

Abstract
β-1,4-Galactosyltransferase 1 (B4GALT1) and ST6 β-galactoside α-2,6-sialyltransferase 1 (ST6GAL1) catalyze the successive addition of terminal β-1,4–linked galactose and α-2,6–linked sialic acid to N-glycans. Their exclusive interaction in the Golgi compartment is a prerequisite for their full catalytic activity, whereas a lack of this interaction is associated with cancers and hypoxia. To date, no structural information exists that shows how glycosyltransferases functionally assemble with each other. Using molecular docking simulations to predict interaction surfaces, along with mutagenesis screens and high-throughput FRET analyses in live cells to validate these predictions, we show here that B4GALT1 and ST6GAL1 interact via highly charged noncatalytic surfaces, leaving the active sites exposed and accessible for donor and acceptor substrate binding. Moreover, we found that the assembly of ST6GAL1 homomers in the endoplasmic reticulum before ST6GAL1 activation in the Golgi utilizes the same noncatalytic surface, whereas B4GALT1 uses its active-site surface for assembly, which silences its catalytic activity. Last, we show that the homomeric and heteromeric B4GALT1/ST6GAL1 complexes can assemble laterally in the Golgi membranes without forming cross-cisternal contacts between enzyme molecules residing in the opposite membranes of each Golgi cisterna. Our results provide detailed mechanistic insights into the regulation of glycosyltransferase interactions, the transitions between B4GALT1 and ST6GAL1 homo- and heteromers in the Golgi, and cooperative B4GALT1/ST6GAL1 function in N-glycan synthesis.
Keywords: glycosyltransferase; galactosyltransferase; β-galactoside α-2,6-sialyltransferase 1 (ST6GAL1); protein complex; protein–protein interaction; molecular interplay
Introduction
Glycosyltransferases and glycosidases in the endoplasmic reticulum (ER)2 and the Golgi apparatus are responsible for enzymatic addition and removal of sugar moieties to generate linear or branched glycan chains found in proteins and lipids (1). Most glycosyltransferases are type II transmembrane proteins with three distinct structural and functional domains: a transmembrane domain preceded by an N-terminal cytoplasmic tail, a stem domain (STEM) oriented toward the lumen of the Golgi, and a globular catalytic domain (CAT) (2). The presence of the catalytic domains in the Golgi lumen allows for the processing of glycan chains along their passage through the secretory pathway (3).
Recent evidence has shown that many glycosyltransferases exist and function in cells either as homomers or heteromers rather than as monomers, as thought previously (4). Their oligomerization state depends on several factors; for instance, glycosyltransferases acting on N-glycans assemble into less active homomeric complexes in the ER but form functionally relevant and fully active heteromers in the Golgi (5–7). This transition between homomers and heteromers is key for spatial and temporal regulation of glycosylation, as glycan addition and removal are sequential and do not use any template, unlike synthesis of mRNA and proteins. Such complexes have been identified so far in all glycosylation pathways, i.e. in the synthesis of N- and O-glycans, glycosaminoglycans, and glycolipids. For instance, in the case of glycosaminoglycans, complexes termed GAGosomes (8) are involved in polymerization of heparan sulfate and needed for catalytic activity of EXT1 and EXT2 and their correct localization in the Golgi (9). The ER and Golgi microenvironments have primary roles in governing the type of assembly made, in addition to contributing to the preservation of the Golgi structure and ER–Golgi trafficking (10, 11). Accordingly, the redox state difference between these two compartments regulates the interaction between B4GALT1 and ST6GAL1, whereas the pH gradient (pH 7 versus pH 6.3) affects B4GALT1 and ST3GAL3 interactions as well as interactions between glycosyltransferases that act on O-glycans (6, 12). Changes in Golgi redox potential in hypoxic cells correlates with loss of α-2,6 sialylation and inactivation of ST6GAL1 and its inability to bind B4GALT1. These events have been shown to be coordinated by formation of two disulfide bonds on two distinct surface areas within the catalytic domain of the enzyme (12). On the other hand, the first human B4GALT1 dimeric structure indicates that the homodimeric interaction takes place via the surface surrounding the active site, and two mutations there (M340H and H343A) prevented dimer formation, most likely by changing the affinity to the Mn2+ binding and by altering the active site's loop dynamicity (13, 14).
To decipher how the B4GALT1 and ST6GAL1 glycosyltransferases interact and function at the molecular and cellular levels, we utilized molecular modeling and docking, mutagenesis screens, domain truncations, and validation with high-throughput FRET analyses. Our data unveil, for the first time, interaction surfaces important for assembly of both homomeric and heteromeric complexes between B4GALT1 and ST6GAL1. Our findings also show that the complexes can interact laterally, i.e. without necessarily having contacts across the cisternal space. The results also provide mechanistic details that facilitate transitions between enzyme homo- and heteromers in the Golgi in addition to providing a rationale for having such an intricate transition system to regulate glycosylation.
Results
B4GALT1 uses its noncatalytic surface for interaction with ST6GAL1
Recently, we presented the first human homodimeric crystal structure of B4GALT1 and showed that the interaction takes place at the surface surrounding the active site of the enzyme through concerted loop movements involving amino acids Met340 and His343 (13). To determine possible heterodimer interaction sites in the B4GALT1 catalytic domain, we first utilized molecular docking predictions using the ClusPro server (15) and obtained several potential residues that can mediate the interaction with ST6GAL1. We created single mutations of selected amino acids (Fig. 1A) and screened their effects on the interaction by using the FRET system. Enzymes tagged with mVenus (donor) and mCherry (acceptor) were expressed in COS-7 cells, and FRET signals were acquired strictly from Golgi regions of cells by image segmentation and fine adjustments (7). The active-site mutants M340H, M340E, and H343A did not affect the heteromeric interaction, as no FRET signal loss was observed (Fig. 1D). Based on the crystal structure (13), the Cys172 residue forms a disulfide bridge with Cys130 that restricts the conformational flexibility of a semi-disordered loop (Cys130 to Cys172) in the catalytic domain of B4GALT1 opposite the active site (Fig. 1B). Therefore, we mutated C172A and nearby Q206A. The mutations had a significant effect on heteromers, with 67% and 34% loss of FRET signal, respectively (Fig. 1D). No such loss was detected with B4GALT1 homodimers (Fig. 1C), indicating the specificity of the C172A and Q206A mutations.
Figure 1.

Identification of the B4GALT1 interaction surfaces. A, structure of the B4GALT1 monomer (PDB code 4EE4) in surface representation, showing single mutations in purple (chosen using ClusPro docking predictions) and the active site surface in red. B, B4GALT1 structure in cartoon representation, showing the C130-C172 disulfide bond in purple and the interaction surface in green. The active-site surface is shown in red. C, homomeric FRET interactions between B4GALT1 mutants. COS-7 cells were transfected with the depicted mVenus- and mCherry-tagged FRET enzyme constructs, fixed 24 h later, and quantified with the Operetta High Content Imaging system. FRET efficiencies were calculated from 10,000 to 20,000 cells and expressed as percentages of control values (mean ± S.D., n = 3) after subtracting the FRET efficiencies of negative control (MGAT1–B4GALT1) values. D, heteromeric FRET interactions. B4GALT1.mVenus mutants and ST6GAL1.mCherry WT were coexpressed, and FRET signals were determined as above. E, B4GALT1 structure showing the predicted heteromeric interaction surface for ST6GAL1in green, as revealed by docking simulations with Haddock. Blue spheres show positively charged residues (Lys156, Lys162, Arg170, and Arg204), and red spheres show negatively charged ones (Glu144, Glu152, and Asp171). The charged residues are situated on the binding interface. F, B4GALT1 structure showing in green the predicted interaction surface for ST6GAL1, as revealed by docking simulations. The yellow residues depict the mutated residues on that surface (D150S, E152S, K156S, K162S, R166S, and D171S). The mutant was named B4GALT1.Ncat. G, B4GALT1.Ncat homomeric and heteromeric (with ST6GAL1) FRET assays. Constructs were coexpressed in COS-7 cells, and FRET efficiencies were calculated as mentioned above. H, B4GALT1.Ncat heteromeric FRET assay with ST3GAL3 and B4GALT1–ST6GAL1 inhibition assay by ST3GAL3-HA (triple cotransfection). Constructs were coexpressed in COS-7 cells, and FRET efficiencies were calculated as mentioned above. Statistically significant changes relative to the control samples (gray columns) are indicated (*, p < 0.05; **, p < 0.01; ***, p < 0.001).
Interestingly, directed docking of ST6GAL1 to this interface of B4GALT1 with the Haddock server (16) highlighted the same region as a potential contact site between the two enzymes. Based on the docked model, seven charged residues (Glu144, Glu152, Lys156, Lys162, Arg170, Asp171, and Arg204) involved in salt bridges with ST6GAL1 were identified as being important for the heteromeric interaction (Fig. 1E). Of these, we selected six to mutate into serine residues (D150S, E152S, K156S, K162S, R166S, and D171S). The mutant construct (B4GALT1.Ncat, Fig. 1F) lost its ability to bind ST6GAL1 but preserved its ability to bind to itself (Fig. 1G). These data provide strong support for this surface to mediate B4GALT1 binding to ST6GAL1.
We then tested whether ST3GAL3 binding is similarly affected by the mutations. ST3GAL3 is an enzyme that competes with ST6GAL1 on binding to B4GALT1 (5) and can use both Galβ1,4GlcNAc and Galβ1,3GlcNAc as its acceptor substrate. To our surprise, ST3GAL3 still interacted with the mutant B4GALT1.Ncat protein (Fig. 1H). This suggests that, even though the two sialyltransferases compete for binding to B4GALT1 (Fig. 1H), they use different but mutually exclusive interaction surfaces on B4GALT1.
ST6GAL1 interaction surfaces
The same methodology was applied to determine the ST6GAL1 interaction surfaces, which effectuate both homodimerization as well as heterodimerization to B4GALT1. Only the monomeric crystal structure for the rat and human ST6GAL1 is currently available (17, 18). Molecular docking on the ClusPro server revealed residues that can modulate the interaction of B4GALT1 with ST6GAL1 (Fig. S1A) in two distinct surfaces, one being the active site and the other one residing opposite the active site. Point mutations of the depicted residues on ST6GAL1 significantly inhibited both homomeric and heteromeric interactions. For example, the H263A mutation inhibited homomer formation by 50% and heteromer formation by 15% (Fig. S1, B and C), whereas R145A and H405A each inhibited only homomer formation by 16% and 40%, respectively. All of these residues are situated opposite the active site (Fig. S1A).
Molecular docking of ST6GAL1 with itself using the SymmDock server (19, 20) (Fig. 2A) or with B4GALT1 using the Haddock server (Fig. 2B) revealed potential interacting residues in the noncatalytic surface for both interactions. The three residues (Arg145, His263, and His405) mentioned above are part of this same surface. The heterodimer and homodimer docking models were overlapping and involved several shared salt bridges on the same surface (Fig. 2C). Based on these models, a mutated construct was designed (Fig. 2D) to remove seven charged amino acids. The following mutations were included: D146S, D157S, K171S, H263S, E310S, D336S, and R402S (yielding the mutant ST6GAL1.Ncat, Fig. 2D). FRET measurements showed that the mutations significantly inhibited both interactions (homomers by 68% and heteromers by 87%). Based on one of the docking models, the active-site area may participate in ST6GAL1 homodimer formation (similar to B4GALT1). Therefore, we also tested this possibility and prepared another mutant construct, ST6GAL1.Cat, in which the lysine and arginine residues were mutated to glutamic acid residues (K111E, K241E, R242E, and K358E; Fig. S1D). FRET measurements revealed that the mutations did not affect homomeric interactions, but they inhibited the formation of heteromers by 36% (Fig. S1A, right panel). Because of this, we constructed several other mutations within the same active-site area, which, however, did not fold and/or localize correctly in the Golgi membranes (data not shown).
Figure 2.

Identification of the ST6GAL1 interaction surfaces. A, docking model of the ST6GAL1 homodimer from SymmDock. The active-site surface in both enzymes is presented in dark red, and the green surface indicates the interaction interface. Red spheres depict negatively charged residues (Asp146, Glu154, Asp157, Glu166, Glu303, and Asp336), blue spheres indicate positively charged residues (Lys127, His147, Arg175, His263, Lys297, and His405) of human ST6GAL1, and black lines denote to residues that interact with each other. Hydrophobic amino acids forming hydrophobic contacts between the two ST6GAL1 monomers on the interface are shown as light brown spheres. B, docking model of B4GALT1 and ST6GAL1 heterodimeric interaction from Haddock. The active-site surface in both enzymes is presented in dark red, and the green surface indicates the interaction interface. Red spheres depict negatively charged residues (B4GALT1: Glu144, Glu152, and Asp171; ST6GalT1: Asp146, Asp157, Glu310, and Asp336), and blue spheres indicate positively charged residues (B4GALT1: Lys156, Lys162, Arg170, and Arg204; ST6GAL1: Lys171, His263, and Arg402) in both enzymes. Light brown spheres show hydrophobic amino acids forming hydrophobic contacts between the two enzymes. C, ST6GAL1 interaction interface. In red and blue, we show negatively and positively charged residues, respectively, and in yellow shared residues between the homodimeric (model I) and the heterodimeric (model II) surface. D, model showing in yellow the mutations performed on ST6GAL1 (named ST6GAL1.Ncat: D146S, D157S, K171S, H263S, E310S, D336S, and R402S). The graph shows the ST6GAL1.Ncat homomeric and heteromeric (with B4GALT1) FRET assays. COS-7 cells were transfected with the depicted mVenus-tagged (for B4GALT1) and mCherry-tagged (for ST6GAL1) FRET enzyme constructs, fixed 24 h later and quantified with the Operetta High Content Imaging system. FRET efficiencies were calculated from 10,000 to 20,000 cells and expressed as percentages of control values (mean ± S.D., n = 3) after subtracting the FRET efficiencies of negative control (MGAT1–B4GALT1) values. Statistically significant changes relative to the control samples (gray columns) are indicated (***, p < 0.001).
An important factor to consider is the presence of one N-glycosylated residue, Asn149 (17), and another potential glycosylation site in residue Asn161. They both reside in the identified interaction surface area of ST6GAL1 (Fig. 3A). To examine whether they contribute to the interactions, we carried out FRET measurements in cells treated with swainsonine or tunicamycin. Swainsonine is a known mannosidase inhibitor and halts processing of high-mannose N-glycans to complex N-glycans. Tunicamycin, in turn, depletes all N-glycans from newly synthetized proteins by inhibiting the formation of N-acetylglucosaminylpyrophosphoryldolichol, a necessary acceptor substrate for N-glycan precursor synthesis. FRET measurements in swainsonine-treated cells showed no loss of the FRET signal between ST6GAL1 homomers and between B4GALT1/ST6GAL1 heteromers, indicating that the interactions are not dependent on complex N-glycans (Fig. 3B). Intriguingly, tunicamycin treatment resulted in a 2-fold increase in the formation of B4GALT1/ST6GAL1 heteromers (Fig. 3C). The reason for this is not clear but may relate either to steric hindrance of the interaction by the existing N-glycan, tunicamycin-induced oxidative stress in the ER and the Golgi compartments (21), or both. In line with the latter possibility, the more oxidizing environment of the Golgi lumen compared with the ER favors B4GALT1–ST6GAL1 interaction (12).
Figure 3.
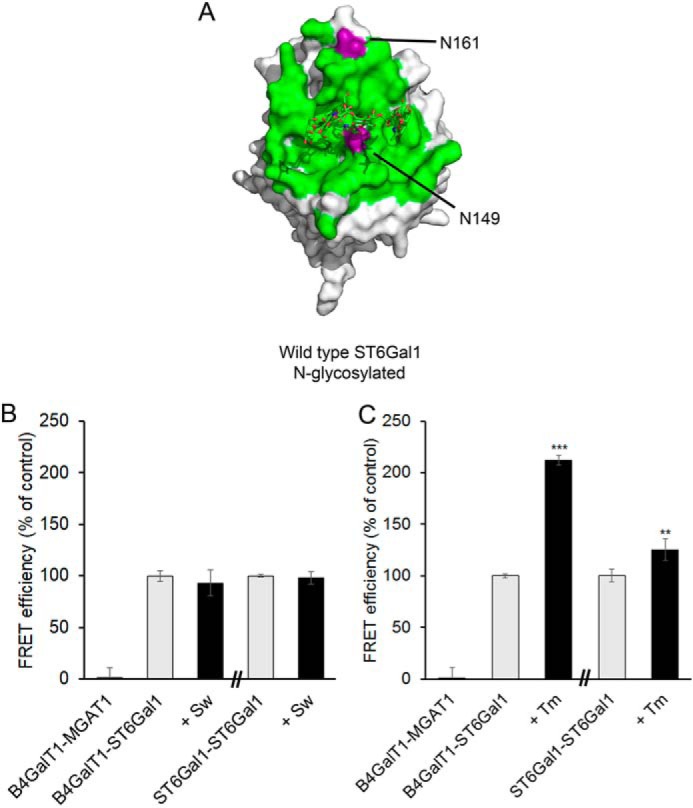
N-glycan effect on the ST6GAL1 interactions. A, crystal structure of N-glycosylated ST6GAL1 (PDB code 4JS2). The structure originally contains one N-glycan at Asn149 only. The green surface represents the predicted ST6GAL1 interaction surface, and Asn149 and Asn161 are shown in purple. B and C, COS-7 cells were transfected with the depicted mVenus-tagged (for B4GALT1) and mCherry-tagged (for ST6GAL1) FRET enzyme constructs, treated with 5 μm swainsonine (Sw) or 5 μm tunicamycin (Tm) 6 h after transfection, fixed 24 h later, and quantified with the Operetta High Content Imaging system. FRET efficiencies were calculated from 10,000 to 20,000 cells and expressed as percentages of control values (mean ± S.D., n = 3) after subtracting the FRET efficiencies of negative control (MGAT1–B4GALT1) values. Statistically significant changes relative to the control samples (gray columns) are indicated (**, p < 0.01; ***, p < 0.001).
Heteromeric assembly regulates the catalytic activity of B4GALT1 and ST6GAL1
To investigate whether the interaction mutants are enzymatically active, we measured their activities first in cellulo by utilizing electroporated Lec20 cells lacking endogenous B4GALT1 activity and staining with rhodamine-conjugated RCA-I- or fluorescein-conjugated SNA lectin. These lectins are used as markers for terminal β-1,4-galactose and α-2,6-sialic acid, respectively. Transfection of cells expressing WT B4GALT1 or the B4GALT1.Ncat mutant alone with RCA-I lectin (Fig. 4, A and B) slightly increased terminal galactose residues in the cells. Cells expressing both enzymes (B4GALT1-WT and ST6GAL1-WT) simultaneously showed 2-fold higher galactose levels in the cells (Fig. 4B). In contrast, cells expressing the interaction mutants (B4GALT1.Ncat and ST6GAL1.Ncat) did not show such an increase, suggesting either that the mutations rendered the B4GALT1 enzyme inactive despite not localizing in the active site or that the low activity may be caused by the loss of its ability to interact with ST6GAL1 (Figs. 1G and 2D), consistent with our previous results (22). SNA lectin staining, on the other hand, showed that cells expressing WT B4GALT1 and ST6GAL1 together displayed 3-fold higher α-2,6-sialic acid levels than cells expressing the mutant B4GALT1.Ncat and ST6GAL1.Ncat (Fig. 4, A and C). Similar results were obtained from in vitro sialylation assays with cell lysates prepared from cells transfected with an empty vector (Mock), with WT ST6GAL1 (ST6GAL1.WT), or with the ST6GAL1.Ncat mutant constructs. The ST6GAL1.Ncat interaction mutant retained only 10% of the activity of WT ST6GAL1 (Fig. 4D). These data suggest that enzyme interactions are necessary for the full catalytic activity of B4GALT1 and ST6GAL1. However, the possibility that the mutations also intrinsically impair the activity of these two enzymes cannot be excluded at the moment.
Figure 4.

Enzymatic activity assays with B4GALT1 and ST6GAL1. A, lectin staining of COS-7 cells expressing WT and mutant B4GALT1 and ST6GAL1. Pictures of stained cells were taken using OperettaTM (PerkinElmer Life Sciences) using blue, green, and red filters. Blue represents nuclear Hoechst stain, green represents bound SNA-fluorescein on α-2,6 sialic acid, and red-orange represents bound RCA-I–rhodamine on β-1,4 galactose. Scale bar = 200 μm. B, RCA-I–rhodamine mean intensities as percent of up to 50,000 Lec20 cells expressing the depicted constructs (mean ± S.D., n = 3). Statistically significant changes relative to control cells (mock-transfected cells) are indicated (***, p < 0.001). C, SNA-fluorescein mean intensities as percent of up to 50,000 Lec 20 cells expressing the depicted constructs (mean ± S.D., n = 3). Statistically significant changes relative to control cells (mock-transfected cells) are indicated (**, p < 0.01; ***, p < 0.001). D, enzymatic activity of the ST6Gal mutants. Cells transfected with the indicated ST6GAL1 constructs were lysed in radioimmune precipitation assay buffer 24 h post-transfection, and then their activities were determined as described under “Experimental procedures” using asialofetuin as an acceptor. The values shown (columns) are expressed as the mean percent of disintegrations per minute (n = 3) after normalizing them against the ST6GAL1 protein present (determined by immunoblotting). Statistically significant changes relative to control cells (mock-transfected cells) are indicated (*, p < 0.05; ***, p < 0.001).
Lateral interactions mediate formation of B4GALT1 and ST6GAL1 complexes
A recent Golgi EM and tomography study suggested the presence of cisternal protein arrays in the trans-Golgi that likely hold cisternal membranes together (23). B4GALT1 and ST6GAL1 are mainly localized in the trans-Golgi cisternae and, in theory, can adopt two possible configurations: cis and trans, where cis represents lateral interactions in the Golgi membrane and the trans configuration denotes transferases that bind to each other while residing in opposite membranes in one cisterna (Fig. 5A). In support of the latter theoretical possibility, the calculated molecular dimensions of the two transferases (15- to 25-nm diameter) potentially exceed the 20-nm diameter of intracisternal space (24). To examine whether B4GALT1 and ST6GAL1 indeed interact via cis or trans and what role the stem and catalytic domains have in the interactions, we deleted them separately, expressed the constructs in COS-7 cells, and measured their effect on the interactions using the FRET system. In the case of B4GALT1 homodimers, we found that deletion of the catalytic domain completely inhibited the interaction, indicating that B4GALT1 interacts with itself mainly via this domain (Fig. 5B). Removal of only the stem domain did not inhibit the interaction. Instead, we detected an even higher FRET signal with the stemless enzyme constructs, confirming that homomers form via catalytic domains (Fig. 5B). As the deletion truncates the enzyme size by 10–15 nm, they cannot reach each other from opposite membranes and can therefore only interact via lateral interactions. In the case of ST6GAL1 homodimer formation, neither removal of the catalytic domain nor of the stem domain had any effect on the interaction (Fig. 5B). This indicates that ST6GAL1 can interact with itself via both the stem and the catalytic domains. The reason for the increased FRET signal is not clear but may involve reduced spatial hindrance of the donor and acceptor fluorophores or reduced rigidity of the construct upon removal of the ST6GAL1 stem domain. Structural predictions of the stem domain revealed two α-helical domains that may make the stem domain more rigid. The fact that removal of the stem domain again did not inhibit formation of ST6GAL1 homomers suggests that ST6GAL1 homodimers assemble via lateral (cis) interactions. To investigate whether B4GALT1/ST6GAL1 heteromers utilize the cis or trans configuration for interaction, we expressed truncated enzyme constructs separately in COS-7 cells and measured the FRET signals for comparison. As expected, removal of the catalytic domain reduced heteromer formation by 90%, indicating that the two enzymes interact only via their catalytic domains. When one stemless construct was expressed with a WT partner (e.g. stemless B4GALT1–WT ST6GAL1 or WT B4GALT1–stemless ST6GAL1), we detected a 20% inhibition of the FRET signal. When both stem domains were removed, we found a cumulative 40% decrease in FRET signal. This loss was rescued when we inserted the B4GALT1 stem domain into the stemless ST6GAL1 construct (Fig. 5B). Because 60% of the stemless–stemless FRET signal still exists, it is our conclusion that heteromers can also interact laterally via cis configuration. However, the data do not exclude the possibility that the complexes also form via trans configuration.
Figure 5.

B4GALT1 and ST6GAL1 complexes in Golgi membranes. A, schematic of the possible configurations (cis, trans) that can be used for binding. Cis represents two enzymes interacting laterally on one side of the Golgi cisternae, whereas the trans configuration represents two enzymes interacting from opposite sides of one cisterna. Red depicts the active-site surface, and green represents the interaction interface. B, FRET assays of COS-7 cells expressing WT and truncated B4GALT1 and ST6GAL1. COS-7 cells were transfected with the depicted mVenus-tagged (for B4GALT1) and mCherry-tagged (for ST6GAL1) FRET enzyme constructs, fixed 24 h later, and quantified with the Operetta High Content Imaging system. FRET efficiencies were calculated from 10,000 to 20,000 cells and expressed as percentages of control values (mean ± S.D., n = 3) after subtracting the FRET efficiencies of negative control (MGAT1–B4GALT1) values. Statistically significant changes relative to the control samples (gray bar) are indicated (*, p < 0.05; **, p < 0.01; ***, p < 0.001).
Discussion
Glycan synthesis in the Golgi is not only dependent on the catalytic activity of the glycosyltransferases but also on their proper localization and their spatial and temporal interactions with each other. In addition, it has been shown recently that Golgi-resident glycosyltransferases also form complexes with nucleotide sugar transporters (7). This functional coupling between enzymes and their transporters is expected to enhance their catalytic activity and glycosylation efficiency, as each sugar residue can be transferred directly from the transporter to the enzyme and the acceptor substrate in the same complex before transferring the acceptor to the next complex.
So far, structural data on Golgi heteromeric assemblies do not exist. This lack of information likely stems from the difficulties in getting such complexes to crystallize in vitro. In live cells, their formation needs special conditions (pH, redox state) provided by the Golgi lumen. In this study, we focused on identifying interaction surfaces needed for assembly of B4GALT1 and ST6GAL1 homomers and their heteromeric complexes. Both enzymes reside in and interact only after their arrival in the Golgi, whereas they both form homomers in the ER (6). Previously, we showed that heteromeric coupling between them depends on the Golgi redox state and, specifically, on the formation of two surface-exposed disulfide bonds in ST6GAL1 (12).
By utilizing molecular docking simulations, directed mutagenesis of both enzymes, and FRET interaction screens, here we report seven salt bridges scattered around the predicted interaction surface and five hydrophobic contacts that are fundamental for the heteromeric interaction between B4GALT1 and ST6GAL1. The surface is surprisingly rich in exposed charged residues. In B4GALT1, the interaction surface consists of a disordered loop with one small charged helix. This loop appears to be stabilized by the Cys130-Cys172 disulfide bond. Mutagenesis of cysteine 172 to alanine significantly affected B4GALT1 binding to ST6GAL1, suggesting that loss of this particular disulfide bond most likely destabilizes the interface and therefore affects its binding affinity or capacity. Removal of the charged residues on B4GALT1 by replacing them with serine completely alleviated the interaction between B4GalT1 and ST6GAL1 without causing loss of enzyme homomers. In addition, B4GALT1 mutants M340H/E and H343A, which have been shown previously to be essential for homomer assembly, did not affect heteromeric interaction with ST6GAL1. Based on these results, we conclude that B4GALT1 possesses two distinct interaction surfaces: one surface in the active site surface for homodimerization and the other noncatalytic surface for heterodimerization. According to the homodimeric structure of B4GALT1, active sites are partially hidden in the homodimer and can explain the lower enzymatic activity of the enzyme homomers. When in the Golgi, B4GALT1 and ST6GAL1 can bind to each other using the heteromeric interaction surface on B4GALT1 and the noncatalytic surface in ST6GAL1, leaving the active sites fully accessible. This molecular arrangement is the first example of how glycosyltransferase activity can be regulated in the cell by modulating enzyme interactions.
In the case of ST6GAL1, both homo- and heterodimeric docking models overlapped at the same highly charged surface area opposite the active site. They have in common four charged residues and two hydrophobic residues. Single mutations of histidine 263 to alanine pointed out the importance of this shared residue in both interactions. In the docking models, His263 binds to Asp171 in B4GALT1 and to Asp157 in itself. Further mutagenesis of the charged amino acids on this surface almost completely abolished both interactions. It remains uncertain how ST6GAL1 homodimers disassemble to allow B4GALT1 binding to the same surface. One possibility may be that the disulfide bond formation on ST6GAL1 triggers certain conformational changes in ST6GAL1, weakening the homodimer interaction while increasing the affinity for B4GALT1. This normally takes place after the enzyme arrives in the Golgi (12). Verification of whether such conformational changes are needed for heteromeric interactions must wait until a combination of atomic structures of these complexes are solved.
Besides increasing enzymatic activity, heterodimerization has another benefit to glycosylation: it can enhance substrate channeling directly from B4GALT1 to ST6GAL1 and thereby increase the rate of glycan synthesis. In support of this view, B4GALT1 and ST6GAL1 coexpression increased cell surface galactose and sialic acid by 2- and 3-fold, respectively, compared with cells expressing B4GALT1 alone or cells expressing the mutant constructs. Nevertheless, it remains unclear whether the apparent inactivity is due to loss of its interactions or indirect consequences on the active site, perhaps via conserved regions such as sialylmotifs, which participate in binding donor and acceptor substrates and in keeping the active conformation of the enzyme (25). For example, the D336S mutation in the ST6GAL1.Ncat construct is part of such a sialylmotif (region 321–343). However, this residue appears not to be conserved in all sialyltransferases, and it also does not seem to interact with other residues, as its side chain orients outward from the catalytic domain. Therefore, it might be irrelevant for ST6GAL1 activity (26). Crystallization of the mutant ST6GAL1 is needed to clarify this point. It is important to note that even a single mutation can inactivate the enzyme, especially when it targets the active site. This does not necessarily mean that it destroys the enzyme's ability to bind to itself or to another enzyme. For instance, the B4GALT1 M340H/E mutant has been shown to be enzymatically dead (14), but it retained its ability to bind ST6GAL1. Therefore, enzyme activity is not a necessary prerequisite for the interaction. Rather, they seem to be independent phenomena.
Regarding whether the B4GALT1 and ST6GAL1 interact laterally or have contact while residing in opposite membranes of the Golgi cisterna, we showed that the truncated stem domain mutants still retained their ability to form homomers or heteromers. These findings suggest that the complexes form via lateral interactions. However, because the dimensions of the two enzymes suffice to span the cisternal space, we cannot exclude the possibility that they can also interact via trans configuration. Moreover, we showed that the catalytic domain truncations attenuated binding of B4GALT1 not only with itself but also with ST6GAL1, confirming that these interactions are mediated mainly via the catalytic domains. In contrast, ST6GAL1 homomers utilize both the catalytic and the stem domains for assembly of homomers. Structural predictions of the ST6GAL1 stem domain (data not shown) suggested the presence of two helices which, if true, could contribute to the rigidity of the stem domain. It is important to note that all truncated enzymes localized correctly to the Golgi (Figs. S2–S5) and were properly folded, excluding the possibility that the stem domain has some role in these phenomena. In contrast, it seems to be important for homodimerization of the enzyme. We also expect that the mutations themselves in the catalytic domain of the enzymes do not affect localization of the constructs within the Golgi stacks, as many fusion proteins tagged with the N terminus of B4GALT1 localize similar to B4GALT1 in the trans side of the Golgi stack.
Taken together, we have shown in cellulo that B4GALT1 uses two distinct interaction surfaces for binding itself or ST6GAL1, whereas ST6GAL1 has overlapping interaction surfaces for binding. In addition, B4GALT1 has another interaction site for ST3GAL3 and for lactalbumin that are distinct from the ones described in this report.3 From the functional point of view, the formation of B4GALT1 homomers via the active site interaction surface serves to keep the enzyme minimally active until it reaches the Golgi compartment, where its binds ST6GAL1 and exposes its active site because of disassembly of the homodimers. In contrast, the ST6GAL1 enzyme is activated only after it reaches the more oxidizing environment of the Golgi lumen, a condition that is necessary for the formation of two surface-exposed disulfide bonds in the enzyme's catalytic domain. This process may occur concomitantly with or may even helped by binding of B4GALT1 to ST6GAL1. We also provide evidence to suggest that the interactions can occur laterally and do not necessarily involve cross-cisternal contacts between the enzymes. Collectively, these findings help understand how the cooperative functioning and activity of glycosyltransferases are regulated at the molecular and cellular levels.
Experimental procedures
Molecular docking and modeling
For the docking predictions, we used the published B4GALT1 (PDB code 4EE4) and ST6GAL1 (PDB code 4JS2) structures. For B4GALT1, we considered residues 126–397 chain A and built a model of the WT structure, mutating residues T337R, T338C, and H340M using the mutation tool Coot (27); version A for the alternative positions of residues 357 and 382 was selected. For ST6GAL1, we considered residues 89–406 chain A. Docking of the heteromer interaction between B4GALT1 and ST6GAL1 was performed with ClusPro (15) with default parameters and with Haddock (16), specifying the noncatalytic surface for B4GALT1 and the entire surface for ST6GAL1 and letting the passive residues be defined automatically around the active residues. The docking of the homodimer ST6GAL1 was computed with SymmDock using a symmetry of order 2 (19, 20). The docking parameter files are available upon request.
Plasmid constructs
All glycosyltransferase expression plasmids were prepared from commercially available full-length cDNA clones (Imagenes GmbH, Berlin, Germany). Golgi-localized pcDNA3-based FRET enzyme constructs possessing C-terminal mVenus or mCherry variants as well as an HA epitope tag were prepared as described previously (22). The glycosyltransferase genes were inserted in-frame with the tags using 5′ HindIII and the 3′ XbaI restriction sites. Detailed information regarding the cloning strategies, primer sequences, and sequences of the constructs is available upon request. All constructs were sequence-verified with the ABI3500xL genetic analyzer. The MGAT1 construct was prepared as described earlier (7).
Site-directed mutagenesis and domain truncations
The B4GALT1 mutants L142D, N158A, C172A, Q206A, M340H, M340E, and H343A and the ST6GAL1 mutants R145A, Q235A, K241A, H263A, N279A, and H405A were generated by site-directed mutagenesis of the WT B4GALT1 and ST6GAL1 plasmid cDNA using the QuikChange Lightning Site-Directed Mutagenesis Kit (Agilent Technologies Inc., Santa Clara, CA). Primers containing 1-bp mutations were used for PCR amplification. The XL10 Gold Escherichia coli strain was used to clone the mutants. The mutated inserts were ligated into the pcDNA3 plasmid containing either C-terminal mVenus, mCherry, or HA tags (22) using the 5′ cloning site (HindIII) and the 3′ cloning site (XbaI). B4GALT1 STEM-truncated (Δ55–125) and CAT-truncated (Δ126–399) and ST6GAL1 STEM-truncated (Δ32–99) and CAT-truncated (Δ89–406) were generated using a similar PCR and ligation protocol. B4GALT1 Ncat mutant (D150S, E152S, K156S, K162S, R166S, and D171S), ST6GAL1 Ncat (D146S, D157S, K171S, H263S, E310S, D336S, and R402S), and Cat (K111E, K241E, R242E, and K358E) constructs in pUC57 were ordered from Genscript and ligated into the pcDNA3 plasmid containing either C-terminal mVenus, mCherry, HA, or stop codon tags. The clones were selected using ampicillin resistance as a marker. All plasmid cDNAs were sequence-verified before use. Correct Golgi localization of the mutants was also verified in transfected cells by using confocal microscopy. The primer sequences used for mutagenesis and domain truncations are available upon request. A list of all mutant constructs is shown in Table S1.
Cell maintenance and transfections
COS-7 cells were obtained from the ATCC, and Lec20 cells (Chinese hamster ovary (CHO)-K1 cell line containing a B4GALT1 loss-of-function mutation (28)) were a generous gift from Dr. Pamela Stanley. Cells were grown in DMEM supplemented with 10% fetal bovine serum, 4 mm l-glutamine, 100 units of penicillin/ml, and 100 μg of streptomycin/ml. Cells were transfected with expression plasmid(s) using either FuGENE 6TM (Promega) or Lonza Electroporation Kit R according to the manufacturer's instructions. When appropriate, cells were treated with 5 μm swainsonine or 5 μm tunicamycin 6 h post-transfection for 18 to 20 h and then fixed with 4% paraformaldehyde for 15 min and washed with PBS (pH 7.4).
Cell staining and fluorescence imaging
Fixed cells were permeabilized with 0.1% saponin and 1% BSA in PBS and stained with anti-GM130 (610822, BD Biosciences) and then Alexa Fluor 594–conjugated anti mouse secondary antibodies (Invitrogen). Cells were mounted and imaged using a Zeiss Observer Z1 microscope equipped with an LSM 700 confocal unit, Zen 2009 software (Carl Zeiss AG, Oberkochen, Germany), a ×63 Plan-Apo oil immersion objective, and appropriate filter sets for each dye. Appropriate filter sets for each fluorophore were used.
FRET analyses
FuGENE-transfected COS-7 cells with FRET plasmids (in the case of heteromeric interactions, B4GALT1 was tagged with mVenus, and ST6GAL1 was tagged with mCherry) were incubated for 18 to 20 h, seeded for 5 h in 96-well plates (Cell Carrier UltraTM, PerkinElmer Life Sciences), fixed with 4% paraformaldehyde for 15 min, and washed with PBS (pH 7.4) (6). Using OperettaTM (PerkinElmer Life Sciences), we measured the FRET signal and analyzed the data via Harmony 4.1 software as described in Ref. 7. Briefly, Golgi regions were carefully selected using mVenus contrast. Donor, acceptor, and FRET intensity raw values were collected from 10,000–20,000 cells, and FRET efficiencies were calculated. Background level intensities and overexpression crowding (negative control) values were subtracted from the raw values. Measurements were performed in at least three independent experiments.
Lectin staining
Electroporated cells with relevant plasmids were incubated for 48 h, seeded for 5 h in 96-well plates, and fixed with 4% paraformaldehyde. Cells were then quenched with 1% BSA for 1 h and stained with SNA lectin tagged with fluorescein (Vector Laboratories, FL-1301) and with RCA-I lectin tagged with rhodamine (Vector Laboratories, FL-1081) for 1 h. Cells were washed and counterstained with Hoechst dye for nuclear staining.
Sialyltransferase activity assays
Each transfected COS-7 cell pellet (100,000 cells) was suspended directly in 35 μl of radioimmune precipitation assay buffer (10 mm Tris HCl, 150 mm NaCl, and 1% Triton X-100 (pH 6.4)). Further mechanical lysis of the cells was achieved by loading the cell lysate into a 1-ml syringe and using a 26-gauge needle. After 5 min of centrifugation (14,000 rpm at 4 °C) the supernatant was used as an enzyme source. Sialylation assays were carried out using desialylated fetuin as an acceptor substrate, prepared as described previously (29). Briefly, the sialylation reaction mixture (30 μl) contained cacodylate buffer (40 mm (pH 6.2)), MnCl2 (4 mm), Triton CF-54 (0.08%, Sigma-Aldrich), CMP-Neu5Ac (50 μm: CMP-[14C]Neu5Ac (22.5 nCi, 50,000 dpm, 13.6 μm) and 36.07 μm cold CMP-Neu5Ac), desialylated fetuin (20 mg/ml), and enzyme source (6 μl), and the mixture was incubated at 37 °C for 4 h. The reaction was stopped by precipitation of glycoproteins with phosphotungstic acid (1 ml, 5% in 2 n HCl), followed by filtration on Whatman GF/A glass microfiber filters. Radiolabeled sialic acid incorporated into glycoproteins was quantified by liquid scintillation counting in UltimaGold (3 ml, PerkinElmer Life Sciences) with a Hidex 300 SL counter.
SDS-PAGE and immunoblotting
COS-7 cell pellets expressing relevant proteins were resuspended with SDS sample buffer, loaded, and run on precast Bio-Rad gels (4%–15%) in 1% SDS electrophoresis buffer. Western blotting analyses were carried out as described elsewhere (30). For immunoblotting, primary antibodies used were rabbit polyclonal anti-B4GALT1 (Sigma, HPA010807), rabbit polyclonal anti-ST6GAL1 (anti-CD75, Acris Antibodies, Herford, Germany, TA314536), and mouse monoclonal anti α-tubulin (Sigma-Aldrich, St. Louis, MO, T6074). The secondary antibodies used were either anti-mouse or anti-rabbit Fab2 fragments conjugated to horseradish peroxidase (1:10,000, Abliance, Compiègne, France). Stained protein bands were visualized using ECL reagent and a GelDoc instrument (Bio-Rad). Quantification of protein bands from the digitalized pictures was done using ImageJ software.
Statistical analyses
Statistical significance was evaluated using two-tailed t test. Each FRET experiment was repeated at least three times with 10,000 to 20,000 cells/experiment (not significant, p ≥ 0.05; *, p < 0.05; **, p < 0.01; ***, p < 0.001).
Author contributions
F. K.-A. and A. H.-L. data curation; F. K.-A., D. H., and A. H.-L. formal analysis; F. K.-A. and A. H.-L. validation; F. K.-A., D. H., G. B., T. G., and S. K. investigation; F. K.-A., D. H., and A. H.-L. visualization; F. K.-A., D. H., A. H.-L., and S. K. methodology; F. K.-A. and S. K. writing-original draft; F. K.-A., D. H., G. B., M. F. L., A. H.-L., and T. G. writing-review and editing; D. H., G. B., and M. F. L. software; M. F. L., T. G., and S. K. supervision; A. H.-L., T. G., and S. K. funding acquisition; T. G. and S. K. conceptualization; S. K. project administration.
Supplementary Material
Acknowledgments
We thank Céline Schulz (UGSF, UMR CNRS 8576, France) for excellent technical assistance with the sialylation assays on the radioactivity platform.
This work was supported by Academy of Finland Grant 285232 and CNRS. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S5 and Tables S1.
F. Khoder-Agha, D. Harrus, G. Brysbaert, M. F. Lensink, A. Hardiun-Lepers, T. Glumoff, and S. Kellokumpu, unpublished data.
- ER
- endoplasmic reticulum
- STEM
- stem domain
- CAT
- catalytic domain
- cDNA
- complementary DNA
- SNA
- Sambucus Nigra Lectin
- RCA-I
- Ricinus Communis Agglutinin
- Neu5Ac
- N-Acetylneuraminic acid.
References
- 1. Varki A., Cummings R.D., Esko J.D., Freeze H.H., Stanley P., Bertozzi C.R., Hart G.W., and Etzler M.E. (2017) in Essentials of Glycobiology, 3rd Ed., Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY: [PubMed] [Google Scholar]
- 2. Harduin-Lepers A. (2013) Sialobiology: Structure, Biosynthesis and Function: Sialic Acid Glycoconjugates in Health and Disease, (Joe T. and Ivan M. D., eds), pp 85–116, Bentham Science Publishers [Google Scholar]
- 3. Harrus D., Kellokumpu S., and Glumoff T. (2018) Crystal structures of eukaryote glycosyltransferases reveal biologically relevant enzyme homooligomers. Cell Mol. Life Sci. 75, 833–848 10.1007/s00018-017-2659-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kellokumpu S., Hassinen A., and Glumoff T. (2016) Glycosyltransferase complexes in eukaryotes: long-known, prevalent but still unrecognized. Cell Mol. Life Sci. 73, 305–325 10.1007/s00018-015-2066-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hassinen A., Rivinoja A., Kauppila A., and Kellokumpu S. (2010) Golgi N-glycosyltransferases form both homo- and heterodimeric enzyme complexes in live cells. J. Biol. Chem. 285, 17771–17777 10.1074/jbc.M110.103184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hassinen A., and Kellokumpu S. (2014) Organizational interplay of Golgi N-glycosyltransferases involves organelle microenvironment-dependent transitions between enzyme homo- and heteromers. J. Biol. Chem. 289, 26937–26948 10.1074/jbc.M114.595058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Khoder-Agha F., Sosicka P., Escriva Conde M., Hassinen A., Glumoff T., Olczak M., and Kellokumpu S. (2019) N-acetylglucosaminyltransferases and nucleotide sugar transporters form multi-enzyme-multi-transporter assemblies in Golgi membranes in vivo. Cell Mol. Life Sci. 76, 1821–1832 10.1007/s00018-019-03032-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Esko J. D., and Selleck S. B. (2002) Order out of chaos: assembly of ligand binding sites in heparan sulfate. Annu. Rev. Biochem. 71, 435–471 10.1146/annurev.biochem.71.110601.135458 [DOI] [PubMed] [Google Scholar]
- 9. McCormick C., Duncan G., Goutsos K. T., and Tufaro F. (2000) The putative tumor suppressors EXT1 and EXT2 form a stable complex that accumulates in the Golgi apparatus and catalyzes the synthesis of heparan sulfate. Proc. Natl. Acad. Sci. U.S.A. 97, 668–673 10.1073/pnas.97.2.668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bräuer P., Parker J. L., Gerondopoulos A., Zimmermann I., Seeger M. A., Barr F. A., and Newstead S. (2019) Structural basis for pH-dependent retrieval of ER proteins from the Golgi by the KDEL receptor. Science 363, 1103–1107 10.1126/science.aaw2859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Soonthornsit J., Yamaguchi Y., Tamura D., Ishida R., Nakakoji Y., Osako S., Yamamoto A., and Nakamura N. (2014) Low cytoplasmic pH reduces ER-Golgi trafficking and induces disassembly of the Golgi apparatus. Exp. Cell Res. 328, 325–339 10.1016/j.yexcr.2014.09.009 [DOI] [PubMed] [Google Scholar]
- 12. Hassinen A., Khoder-Agha F., Khosrowabadi E., Mennerich D., Harrus D., Noel M., Dimova E. Y., Glumoff T., Harduin-Lepers A., Kietzmann T., and Kellokumpu S. (2019) A Golgi-associated redox switch regulates catalytic activation and cooperative functioning of ST6Gal-I with B4GalT-I. Redox. Biol. 24, 101182 10.1016/j.redox.2019.101182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Harrus D., Khoder-Agha F., Peltoniemi M., Hassinen A., Ruddock L., Kellokumpu S., and Glumoff T. (2018) The dimeric structure of wild-type human glycosyltransferase B4GALT1. PLoS ONE 13, e0205571 10.1371/journal.pone.0205571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ramakrishnan B., Boeggeman E., and Qasba P. K. (2004) Effect of the Met344His mutation on the conformational dynamics of bovine β-1,4-galactosyltransferase: crystal structure of the Met344His mutant in complex with chitobiose. Biochemistry 43, 12513–12522 10.1021/bi049007+ [DOI] [PubMed] [Google Scholar]
- 15. Kozakov D., Hall D. R., Xia B., Porter K. A., Padhorny D., Yueh C., Beglov D., and Vajda S. (2017) The ClusPro web server for protein-protein docking. Nat. Protoc. 12, 255–278 10.1038/nprot.2016.169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. van Zundert G. C. P., Rodrigues J. P. G. L. M., Trellet M., Schmitz C., Kastritis P. L., Karaca E., Melquiond A. S. J., van Dijk M., de Vries S. J., Bonvin A. M. J. J. (2016) The HADDOCK2.2 web server: user-friendly integrative modeling of biomolecular complexes. J. Mol. Biol. 428, 720–725 10.1016/j.jmb.2015.09.014 [DOI] [PubMed] [Google Scholar]
- 17. Kuhn B., Benz J., Greif M., Engel A. M., Sobek H., and Rudolph M. G. (2013) The structure of human α-2,6-sialyltransferase reveals the binding mode of complex glycans. Acta Crystallogr. D. Biol. Crystallogr. 69, 1826–1838 10.1107/S0907444913015412 [DOI] [PubMed] [Google Scholar]
- 18. Meng L., Forouhar F., Thieker D., Gao Z., Ramiah A., Moniz H., Xiang Y., Seetharaman J., Milaninia S., Su M., Bridger R., Veillon L., Azadi P., Kornhaber G., Wells L., et al. (2013) Enzymatic basis for N-glycan sialylation: structure of rat α2,6-sialyltransferase (ST6GAL1) reveals conserved and unique features for glycan sialylation. J. Biol. Chem. 288, 34680–34698 10.1074/jbc.M113.519041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schneidman-Duhovny D., Inbar Y., Nussinov R., and Wolfson H. J. (2005) PatchDock and SymmDock: servers for rigid and symmetric docking. Nucleic Acids Res. 33, W363–7 10.1093/nar/gki481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Schneidman-Duhovny D., Inbar Y., Nussinov R., and Wolfson H. J. (2005) Geometry-based flexible and symmetric protein docking. Proteins 60, 224–231 10.1002/prot.20562 [DOI] [PubMed] [Google Scholar]
- 21. Kim S. H., Kwon D. Y., Kwak J. H., Lee S., Lee Y. H., Yun J., Son T. G., and Jung Y. S. (2018) Tunicamycin-induced ER stress is accompanied with oxidative stress via abrogation of sulfur amino acids metabolism in the liver. Int. J. Mol. Sci. 19, E4114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hassinen A., Pujol F. M., Kokkonen N., Pieters C., Kihlström M., Korhonen K., and Kellokumpu S. (2011) Functional organization of Golgi N- and O-glycosylation pathways involves pH-dependent complex formation that is impaired in cancer cells. J. Biol. Chem. 286, 38329–38340 10.1074/jbc.M111.277681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Engel B. D., Schaffer M., Albert S., Asano S., Plitzko J. M., and Baumeister W. (2015) In situ structural analysis of Golgi intracisternal protein arrays. Proc. Natl. Acad. Sci. U.S.A. 112, 11264–11269 10.1073/pnas.1515337112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ladinsky M. S., Mastronarde D. N., McIntosh J. R., Howell K. E., and Staehelin L. A. (1999) Golgi structure in three dimensions: functional insights from the normal rat kidney cell. J. Cell Biol. 144, 1135–1149 10.1083/jcb.144.6.1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Datta A. K. (2009) Comparative sequence analysis in the sialyltransferase protein family: analysis of motifs. Curr. Drug Targets 10, 483–498 10.2174/138945009788488422 [DOI] [PubMed] [Google Scholar]
- 26. Datta A. K., Chammas R., and Paulson J. C. (2001) Conserved cysteines in the sialyltransferase sialylmotifs form an essential disulfide bond. J. Biol. Chem. 276, 15200–15207 10.1074/jbc.M010542200 [DOI] [PubMed] [Google Scholar]
- 27. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lee J., Sundaram S., Shaper N. L., Raju T. S., and Stanley P. (2001) Chinese hamster ovary (CHO) cells may express six β4-galactosyltransferases (β4GalTs): consequences of the loss of functional β4GalT-1, β4GalT-6, or both in CHO glycosylation mutants. J. Biol. Chem. 276, 13924–13934 10.1074/jbc.M010046200 [DOI] [PubMed] [Google Scholar]
- 29. Harduin-Lepers A., Stokes D. C., Steelant W. F., Samyn-Petit B., Krzewinski-Recchi M., Vallejo-Ruiz V., Zanetta J. P., Auge C., and Delannoy P. (2000) Cloning, expression and gene organization of a human Neu5Ac α 2-3Gal β 1-3GalNAc α 2,6-sialyltransferase: hST6GalNAcIV. Biochem. J. 352, 37–48 [PMC free article] [PubMed] [Google Scholar]
- 30. Rivinoja A., Kokkonen N., Kellokumpu I., and Kellokumpu S. (2006) Elevated Golgi pH in breast and colorectal cancer cells correlates with the expression of oncofetal carbohydrate T-antigen. J. Cell Physiol. 208, 167–174 10.1002/jcp.20653 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.