesion to collagen involving integrin α2β1-mediated and other mechanisms: multiple α2β1-recognition sites in collagen type I. Biochem. J. 299, 791–797 10.1042/bj2990791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Baxter N. J., and Williamson M. P. (1997) Temperature dependence of 1H chemical shifts in proteins. J. Biomol. NMR 9, 359–369 10.1023/A:1018334207887 [DOI] [PubMed] [Google Scholar]
- 74. Gopalakrishnan R., Azhagiya Singam E. R., Vijaya Sundar J., and Subramanian V. (2015) Interaction of collagen like peptides with gold nanosurfaces: a molecular dynamics investigation. Phys. Chem. Chem. Phys. 17, 5172–5186 10.1039/C4CP04969A [DOI] [PubMed] [Google Scholar]
- 75. Emsley J., Knight C. G., Farndale R. W., Barnes M. J., and Liddington R. C. (2000) Structural basis of collagen recognition by integrin α2β1. Cell 101, 47–56 10.1016/S0092-8674(00)80622-4 [DOI] [PubMed] [Google Scholar]
- 76. Uitto J., and Lichtenstein J. R. (1976) Defects in the biochemistry of collagen in diseases of connective tissue. J. Invest. Dermatol. 66, 59–79 10.1111/1523-1747.ep12481404 [DOI] [PubMed] [Google Scholar]
- 77. Arseni L., Lombardi A., and Orioli D. (2018) From structure to phenotype: impact of collagen alterations on human health. Int. J. Mol. Sci. 19, E1407 10.3390/ijms19051407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Brodsky B., and Baum J. (2008) Structural biology: modelling collagen diseases. Nature 453, 998–999 10.1038/453998a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Gautieri A., Vesentini S., Redaelli A., and Buehler M. J. (2009) Single molecule effects of osteogenesis imperfecta mutations in tropocollagen protein domains. Protein Sci. 18, 161–168 10.1002/pro.21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Bodian D. L., and Klein T. E. (2009) COLdb, a database linking genetic data to molecular function in fibrillar collagens. Hum. Mutat. 30, 946–951 10.1002/humu.20978 [DOI] [PubMed] [Google Scholar]
- 81. Rauch F., Lalic L., Roughley P., and Glorieux F. H. (2010) Genotype-phenotype correlations in nonlethal osteogenesis imperfecta caused by mutations in the helical domain of collagen type I. Eur. J. Hum. Genet. 18, 642–647 10.1038/ejhg.2009.242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Wallace J. M., Orr B. G., Marini J. C., and Holl M. M. (2011) Nanoscale morphology of type I collagen is altered in the Brtl mouse model of osteogenesis imperfecta. J. Struct. Biol. 173, 146–152 10.1016/j.jsb.2010.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Andriotis O. G., Chang S. W., Vanleene M., Howarth P. H., Davies D. E., Shefelbine S. J., Buehler M. J., and Thurner P. J. (2015) Structure-mechanics relationships of collagen fibrils in the osteogenesis imperfecta mouse model. J. R. Soc. Interface 12, 20150701 10.1098/rsif.2015.0701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Sun X., Chai Y., Wang Q., Liu H., Wang S., and Xiao J. (2015) A natural interruption displays higher global stability and local conformational flexibility than a similar Gly mutation sequence in collagen mimic peptides. Biochemistry 54, 6106–6113 10.1021/acs.biochem.5b00747 [DOI] [PubMed] [Google Scholar]
- 85. Palmer A. G. 3rd, Cavanagh J., Wright P. E., and Rance M. (1991) Sensitivity improvement in proton-detected two-dimensional heteronuclear correlation NMR spectroscopy. J. Magn. Reson. 93, 151–170 10.1016/0022-2364(91)90036-S [DOI] [Google Scholar]
- 86. Kay L., Keifer P., and Saarinen T. (1992) Pure absorption gradient enhanced heteronuclear single quantum correlation spectroscopy with improved sensitivity. J. Am. Chem. Soc. 114, 10663–10665 10.1021/ja00052a088 [DOI] [Google Scholar]
- 87. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., and Bax A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 88. Goddard T. D., and Kneller D. G. (2008) Sparky 3.115, University of California, San Francisco [Google Scholar]
- 89. Rainey J. K., and Goh M. C. (2004) An interactive triple-helical collagen builder. Bioinformatics 20, 2458–2459 10.1093/bioinformatics/bth247 [DOI] [PubMed] [Google Scholar]
- 90. Jorgensen W. L., Chandrasekhar J., Madura J. D., Impey R. W., and Klein M. L. (1983) Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 10.1063/1.445869 [DOI] [Google Scholar]
- 91. Maier J. A., Martinez C., Kasavajhala K., Wickstrom L., Hauser K. E., and Simmerling C. (2015) ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 11, 3696–3713 10.1021/acs.jctc.5b00255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Case D. A., Ben-Shalom I. Y., Brozell S. R., Cerutti D. S., Cheatham T. E. 3rd, Cruzeiro V. W. D., Darden T. A., Duke R. E., Ghoreishi D., Gilson M. K., Gohlke H., Goetz A. W., Greene D., Harris R., Homeyer N., et al. (2018) AMBER 2018, University of California, San Francisco [Google Scholar]
- 93. Darden T., York D., and Pedersen L. (1993) Particle mesh Ewald: an N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092 10.1063/1.464397 [DOI] [Google Scholar]
- 94. Essmann U., Perera L., Berkowitz M. L., Darden T., Lee H., and Pedersen L. G. (1995) A smooth particle mesh Ewald method. J. Chem. Phys. 103, 8577–8593 10.1063/1.470117 [DOI] [Google Scholar]
- 95. Ryckaert J.-P., Ciccotti G., and Berendsen H. J. C. (1977) Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 23, 327–341 10.1016/0021-9991(77)90098-5 [DOI] [Google Scholar]
- 96. Roe D. R., and Cheatham T. E. 3rd. (2013) PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 9, 3084–3095 10.1021/ct400341p [DOI] [PubMed] [Google Scholar]
- 97. Dalgleish R. (1997) The human type I collagen mutation database. Nucleic Acids Res. 25, 181–187 10.1093/nar/25.1.181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Dalgleish R. (1998) The human collagen mutation database. Nucleic Acids Res. 26, 253–255 10.1093/nar/26.1.253 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.