

Abstract
Recoding of UGA codons as selenocysteine (Sec) codons in selenoproteins depends on a selenocysteine insertion sequence (SECIS) in the 3′-UTR of mRNAs of eukaryotic selenoproteins. SECIS-binding protein 2 (SECISBP2) increases the efficiency of this process. Pathogenic mutations in SECISBP2 reduce selenoprotein expression and lead to phenotypes associated with the reduction of deiodinase activities and selenoprotein N expression in humans. Two functions have been ascribed to SECISBP2: binding of SECIS elements in selenoprotein mRNAs and facilitation of co-translational Sec insertion. To separately probe both functions, we established here two mouse models carrying two pathogenic missense mutations in Secisbp2 previously identified in patients. We found that the C696R substitution in the RNA-binding domain abrogates SECIS binding and does not support selenoprotein translation above the level of a complete Secisbp2 null mutation. The R543Q missense substitution located in the selenocysteine insertion domain resulted in residual activity and caused reduced selenoprotein translation, as demonstrated by ribosomal profiling to determine the impact on UGA recoding in individual selenoproteins. We found, however, that the R543Q variant is thermally unstable in vitro and completely degraded in the mouse liver in vivo, while being partially functional in the brain. The moderate impairment of selenoprotein expression in neurons led to astrogliosis and transcriptional induction of genes associated with immune responses. We conclude that differential SECISBP2 protein stability in individual cell types may dictate clinical phenotypes to a much greater extent than molecular interactions involving a mutated amino acid in SECISBP2.
Keywords: selenium, selenocysteine insertion sequence (SECIS), selenoprotein, neurobiology, neuroinflammation, translation, thyroid hormone, Sbp2, selenocysteine, thermal instability, brain, liver, deiodinase, SECIS-binding protein 2 (SECISBP2)
Introduction
Mutations in the SECISBP2 gene have been associated with an atypical resistance to thyroid hormone, which is caused by reduced activity of deiodinase enzymes (1). Deiodinases belong to the class of selenoproteins, proteins carrying the 21st proteinogenic amino acid, selenocysteine (Sec)3 (2). Because Sec is encoded by a UGA codon, recoding as a sense codon depends on the presence of a selenocysteine insertion sequence (SECIS) in the 3′-UTR of the mRNA, which is recognized by SECIS-binding protein 2 (SECISBP2) (3, 4). Selenoprotein expression is generally believed to be highly dependent on SECISBP2, and gene targeting in mice revealed a significantly reduced level of selenoprotein translation in vivo in the absence of functional Secisbp2 (5, 6). Some of the sensitivity toward the lack of Secisbp2 may be due to mRNA degradation following slow decoding or termination at Sec/UGA codons. A number of patients carrying mutations in SECISBP2 have been described and reveal a conspicuous phenotypic heterogeneity among their clinical presentations (7). Several domains in SECISBP2 have been identified that may mediate interactions with the SECIS element or interact with the ribosome and/or elongation factor eEFSEC. Thus, it might be possible that mutations that affect specific functions of SECISBP2 might cause the phenotypic heterogeneity in patients. For example, the first missense mutation identified in homozygosity, R540Q, leads to a rather mild clinical phenotype (1). The changed amino acid lies in the so-called selenocysteine insertion domain of SECISBP2 (8, 9). The mutation has been shown to differentially affect translation of selenoprotein mRNAs (10). Another mutation, C691R, is located in the RNA-binding domain (11). The patient presented with a more severe clinical picture but was compound heterozygous for a presumably hypomorphic splice site mutation. There are several nonsense or frameshift mutations in the N-terminal part of the protein, and patients carrying SECISBP2 mutations R120X/R770X, R128X/R128X, and K276Kfs*2/K276Kfs*2 show phenotypes of various severity (12–15). It is generally believed that reinitiation 3′ of the premature termination codons (e.g. at Met-300) rescues some SECISBP2 function in these patients (14, 15).
It was suggested that the affinity of SECISBP2 for individual SECIS elements establishes the observed hierarchy among selenoproteins (16). A systematic study comparing SECISBP2:SECIS binding and SECIS-dependent Sec incorporation in a luciferase reporter system reported that there is no such correlation (17). Because the R540Q mutation differentially affected selenoprotein expression and SECIS binding in vitro (10), we wanted to more systematically assess how this mutation impinges on the translation of different selenoprotein mRNAs in vivo. Using ribosome profiling (18), we aimed to specifically probe the efficiency of UGA/Sec recoding in mice in the same transcript-specific way as we have assessed it previously in Secisbp2-knockout mice (19).
To this end, we have generated mouse models for the R540Q mutation (Secisbp2R543Q) in the selenocysteine incorporation domain and for the C691R mutation (Secisbp2C696R) in the RNA-binding L7Ae domain. We assumed that any residual selenoprotein expression in Secisbp2C696R/− that exceeds the levels in the Secisbp2 knockout may hint toward a potential SECIS binding–independent function of Secisbp2.
Because both homozygous mutations turned out to be embryonic lethal in mice, we combined the missense mutant alleles with a conditional allele and chose to focus on hepatocytes and neurons, the two cell types in which we analyzed selenoprotein expression in the absence of Secisbp2 before (5, 6). Whereas the C696R mutation apparently disrupts RNA-binding activity of Secisbp2 and acts as a complete null in vitro and in vivo, R543Q exhibits thermal instability. Interestingly, while acting as a functional null in liver, it rescues the severe neurological phenotype of neuron-specific Secisbp2 knockout mice (6). The differential activity of the Secisbp2R543Q protein in liver and neurons suggests that the potential function of amino acid 543 within the selenocysteine incorporation domain is being obscured by thermal instability of the protein in vivo. We assessed the effect of the Secisbp2R543Q mutation on selenoprotein translation in detail by ribosome profiling in the cerebral cortex. Despite the general down-regulation of selenoprotein translation, no behavioral phenotype was detected. However, we found a gene expression pattern indicative of a mild inflammatory response in the mutant mouse cortex that was accompanied by obvious astrogliosis (a cellular stress response of astrocytes involving gene expression and shape changes that is readily detected by the up-regulation of the intermediate filament glial fibrillary acidic protein (GFAP)). Thus, different phenotypes of patients carrying different SECISBP2 alleles may not necessarily reflect the functions of individual amino acids in SECISBP2, but may be caused by differences in stability in different cellular contexts.
Results
Generation of mice carrying pathogenic missense mutations in the Secisbp2 gene
Point mutations C696R and R543Q (C691R and R540Q in human SECISBP2, respectively) were introduced into the Secisbp2 locus using recombineering in Escherichia coli and double homologous recombination in embryonic stem cells as detailed under “Experimental procedures.” For the R543Q allele, the mutation was combined with a silent G→A mutation in Leu-540 to produce a diagnostic DraI restriction site (Fig. 1A). The C696R mutation was introduced in exon 14 combined with a silent G>T mutation in Leu-694, creating a diagnostic AflII restriction site (Fig. 1B). Correct homologous recombination of the modified alleles was ensured by Sanger sequencing over the targeted sequence (Fig. 1, A and B) and Southern blotting (Fig. 1, C and D). After germline transmission, the FLP recognition target (FRT)-flanked neomycin resistance cassettes were removed in vivo using FLPe expression in the germline, and deletion of the selection cassette was verified by PCR.
Figure 1.

Generation of Secisbp2 alleles in mice carrying pathogenic mutations found in patients. A, strategy to build the targeting vector containing a R543Q mutation in exon 12 and a FRT-flanked neo cassette in intron 12 for double homologous recombination in embryonic stem cells. After germline transmission, the neo cassette is removed by germline FLPe expression. Sanger sequencing confirmed the heterozygous base substitutions in the Arg-543 codon and in the Leu-540 codon, creating a diagnostic DraI restriction site. DTA refers to a diphtheria toxin A chain used for negative selection against off-site integration. B, strategy to build the targeting vector containing a C696R mutation in exon 14 and a FRT-flanked neo cassette in intron 14 for double homologous recombination in embryonic stem cells. After germline transmission, the neo cassette is removed by germline FLPe expression. Sanger sequencing confirmed the heterozygous base substitutions in the Cys-696 codon and in the Leu-694 codon creating a diagnostic AflII restriction site. C and D, verification of correct gene targeting by Southern blotting using hybridization probes located outside of the targeting vector sequence. C, Secisbp2 R543Q allele. The SpeI digestion was combined with the 3′ probe and yielded the expected fragments of 12,148 bp (WT) and 14,048 bp (RQ). The EcoRI digestion was combined with the 5′ probe and yielded fragments of 9129 bp (WT) and 4209 bp (RQ). D, Secisbp2 C696R allele. The BglII digestion was combined with the 5′ probe and yielded the expected fragments of 8328 bp (WT) and 10,228 bp (CR). The HindIII digestion was combined with the 3′ probe and yielded the expected fragments of 10,177 bp (WT) and 7340 bp (CR). DNA marker sizes in bp are indicated.
Heterozygous matings of Secisbp2R543Q/+ or Secisbp2C696R/+ mice did not result in viable homozygous offspring (Table 1), but were embryonic lethal around E7, the same time as Secisbp2−/−. We thus chose to combine the missense alleles with the conditional Secisbp2fl allele described before (5, 6) and generated mice in which the missense Secisbp2 allele produced the only translated Secisbp2 mRNA in a cell type–specific manner.
Table 1.
Number of mice born from heterozygous Secisbp2 mutant cross-breeding
| Genotype | Number |
|---|---|
| Heterozygous Secisbp2C696R/+ cross-breeding | |
| Secisbp2+/+ | 30 |
| Secisbp2C696R/+ | 80 |
| Secisbp2C696R/C696R | 0 |
| Heterozygous Secisbp2R543Q/+ cross-breeding | |
| Secisbp2+/+ | 43 |
| Secisbp2R543Q/+ | 66 |
| Secisbp2R543Q/R543Q | 0 |
Selenoprotein expression in liver
Liver-specific Secisbp2 mutant mice were generated for both missense alleles by breeding with albumin-Cre transgenic mice: Alb-Cre; Secisbp2R543Q/fl (called subsequently Alb-RQ) and Alb-Cre; Secisbp2C696R/fl (Alb-CR) mice are the desired mutants, and Alb-Cre; Secisbp2fl/+ and Secisbp2mut/fl mice served as controls (WT). In most analyses, the missense mutant mice were compared with Alb-Cre, Secisbp2fl/fl conditional knockout (Alb-KO) mice. Reverse transcription semiquantitative PCR analysis revealed that in the Secisbp2 missense mutants, all hepatic selenoprotein mRNAs analyzed were reduced to the same level and indistinguishable from the Alb-KO (Fig. 2A). Likewise, on the protein level, Gpx1, Gpx4, Selenot, and Sephs2 were reduced as in the Alb-KO (Fig. 2B). Txnrd1 protein abundance was not reduced in the absence of functional Secisbp2, because the Sec occupies the penultimate position close to the C terminus. Because we were surprised that both mutants behaved apparently like the Alb-KO, we performed Western blot analysis for Secisbp2 and found that both mutant Secisbp2 proteins were virtually undetectable (Fig. 2C). Thus, both missense mutant proteins are functional nulls in vivo.
Figure 2.

Selenoprotein expression in liver-specific Secisbp2 mutant mice. Control mice (WT) were compared with Alb-Cre; Secisbp2fl/fl (KO), Alb-Cre; Secisbp2C696R/fl (CR), and Alb-Cre; Secisbp2R543Q/fl (RQ) mice. A, semiquantitative RT-PCR analysis for selected selenoprotein mRNAs. ΔΔCt values are calculated in relation to 18S rRNA. Means are given ± S.D. (error bars), n = 2. B, Western blot analysis for selected selenoproteins. Two individual liver extracts are analyzed for each genotype. C, Western blot analysis for Secisbp2. The nonspecific bands demonstrate equal loading. Images were copied directly from the camera output files and displayed without modifications. The complete image files are available as Fig. S2.
In vitro activity of recombinantly expressed Secisbp2 mutants
We speculated that the similar levels of Secisbp2 protein in the liver of conditional missense and null mutants and the apparent complete loss of function in the liver of missense mutants might be caused by protein instability and degradation in vivo. We therefore recombinantly expressed mouse Secisbp2 as WT, R543Q, and C696R variants in E. coli, purified the protein by nickel-nitrilotriacetic acid affinity chromatography using a C-terminal hexahistidine tag, and included the recombinant protein in the SECIS-dependent luciferase reporter in which the codon for Cys-258 is replaced by UGA/Sec (20). To create a consistent experimental system, we cloned several mouse SECIS elements into the reporter plasmid. In a first experiment, we used 100 ng of reporter mRNA (about 13 nm) and a 160 nm concentration of each Secisbp2 protein (Fig. 3A). There was no appreciable difference in luciferase activity between WT and R543Q protein irrespective of the SECIS element, whereas C696R was completely inactive, comparable with negative controls (no Secisbp2 added or mutation in the AUGA quartet of SECIS).
Figure 3.

In vitro functional analysis of recombinantly expressed mouse Secisbp2 R543Q and C696R. Reporter constructs containing a Sec-dependent luciferase cDNA and SECIS elements cloned into the 3′ UTR as given are in vitro translated, and the amount of luciferase produced was measured by an activity assay. A, Secisbp2 R543Q is functional, whereas C696R is not functional at all. Recombinant Secisbp2 at 160 nm was incubated with constructs containing murine Gpx1, Gpx4, and Txnrd1 SECIS elements, and luciferase activity was measured. A rat Gpx4 SECIS element lacking the AUGA sequence of the essential kink-turn served as negative control. Further negative controls were no mRNA added, no Secisbp2 protein added, and a luciferase carrying a UAA codon at the position of the UGA/Sec codon. A log2 scale is used to better appreciate the small increment of luciferase activity in the controls lacking Secisbp2 or the SECIS quartet over lack of mRNA or a definite stop codon indicating a small SECIS/Secisbp2-independent UGA read-through. B, kinetics of Secisbp2R543Q:SECIS binding compared with Secisbp2WT for several murine SECIS elements. The concentration of recombinant Secisbp2 was 80 nm, and 0.1–20 nm mRNA was used. The apparent KD of Secisbp2R543Q was significantly reduced for Dio1 and Gpx1 SECIS only, and the maximal amount of luciferase was decreased when using the mutant as compared with Secisbp2WT. C, thermal instability of Secisbp2R543Q in vitro. Recombinant Secisbp2 protein at 160 nm concentration was incubated for 30 min on ice, at 37 °C, and at 40.5 °C and then used for in vitro translation with the luciferase/Gpx4 SECIS construct. The luciferase activity values of the respective Secisbp2WT incubated on ice was defined as 100%. The experiment was done twice in triplicates. Error bars, S.D.
We reasoned that an excess of Secisbp2 protein might have obscured moderate effects of the R543Q mutation. Further, it was difficult to assess what proportion of the recombinant protein was active in this experiment. We therefore titrated SECIS-containing mRNAs (0.1–20 nm) against a fixed concentration of Secisbp2 (80 nm) and reasoned that under our experimental conditions, the EC50 concentration could represent a measure for the SECIS:Secisbp2 interaction (Fig. 3B). The C696R mutant was not included in these experiments, as it was unable to mediate SECIS-dependent luciferase activity. Comparison of WT and R543Q mutant protein demonstrated that in the presence of the mutant protein, consistently less luciferase activity was produced from SECIS-dependent reporters carrying the SECIS elements of Gpx1, Gpx4, Txnrd1, and Dio1. The EC50 constants of the R543Q mutant were unchanged for Gpx4 but significantly higher for Txnrd1 and Dio1. The expected reduction of the Gpx1 EC50 constant was observed but did not reach statistical significance (Fig. 3C). Thus, we observed two independent changes: 1) the R543Q mutation reduces the UGA-recoding in response to a subset of SECIS elements (i.e. Txnrd1 and Dio1 SECIS), and 2) in addition, the R543Q mutation was less active even when the binding affinity was unchanged (i.e. Gpx4). This suggested that the fraction of active protein was reduced in the R543Q mutant.
This observation raised the question of whether the mutant protein has reduced thermal stability. In order to test this, we preincubated the recombinant protein for 30 min on ice, at 37 or at 40.5 °C, before the luciferase assay (which is performed at 30 °C). The R543Q mutant protein was clearly more heat-labile than WT protein during incubation at elevated temperatures (Fig. 3D). This explains why the R543Q mutant protein was virtually undetectable in the liver of mutant mice and suggests that the C696R mutant, which is also undetectable in liver, might also be thermally unstable.
Selenoprotein expression in neurons
Judging from Western blotting experiments, selenoprotein expression is reduced in CamK-Cre; Secisbp2R543Q/fl (CamK-RQ) and CamK-Cre; Secisbp2C696R/fl (CamK-CR) mutants, but not above the level in CamK-Cre; Secisbp2fl/fl (CamK-KO) cortex (Fig. 4A). In line with the above results showing the inactivity of Secisbp2C696R in vitro and in the liver, mice with CamK-KO and CamK-CR mutants show indistinguishable phenotypes (6). On P16–P18, the mean body masses were 7.27 ± 0.56 and 3.82 ± 0.60 g for the control and CamK-CR mice, respectively. Furthermore, CamK-CR mice died before weaning (P16–P18) and showed astrogliosis in the cerebral cortex at P16 similar to CamK-KO mice (not shown). As expected, selenoprotein levels were clearly reduced in the brains of these mice (Fig. 4B). Likewise, mRNAs of sensitive selenoproteins (e.g. Gpx1 and Selenow) were reduced (Fig. 4C). The increase of Gpx4 may be related to the astrocytic response, and likewise increased Selenot in nonneuronal cells may compensate for a reduction in neurons. In addition, CR mice showed a reduced number of parvalbumin-positive (Pvalb+) GABAergic interneurons in the somatosensory cortex at age P16 (Table 2).
Figure 4.

Selenoprotein expression in neuron-specific Secisbp2 mutant mice. A, Western blotting against Secisbp2. 100 μg of total cortical protein from two individual mice of each genotype. B, Western blotting against selenoproteins in the cortex of CamK-Cre; Secisbp2C696R/fl (CR) and littermate control mice. C, semiquantitative RT-PCR analysis of selected selenoprotein mRNAs. n = 2; ***, p < 0.001, Student's t test. D, Western blotting against selenoproteins in the cortex of CamK-Cre; Secisbp2R543Q/fl (RQ) and litter mate control mice. E, semiquantitative RT-PCR analysis of selected selenoprotein mRNAs. n = 3; ***, p < 0.001, Student's t test. Images were copied directly from the camera output files and displayed without modifications. The complete image files are available as Fig. S3. Error bars, S.E.
Table 2.
Parvalbumin-positive interneuron density in the primary somatosensory barrel field cortex (S1BF)
Given are means ± S.D. of Pvalb+ interneurons per mm2 in n samples. **, p < 0.01, Student's t test, one-sided.
| Control | Mutant | Age at analysis | |
|---|---|---|---|
| CamK-Cre; Secisbp2C696R mice | 107 ± 9 (3) | 75 ± 11 (3)** | P16 |
| CamK-Cre; Secisbp2R543Q mice | 197 ± 18 (4) | 215 ± 9 (4) | P35 |
Despite its thermal instability and impaired functionality, the low expression of the mutant Secisbp2 protein, CamK-RQ mice showed an apparently normal phenotype without signs of neurological dysfunction. The number of Pvalb+ interneurons was not reduced in the somatosensory cortex at age P35 (Table 2). Seizures or a movement phenotype were not observed.
Expression of all tested selenoproteins was reduced in the cerebral cortex of CamK-RQ mice, with the exception of Txnrd1, which carries the Sec residue at the penultimate position at the C terminus (Fig. 4D). A low but significant functionality of Secisbp2R543Q is suggested by the fact that Selenom and Selenow proteins were present in the cortex at higher levels than in CamK-CR mice. However, semiquantitative RT-PCR demonstrated that Selenow message is reduced in CamK-RQ mice to a similar extent as in CamK-CR mice. RNA-Seq supported this result and showed on a genome-wide level that selenoprotein mRNA amounts were generally reduced in CamK-RQ mutant brains (Figs. 4E and 5A). We reasoned that this apparent discrepancy between mRNA and protein abundance could be related to differential activity of the Secisbp2 mutants during translational redefinition of the UGA/Sec codon.
Figure 5.

Selenoprotein expression in neuron-specific Secisbp2R543Q mice assessed by ribosomal profiling and RNA-Seq in cerebral cortex. A, relative abundance of selenoprotein-related reads in Ribo-Seq (RPF) and RNA-Seq (RNA) in RQ mice compared with controls. n = 2 per genotype. *, q < 0.05, BH correction. Significant changes are highlighted in red. B, RPF coverage of selected selenoprotein mRNAs. The position of the Sec/UGA codon is indicated by a red bar. Reads are plotted in blue for controls (Ctl) and in orange for CamK-Cre; Secisbp2R543Q/fl (RQ) mice. C, URE calculated for selenoproteins with UGA/Sec far from the termination codon. URE is calculated as (3′RPFmutant/5′RPFmutant)/(3′RPFcontrol/5′RPFcontrol). URE is not a good measure if mRNA levels or initiation rates change. D, 3′ RPM (reads 3′ of UGA/Sec per million mapped reads) calculated for selenoproteins. This measure gives a measure for the actual translation of full-length selenoproteins. E, 3′ RPM/mRNA is the same measure as in D, but normalized to mRNA abundance. C–E, *, p < 0.05, Student's t test. Error bars, S.D.
UGA recoding in brain assessed by ribosomal profiling
To directly assess the functionality of mutant Secisbp2 during selenoprotein biosynthesis, we probed selenoprotein expression in the brains of CamK-RQ mice by ribosomal profiling. Compared with mRNA abundance, ribosome profiling revealed a more pronounced reduction of ribosome protected fragments (RPFs) associated with selenoproteins, indicating that selenoprotein translation was specifically affected in the mutant brains (Fig. 5A). This was particularly evident for Gpx4, where mRNA abundance did not significantly change in the mutant, but RPF levels dropped more than 2-fold. Plotting the distribution of RPFs over the ORF revealed that 5′ of the UGA/Sec codon, the coverage of Gpx4 was unaltered, whereas 3′ of the UGA/Sec, the ribosomal density (i.e. RPFs/nucleotide mRNA) was significantly decreased (Fig. 5B). Thus, read-through of the UGA/Sec codon is inefficient in Secisbp2R543Q mutants. A similar observation was made for Selenof. In the case of Selenow, ribosomal coverage appears unchanged, but this may be due to degradation of the mRNA whenever UGA/Sec recoding fails. Gpx1 shows reduction of RPFs along the entire length of the ORF, but the reduced read-through of the UGA/Sec codon was nevertheless obvious.
We have previously defined UGA redefinition efficiency (URE) as ribosomal density 3′ of the UGA/Sec divided by ribosomal density 5′ of the UGA. Fig. 5C shows that URE is reduced for several selenoproteins that carry the UGA/Sec codon far away from the termination codon. The URE, however, does not account for a reduction of the absolute number of RPFs covering a certain message. To reflect also absolute changes of UGA/Sec read-through, the number of RPFs 3′ of the UGA/Sec per million mapped reads (3′ RPM) was calculated (Fig. 5D). Using this measure, the impairment of selenoprotein translation in Secisbp2R543Q mice was evident. Interestingly, Selenot was little affected, probably because this selenoprotein is significantly expressed in astrocytes that are not targeted in our CamK-Cre mouse model. Since 3′ RPM is a measure that reflects both translation and mRNA abundance, we also normalized 3′ RPM by mRNA abundance (Fig. 5E).
Taken together, ribosomal profiling reveals that Secisbp2R543Q does not fully support faithful UGA/Sec recoding and selenoprotein biosynthesis in neurons in vivo.
A mild inflammatory response in the brain
Transcriptomic analysis confirmed the significant reduction (20–60%) of selenoprotein mRNA levels, including Selenow, Gpx1, Selenom, Gpx3, and Selenof. Apart from selenoproteins, among the genes up-regulated on the transcriptional (RNA-Seq) or translational (Ribo-Seq) level in Secisbp2R543Q mice, we found a clear signature of genes of inflammatory response pathways. Typically, the up-regulated mRNA levels correlated with a higher translation (RPFs) of the corresponding proteins (Fig. 6A). In particular, we observed an increase in the expression and/or translation of immune cell markers (Itgax, Itgam, Mpeg1, Cd48, Cd180, Cx3cr1, Ly6e, Ly86, Lag3, Trem2, Adgre1, Tyrobp, and Tlr2), chemokines (Ccl3 and Ccl4), complement (C1qa and C4b), and proteins associated with lysosomes (Lamp2, Lyz1, Lyz2, Laptm5, Hexb, Ctss, Ctsd, and Ctsz) and cystatins (Cst3 and Cst7) as well as the antiviral or antimicrobial proteins Bst2, B2m, Vcp, Chil1, and Atf3 (Fig. 6B). Among the down-regulated genes, we observed mainly transcripts of selenoprotein mRNAs (indicated in green) as well as Capn5, a protease located in synapses, and Cdx4, a transcription factor known to act both in the hematopoietic and neural lineage.
Figure 6.

Mild inflammatory response in neuron-specific Secisbp2R543Q mice. A and B, brain transcriptomic analysis reveals induction of immunity-related genes (red) on mRNA (A) and RPF levels (B). Selenoproteins are labeled in green and neuronal genes in blue. Only significant regulated genes are shown. n = 2 animals/genotype. *, q < 0.05, BH correction. C, pathway analysis shows that immunity-related pathways were induced on the transcriptional level. D, astrogliosis assessed by GFAP staining in the somatosensory cortex. E, Iba1 staining in the somatosensory cortex. Mean cell density was 375 and 378 cells/mm2 for control and CamK-RQ mice, respectively.
The changes in the gene expression observed are indicative of immune infiltration of the brain or, alternatively, induction of these genes in cells present in the brain or brain circulation. Because the astrocytic markers Gfap and Aqp4 were among the up-regulated genes, we performed immunohistochemistry against GFAP and found widespread astrogliosis in CamK-RQ mice, which was limited to lower cortical layers (Fig. 6C). We then asked whether the increase in microglial marker genes (Ctss, Itgam, Cx3cr1, and Gpr34) was related to microglial invasion of the cortex. Immunohistochemical staining for Iba1 did not show any differences in cell density, suggesting that the increased microglial marker gene expression was related to up-regulation of genes in activated microglia (Fig. 6D).
Several neuronal genes were deregulated, including the neuropeptides Penk, Tac1, Oxt, and Pdyn (Fig. 6A). Genes generally associated with inhibitory interneurons like Gad1, Gad2, GABA transporters, and GABA receptors were not among the significantly deregulated genes, whereas, interestingly, Pvalb and Gpx3 were reduced on the mRNA level, possibly indicating that cells in lower cortical layers are particularly affected (21). Furthermore, Penk and Mgp, markers of layer 6a glutamatergic neurons, were up- and down-regulated, respectively. Finally, astrogliosis was more prominent in deeper cortical layers. Taken together, these gene regulation data suggest that there is a pathophysiological process associated with the dysregulation of selenoprotein expression that starts in lower cortical layers.
Discussion
Choice and evaluation of models
Which function the N-terminal domain of Secisbp2 may have remains an open question. Because the N terminus of Secisbp2 is intrinsically disordered (22), this part of the protein is usually omitted when expressing Secisbp2 recombinantly. In vitro experiments pioneered by Paul Copeland and adapted here for murine Secisbp2 and SECIS elements show that selenoprotein translation can be supported by the so-called C-terminal part of Secisbp2, which was as effective as the full-length recombinant protein (20). In addition, pathogenic SECISBP2 mutations in humans that involve premature termination codons sometimes lead to a relatively mild clinical phenotype that has been explained by the possibility of initiation of translation at methionine codons 139, 233, and 300 (14, 15).
We have therefore designed the C696R mutation in the mouse to specifically disrupt the L7Ae RNA-binding domain (23). By homology with the crystal structure of the spliceosomal 15.5-kDa protein (24), the Cys is predicted to reside in the central β-sheet of the L7Ae domain, and insertion of a bulky and charged amino acid, as in the pathogenic C691R mutation (11), is expected to fundamentally disrupt protein structure. In fact, no SECIS binding of recombinant Secisbp2C696R was detected in our in vitro experiments, which is compatible with a previous report using rat Secisbp2 (25). The phenotype of CamK-Cre; Secisbp2C696R/fl mice was indistinguishable from the phenotype of CamK-Cre; Secisbp2fl/fl mice described before (6). Similar to PGK-Cre; Secisbp2fl/fl mice, Secisbp2C696R/C696R mice died during embryogenesis around E7. Finally, Alb-CR mice did not support any selenoprotein expression in liver beyond the levels observed before in the Alb-KO mice (5). We thus conclude that we did not detect in vivo any evidence for a function of Secisbp2 (and its unaltered N terminus), which was independent of the RNA-binding capacity of the L7Ae domain.
The mutant Secisbp2R543Q was designed as a model for the human SECISBP2 mutation R540Q (1), which leads to a relatively mild phenotype in homozygous patients. Interestingly, Secisbp2R543Q/R543Q embryos died in utero like the knockout and homozygous C696R embryos, suggesting that the mutant protein is not fully functional during mouse development. This species difference is reminiscent of the observation that Gpx4-knockout embryos die in utero, whereas a patient with a homozygous nonsense mutation in GPX4 lived 4 months after birth (26). The R543Q mutation is apparently partially functional in neurons, because clear-cut phenotypes of neuronal selenoprotein deficiency (i.e. loss of Pvalb-positive interneurons and movement disorders) are not observed (6, 27–31). Yet, Ribo-Seq and RNA-Seq revealed a decreased expression of Pvalb and Satb1, a transcription factor specific for Pvalb+ interneurons. Such a partial deficiency of Secisbp2R543Q is also evident from a reduction by half of RPFs and an altered ribosomal coverage of Secisbp2R543Q (Fig. 7). Whether the altered ribosomal coverage is caused by the R543Q mutation or, inadvertently, by the silent third position base change in the codon for Leu-540 cannot be resolved at present. However, it is known that missense mutations, and even silent base changes, can affect the kinetics and efficiency of translation and cause pathological results (32, 33).
Figure 7.
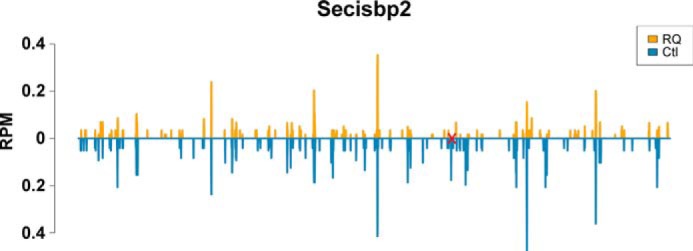
Ribosomal coverage of Secisbp2 in cortex. The site of the R543Q mutation is marked in red. Although the ribosomal coverage pattern is similar in mutant and control, it is obvious that the mutant is translated at a lower level, in particular 3′ of the site of the mutation indicated in red.
The fact that Secisbp2R543Q is functional in neurons but apparently unstable in hepatocytes suggests that the cellular environment determines the stability of a mutant protein and thus strongly affects its activity. With respect to interpreting the genotype:phenotype relationships in SECISBP2-deficient patients, this means that we cannot easily interrogate, as initially attempted in the present work, the functions of single parts of a protein using point mutations in vivo. Hence, other pathogenic missense mutations not studied here may also depend on the stability of the protein in vivo rather than revealing their immediate impact on a particular binding partner. Second, because the cellular environment determines the activity of a protein, the phenotype in different organs/cell types may be more informative about the variant protein's stability in different organs than about the specific role of the amino acid. Again, such an unexpected cell type–specific effect is reminiscent of a “silent” pathogenic mutation in cystic fibrosis transmembrane conductance regulator, which is read by a specific tRNA species that is expressed at low levels in airway epithelium but abundant in other tissues (34). Similarly, recently, a polymorphism found in the human DIO2 gene was modeled in a mouse model. Whereas kinetic studies had previously shown only moderate changes in catalytic activity, the mouse model clearly showed a brain-specific ER stress provoked by the missense mutation (35).
In vitro studies provide valuable additional information in such situations. Our Secisbp2:SECIS interaction assays suggest a decreased affinity of R543Q for several mouse SECIS elements. The concentration of SECIS associated with half-maximal luciferase activity may be interpreted as an apparent KD value if no other component in the system is limiting. We found an apparent EC50 in the range of 3–4 nm for the WT protein and lower EC50 of up to 8 nm (for Dio1 SECIS) for the R543Q mutant protein. These values are close to the rat Secisbp2:rat Gpx4 SECIS KD of 17 nm determined in the Driscoll laboratory by rEMSA (10) and the nanomolar KD for human SelX (MSRB1) and GPX3 SECIS (17). Our assay detected differences in SECIS affinity but did not show a loss of SECIS binding in the case of Gpx1 and Dio1 as was previously reported (7). The differences between the ability of mouse and rat Secisbp2 R531Q mutants to interact with Dio1 and Gpx1 SECIS may represent a species difference, be related to different methods used for the analysis, or reflect the mutant protein's instability.
Selenoprotein expression
The relative stability of the Secisbp2R543Q mutant protein in neurons allowed us to assess the impact of the mutation on UGA/Sec recoding by ribosomal profiling (Ribo-Seq). Neurons express a large number of selenoproteins and thus are ideally suited to address this question (36). The Secisbp2R543Q protein retains sufficient function to avoid an acute neurodegeneration. We observed that 196 and 107 genes were significantly deregulated on the RNA or RPF level, respectively. Among the deregulated genes was Pvalb, a marker of a subset of GABAergic interneurons that is known to be exquisitely sensitive to impaired selenoprotein expression. In general, selenoprotein expression was reduced in the CamK-RQ mutant mice, supporting a role for Secisbp2 in UGA/Sec recoding. More specific insights regarding the UGA/Sec recoding event can be gained from Ribo-Seq. In the case of Gpx4, a selenoprotein mRNA stable even in the absence of available selenium or Secisbp2, the impairment of UGA/Sec recoding is evident, irrespective of the mode of analysis; the coverage plot shows that the pattern of RPFs 5′ to the UGA/Sec codon is similar in CamK-RQ and control cortex, whereas 3′ there are far fewer RPFs in the CamK-RQ mutant. Similar patterns can also be seen in other selenoproteins, but if their mRNAs are unstable, the messages are degraded upon failure to translate the UGA/Sec codon, and thus no signals are recorded. Hence, the URE values obtained for messages like Gpx1, Selenow, Selenom, and Selenoh appear less affected. Whereas the interpretation that a mutation in the selenocysteine insertion domain of Secisbp2 is compatible with previous reports (8, 9), our data demonstrate that Secisbp2R543Q levels are reduced, as assessed by Ribo-Seq and Western blotting. Thus, several mechanisms (i.e. protein abundance, stability, and SECIS affinity) may work together and impair selenoprotein translation.
Inflammatory response
The induction of astrogliosis has been reported in several mouse models of selenoprotein deficiency, also if the selenoprotein deficiency was limited to neurons (28), which is supported by our observations here. Thus, a astrogliosis represents a general response to neuronal selenoprotein deficiency. Interestingly, a similar limitation of astrogliosis to deeper cortical layers was observed in the tRNA[Ser]Sec hypomorph mouse model (29). The primary target cells of selenoprotein deficiency might be Pvalb-positive GABAergic interneurons (28), and our transcriptomic analysis shows a down-regulation of Pvalb and Satb1. In the CamK-RQ model, the dysfunction is not sufficiently severe for a loss of these cells, in contrast to the CamK-CR or CamK-KO models. We also observed a reduction of the expression of several nuclear encoded genes for components of the mitochondrial electron transport chain. Why these genes are down-regulated is unclear, but Gpx4 and Txnrd2 are known to be present in mitochondria, and their reduced activity may lead to mitochondrial damage. We did not observe an increase in Iba1-positive cells, indicating that no invasion of microglial Iba1-positive cells occurred in the CamK-RQ mice. The increased expression of a wide range of immunity-related genes indicates that a mild inflammatory response occurs in CamK-RQ mice. The set of up-regulated immunity-related genes in our experiment is very similar to the genes induced in Npc1 mutant mice (37). Deficiency of NPC1 causes Niemann–Pick disease type C, a lysosomal storage disorder, in which cholesterol accumulates in lysosomes that are unable to export cholesterol. In humans and mice, this disorder leads to neurodegeneration. In our mice, the induction of inflammatory genes is lower than in the Npc1 model, a finding that is compatible with a lack of obvious neurodegeneration in CamK-Cre-RQ mice. Neurodegeneration may occur during aging of our mice but was not the focus of the present study.
Experimental procedures
Construction of targeting vectors by homologous recombination in bacteria
Chloramphenicol-resistant E. coli DH10B bacteria clones containing mouse Secisbp2 gene into bacterial artificial chromosome (BAC) vectors were purchased from Source Bioscience. pDTA contains an ampicillin resistance gene and a diphtheria toxin A fragment (DTA) for positive and negative selection in bacteria and ES cells, respectively. pFRT Dual Neo vector includes a neomycin phosphotransferase gene for ES cell selection, flanked by two FRT sites that allow the cassette removal. 5′ (exon 10) and 3′ (exon 16) homologous arms were amplified by PCR using a BAC vector as template and were subcloned into pGEMT-Easy vector (Promega). See Table S1 for primer details. Restriction sites were added by PCR (XhoI and HindIII for exon 10 and HindIII and XbaI for exon 16). Both homologous arms were subsequently cloned stepwise into pDTA vector.
Homologous arms subcloned first into pGEMT-Easy and posteriorly into pFRT Dual Neo vector were as follows: exon 12 (5′ arm where the R543Q mutation is found) and part of intron 12 (3′ arm) and exon 14 (5′ arm where the C696R mutation is found) and part of intron 14 (3′ arm) of the Secisbp2 gene. These arms were PCR-amplified using BAC vector as template. The following restriction sites were added by PCR to facilitate cloning: XhoI and HindIII for exon 12 and exon 14; BamHI and NotI for intron 12 and intron 14, respectively. After subcloning in pGEMT-Easy, the corresponding mutations, R543Q in exon 12 and C969R in exon 14, were introduced, respectively, by site-directed mutagenesis following the manufacturer's instructions (QuikChange II site-directed mutagenesis kit, Agilent Technologies). To facilitate the posterior screening, DraI or AflII restriction sites were also created upstream of the corresponding Secisbp2 mutation (R543Q or C696R) by site-directed mutagenesis. In both cases, nucleotide sequence was modified creating a silent mutation at Leu-540 and Leu-694, respectively. Finally, these arms were cloned stepwise into pFRT Dual Neo vector to create pFRT Dual Neo_R543Q and pFRT Dual Neo_C696R vectors, respectively.
A defective bacteriophage λ (mini λ Tet) provides a temperature-inducible system, which expresses transiently the phage recombination genes and contains a tetracycline resistance gene (38). After induction at 42 °C of bacteria containing mini λ Tet plasmid, excised plasmid particles were purified by the PureYieldTM Plasmid Midiprep System kit (Promega). Electrocompetent DH10B bacteria containing Secisbp2 BAC were electroporated with mini λ plasmid. Selected tetracycline- and chloramphenicol-resistant bacteria were induced at 42 °C and made electrocompetent. These bacteria were electroporated with HindIII-linearized pDTA vector. A first homologous recombination event rescued the Secisbp2 gene sequence between exon 10 and 16 into the BAC vector, which was introduced in pDTA vector. See Fig. S1. Positive colonies were confirmed by restriction analysis and exon PCR. Electrocompetent DH10B bacteria containing pDTA_e10-e16 and mini λ vectors were induced and electroporated with XhoI-NotI linearized pFRT Dual Neo_C696R or pFRT Dual Neo_R540Q, respectively. A second homologous recombination step between homologous sequences (exon 12 and intron 12 for R543Q and exon 14 and intron 14 for C696R) introduced the corresponding point mutation and the neo-cassette into pDTA_e10-e16 vector. After confirmation of correct vector sequence by restriction analysis and sequencing, these targeting vectors (pDTA_e10-e16_FRT Dual Neo_R543Q and pDTA_e10-e16_FRT Dual Neo_C696R) were linearized by NotI and used to create the corresponding mouse models.
Generation of mouse models
Mouse studies were approved by the authorities in Berlin and Nordrhein-Westfalen: approval numbers G 0468/09 and T0458/09 and approval number 84-02.04.2012.A146, respectively. 30 μg of linearized targeting vectors, pDTA_e10-e16_FRT Dual Neo_R543Q and pDTA_e10-e16_FRT Dual Neo_C696R, were used for homologous recombination in 126 Ola Hsd ES cells and HM1, respectively. After electroporation, 50 neomycin-resistant ES clones were grown and posteriorly analyzed for correct insertion by Southern blot. Two ESC positive clones, which showed an acceptable karyotype, were microinjected into blastocysts from the C57Bl/6 mouse strain and transferred to a foster mother. Male chimeras were back-crossed with C57Bl/6 females for germline transmission, which was confirmed by PCR. Heterozygous mice for the transgene were backcrossed with the FLP-deleter mouse to remove the selection cassette. Homozygous mice for both transgenes were never born from transgene heterozygous mouse matings (Table 1). To analyze these Secisbp2 mutant proteins specifically in hepatocytes or neurons, heterozygous mutant (Secisbp2R543Q/+ or Secisbp2C696R/+) mice were bred with cell-specific Secisbp2fl/+ (Alb-Cre; Secisbp2fl/+ or CamKII-Cre; Secisbp2fl/+) mice (5, 6). Thus, Alb-Cre; Secisbp2mut/fl or CamK-Cre; Secisbp2mut/fl mice were considered as mutant mice, and their Secisbp2mut/fl and Alb-Cre; Secisbp2fl/+ or CamK-Cre; Secisbp2fl/+ littermates as control animals for the experiments. Animals were killed for experiments at different ages: Alb-Cre; Secisbp2mut/fl mice were killed 7 weeks after birth; CamK-Cre; Secisbp2C696R/fl mice were killed after 16 days, and CamK-Cre; Secisbp2R543Q/fl mice were killed 5 weeks after birth, respectively.
Southern blot
Ten μg of genomic DNA was extracted from different ESC clones using phenol/chloroform/isoamyl alcohol (Sigma). ESC clones were subjected to restriction enzyme digestion and then to electrophoresis in 0.8% agarose gel. Depurination followed by denaturation and neutralization steps were performed before capillary transfer overnight using a positively charged nylon membrane (Hybond-N+, GE Healthcare). Membrane was UV-cross-linked and prehybridized with Church buffer (0.25 m sodium phosphate buffer, pH 7.2, 1 mm EDTA, 7% SDS) at 65 °C for 1 h. Probes were labeled with 50 μCi of [α-32P]dCTP (PerkinElmer Life Sciences) using the Prime-It RmT random primer labeling kit (Agilent Technologies). Illustra microspin G-50 columns (GE Healthcare) were used to remove unincorporated labeled nucleotides. Labeled probes were combined with 10 μg of unlabeled genomic DNA and Church buffer, denatured at 99 °C, and incubated at 65 °C for 1 h. Membrane hybridization was performed overnight at 65 °C. Washing steps (30 min at 65 °C) using buffer I (1× SSC and 1% SDS), buffer II (1× SSC and 0.1% SDS), and buffer III (0.5× SSC and 0.1% SDS) were carried out. The membrane was exposed for 2 days and visualized using a BAS 1800 II PhosphorImager (Fujifilm).
Mouse genotyping
Primers used for genotyping Secisbp2R543Q/+ and Secisbp2C696R/+ mice are displayed in Table S1. Alb-Cre; Secisbp2fl/fl or CamK-Cre; Secisbp2fl/fl were genotyped as described (5, 6).
RT-PCR
Real-time PCR was carried out as described before (5, 6) with minor modifications. RNA was extracted from mouse tissue (liver or cortex) according to the instructions for TRIzol reagent (Invitrogen). cDNA was prepared following the protocol of the iScript cDNA synthesis kit (Bio-Rad). Real-time PCR was performed on a Mastercycler epgradient S realplex (Eppendorf) using Absolute qPCR SYBR Green Fluorescein Mix (Thermo Fisher Scientific). Primers and PCR conditions were as described previously (5, 6). 18S rRNA was used as a housekeeping gene for normalization.
Western blotting
50 μg of liver or 100 μg of cortex protein in radioimmune precipitation buffer were electrophoresed in a SDS-polyacrylamide gel and transferred onto a nitrocellulose membrane (GE Healthcare). After Ponceau staining, membranes were blocked in 5% skim milk in TBST. Antibodies and dilutions used for this study were already published before (5, 6) with the exception of rabbit polyclonal anti-Sepsh2 (Rockland) used at 1:2000. Detection was performed by Fusion Solo imaging system (Vilber Lourmat Deutschland GmbH) using horseradish peroxidase–conjugated anti-rabbit or anti-mouse antibodies (Jackson Immunotech) and the enhanced horseradish peroxidase chemiluminescence substrate SuperSignalTM West Dura (Thermo Fisher Scientific).
Inmunohistochemistry
Mice were perfused with 4% paraformaldehyde in 1× PBS. After dissection, brains were post-fixed in 4% paraformaldehyde overnight and washed in 1× PBS before sectioning. 70-μm brain sections were cut using a vibratome VT 1000S (Leica). After blocking in 5% BSA, staining of free-floating sections was performed with the indicated antibodies at 4 °C overnight. Anti-Pvalb and anti-GFAP antibody dilutions were published before (5, 6). Rabbit anti-Iba1 (Wako) was used at 1:1000 dilution. Fluorescence was detected using Alexa 488 anti-mouse (Thermo Fisher Scientific) or Cy3 anti-rabbit (Jackson Immunotech) secondary antibodies. The images were captured at a Zeiss Axioplan 2 microscope operated with Axiovision software (Carl Zeiss). Pvalb+ interneurons were counted using ImageJ and quantified as cells/area (mm2).
Cloning, expression, and purification of CSecisbp2
We followed the procedures described previously (39) with minor modifications. The C terminus of the mouse Secisbp2 gene was cloned into pTrcHis2 vector (Invitrogen) using BstBI and NcoI restriction sites. The R543Q and C696R mutations were introduced into the pTrcHis2-CSecisbp2 vector using the QuikChange II site-directed mutagenesis kit (Agilent Technologies). Sanger sequencing was used to check for success. pTrcHis2-CSecisbp2 plasmids were transformed into BL21 star E. coli. Bacteria grown in lysogeny broth medium to a density of ∼1.0 A600 were induced with 1 mm isopropyl 1-thio-β-d-galactopyranoside. Bacteria pellets were collected by centrifugation, resuspended in buffer XH1 (50 mm sodium phosphate, pH 8, 1 m NaCl, 1% Tween 20, 20 mm imidazole, 1× cOmplete EDTA-free protease inhibitor (Roche Applied Science)), and lysed by sonication. After centrifugation, the supernatants were mixed with 2 ml of equilibrated nickel-nitrilotriacetic acid agarose beads (Qiagen) into gravity flow columns (Bio-Rad) and incubated for 2 h on a shaker at 4 °C. After allowing the flow-through to run out of the column, the beads were washed three times with 5 ml of buffer XH1 and once with 5 ml of buffer XH2 (same as XH1 with 50 mm imidazole). Proteins were eluted with 3 ml of elution buffer XH3 (500 mm imidazole). All fractions were collected for analysis by SDS-PAGE. Elution fractions were concentrated using an Amicon Ultra 2 centrifugal filter unit (Millipore). Buffer was exchanged to storage buffer (50 mm sodium phosphate buffer, pH 7.2, 200 mm NaCl, 10% glycerol, 1 mm DTT), and protein concentration was measured using the Pierce BCA protein assay kit (Thermo Fisher Scientific).
Cloning SECIS elements and in vitro transcription
RNA was extracted from mouse liver using TRIzol following the manufacturer's protocol. cDNA was then prepared using Superscript III Reverse Transcriptase kit (Invitrogen) following the manufacturer's protocol too. Genomic DNA was extracted from mouse liver. Gpx1, Gpx4, Txnrd1, and Dio1 SECIS elements were cloned from cDNA and Dio2 SECIS from genomic DNA by PCR introducing PacI and NotI restriction sites. Dio3 SECIS was cloned from annealing long primers that covered the whole sequence. These SECIS elements were cloned into the pcDNA3.1_Luc_C258U plasmid (gift from Paul Copeland). These plasmids and negative control plasmids (gifts from Paul Copeland) were linearized with XhoI, in vitro transcribed using mMESSAGE mMACHINE T7 ULTRA (Thermo Fisher Scientific) following the manufacturer's protocol but omitting the poly(A)-tailing step. Subsequently, the mRNA was extracted with phenol/chloroform (5:1, pH 4.5) followed by precipitation with 7.5 m ammonium acetate.
Sec incorporation assays
The incorporation assays were performed as described (20) with minor modifications. Reactions consisted of 6.5 μl of rabbit reticulocyte lysate (Promega), 100 ng of reporter mRNA, 0.02 mm amino acid mixture, 40 units of RiboLock (Thermo Fisher Scientific), and recombinant CSecisbp2 (diluted in 20 mm Tris acetate, pH 7.2, to concentrations shown in the figures) in a total volume of 12.5 μl. After incubation at 30 °C for 1 h, 2 μl of the mixture (6 μl in titration experiments) were added to 50 μl of 1× PBS. Luminescence was measured after adding 50 μl of luciferase assay reagent (Promega) using an Infinite M200 Pro plate reader (Tecan). For testing the temperature stability of CSecisbp2, WT and RQ protein (160 nm final concentration) was incubated for 30 min on ice or at 37/40.5 °C before in vitro translation. For titration experiments, reporter mRNA was diluted in nuclease-free water to obtain nine different concentrations shown in the figure before in vitro translation.
Ribosome profiling and RNA-Seq
Treatment of the samples was performed as described previously (19) with the exception of the RNase I incubation time, which was reduced to 20 min for the cortex tissue. Raw sequence data and raw counts can be obtained from the NCBI GEO repository entry GSE119681.
Quality control and preprocessing of deep-sequence data
Quality of sequencing data was controlled via FastQC version 0.11.8 (40) before and after trimming and rRNA removal. Adapter sequences and low-quality bases were trimmed using TrimGalore 0.4.3 with cutadapt 1.12 (41, 42). Sequences from ribosome profiling data were first aligned with bowtie2 2.3.4.3 (43) (maximum of 2 mismatches allowed) against rRNA and tRNA sequences obtained from the UCSC Genome Browser via the Table Browser tool (43–45). Unaligned reads (reads that are neither tRNA nor rRNA) were then aligned with bowtie2 against a UCSC mm10 RefSeq mouse transcriptome, containing the longest isoform of corresponding genes with 5′-UTR, CDS, and 3′UTR regions. Results were filtered after unique reads with samtools 1.9 (−F 4 −q 4) for downstream analysis (46). The size distribution was calculated, and all reads with length within mean ± S.D. were used for further analysis. Trimmed sequences of the RNA-Seq data were aligned with STAR aligner 2.6.0 against the GRCm38 (release 95) mouse genome retrieved from the Ensembl database via the biomaRt tool (47–49). Samtools was used to sort and index the resulting files containing the aligned sequences. The amount of sequences aligned to transcripts was counted with HTSeq 0.6.0 for following differential analysis with the DESeq2-package version 1.14.1 in R 3.3.1 (50–52). For ribosome profiling, reads with length between 28 and 32 nucleotides were used for differential expression analysis. GO enrichment analysis for significant overexpressed genes was done with PantherDB 14.1 (53–55).
Offset to ribosome P-site
Distribution of read sizes was calculated, and the sizes in the area of the mean ± S.D. (28–32 nucleotides for all samples) were used for downstream analysis. Offsets from 5′ ends of the reads to corresponding ribosome P-sites were calculated length- and sample-specific. Therefore, reads in an area of 20 nucleotides 5′ of start codons to 150 nucleotides 3′ of start codons were extracted, and the relative positions of the 5′ ends in this area, according to the start codon, were set. The highest peak 5′ of the start codon shows the highest density of ribosomes at the translation initiation codon. This peak was set as preliminary offset to P-site, and the amount of ribosomes within this periodicity was calculated for the first 50 codons. It was done in the same way for the positions before and after this peak. The position with the highest periodicity value was set as offset to the P-site. Size-specific offsets were the same in all samples. Sizes 28, 29, and 30 had an offset of 12 nucleotides, whereas sizes 31 and 32 showed an offset of 13 nucleotides.
Statistics
Means are reported with S.D. unless otherwise indicated. The numbers of biological replicates are given in the figure legends. Statistical tests employed were usually Student's t test. Statistics and curve fittings were calculated using GraphPad Prism. In RNA-Seq experiments, Benjamini–Hochberg (BH) correction has been applied. Throughout this work, we have adhered to the new, systematic nomenclature for selenoproteins (56).
Author contributions
W. Z., S. B., H. S., S. S., S. A., U. R., N. F.-V., and U. S. data curation; W. Z., S. B., H. S., M. T. H., U. R., N. F.-V., and U. S. formal analysis; W. Z., S. B., H. S., S. S., M. T. H., D. B., U. R., H. W., C. B., N. F.-V., and U. S. investigation; W. Z., S. B., D. B., N. F.-V., and U. S. visualization; W. Z., S. B., H. S., S. S., M. T. H., S. A., U. R., H. W., N. F.-V., and U. S. methodology; W. Z., C. B., N. F.-V., and U. S. writing-original draft; W. Z., S. B., H. S., S. S., M. T. H., D. B., S. A., U. R., H. W., C. B., N. F.-V., and U. S. writing-review and editing; S. B., M. T. H., C. B., N. F.-V., and U. S. conceptualization; S. B. software; S. B. and U. S. validation; H. W., C. B., and U. S. resources; N. F.-V. and U. S. project administration; U. S. supervision; U. S. funding acquisition.
Supplementary Material
Acknowledgments
We thank Magdalena Antes, who established recombinant mouse C-terminal Secisbp2 expression and purification and the luciferase assay as part of a bachelor's thesis in Molecular Medicine at the University of Bonn. We thank Paul Copeland for providing the luciferase reporter and control plasmids.
This work was supported by Deutsche Forschungsgemeinschaft Grants Schw914/2 and Schw914/5 and Universitätsklinikum Bonn. The authors declare that they have no conflicts of interest with the contents of this article.
This article was selected as one of our Editors' Picks.
This article contains Table S1 and Figs. S1–S3.
Raw and processed original data have been deposited in the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO) under accession number GSE119681.
- Sec
- selenocysteine
- SECIS
- selenocysteine insertion sequence
- SECISBP2
- SECIS-binding protein 2
- RPM
- per million mapped reads
- 3′ RPM
- reads 3′ of UGA/Sec per million mapped reads
- GFAP
- glial fibrillary acidic protein
- E
- embryonic day
- P
- postnatal day
- RPF
- ribosome protected fragment
- URE
- UGA redefinition efficiency
- BAC
- bacterial artificial chromosome
- DTA
- diphtheria toxin A fragment
- ES cell or ESC
- embryonic stem cell
- FRT
- FLP recognition target
- BH
- Benjamini–Hochberg.
References
- 1. Dumitrescu A. M., Liao X. H., Abdullah M. S., Lado-Abeal J., Majed F. A., Moeller L. C., Boran G., Schomburg L., Weiss R. E., and Refetoff S. (2005) Mutations in SECISBP2 result in abnormal thyroid hormone metabolism. Nat. Genet. 37, 1247–1252 10.1038/ng1654 [DOI] [PubMed] [Google Scholar]
- 2. Schweizer U., and Fradejas-Villar N. (2016) Why 21? The significance of selenoproteins for human health revealed by inborn errors of metabolism. FASEB J. 30, 3669–3681 10.1096/fj.201600424 [DOI] [PubMed] [Google Scholar]
- 3. Copeland P. R., Fletcher J. E., Carlson B. A., Hatfield D. L., and Driscoll D. M. (2000) A novel RNA binding protein, SBP2, is required for the translation of mammalian selenoprotein mRNAs. EMBO J. 19, 306–314 10.1093/emboj/19.2.306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fagegaltier D., Hubert N., Yamada K., Mizutani T., Carbon P., and Krol A. (2000) Characterization of mSelB, a novel mammalian elongation factor for selenoprotein translation. EMBO J. 19, 4796–4805 10.1093/emboj/19.17.4796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Seeher S., Atassi T., Mahdi Y., Carlson B. A., Braun D., Wirth E. K., Klein M. O., Reix N., Miniard A. C., Schomburg L., Hatfield D. L., Driscoll D. M., and Schweizer U. (2014) Secisbp2 is essential for embryonic development and enhances selenoprotein expression. Antioxid. Redox Signal. 21, 835–849 10.1089/ars.2013.5358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Seeher S., Carlson B. A., Miniard A. C., Wirth E. K., Mahdi Y., Hatfield D. L., Driscoll D. M., and Schweizer U. (2014) Impaired selenoprotein expression in brain triggers striatal neuronal loss leading to co-ordination defects in mice. Biochem. J. 462, 67–75 10.1042/BJ20140423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dumitrescu A. M., Di Cosmo C., Liao X. H., Weiss R. E., and Refetoff S. (2010) The syndrome of inherited partial SBP2 deficiency in humans. Antioxid. Redox Signal. 12, 905–920 10.1089/ars.2009.2892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Donovan J., Caban K., Ranaweera R., Gonzalez-Flores J. N., and Copeland P. R. (2008) A novel protein domain induces high affinity selenocysteine insertion sequence binding and elongation factor recruitment. J. Biol. Chem. 283, 35129–35139 10.1074/jbc.M806008200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Takeuchi A., Schmitt D., Chapple C., Babaylova E., Karpova G., Guigo R., Krol A., and Allmang C. (2009) A short motif in Drosophila SECIS binding protein 2 provides differential binding affinity to SECIS RNA hairpins. Nucleic Acids Res. 37, 2126–2141 10.1093/nar/gkp078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bubenik J. L., and Driscoll D. M. (2007) Altered RNA binding activity underlies abnormal thyroid hormone metabolism linked to a mutation in selenocysteine insertion sequence-binding protein 2. J. Biol. Chem. 282, 34653–34662 10.1074/jbc.M707059200 [DOI] [PubMed] [Google Scholar]
- 11. Schoenmakers E., Agostini M., Mitchell C., Schoenmakers N., Papp L., Rajanayagam O., Padidela R., Ceron-Gutierrez L., Doffinger R., Prevosto C., Luan J., Montano S., Lu J., Castanet M., Clemons N., et al. (2010) Mutations in the selenocysteine insertion sequence-binding protein 2 gene lead to a multisystem selenoprotein deficiency disorder in humans. J. Clin. Invest. 120, 4220–4235 10.1172/JCI43653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Azevedo M. F., Barra G. B., Naves L. A., Ribeiro Velasco L. F., Godoy Garcia Castro P., de Castro L. C., Amato A. A., Miniard A., Driscoll D., Schomburg L., and de Assis Rocha Neves F. (2010) Selenoprotein-related disease in a young girl caused by nonsense mutations in the SBP2 gene. J. Clin. Endocrinol. Metab. 95, 4066–4071 10.1210/jc.2009-2611 [DOI] [PubMed] [Google Scholar]
- 13. Hamajima T., Mushimoto Y., Kobayashi H., Saito Y., and Onigata K. (2012) Novel compound heterozygous mutations in the SBP2 gene: characteristic clinical manifestations and the implications of GH and triiodothyronine in longitudinal bone growth and maturation. Eur. J. Endocrinol. 166, 757–764 10.1530/EJE-11-0812 [DOI] [PubMed] [Google Scholar]
- 14. Di Cosmo C., McLellan N., Liao X. H., Khanna K. K., Weiss R. E., Papp L., and Refetoff S. (2009) Clinical and molecular characterization of a novel selenocysteine insertion sequence-binding protein 2 (SBP2) gene mutation (R128X). J. Clin. Endocrinol. Metab. 94, 4003–4009 10.1210/jc.2009-0686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Çatli G., Fujisawa H., Kirbiyik Ö., Mimoto M. S., Gençpinar P., Özdemir T. R., Dündar B. N., and Dumitrescu A. M. (2018) A Novel homozygous selenocysteine insertion sequence binding protein 2 (SECISBP2, SBP2) gene mutation in a Turkish boy. Thyroid 28, 1221–1223 10.1089/thy.2018.0015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Squires J. E., Stoytchev I., Forry E. P., and Berry M. J. (2007) SBP2 binding affinity is a major determinant in differential selenoprotein mRNA translation and sensitivity to nonsense-mediated decay. Mol. Cell. Biol. 27, 7848–7855 10.1128/MCB.00793-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Latrèche L., Jean-Jean O., Driscoll D. M., and Chavatte L. (2009) Novel structural determinants in human SECIS elements modulate the translational recoding of UGA as selenocysteine. Nucleic Acids Res. 37, 5868–5880 10.1093/nar/gkp635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ingolia N. T., Ghaemmaghami S., Newman J. R., and Weissman J. S. (2009) Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 324, 218–223 10.1126/science.1168978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fradejas-Villar N., Seeher S., Anderson C. B., Doengi M., Carlson B. A., Hatfield D. L., Schweizer U., and Howard M. T. (2017) The RNA-binding protein Secisbp2 differentially modulates UGA codon reassignment and RNA decay. Nucleic Acids Res. 45, 4094–4107 10.1093/nar/gkw1255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mehta A., Rebsch C. M., Kinzy S. A., Fletcher J. E., and Copeland P. R. (2004) Efficiency of mammalian selenocysteine incorporation. J. Biol. Chem. 279, 37852–37859 10.1074/jbc.M404639200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tasic B., Menon V., Nguyen T. N., Kim T. K., Jarsky T., Yao Z., Levi B., Gray L. T., Sorensen S. A., Dolbeare T., Bertagnolli D., Goldy J., Shapovalova N., Parry S., Lee C., et al. (2016) Adult mouse cortical cell taxonomy revealed by single cell transcriptomics. Nat. Neurosci. 19, 335–346 10.1038/nn.4216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Oliéric V., Wolff P., Takeuchi A., Bec G., Birck C., Vitorino M., Kieffer B., Beniaminov A., Cavigiolio G., Theil E., Allmang C., Krol A., and Dumas P. (2009) SECIS-binding protein 2, a key player in selenoprotein synthesis, is an intrinsically disordered protein. Biochimie 91, 1003–1009 10.1016/j.biochi.2009.05.004 [DOI] [PubMed] [Google Scholar]
- 23. Allmang C., Carbon P., and Krol A. (2002) The SBP2 and 15.5 kD/Snu13p proteins share the same RNA binding domain: identification of SBP2 amino acids important to SECIS RNA binding. RNA 8, 1308–1318 10.1017/S1355838202020034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Vidovic I., Nottrott S., Hartmuth K., Lührmann R., and Ficner R. (2000) Crystal structure of the spliceosomal 15.5kD protein bound to a U4 snRNA fragment. Mol. Cell 6, 1331–1342 10.1016/S1097-2765(00)00131-3 [DOI] [PubMed] [Google Scholar]
- 25. Caban K., Kinzy S. A., and Copeland P. R. (2007) The L7Ae RNA binding motif is a multifunctional domain required for the ribosome-dependent Sec incorporation activity of Sec insertion sequence binding protein 2. Mol. Cell. Biol. 27, 6350–6360 10.1128/MCB.00632-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Smith A. C., Mears A. J., Bunker R., Ahmed A., MacKenzie M., Schwartzentruber J. A., Beaulieu C. L., Ferretti E., FORGE Canada Consortium, Majewski J., Bulman D. E., Celik F. C., Boycott K. M., and Graham G. E. (2014) Mutations in the enzyme glutathione peroxidase 4 cause Sedaghatian-type spondylometaphyseal dysplasia. J. Med. Genet. 51, 470–474 10.1136/jmedgenet-2013-102218 [DOI] [PubMed] [Google Scholar]
- 27. Seiler A., Schneider M., Förster H., Roth S., Wirth E. K., Culmsee C., Plesnila N., Kremmer E., Rådmark O., Wurst W., Bornkamm G. W., Schweizer U., and Conrad M. (2008) Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab 8, 237–248 10.1016/j.cmet.2008.07.005 [DOI] [PubMed] [Google Scholar]
- 28. Wirth E. K., Conrad M., Winterer J., Wozny C., Carlson B. A., Roth S., Schmitz D., Bornkamm G. W., Coppola V., Tessarollo L., Schomburg L., Köhrle J., Hatfield D. L., and Schweizer U. (2010) Neuronal selenoprotein expression is required for interneuron development and prevents seizures and neurodegeneration. FASEB J. 24, 844–852 10.1096/fj.09-143974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Carlson B. A., Schweizer U., Perella C., Shrimali R. K., Feigenbaum L., Shen L., Speransky S., Floss T., Jeong S. J., Watts J., Hoffmann V., Combs G. F., Gladyshev V. N., and Hatfield D. L. (2009) The selenocysteine tRNA STAF-binding region is essential for adequate selenocysteine tRNA status, selenoprotein expression and early age survival of mice. Biochem. J. 418, 61–71 10.1042/BJ20081304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pitts M. W., Raman A. V., Hashimoto A. C., Todorovic C., Nichols R. A., and Berry M. J. (2012) Deletion of selenoprotein P results in impaired function of parvalbumin interneurons and alterations in fear learning and sensorimotor gating. Neuroscience 208, 58–68 10.1016/j.neuroscience.2012.02.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wirth E. K., Bharathi B. S., Hatfield D., Conrad M., Brielmeier M., and Schweizer U. (2014) Cerebellar hypoplasia in mice lacking selenoprotein biosynthesis in neurons. Biol. Trace Elem. Res. 158, 203–210 10.1007/s12011-014-9920-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kirchner S., Cai Z., Rauscher R., Kastelic N., Anding M., Czech A., Kleizen B., Ostedgaard L. S., Braakman I., Sheppard D. N., and Ignatova Z. (2017) Alteration of protein function by a silent polymorphism linked to tRNA abundance. PLoS Biol. 15, e2000779 10.1371/journal.pbio.2000779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yu C. H., Dang Y., Zhou Z., Wu C., Zhao F., Sachs M. S., and Liu Y. (2015) Codon usage influences the local rate of translation elongation to regulate co-translational protein folding. Mol. Cell 59, 744–754 10.1016/j.molcel.2015.07.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kirchner S., and Ignatova Z. (2015) Emerging roles of tRNA in adaptive translation, signalling dynamics and disease. Nat. Rev. Genet. 16, 98–112 10.1038/nrg3861 [DOI] [PubMed] [Google Scholar]
- 35. Jo S., Fonseca T. L., Bocco B., Fernandes G. W., McAninch E. A., Bolin A. P., Da Conceição R. R., Werneck-de-Castro J. P., Ignacio D. L., Egri P., Nemeth D., Fekete C., Bernardi M. M., Leitch V. D., Mannan N. S., et al. (2019) Type 2 deiodinase polymorphism causes ER stress and hypothyroidism in the brain. J. Clin. Invest. 129, 230–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhang Y., Zhou Y., Schweizer U., Savaskan N. E., Hua D., Kipnis J., Hatfield D. L., and Gladyshev V. N. (2008) Comparative analysis of selenocysteine machinery and selenoproteome gene expression in mouse brain identifies neurons as key functional sites of selenium in mammals. J. Biol. Chem. 283, 2427–2438 10.1074/jbc.M707951200 [DOI] [PubMed] [Google Scholar]
- 37. Alam M. S., Getz M., Safeukui I., Yi S., Tamez P., Shin J., Velázquez P., and Haldar K. (2012) Genomic expression analyses reveal lysosomal, innate immunity proteins, as disease correlates in murine models of a lysosomal storage disorder. PLoS One 7, e48273 10.1371/journal.pone.0048273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Court D. L., Swaminathan S., Yu D., Wilson H., Baker T., Bubunenko M., Sawitzke J., and Sharan S. K. (2003) Mini-lambda: a tractable system for chromosome and BAC engineering. Gene 315, 63–69 10.1016/S0378-1119(03)00728-5 [DOI] [PubMed] [Google Scholar]
- 39. Kinzy S. A., Caban K., and Copeland P. R. (2005) Characterization of the SECIS binding protein 2 complex required for the co-translational insertion of selenocysteine in mammals. Nucleic Acids Res. 33, 5172–5180 10.1093/nar/gki826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Andrews S. (2010) FastQC: a quality control tool for high throughput sequence data, Babraham Institute, Babraham, UK [Google Scholar]
- 41. Krueger F. (2012) Trim Galore! A wrapper tool around Cutadapt and FastQC, Babraham Institute, Babraham, UK [Google Scholar]
- 42. Martin M. (2011) Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 17, 10.14806/ej.17.1.200 10.14806/ej.17.1.200 [DOI] [Google Scholar]
- 43. Langmead B., and Salzberg S. L. (2012) Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kent W. J., Sugnet C. W., Furey T. S., Roskin K. M., Pringle T. H., Zahler A. M., and Haussler D. (2002) The human genome browser at UCSC. Genome Res. 12, 996–1006 10.1101/gr.229102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Karolchik D., Hinrichs A. S., Furey T. S., Roskin K. M., Sugnet C. W., Haussler D., and Kent W. J. (2004) The UCSC Table Browser data retrieval tool. Nucleic Acids Res. 32, D493–D496 10.1093/nar/gkh103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., and 1000 Genome Project Data Processing Subgroup (2009) The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Dobin A., Davis C. A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., and Gingeras T. R. (2013) STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zerbino D. R., Achuthan P., Akanni W., Amode M. R., Barrell D., Bhai J., Billis K., Cummins C., Gall A., Girón C. G., Gil L., Gordon L., Haggerty L., Haskell E., Hourlier T., et al. (2018) Ensembl 2018. Nucleic Acids Res. 46, D754–D761 10.1093/nar/gkx1098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Durinck S., Spellman P. T., Birney E., and Huber W. (2009) Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc. 4, 1184–1191 10.1038/nprot.2009.97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Anders S., Pyl P. T., and Huber W. (2015) HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 10.1093/bioinformatics/btu638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Love M. I., Huber W., and Anders S. (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. R Core Team (2018) R: A Language and Environment for Statistical Computing, R Foundation for Statistical Computing, Vienna, Austria [Google Scholar]
- 53. Thomas P. D., Campbell M. J., Kejariwal A., Mi H., Karlak B., Daverman R., Diemer K., Muruganujan A., and Narechania A. (2003) PANTHER: a library of protein families and subfamilies indexed by function. Genome Res. 13, 2129–2141 10.1101/gr.772403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Mi H., Dong Q., Muruganujan A., Gaudet P., Lewis S., and Thomas P. D. (2010) PANTHER version 7: improved phylogenetic trees, orthologs and collaboration with the Gene Ontology Consortium. Nucleic Acids Res. 38, D204–D210 10.1093/nar/gkp1019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Thomas P. D., Kejariwal A., Guo N., Mi H., Campbell M. J., Muruganujan A., and Lazareva-Ulitsky B. (2006) Applications for protein sequence-function evolution data: mRNA/protein expression analysis and coding SNP scoring tools. Nucleic Acids Res. 34, W645–W650 10.1093/nar/gkl229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Gladyshev V. N., Arnér E. S., Berry M. J., Brigelius-Flohe R., Bruford E. A., Burk R. F., Carlson B. A., Castellano S., Chavatte L., Conrad M., Copeland P. R., Diamond A. M., Driscoll D. M., Ferreiro A., Flohé L., et al. (2016) Selenoprotein gene nomenclature. J. Biol. Chem. 291, 24036–24040 10.1074/jbc.M116.756155 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.