

Abstract
Hajdu Cheney syndrome (HCS) is characterized by craniofacial developmental abnormalities, acro-osteolysis, and osteoporosis and is associated with gain–of–NOTCH2 function mutations. A mouse model of HCS termed Notch2tm1.1Ecan harboring a mutation in exon 34 of Notch2 replicating the one found in HCS was used to determine whether the HCS mutation sensitizes the skeleton to the osteolytic effects of tumor necrosis factor α (TNFα). TNFα injected over the calvarial vault caused a greater increase in osteoclast number, osteoclast surface, and eroded surface in Notch2tm1.1Ecan mice compared with littermate WT controls. Accordingly, the effect of TNFα on osteoclastogenesis was greatly enhanced in cultures of bone marrow–derived macrophages (BMMs) from Notch2tm1.1Ecan mice when compared with the activity of TNFα in control cultures. TNFα induced the expression of Notch2 and Notch2 mutant mRNA by ∼2-fold, possibly amplifying the NOTCH2-dependent induction of osteoclastogenesis. The effect of TNFα on osteoclastogenesis in Notch2tm1.1Ecan mutants depended on NOTCH2 activation because it was reversed by anti-NOTCH2 negative regulatory region and anti-jagged 1 antibodies. The inactivation of Hes1 prevented the TNFα effect on osteoclastogenesis in the context of the Notch2tm1.1Ecan mutation. In addition, the induction of Il1b, but not of Tnfa and Il6, mRNA by TNFα was greater in Notch2tm1.1Ecan BMMs than in control cells, possibly contributing to the actions of TNFα and NOTCH2 on osteoclastogenesis. In conclusion, the HCS mutation enhances TNFα-induced osteoclastogenesis and the inflammatory bone-resorptive response possibly explaining the acro-osteolysis observed in affected individuals.
Keywords: cytokine, inflammation, Notch receptor, osteoclast, tumor necrosis factor (TNF), Hajdu Cheney syndrome, osteolysis
Introduction
NOTCH receptors 1–4 are single-pass type I transmembrane proteins that play a central role in cell fate determination and function (1, 2). In the skeleton, Notch signaling regulates development and homeostasis by controlling the differentiation and function of bone cells, including osteoblasts, osteoclasts, chondrocytes, and osteocytes (3–9). In mammals, there are five ligands for the Notch receptors: namely jagged (JAG)1, JAG2, delta-like (DLL)1, DLL3, and DLL4 (10). Activation of NOTCH receptors follows their interactions with ligands on adjacent cells, resulting in the cleavage of NOTCH by a disintegrin and metalloprotease (ADAM) and the γ-secretase complex and the release of the NOTCH intracellular domain (NICD)2 (11, 12). The NICD translocates into the nucleus to form a complex with mastermind-like and recombination signal-binding protein for the immunoglobulin κ region (RBPJκ) and induce the expression of its target genes hairy enhancer of split (Hes) and HES-related with YRPW motif (Hey) (2, 13, 14 Although NOTCH receptors share structural and some biological functions, it is important to note that each NOTCH receptor exerts distinct effects on the skeleton; these are in part related to specific patterns of cellular expression of each receptor (11).
Hajdu Cheney syndrome (HCS) is a rare inherited disease characterized by craniofacial developmental abnormalities, acro-osteolysis, short stature, and osteoporosis (15–17). HCS is caused by point mutations or short deletions in exon 34 of NOTCH2 that lead to the creation of a stop codon upstream of the proline (P), glutamic acid (E), serine (S), and threonine (T)-rich (PEST) domain (18–22). The PEST domain is recognized by the E3 ligase complex for ubiquitin-mediated degradation of NOTCH2. Therefore, the mutations result in the translation of a truncated NOTCH2 protein resistant to ubiquitin-dependent degradation and a gain–of–NOTCH2 function (23). To investigate the mechanisms responsible for HCS, we created a mouse model termed Notch2tm1.1Ecan harboring a point mutation (6955C→T) in exon 34 of Notch2 upstream of the PEST domain. Heterozygous Notch2tm1.1Ecan mice exhibit cancellous and cortical bone osteopenia due to increased osteoclast number and bone resorption (5). Notch2tm1.1Ecan mice also display a reallocation of B cells to the marginal zone of the spleen, shortening of the limbs, and sensitization to the development of osteoarthritis in destabilized joints (24, 25). This is possibly because of increased expression of interleukin (IL) 6, revealing a propensity to an enhanced inflammatory response (24). Notch2tm1.1Ecan does not exhibit apparent acro-osteolysis, and homozygous mice display craniofacial dysmorphism and newborn lethality.3 The skeletal phenotype of Notch2tm1.1Ecan reproduces the human syndrome, and iliac crest biopsies from humans afflicted by HCS reveal osteopenia and trabecularization of cortical bone (26).
Histological examination of biopsies from the acro-osteolysis lesions in subjects with HCS reveal the presence of an inflammatory process and neovascularization, but the mechanisms responsible for the bone lysis are not known (17, 27–29). Tumor necrosis factor α (TNFα) is a proinflammatory cytokine primarily produced by activated macrophages. TNFα induces gene expression of Il6 and Il1b as well as its own expression (30, 31). TNFα, IL6, and IL1β enhance the differentiation of cells of the myeloid lineage toward osteoclasts and increase the bone-resorbing activity of mature osteoclasts (32–39).
The excessive release of TNFα, IL6, and IL1β during the inflammatory response perturbs bone homeostasis and promotes pathologic bone erosion and may be mechanistically involved in the acro-osteolysis of HCS (40–42). Therefore, we asked the question whether the HCS mutation sensitizes the skeleton to the osteolytic actions of TNFα. To this end, we examined the effects of TNFα on bone resorption in vivo and on osteoclastogenesis in vitro in Notch2tm1.1Ecan mice and mechanisms responsible. Because we have found no differences in phenotypic manifestations between male and female Notch2tm1.1Ecan mice, the studies were conducted in male mice and sex-matched littermate controls.
Results
Hajdu Cheney Notch2tm1.1Ecan mutation enhances TNFα-induced osteolysis in calvarial bone
To examine whether the Hajdu Cheney mutation sensitizes mice to the osteolytic actions of TNFα, Notch2tm1.1Ecan mice and control littermates were administered TNFα or PBS as a vehicle control by subcutaneous injection over the calvarial vault once a day for 4 days. Tartrate-resistant acid phosphatase (TRAP)/hematoxylin-stained calvarial sections revealed that TNFα administration increased the number of TRAP-positive multinucleated cells and osteolysis in Notch2tm1.1Ecan and littermate control mice. The effect was more pronounced in Notch2tm1.1Ecan mice, and osteoclast number, osteoclast surface, and eroded surface were 1.7-fold higher in Notch2tm1.1Ecan calvarial bones than in controls (Fig. 1).
Figure 1.
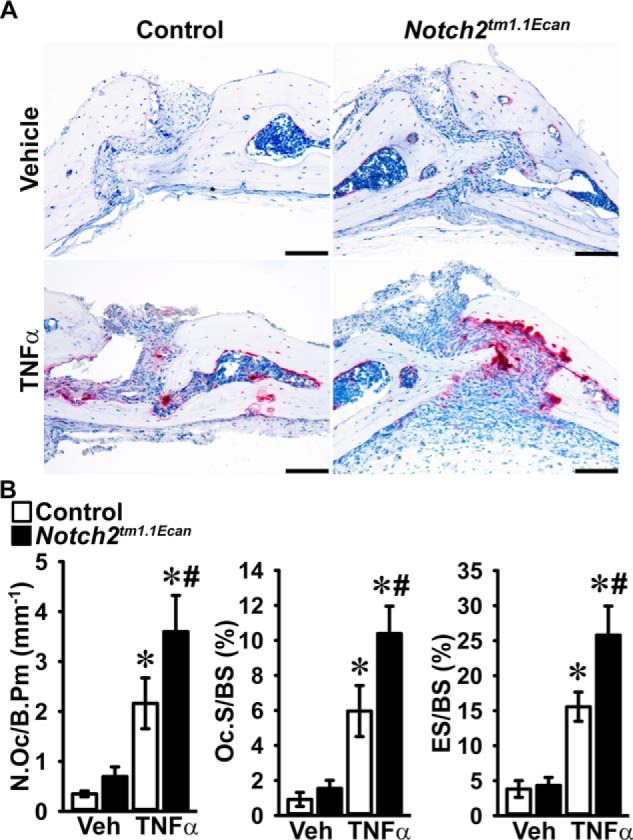
Hajdu Cheney mice display greater osteoclast number and eroded surface than littermate controls following TNFα treatment. Calvarial bones of Notch2tm1.1Ecan and sex-matched littermate control mice were administered 2 μg TNFα or PBS (vehicle, Veh) once a day for 4 days over the calvarial vault by subcutaneous injection. A, representative images of histological sections of calvariae stained with TRAP and hematoxylin showing increased number of osteoclasts and eroded surface in TNFα-treated Notch2tm1.1Ecan mice. The scale bars in the right corner represent 100 μm. B, bone histomorphometric analysis of the calvarial bones from Notch2tm1.1Ecan (black bars) mutant mice and littermate controls (white bars). Values are means ± S.D.; vehicle n = 4 and TNFα n = 8 biological replicates for control and Notch2tm1.1Ecan mice, respectively. Parameters shown are as follows: number of osteoclasts/bone perimeter (N.Oc/B.Pm); osteoclast surface/bone surface (Oc.S/BS); eroded surface/bone surface (ES/BS). *, significantly different between TNFα and vehicle, p < 0.05. #, significantly different between Notch2tm1.1Ecan and control, p < 0.05.
Hajdu Cheney mutation enhances TNFα-induced osteoclastogenesis in vitro
TNFα acts directly and indirectly to induce osteoclastogenesis by promoting the osteoclastogenic potential of osteoclast precursors and by increasing receptor activator of NF-κB (NF-κB) ligand (RANKL) expression in osteoblasts (43, 44). To confirm a direct effect of TNFα on osteoclastogenesis in the context, or not, of the Hajdu Cheney mutation, bone marrow-derived macrophages (BMMs) from Notch2tm1.1Ecan and control littermates were cultured in the presence of macrophage colony-stimulating factor (M-CSF) and TNFα. The effect of TNFα on osteoclastogenesis was enhanced in cultures of Notch2tm1.1Ecan BMMs compared with the effects of TNFα in control cultures (Fig. 2). Although Notch2tm1.1Ecan BMMs were sensitized to the action of TNFα, there was no difference in Tnfr1 and Tnfr2 mRNA expression between Notch2tm1.1Ecan and control mice either in vivo in calvariae or in vitro in BMM cultures (Fig. 3). The effect of TNFα on early signal activation of mitogen-activated protein kinases and IκBα was comparable between Notch2tm1.1Ecan and control BMMs, although a greater induction of AKT phosphorylation was observed in Notch2tm1.1Ecan cultures than in control cultures treated with TNFα (Fig. 3). TNFα treatment induced NF-κB activation in BMMs of both genotypes, as defined by enhanced NF-κB binding to consensus DNA sequences, but there was no difference in NF-κB activation between Notch2tm1.1Ecan and control BMM cultures (Fig. 3). The results suggest that the enhanced osteoclastogenic response of Notch2tm1.1Ecan cells to TNFα was independent of NF-κB activation and possibly related to enhanced AKT phosphorylation.
Figure 2.
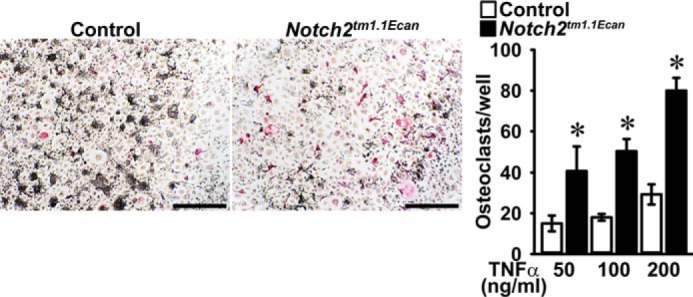
Hajdu Cheney mutant BMMs are sensitized to the action of TNFα on osteoclastogenesis. BMMs derived from 2-month-old Notch2tm1.1Ecan mice and control littermates were cultured in the presence of M-CSF at 30 ng/ml and of TNFα at 50, 100, and 200 ng/ml for 6 days. Cells were stained with TRAP, and representative images of TRAP-stained multinucleated cells are shown. The scale bar in the right corner represents 500 μm. TRAP-positive cells with more than three nuclei were considered osteoclasts, and values are means ± S.D.; n = 4 biological replicates for control (white bars) and Notch2tm1.1Ecan (black bars) cells. *, significantly different between Notch2tm1.1Ecan and control, p < 0.05.
Figure 3.

TNFα receptor expression and TNFα-induced early signal activation are not altered in Hajdu Cheney mutants. A, total RNA was extracted from calvarial bones (left) or BMMs (right) of Notch2tm1.1Ecan and sex-matched littermate control mice and examined for relative Tnfr1 and Tnfr2 gene expression by qRT-PCR, corrected for Rpl38 copy number. Values are means ± S.D.; n = 4 biological replicates for control (white bars) and Notch2tm1.1Ecan (black bars) calvarial bones or BMMs. B–D, BMMs derived from 2-month-old Notch2tm1.1Ecan mice and control littermates were cultured for 2 h in the absence of serum and exposed to TNFα at 200 ng/ml for the indicated periods of time, and whole-cell lysates (35 μg of total protein except panel C, 15 μg of total protein for C) were examined by immunoblotting. B, using anti-p-p38, p-ERK, and p-JNK antibodies, stripped and reprobed with anti-p38, ERK, and JNK antibodies. C, using anti-p-AKT antibodies and reprobed with anti-AKT antibodies. D, using anti-p-IκBα or β-Actin antibodies, stripped and reprobed with IκBα antibodies. The band intensity was quantified by ImageLabTM software (version 5.2.1), and the numerical ratios of phosphorylated/unphosphorylated signal in B and C or of p-IκBα/β-Actin and of IκBα/β-Actin in D are shown below each blot. Control ratios at day 0 are normalized to 1. E, BMMs from 2-month-old Notch2tm1.1Ecan mice and control littermates were cultured for 2 h in the absence of serum and exposed to TNFα at 200 ng/ml for 1 h. 20 μg of nuclear extracts for each sample were examined by TransAMTM Flexi NF-κB p65 activation assay kit, and colorimetric changes were measured at 450 nm. Values are means ± S.D.; n = 3 technical replicates for control (white bars) and Notch2tm1.1Ecan (black bars) BMMs. *, significantly different compared with control without TNFα, p < 0.05.
TNFα promotes the expression of Notch2 and proinflammatory cytokines
To test for the acute effect of TNFα on gene expression, BMMs from Notch2tm1.1Ecan mice and control littermates were treated with TNFα for 6 and 18 h. TNFα induced the expression of Notch2 mRNA in Notch2tm1.1Ecan and control BMMs. Notch2 mutant (Notch26955C→T) transcripts were detected only in Notch2tm1.1Ecan cells, and their expression was enhanced by TNFα. Hes1 mRNA levels were significantly increased in Notch2tm1.1Ecan BMMs, but they were not affected by treatment with TNFα. The expression of Tnfa, Il6, and Il1b was significantly increased by TNFα, but only the induction of Il1b was greater in Notch2tm1.1Ecan BMMs than in control cultures (Fig. 4). To examine for changes in gene expression during TNFα-induced osteoclast differentiation, Notch2tm1.1Ecan and control BMMs were cultured in the presence of M-CSF and TNFα for 3 and 6 days. TNFα induced Notch2 and Notch26955C→T transcripts by up to 2-fold. Hes1 mRNA expression was increased in Notch2tm1.1Ecan cells but was not altered by TNFα (Fig. 5). TNFα induced Tnfa, Il6, and Il1b in both Notch2tm1.1Ecan and control osteoclasts, but only Il1b was increased in Notch2tm1.1Ecan osteoclasts to a greater extent than in control cells (Fig. 5). Osteoclastogenic gene markers, such as Acp5 and Ctsk, were up-regulated during TNFα-induced osteoclastogenesis and were significantly greater in Notch2tm1.1Ecan osteoclasts than in control cells (Fig. 5). The NF-κB–dependent Nfatc1 gene was up-regulated by TNFα; but in accordance with the results on NF-κB activation, its induction was of equal magnitude in control and Notch2tm1.1Ecan cells.
Figure 4.

TNFα enhances the expression of Notch2 and proinflammatory cytokines in Hajdu Cheney mutant and control BMMs. BMMs derived from 2-month-old Notch2tm1.1Ecan mice and control littermates were cultured for the indicated periods of time in the presence of M-CSF at 30 ng/ml and TNFα at 200 ng/ml. Total RNA was extracted, and gene expression was determined by qRT-PCR. Data are expressed as Notch26955C→T, Notch2, Hes1, Tnfa, Il6, and Il1b, corrected for Rpl38 copy number. Values are means ± S.D.; n = 4 biological replicates for control (white bars) and Notch2tm1.1Ecan (black bars) BMMs. *, significantly different compared with time 0, p < 0.05. #, significantly different between Notch2tm1.1Ecan and control, p < 0.05.
Figure 5.

Expression of Notch2 and proinflammatory cytokines is increased during TNFα-induced osteoclastogenesis. BMMs derived from 2-month-old Notch2tm1.1Ecan mice and control littermates were cultured in the presence of M-CSF at 30 ng/ml and TNFα at 200 ng/ml for 6 days. Total RNA was extracted, and gene expression was determined by qRT-PCR. Data are expressed as Notch26955C→T, Notch2, Hes1, Tnfa, Il6, Il1b, Nfatc1, Acp5, and Ctsk, corrected for Rpl38 copy number. Values are means ± S.D.; n = 4 biological replicates for control (white bars) and Notch2tm1.1Ecan (black bars) cells. *, significantly different compared with day 0, p < 0.05. #, significantly different between Notch2tm1.1Ecan and control, p < 0.05.
TNFα accelerates NOTCH2 signal activation and increases JAG1 expression
NOTCH signaling is activated following interactions with ligands of the JAG and DLL families. In previous work, we found that Jag1, but not Jag2 or Dll1, Dll3, and Dll4 transcripts, is expressed as BMMs differentiate toward osteoclasts (11). Jag1 mRNA and JAG1 protein levels were increased about 1.4- and 3-fold, respectively, during TNFα-induced osteoclastogenesis, but the induction was of equal magnitude in Notch2tm1.1Ecan and control osteoclasts (Fig. 6). In accordance with the increase in Notch2 mRNA during osteoclastogenesis, the levels of NOTCH2 were increased as BMMs matured as osteoclasts in the presence of TNFα. The NOTCH2 intracellular domain (N2ICD), representative of NOTCH2 signal activation and cleavage of NOTCH2, was increased in Notch2tm1.1Ecan and control osteoclasts following TNFα treatment. Although N2ICD was increased in both Notch2tm1.1Ecan and control cells, the truncated form of NOTCH2, lacking the PEST domain (N2ICDΔPEST), was only detected in Notch2tm1.1Ecan cells and increased during differentiation. Therefore, the total levels of N2ICD, intact and truncated, were 2-fold greater in Notch2tm1.1Ecan cells than in control cells (Fig. 6). HES1 levels were 2-fold greater in Notch2tm1.1Ecan cultures, but NFATc1 was increased to an equal extent in Notch2tm1.1Ecan and control cultures as they differentiated toward osteoclasts in the presence of TNFα (Fig. 6).
Figure 6.

TNFα accelerates NOTCH2 signal activation during osteoclast differentiation and enhances JAG1 expression. BMMs derived from 2-month-old Notch2tm1.1Ecan mice and control littermates were cultured in the presence of M-CSF at 30 ng/ml and of TNFα at 200 ng/ml for 6 days. A, total RNA was extracted, and gene expression was determined by qRT-PCR. Data are expressed as Jag1, corrected for Rpl38 copy number. Values are means ± S.D.; n = 4 biological replicates for control (white bars) and Notch2tm1.1Ecan (black bars) cells. *, significantly different compared with day 0, p < 0.05. B, whole-cell lysates (35 μg of total protein) were examined by immunoblotting using anti-JAG1, NOTCH2 and N2ICD, HES1, NFATc1, and β-Actin antibodies. The band intensity was quantified by ImageLabTM software (version 5.2.1), and the numerical ratio of JAG1/β-Actin, NOTCH2/β-Actin, N2ICD (including N2ICDΔPEST)/β-Actin, HES1/β-Actin, and NFATc1/β-Actin is shown below each blot. All control ratios at day 0 are normalized to 1.
Preventing NOTCH2 signaling reverses the sensitizing effect of the Hajdu Cheney mutation on TNFα-induced osteoclastogenesis
To determine whether preventing NOTCH2 signal activation can reverse the effect of the Notch2tm1.1Ecan mutation on TNFα-induced osteoclastogenesis, BMMs from Notch2tm1.1Ecanmice and control littermates were cultured in the presence of M-CSF and TNFα with antibodies directed to the NRR of NOTCH2 or with anti-JAG1 antibodies (45–47). ΤΝFα induced osteoclastogenesis in Notch2tm1.1Ecan BMMs by ∼1.6–1.7-fold, an effect that was reversed by anti-NOTCH2 NRR and by anti-JAG1 antibodies (Fig. 7). Moreover, anti-JAG1 antibodies reduced osteoclast differentiation in control as well as in Notch2tm1.1Ecan cultures treated with TNFα, demonstrating that NOTCH signal activation is a requirement for TNFα-dependent osteoclastogenesis (Fig. 7).
Figure 7.
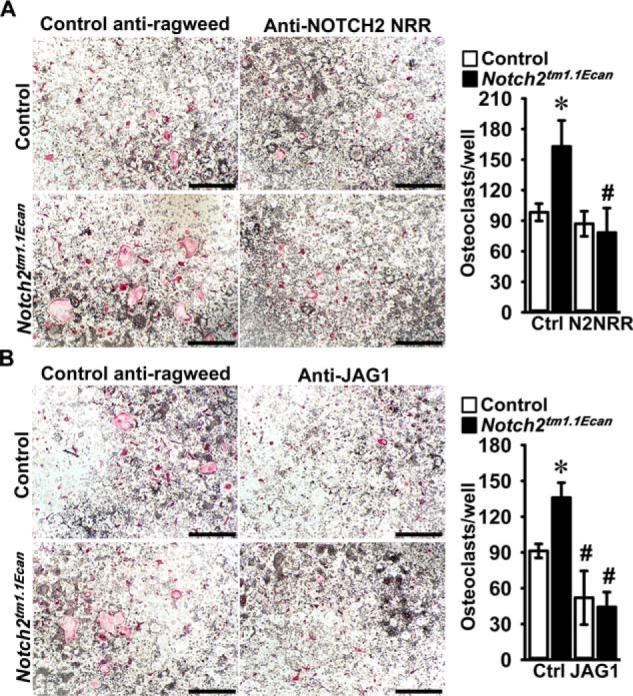
Preventing NOTCH2 signal activation reverses the effect of the Hajdu Cheney mutation on TNFα-induced osteoclastogenesis. BMMs derived from 2-month-old Notch2tm1.1Ecan mice and control littermates were cultured with M-CSF at 30 ng/ml and TNFα at 200 ng/ml in the presence of control anti-ragweed at 10 or 20 μg/ml (Ctrl) or anti-NOTCH2 NRR (N2NRR) at 10 μg/ml (A), or anti-JAG1 (JAG1) at 20 μg/ml (B) for 6 days. A and B, representative images of TRAP-stained multinucleated cells obtained after 6 days of culture are shown. The scale bars in the right corners represent 500 μm. TRAP-positive cells with more than three nuclei were considered as osteoclasts, and values are means ± S.D. (top and bottom right); n = 4 biological replicates for control (white bars) and Notch2tm1.1Ecan (black bars). *, significantly different between Notch2tm1.1Ecan and control, p < 0.05. #, significantly different between anti-NOTCH2 NRR or anti-JAG1 and control anti-ragweed antibodies, p < 0.05.
Inactivation of Hes1 reverses the sensitizing effect of the Hajdu Cheney mutation on TNFα-induced osteoclastogenesis
In preliminary experiments, we demonstrated that Hes1 is expressed in BMMs, and its expression increases during osteoclastogenesis, whereas Hey1, Hey2, and HeyL transcripts are not detected in this cell lineage (11). To examine the effect of HES1 on osteoclastogenesis in Notch2tm1.1Ecan cells, osteoclast precursors from Notch2tm1.1Ecan;Hes1loxP/loxP and Hes1loxP/loxP littermate controls were transduced with adenoviruses carrying CMV-Cre (Ad-Cre) or GFP (Ad-GFP) control vectors. Hes1 mRNA levels were decreased by 55–80% in Notch2tm1.1Ecan;Hes1Δ/Δ and Hes1Δ/Δ osteoclasts transduced with Ad-Cre compared with Notch2tm1.1Ecan;Hes1loxP/loxP and Hes1loxP/loxP cells transduced with Ad-GFP. Notch2 and Notch26955C→T mutant transcripts were not affected by the Hes1 inactivation, whereas the down-regulation of Hes1 decreased the Il1b induction observed in Notch2tm1.1Ecan cells (Fig. 8). Notch2tm1.1Ecan;Hes1loxP/loxP osteoclast precursors treated with TNFα exhibited a 1.5-fold increase in osteoclast number compared with Hes1loxP/loxP cells. Osteoclast number was decreased by 60% in Notch2tm1.1Ecan;Hes1Δ/Δ and decreased by about 30% in Hes1Δ/Δ cells so that the Hes1 inactivation reversed the TNFα effect on osteoclastogenesis in the context of the Notch2tm1.1Ecan mutation and reduced the effect of TNFα in control cultures (Fig. 8).
Figure 8.
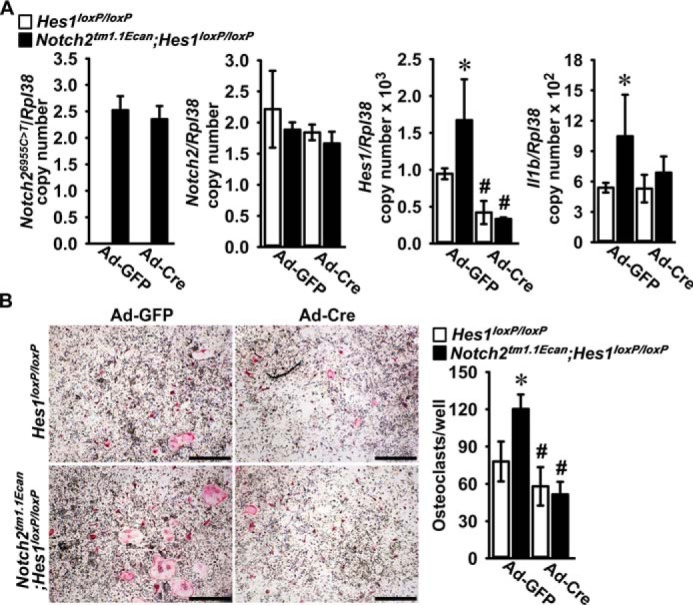
Hes1 inactivation reverses the effect of the Hajdu Cheney mutation on TNFα-induced osteoclastogenesis. Osteoclast precursors derived from 2-month-old Notch2tm1.1Ecan;Hes1loxP/loxP and Hes1loxP/loxP littermate controls were transduced with adenoviruses carrying CMV-Cre (Ad-Cre) or GFP (Ad-GFP) as control at m.o.i. 100 and cultured with M-CSF at 30 ng/ml and TNFα at 200 ng/ml for 3 days until the formation of multinucleated TRAP-positive cells. A, total RNA was extracted, and gene expression was determined by qRT-PCR. Data are expressed as Notch26955C→T, Notch2, Hes1, and Il1b, corrected for Rpl38 copy number. Values are means ± S.D.; n = 4 technical replicates for Hes1loxP/loxP (white bars) and Notch2tm1.1Ecan;Hes1loxP/loxP (black bars) cells transduced with Ad-Cre or Ad-GFP. B, representative images of TRAP-stained multinucleated cells are shown. The scale bars in the right corner represent 500 μm. TRAP-positive cells with more than three nuclei were considered osteoclasts, and values are means ± S.D.; n = 4 technical replicates for Hes1loxP/loxP (white bars) and Notch2tm1.1Ecan;Hes1loxP/loxP (black bars) transduced with Ad-Cre or Ad-GFP. *, significantly different between Notch2tm1.1Ecan;Hes1loxP/loxP and Hes1loxP/loxP control, p < 0.05. #, significantly different between Ad-Cre and Ad-GFP, p < 0.05.
Preventing NOTCH2 signaling reverses the sensitizing effect of the Hajdu Cheney mutation on TNFα-induced osteolysis
To examine whether preventing NOTCH2 signal activation can reverse the effect of the Notch2tm1.1Ecan mutation on TNFα-induced osteolysis, Notch2tm1.1Ecan mice and control littermates were administered anti-NOTCH2 NRR or control anti-ragweed antibodies with TNFα by subcutaneous injection over the calvarial vault once a day for 4 days. TRAP/hematoxylin-stained calvarial sections revealed that osteoclast number, osteoclast surface, and eroded surface were 2-fold higher in TNFα-treated Notch2tm1.1Ecan calvarial bones than in TNFα-treated WT controls. The effect of the Notch2tm1.1Ecan mutation was reversed by the administration of anti-NOTCH2 NRR antibodies, and osteoclast number, osteoclast surface, and eroded surface were significantly reduced compared with anti-ragweed–treated Notch2tm1.1Ecan mice (Fig. 9). As a consequence, osteoclast number and surface were no longer different between TNFα-treated Notch2tm1.1Ecan and control mice; the anti-NOTCH2 NRR antibody also reduced eroded surface in control mice (Fig. 9).
Figure 9.
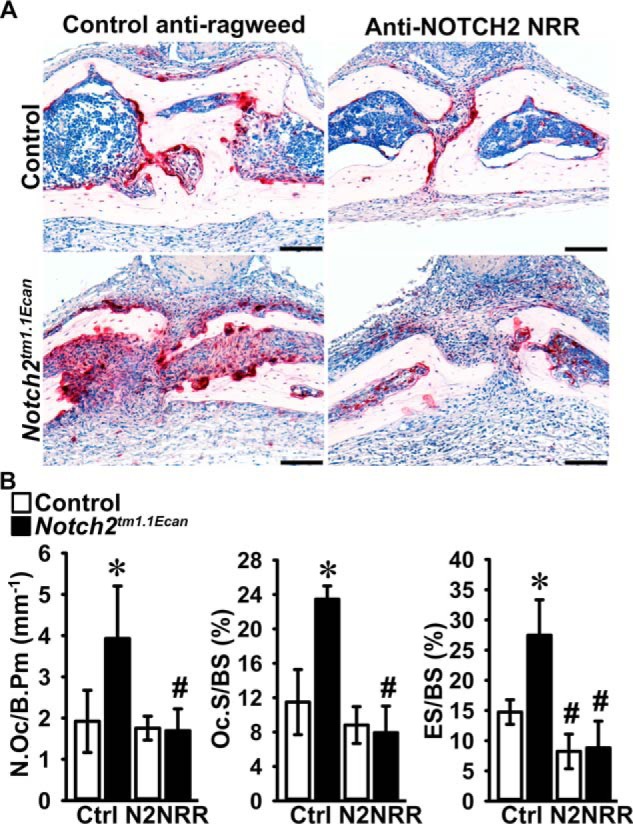
Preventing NOTCH2 signal activation reverses the effect of the Hajdu Cheney mutation on TNFα-induced osteolysis. Calvarial bones from Notch2tm1.1Ecan and sex-matched littermate control mice were administered 2 μg of TNFα with anti-NOTCH2 NRR or control anti-ragweed antibodies at a dose of 10 mg/kg once a day for 4 days over the calvarial vault by subcutaneous injection. A, representative images of histological sections of calvariae stained with TRAP and hematoxylin showing reversal of the TNFα-induced osteolysis in Notch2tm1.1Ecan mice by anti-NOTCH2 NRR antibodies. Scale bars in the right corner represent 100 μm. B, bone histomorphometric analysis of calvarial bones from Notch2tm1.1Ecan (black bars) mutant mice and littermate controls (white bars). Values are means ± S.D.; control anti-ragweed antibody (Ctrl) n = 4–5 and anti-NOTCH2 NRR (N2NRR) n = 5 biological replicates for control and Notch2tm1.1Ecan mice, respectively. Parameters shown are as follows: number of osteoclasts/bone perimeter (N.Oc/B.Pm); osteoclast surface/bone surface (Oc.S/BS); and eroded surface/bone surface (ES/BS). *, significantly different between Notch2tm1.1Ecan and control, p < 0.05. #, significantly different between anti-NOTCH2 NRR and control anti-ragweed antibodies, p < 0.05.
Notch2tm1.1Ecan mice have normal serum TNFα levels
To determine whether mice harboring the Notch2tm1.1Ecan mutation have altered serum levels of TNFα, serum was obtained from Notch2tm1.1Ecan and control littermates. At 2 months of age, serum TNFα was (means ± S.D.; n = 3–4) 15.5 ± 0.2 pg/ml in control and 15.3 ± 0.1 pg/ml in Notch2tm1.1Ecan mice; at 12 months of age TNFα was 15.8 ± 0.4 pg/ml in control and 15.6 pg/ml in Notch2tm1.1Ecan mice (both p > 0.05).
Discussion
In this study, we demonstrated that the TNFα-induced osteoclastogenesis and the inflammatory bone-resorptive response to TNFα are enhanced in a mouse model of HCS (Fig. 10). The effect of TNFα required the activation of NOTCH2 signaling because it was reversed in vitro and in vivo by anti-NOTCH2 NRR and by anti-JAG1 antibodies. Moreover, anti-JAG1 antibodies inhibited control and Notch2tm1.1Ecan mutant-dependent osteoclastogenesis demonstrating that NOTCH activation is necessary for optimal osteoclast differentiation secondary to TNFα.
Figure 10.

Schematic model to show the effects of the Hajdu Cheney mutation on TNFα-induced osteolysis. The blue arrows indicate signal activation. The red arrows and boxes in bold represent increased signal activation and expression in Hajdu Cheney mutants.
A limitation of the in vivo experiments is that they were conducted in male Notch2tm1.1Ecan and sex-matched controls, and as a consequence caution should be exerted before extrapolating the results to female mice. Recently, an alternative mouse model of HCS with a 6272delT in exon 34 of Notch2 was reported, and mice were studied up to 12 months of age (48). Like Notch2tm1.1Ecan mutants, these mice developed osteopenia secondary to increased bone resorption; the major difference between Notch2tm1.1Ecan and mice harboring the 6272delT mutation is that the latter exhibit increased bone formation and high bone turnover (48). Neither mouse model exhibited acro-osteolysis. This could suggest that environmental factors or vascular injury are required in addition to the inflammatory component for the development of acro-osteolysis. These additional factors do not seem to occur in the available mouse models of the disease that are suitable to examine the inflammatory component of the syndrome but not the fully established acro-osteolysis.
Our results are in contrast to previous work demonstrating that RBPJκ, a component of Notch canonical signaling, inhibits TNFα-induced osteoclastogenesis by suppressing Nfatc1 (49). It is possible that RBPJκ acts directly on osteoclastogenesis and independently of Notch signaling or that the effects of NOTCH2 and HES1 on osteoclastogenesis are independent of canonical Notch signaling. However, we and others have consistently demonstrated a stimulatory effect of NOTCH2 on osteoclastogenesis that is congruent with the results observed in this work (5, 7, 23).
NOTCH activation results in the induction of Hes and Heys, and cells of the osteoclast lineage express Hes1 and low levels of Hes3 and Hes5 mRNA but do not express Hey1, Hey2, or HeyL transcripts (11). Hes1 expression levels were increased in Notch2tm1.1Ecan mutant cells and played a role in the TNFα-mediated osteoclastogenic effect because this was no longer detected following the inactivation of Hes1. Whereas HES1 plays an inhibitory role in osteoblast differentiation, and its overexpression in osteoblasts causes osteopenia, there is virtually no knowledge regarding its function in osteoclast differentiation or function (50). It is likely that HES1 plays a critical role in osteoclastogenesis and that its function is not limited to the osteoclastogenesis occurring during an inflammatory state.
It has been reported that toll-like receptor signaling and proinflammatory cytokines, such as TNFα and IL1β, induce gene expression of NOTCH receptors and ligands as well as signal activation of NOTCH in several cells and tissues (51). TNFα increased the expression of JAG1 and NOTCH2 during osteoclast differentiation to a similar extent in Notch2tm1.1Ecan and control cells. However, only Notchtm1.1Ecan mutant cells synthesized the truncated form of N2ICD (N2ICDΔPEST) and the intact N2ICD. The summation of the intact and truncated forms of N2ICD resulted in an ∼2-fold greater expression of N2ICD in Notch2tm1.1Ecan mutants than in control cells. The N2ICDΔPEST is more stable than WT N2ICD because it is resistant to ubiquitin-mediated degradation, explaining the gain–of–NOTCH2 function and the Hes1 induction in Notch2tm1.1Ecan cells (23, 52). The direct effects of NOTCH2 signaling and HES1 on TNFα-induced osteolysis and osteoclast differentiation in the context of the Notch2tm1.1Ecan mutation were reversed by treatment with anti-NOTCH2 NRR and anti-JAG1 antibodies and by the Hes1 inactivation. Moreover, anti-JAG1 antibodies and the down-regulation of Hes1 tempered the effects of TNFα in control cultures. This indicates that the effects of TNFα on osteoclastogenesis require NOTCH signal activation and HES1 expression.
Notch2tm1.1Ecan mutant cells displayed greater Il1b mRNA levels than control cells. The induction of Il1b was HES1-dependent, and this is in agreement with observations in alternative cellular systems (53). Notch signaling and HES1 inhibit the phosphatase and tensin homolog (PTEN) and as a consequence up-regulate the PI3K–AKT signaling pathway, and PI3K–AKT signaling enhances IL1β expression (54–56). In this study, we found greater induction of phospho-AKT by TNFα in Notch2tm1.1Ecan BMM cultures. Therefore, it is possible that HES1 signals through the PI3K–AKT pathway to enhance osteoclastogenesis and induce IL1β (57). IL1β is involved in the bone-resorbing activity of osteoclasts and in osteoclast formation (37). IL1β might accelerate TNFα-induced osteolysis by increasing the bone-resorbing activity of osteoclasts in Notch2tm1.1Ecan mice in vivo and contribute to the induction of Hes1 mRNA, as was reported in chondrocytes (58).
RANKL and TNFα signaling activate transcription factor NF-κB. It has been reported that RANKL-induced N2ICD associates with p65 subunit of NF-κB to enhance the transcriptional activity of Nfatc1 (7). We confirmed that TNFα induced NF-κB activation; however, this was not different between Notch2tm1.1Ecan and control cultures. In accordance with this finding, the expression of target genes dependent on NF-κB activation, including Tnfa, Il6, and Nfatc1, but not Il1b, was not different between Notch2tm1.1Ecan and control cultures treated with TNFα. This would suggest that mechanisms independent of NF-κB activation are responsible for the induction of IL1β as well as for the enhanced osteoclastogenic response of Notch2tm1.1Ecan mice to TNFα. This could entail either direct effects of the N2ICD or effects of HES1 on osteoclastogenesis, possibly by inducing AKT phosphorylation as a result of an inhibition of PTEN.
Serum levels of TNFα were not different between Notch2tm1.1Ecan and control mice. In addition, the serum from Notch2tm1.1Ecan and control mice was examined by Proteome Profiler Mouse Cytokine Array (R&D Systems, Minneapolis, MN) to address whether other proinflammatory cytokines were up-regulated in the systemic circulation of Notch2tm1.1Ecan mice. Few cytokines, including CXC motif chemokine ligand 13, complement component 5a, CD54, M-CSF, and stromal cell-derived factor 1, were detected in both Notch2tm1.1Ecan and control serum, although there was no significant difference between genotypes (data not shown). These findings coincide with the RNA analysis of Notch2tm1.1Ecan BMMs, where the induction of inflammatory cytokines was comparable between Notch2tm1.1Ecan and control cells and observed only after TNFα stimulation. These observations suggest that TNFα is required for Notch2tm1.1Ecan mice to exhibit an inflammatory response. However, the circumstances leading to a possible increase in local or systemic TNFα in subjects afflicted by HCS are not known.
It has been an issue of controversy whether TNFα has direct effects on osteoclastogenesis or whether it requires RANKL to exert its actions (59). In this work, we did not detect Tnfsf11 (encoding RANKL) in BMM cultures, treated or not, with TNFα (data not shown) suggesting that the effects observed were secondary to the direct actions of TNFα and not mediated by RANKL. Moreover, there was no difference on the induction of Tnfsf11 by TNFα in Notch2tm1.1Ecan and control osteoblasts (data not shown) so that a differential expression of RANKL does not explain the phenotypic changes observed in Notch2tm1.1Ecan mutant mice under the influence of TNFα.
In conclusion, Notch2tm1.1Ecan mice are sensitized to the actions of TNFα on osteoclastogenesis and bone resorption, possibly explaining the acro-osteolysis observed in individuals affected by HCS.
Experimental procedures
Mice and TNFα-induced osteolysis in vivo
Notch2tm1.1Ecan mice harboring a 6955C→T substitution in the Notch2 locus have been characterized in previous studies (5, 25, 46). Genotyping was conducted in tail DNA extracts by PCR using forward primer Nch2Lox gtF 5′-CCCTTCTCTCTGTGCGGTAG-3′ and reverse primer Nch2Lox gtR 5′-CTCAGAGCCAAAGCCTCACTG-3′ (Integrated DNA Technologies; IDT, Coralville, IA). Hes1loxP/loxP mice, where loxP sequences are knocked into the first intron and downstream of the 3′UTR of Hes1 alleles, were obtained from RIKEN (Wako Saitama, Japan) (60). Genotyping was performed using forward primer 5′-CAGCCAGTGTCAACACGACACCGGACAAAC-3′ and reverse primer 5′-TCGCCTTCGCCTCTTCTCCATGATA-3′ (IDT).
Two-month-old heterozygous male Notch2tm1.1Ecan mice in a C57BL/6 background and control sex-matched littermates were administered TNFα, at a dose of 2 μg, or PBS by injection in the subcutaneous space over the calvarial vault once a day for 4 consecutive days and sacrificed 24 h after the last injection as reported previously (61). TNFα cDNA and expression vector were obtained from S. Lee (Farmington, CT), and TNFα was purified using nickel-nitrilotriacetic acid–agarose columns (Qiagen, Germantown, MD), in accordance with manufacturer's instructions. To test whether the effect of TNFα on osteolysis in Notch2tm1.1Ecan calvariae can be reversed by blocking NOTCH2 activation, antibodies directed to the NRR of NOTCH2 (anti-NOTCH2 NRR) or control anti-ragweed antibodies at a dose of 10 mg/kg (all from Genentech, South San Francisco, CA) (46) were injected with TNFα at a dose of 2 μg over the calvarial vault of male Notch2tm1.1Ecan mice and sex-matched littermate controls. All animal experiments were approved by the Institutional Animal Care and Use Committee of UConn Health.
Bone histomorphometry
Calvariae were excised and fixed in 10% formalin for 3 days, decalcified in 14% EDTA (pH 7.2) for 7 days, and embedded in paraffin. Histomorphometry of the medial aspect of each calvaria was carried out in 7-μm-thick sections stained with TRAP and hematoxylin (Thermo Fisher Scientific, Waltham, MA). TRAP enzyme histochemistry was conducted using a commercial kit (Sigma), in accordance with manufacturer's instructions. Stained sections were used to outline bone tissue area and to measure osteoclast number and surface as well as eroded surface at a magnification of ×100 using an OsteoMeasure morphometry system (Osteometrics, Atlanta, GA) (62).
BMM, adenovirus-Cre-mediated gene deletion, and osteoclast formation
To obtain BMMs, the marrow from heterozygous male Notch2tm1.1Ecan mutant and control sex-matched littermate mice was removed by flushing with a 26-gauge needle, and erythrocytes were lysed in 150 mm NH4Cl, 10 mm KHCO3 and 0.1 mm EDTA (pH 7.4), as described previously (46). Cells were centrifuged, and the sediment was suspended in α-minimum essential medium (α-MEM) in the presence of 10% fetal bovine serum (FBS; both from Thermo Fisher Scientific) and recombinant human M-CSF at 30 ng/ml. M-CSF cDNA and expression vector were obtained from D. Fremont (St. Louis, MO), and M-CSF was purified as reported previously (63). Cells were seeded on plastic Petri dishes at a density of 300,000 cells/cm2 and cultured for 3 days.
For osteoclast formation, cells were collected following treatment with 0.25% trypsin/EDTA for 5 min and seeded on tissue culture plates at a density of 62,500 cells/cm2 in α-MEM with 10% FBS, M-CSF at 30 ng/ml, and TNFα at 50, 100, or 200 ng/ml, respectively. Cultures were carried out until the formation of multinucleated TRAP-positive cells. TRAP-positive cells containing more than three nuclei were considered osteoclasts. To test whether the effect of TNFα on osteoclastogenesis in Notch2tm1.1Ecan BMMs depended on NOTCH2 activation, anti-NOTCH2 NRR at 10 μg/ml, anti-JAG1 antibodies at 20 μg/ml, or control anti-ragweed antibodies at 10 or 20 μg/ml (all from Genentech) were added directly to the culture medium (45–47).
To inactivate Hes1 in osteoclast precursors in the context of the Notch2tm1.1Ecan mutation, Hes1loxP/loxP alleles were introduced into Notch2tm1.1Ecan mice to create heterozygous Notch2tm1.1Ecan;Hes1loxP/loxP mice. Notch2tm1.1Ecan;Hes1loxP/loxP mice were crossed with Hes1loxP/loxP mice to obtain Notch2tm1.1Ecan;Hes1loxP/loxP and Hes1loxP/loxP control littermates for study. BMMs from both cohorts were cultured in the presence of M-CSF at 30 ng/ml and TNFα at 200 ng/ml for 3 days. The cells were transduced with adenoviruses carrying cytomegalovirus (CMV)-Cre or CMV-GFP as control at a multiplicity of infection (m.o.i.) of 100 and cultured with M-CSF and TNFα for three additional days until formation of multinucleated TRAP-positive cells.
Quantitative RT-PCR (qRT-PCR)
Total RNA was extracted from osteoclasts with the RNeasy kit (Qiagen, Valencia, CA) and from homogenized calvarial bones with the micro-RNeasy kit (Qiagen), in accordance with manufacturer's instructions. The integrity of the RNA extracted from bones was assessed by microfluidic electrophoresis on an Experion system (Bio-Rad), and RNA with a quality indicator number equal to or higher than 7.0 was used for subsequent analysis. Equal amounts of RNA were reverse-transcribed using the iScript RT-PCR kit (Bio-Rad) and amplified in the presence of specific primers (all primers were from IDT; Table 1) with the SsoAdvancedTM Universal SYBR Green Supermix (Bio-Rad) at 60 °C for 40 cycles. Transcript copy number was estimated by comparison with a serial dilution of cDNA for Acp5, Ctsk, Il1b, Il6, Jag1, Notch2, and Tnfa (all from Thermo Fisher Scientific), Hes1 (American Type Culture Collection (ATCC), Manassas, VA), and Nfatc1 (Addgene plasmid 11793 created by A. Rao, La Jolla, CA).
Table 1.
Primers used for qRT-PCR determinations
GenBankTM accession numbers identify transcript recognized by primer pairs.
| Gene | Strand | Sequence | GenBankTM accession no. |
|---|---|---|---|
| Acp5 | Forward | 5′-GACAAGAGGTTCCAGGAGAC-3′ | NM_001102404; NM_001102405; NM_007388 |
| Reverse | 5′-TTCCAGCCAGCACATACC-3′ | ||
| Ctsk | Forward | 5′-AGATATTGGTGGCTTTGGAA-3′ | NM_007802 |
| Reverse | 5′-AACGAGAGGAGAAATGAAACA-3′ | ||
| Hes1 | Forward | 5′-ACCAAAGACGGCCTCTGAGCACAGAAAGT-3′ | NM_008235 |
| Reverse | 5′-ATTCTTGCCCTTCGCCTCTT-3′ | ||
| Il1b | Forward | 5′-GGACAGAATATCAACCAACAAGTG-3′ | NM_008361 |
| Reverse | 5′-TCGTTGCTTGGTTCTCCTT-3′ | ||
| Il6 | Forward | 5′-CGGCCTTCCCTACTTCACAAGTCCG-3′ | NM_001314054; NM_031168 |
| Reverse | 5′-CAGGTCTGTTGGGAGTGGTATCC-3′ | ||
| Jag1 | Forward | 5′-TGGGAACTGTTGTGGTGGAGTCCG-3′ | NM_013822 |
| Reverse | 5′-GTGACGCGGGACTGATACTCCT-3′ | ||
| Nfatc1 | Forward | 5′-GCGCAAGTACAGTCTCAATGGCC-3′ | NM_198429; NM_001164110; NM_001164111; NM_001164112; NM_00116641091; NM_016791 |
| Reverse | 5′-GGATGGTGTGGGTGAGTGGT-3′ | ||
| Notch2 | Forward | 5′-TGACGTTGATGAGTGTATCTCCAAGCC-3′ | NM_010928 |
| Reverse | 5′-GTAGCTGCCCTGAGTGTTGTGG-3′ | ||
| Rpl38 | Forward | 5′-AGAACAAGGATAATGTGAAGTTCAAGGTTC-3′ | NM_001048057; NM_001048058; NM_023372 |
| Reverse | 5′-CTGCTTCAGCTTCTCTGCCTTT-3′ | ||
| Tnfa | Forward | 5′-CCACCATCAAGGACTCAAATGG-3′ | NM_001278601; NM_013693 |
| Reverse | 5′-CCTTTGCAGAACTCAGGAATGGACATTCG-3′ | ||
| Tnfr1 | Forward | 5′-GGTCTGCTGATGTTAGGA-3′ | NM_011609 |
| Reverse | 5′-CTTGGCATCTCTTTGTAGG-3′ | ||
| Tnfr2 | Forward | 5′-TGTTCTTGTCTCAGTTTGTAGGG-3′ | NM_011610 |
| Reverse | 5′-AGTCGTCCTTCTCACCTCTT-3′ |
The level of Notch26955C→T mutant transcript was measured as described previously (5). Total RNA was reverse-transcribed with Moloney murine leukemia virus reverse transcriptase in the presence of reverse primers for Notch2 and Rpl38 (Table 1). Notch2 cDNA was amplified by qPCR in the presence of TaqMan gene expression assay mix, including specific primers (5′-CATCGTGACTTTCCA-3′ and 5′-GGATCTGGTACATAGAG-3′) and a 6-carboxyfluorescein–labeled DNA probe of sequence 5′-CATTGCCTAGGCAGC-3′ covalently attached to a 3′-minor groove binder quencher (Thermo Fisher Scientific), and SsoAdvanced Universal Probes Supermix (Bio-Rad) at 60 °C for 45 cycles (59). Notch26955C→T transcript copy number was estimated by comparison with a serial dilution of a synthetic DNA fragment (IDT) containing ∼200 bp surrounding the 6955C→T mutation in the Notch2 locus, and cloned into pcDNA3.1(−) (Thermo Fisher Scientific) by isothermal single reaction assembly using commercially available reagents (New England Biolabs, Ipswich, MA) (60).
Amplification reactions were conducted in CFX96 qRT-PCR detection systems (Bio-Rad), and fluorescence was monitored during every PCR cycle at the annealing step. Data are expressed as copy number or relative expression corrected for Rpl38 expression estimated by comparison with a serial dilution of cDNA for Rpl38 (ATCC) (64).
Immunoblotting
TNFα-treated BMMs or osteoclasts from control or Notch2tm1.1Ecan mice were extracted in buffer containing 25 mm Tris-HCl (pH 7.5), 150 mm NaCl, 5% glycerol, 1 mm EDTA, 0.5% Triton X-100, 1 mm sodium orthovanadate, 10 mm NaF, 1 mm phenylmethylsulfonyl fluoride and a protease inhibitor mixture (all from Sigma). Quantified total cell lysates (35 μg of total protein) were separated by SDS-PAGE in 8 or 10% polyacrylamide gels and transferred to Immobilon-P membranes (Millipore, Billerica, MA). The blots were probed with anti-p-IκBα (9246), IκBα (9242), p-p38 (9211), p38 (9212), p-ERK (9101), ERK (9102), p-JNK (4668), JNK (9252), p-AKT (9271), AKT (9272) HES1 (11988), and β-Actin (3700) antibodies (all from Cell Signaling Technology, Danvers, MA). Anti-NOTCH2 (C651.6DbHN) and anti-JAG1 (TS1.15H) antibodies were obtained from Developmental Studies Hybridoma Bank (DSHB C651.6DbHN, University of Iowa, Iowa City). Anti-NFATc1 antibody (556602) was obtained from BD Biosciences. The blots were exposed to anti-rabbit IgG, anti-rat IgG, or anti-mouse IgG conjugated to horseradish peroxidase (Sigma) and incubated with a chemiluminescence detection reagent (Bio-Rad). Chemiluminescence was detected by ChemiDocTM XSR+ molecular imager (Bio-Rad) with Image LabTM software (version 5.2.1) (65), and the amount of protein in individual bands was quantified.
NF-κB activation assay
TNFα-treated BMMs from control or Notch2tm1.1Ecan mice were lysed prior to nuclear extraction using the nuclear extract kit (Active Motif, Inc., Carlsbad, CA). To detect and quantify NF-κB activation, 20 μg of nuclear extract samples were examined using a commercial ELISA-based kit (TransAMTM Flexi NF-κB p65, Active Motif, Inc.) (66), in accordance with manufacturer's instructions. Briefly, nuclear extracts were incubated with a biotinylated consensus NF-κB–binding sequence (5′-GGGACTTTCC-3′) (1 pmol/well), and the reaction mixtures were transferred into assay wells. Subsequently, samples were incubated with anti-NF-κB p65 antibody, and anti-rabbit IgG was conjugated to horseradish peroxidase and developed, and colorimetric changes were measured in an iMarkTM Microplate Absorbance Reader (Bio-Rad) at 450 nm with a reference wavelength of 655 nm. To assess the specificity of NF-κB binding to the biotinylated probe, unlabeled WT or mutated consensus NF-κB binding oligonucleotide was added in excess (10 pmol/well) to the reaction mixture.
Serum TNFα
Serum levels of TNFα were measured in 2- and 12-month-old Notch2tm1.1Ecan male mice and control littermates using a mouse TNFα-uncoated enzyme-linked immunosorbent assay kit in accordance with manufacturer's instructions (Thermo Fisher Scientific; catalogue 88-7324).
Statistics
Data are expressed as means ± S.D. Statistical differences were determined by Student's t test or two-way analysis of variance with Holm-Šídák post hoc analysis for pairwise or multiple comparisons, respectively.
Author contributions
J. Y. and E. C. conceptualization; J. Y. formal analysis; J. Y. and E. C. methodology; J.Y. writing-original draft; E. C. supervision; E. C. funding acquisition; E. C. project administration; E. C. writing-review and editing.
Acknowledgments
We thank Genentech for anti-NOTCH2 NRR, anti-JAG1, and control anti-ragweed antibodies; S. Lee for TNFα cDNA; D. Fremont for M-CSF cDNA; M. Glogauer for Tnfsf11 cDNA; A. Rao for Nfatc1 cDNA; Lauren Schilling and Tabitha Eller for technical assistance; and Mary Yurczak for secretarial support.
This work was supported by National Institutes of Health Grant AR068160 from NIAMS and Grant DK045227 from NIDDK. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
E. Canalis, unpublished observations.
- NICD
- NOTCH intracellular domain
- α-MEM
- α-minimum essential medium
- BMM
- bone marrow-derived macrophage
- CMV
- cytomegalovirus
- ES/BS
- eroded surface/bone surface
- FBS
- fetal bovine serum
- HCS
- Hajdu Cheney syndrome
- IL
- interleukin
- M-CSF
- macrophage colony-stimulating factor
- m.o.i.
- multiplicity of infection
- NF-κB
- nuclear factor-κB
- NRR
- negative regulatory region
- N.Oc/B.Pm
- number of osteoblasts/bone perimeter
- Oc.S/BS
- osteoclast surface/bone surface
- PEST
- proline (P), glutamic acid (E), serine (S), and threonine (T)-rich
- qRT-PCR
- quantitative reverse transcription-PCR
- RANKL
- receptor activator of NF-κB ligand
- RBPJκ
- recombination signal-binding protein for immunoglobulin κ region
- TNFα
- tumor necrosis factor α
- TRAP
- tartrate-resistant acid phosphatase
- Veh
- vehicle control
- PI3K
- phosphoinositol 3-kinase.
References
- 1. Siebel C., and Lendahl U. (2017) Notch signaling in development, tissue homeostasis, and disease. Physiol. Rev. 97, 1235–1294 10.1152/physrev.00005.2017 [DOI] [PubMed] [Google Scholar]
- 2. Zanotti S., and Canalis E. (2016) Notch signaling and the skeleton. Endocr. Rev. 37, 223–253 10.1210/er.2016-1002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bai S., Kopan R., Zou W., Hilton M. J., Ong C. T., Long F., Ross F. P., and Teitelbaum S. L. (2008) NOTCH1 regulates osteoclastogenesis directly in osteoclast precursors and indirectly via osteoblast lineage cells. J. Biol. Chem. 283, 6509–6518 10.1074/jbc.M707000200 [DOI] [PubMed] [Google Scholar]
- 4. Canalis E., Parker K., Feng J. Q., and Zanotti S. (2013) Osteoblast lineage-specific effects of notch activation in the skeleton. Endocrinology 154, 623–634 10.1210/en.2012-1732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Canalis E., Schilling L., Yee S. P., Lee S. K., and Zanotti S. (2016) Hajdu Cheney mouse mutants exhibit osteopenia, increased osteoclastogenesis and bone resorption. J. Biol. Chem. 291, 1538–1551 10.1074/jbc.M115.685453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Canalis E., Yu J., Schilling L., Yee S. P., and Zanotti S. (2018) The lateral meningocele syndrome mutation causes marked osteopenia in mice. J. Biol. Chem. 293, 14165–14177 10.1074/jbc.RA118.004242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fukushima H., Nakao A., Okamoto F., Shin M., Kajiya H., Sakano S., Bigas A., Jimi E., and Okabe K. (2008) The association of Notch2 and NF-κB accelerates RANKL-induced osteoclastogenesis. Mol. Cell. Biol. 28, 6402–6412 10.1128/MCB.00299-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hilton M. J., Tu X., Wu X., Bai S., Zhao H., Kobayashi T., Kronenberg H. M., Teitelbaum S. L., Ross F. P., Kopan R., and Long F. (2008) Notch signaling maintains bone marrow mesenchymal progenitors by suppressing osteoblast differentiation. Nat. Med. 14, 306–314 10.1038/nm1716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zanotti S., and Canalis E. (2017) Parathyroid hormone inhibits Notch signaling in osteoblasts and osteocytes. Bone 103, 159–167 10.1016/j.bone.2017.06.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lindsell C. E., Boulter J., diSibio G., Gossler A., and Weinmaster G. (1996) Expression patterns of Jagged, Delta1, Notch1, Notch2, and Notch3 genes identify ligand-receptor pairs that may function in neural development. Mol. Cell. Neurosci. 8, 14–27 10.1006/mcne.1996.0040 [DOI] [PubMed] [Google Scholar]
- 11. Canalis E. (2018) Notch in skeletal physiology and disease. Osteoporos. Int. 29, 2611–2621 10.1007/s00198-018-4694-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sanchez-Irizarry C., Carpenter A. C., Weng A. P., Pear W. S., Aster J. C., and Blacklow S. C. (2004) Notch subunit heterodimerization and prevention of ligand-independent proteolytic activation depend, respectively, on a novel domain and the LNR repeats. Mol. Cell. Biol. 24, 9265–9273 10.1128/MCB.24.21.9265-9273.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Iso T., Kedes L., and Hamamori Y. (2003) HES and HERP families: multiple effectors of the Notch signaling pathway. J. Cell. Physiol. 194, 237–255 10.1002/jcp.10208 [DOI] [PubMed] [Google Scholar]
- 14. Iso T., Sartorelli V., Poizat C., Iezzi S., Wu H. Y., Chung G., Kedes L., and Hamamori Y. (2001) HERP, a novel heterodimer partner of HES/E(spl) in Notch signaling. Mol. Cell. Biol. 21, 6080–6089 10.1128/MCB.21.17.6080-6089.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hajdu N., and Kauntze R. (1948) Cranio-skeletal dysplasia. Br. J. Radiol. 21, 42–48 10.1259/0007-1285-21-241-42 [DOI] [PubMed] [Google Scholar]
- 16. Cheney W. D. (1965) Acro-Osteolysis. Am. J. Roentgenol. Radium. Ther. Nucl. Med. 94, 595–607 [PubMed] [Google Scholar]
- 17. Canalis E., and Zanotti S. (2014) Hajdu-Cheney syndrome: a review. Orphanet J. Rare Dis. 9, 200 10.1186/s13023-014-0200-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gray M. J., Kim C. A., Bertola D. R., Arantes P. R., Stewart H., Simpson M. A., Irving M. D., and Robertson S. P. (2012) Serpentine fibula polycystic kidney syndrome is part of the phenotypic spectrum of Hajdu-Cheney syndrome. Eur. J. Hum. Genet. 20, 122–124 10.1038/ejhg.2011.125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Isidor B., Lindenbaum P., Pichon O., Bézieau S., Dina C., Jacquemont S., Martin-Coignard D., Thauvin-Robinet C., Le Merrer M., Mandel J. L., David A., Faivre L., Cormier-Daire V., Redon R., and Le Caignec C. (2011) Truncating mutations in the last exon of NOTCH2 cause a rare skeletal disorder with osteoporosis. Nat. Genet. 43, 306–308 10.1038/ng.778 [DOI] [PubMed] [Google Scholar]
- 20. Majewski J., Schwartzentruber J. A., Caqueret A., Patry L., Marcadier J., Fryns J. P., Boycott K. M., Ste-Marie L. G., McKiernan F. E., Marik I., Van Esch H., FORGE Canada Consortium, Michaud J. L., and Samuels M. E. (2011) Mutations in NOTCH2 in families with Hajdu-Cheney syndrome. Hum. Mutat. 32, 1114–1117 10.1002/humu.21546 [DOI] [PubMed] [Google Scholar]
- 21. Simpson M. A., Irving M. D., Asilmaz E., Gray M. J., Dafou D., Elmslie F. V., Mansour S., Holder S. E., Brain C. E., Burton B. K., Kim K. H., Pauli R. M., Aftimos S., Stewart H., Kim C. A., et al. (2011) Mutations in NOTCH2 cause Hajdu-Cheney syndrome, a disorder of severe and progressive bone loss. Nat. Genet. 43, 303–305 10.1038/ng.779 [DOI] [PubMed] [Google Scholar]
- 22. Zhao W., Petit E., Gafni R. I., Collins M. T., Robey P. G., Seton M., Miller K. K., and Mannstadt M. (2013) Mutations in NOTCH2 in patients with Hajdu-Cheney syndrome. Osteoporos. Int. 24, 2275–2281 10.1007/s00198-013-2298-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fukushima H., Shimizu K., Watahiki A., Hoshikawa S., Kosho T., Oba D., Sakano S., Arakaki M., Yamada A., Nagashima K., Okabe K., Fukumoto S., Jimi E., Bigas A., Nakayama K. I., et al. (2017) NOTCH2 Hajdu-Cheney mutations Escape SCF(FBW7)-dependent proteolysis to promote osteoporosis. Mol. Cell 68, 645–658.e5 10.1016/j.molcel.2017.10.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zanotti S., Yu J., Bridgewater D., Wolf J. M., and Canalis E. (2018) Mice harboring a Hajdu Cheney syndrome mutation are sensitized to osteoarthritis. Bone 114, 198–205 10.1016/j.bone.2018.06.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yu J., Zanotti S., Walia B., Jellison E., Sanjay A., and Canalis E. (2018) The Hajdu Cheney mutation is a determinant of B-cell allocation of the splenic marginal zone. Am. J. Pathol. 188, 149–159 10.1016/j.ajpath.2017.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sakka S., Gafni R. I., Davies J. H., Clarke B., Tebben P., Samuels M., Saraff V., Klaushofer K., Fratzl-Zelman N., Roschger P., Rauch F., and Högler W. (2017) Bone structural characteristics and response to bisphosphonate treatment in children with Hajdu-Cheney syndrome. J. Clin. Endocrinol. Metab. 102, 4163–4172 10.1210/jc.2017-01102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Blumenauer B. T., Cranney A. B., and Goldstein R. (2002) Acro-osteolysis and osteoporosis as manifestations of the Hajdu-Cheney syndrome. Clin. Exp. Rheumatol. 20, 574–575 [PubMed] [Google Scholar]
- 28. Brown D. M., Bradford D. S., Gorlin R. J., Desnick R. J., Langer L. O., Jowsey J., and Sauk J. J. (1976) The acro-osteolysis syndrome: morphologic and biochemical studies. J. Pediatr. 88, 573–580 10.1016/S0022-3476(76)80009-1 [DOI] [PubMed] [Google Scholar]
- 29. Udell J., Schumacher H. R. Jr, Kaplan F., and Fallon M. D. (1986) Idiopathic familial acroosteolysis: histomorphometric study of bone and literature review of the Hajdu-Cheney syndrome. Arthritis Rheum. 29, 1032–1038 10.1002/art.1780290815 [DOI] [PubMed] [Google Scholar]
- 30. Gu Q., Yang H., and Shi Q. (2017) Macrophages and bone inflammation. J. Orthop. Translat. 10, 86–93 10.1016/j.jot.2017.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kwan Tat S., Padrines M., Théoleyre S., Heymann D., and Fortun Y. (2004) IL-6, RANKL, TNF-α/IL-1: interrelations in bone resorption pathophysiology. Cytokine Growth Factor Rev. 15, 49–60 10.1016/j.cytogfr.2003.10.005 [DOI] [PubMed] [Google Scholar]
- 32. Azuma Y., Kaji K., Katogi R., Takeshita S., and Kudo A. (2000) Tumor necrosis factor-α induces differentiation of and bone resorption by osteoclasts. J. Biol. Chem. 275, 4858–4864 10.1074/jbc.275.7.4858 [DOI] [PubMed] [Google Scholar]
- 33. Komine M., Kukita A., Kukita T., Ogata Y., Hotokebuchi T., and Kohashi O. (2001) Tumor necrosis factor-α cooperates with receptor activator of nuclear factor κB ligand in generation of osteoclasts in stromal cell-depleted rat bone marrow cell culture. Bone 28, 474–483 10.1016/S8756-3282(01)00420-3 [DOI] [PubMed] [Google Scholar]
- 34. Kobayashi K., Takahashi N., Jimi E., Udagawa N., Takami M., Kotake S., Nakagawa N., Kinosaki M., Yamaguchi K., Shima N., Yasuda H., Morinaga T., Higashio K., Martin T. J., and Suda T. (2000) Tumor necrosis factor α stimulates osteoclast differentiation by a mechanism independent of the ODF/RANKL-RANK interaction. J. Exp. Med. 191, 275–286 10.1084/jem.191.2.275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wei S., Kitaura H., Zhou P., Ross F. P., and Teitelbaum S. L. (2005) IL-1 mediates TNF-induced osteoclastogenesis. J. Clin. Invest. 115, 282–290 10.1172/JCI200523394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lee Y. M., Fujikado N., Manaka H., Yasuda H., and Iwakura Y. (2010) IL-1 plays an important role in the bone metabolism under physiological conditions. Int. Immunol. 22, 805–816 10.1093/intimm/dxq431 [DOI] [PubMed] [Google Scholar]
- 37. Shiratori T., Kyumoto-Nakamura Y., Kukita A., Uehara N., Zhang J., Koda K., Kamiya M., Badawy T., Tomoda E., Xu X., Yamaza T., Urano Y., Koyano K., and Kukita T. (2018) IL-1β induces pathologically activated osteoclasts bearing extremely high levels of resorbing activity: a possible pathological subpopulation of osteoclasts, accompanied by suppressed expression of Kindlin-3 and Talin-1. J. Immunol. 200, 218–228 10.4049/jimmunol.1602035 [DOI] [PubMed] [Google Scholar]
- 38. Axmann R., Böhm C., Krönke G., Zwerina J., Smolen J., and Schett G. (2009) Inhibition of interleukin-6 receptor directly blocks osteoclast formation in vitro and in vivo. Arthritis Rheum. 60, 2747–2756 10.1002/art.24781 [DOI] [PubMed] [Google Scholar]
- 39. Kotake S., Sato K., Kim K. J., Takahashi N., Udagawa N., Nakamura I., Yamaguchi A., Kishimoto T., Suda T., and Kashiwazaki S. (1996) Interleukin-6 and soluble interleukin-6 receptors in the synovial fluids from rheumatoid arthritis patients are responsible for osteoclast-like cell formation. J. Bone Miner. Res. 11, 88–95 10.1002/jbmr.5650110113 [DOI] [PubMed] [Google Scholar]
- 40. Osta B., Benedetti G., and Miossec P. (2014) Classical and paradoxical effects of TNF-α on bone homeostasis. Front. Immunol. 5, 48 10.3389/fimmu.2014.00048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mbalaviele G., Novack D. V., Schett G., and Teitelbaum S. L. (2017) Inflammatory osteolysis: a conspiracy against bone. J. Clin. Invest. 127, 2030–2039 10.1172/JCI93356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhao B. (2017) TNF and bone remodeling. Curr. Osteoporos. Rep. 15, 126–134 10.1007/s11914-017-0358-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kim J. H., Kim A. R., Choi Y. H., Jang S., Woo G. H., Cha J. H., Bak E. J., and Yoo Y. J. (2017) Tumor necrosis factor-α antagonist diminishes osteocytic RANKL and sclerostin expression in diabetes rats with periodontitis. PLoS ONE 12, e0189702 10.1371/journal.pone.0189702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Park H. J., Baek K., Baek J. H., and Kim H. R. (2017) TNFalpha increases RANKL expression via PGE(2)-induced activation of NFATc1. Int. J. Mol. Sci. 18, E495 10.3390/ijms18030495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wu Y., Cain-Hom C., Choy L., Hagenbeek T. J., de Leon G. P., Chen Y., Finkle D., Venook R., Wu X., Ridgway J., Schahin-Reed D., Dow G. J., Shelton A., Stawicki S., Watts R. J., et al. (2010) Therapeutic antibody targeting of individual Notch receptors. Nature 464, 1052–1057 10.1038/nature08878 [DOI] [PubMed] [Google Scholar]
- 46. Canalis E., Sanjay A., Yu J., and Zanotti S. (2017) An antibody to Notch2 reverses the osteopenic phenotype of Hajdu-Cheney mutant male mice. Endocrinology 158, 730–742 10.1210/en.2016-1787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lafkas D., Shelton A., Chiu C., de Leon Boenig G., Chen Y., Stawicki S. S., Siltanen C., Reichelt M., Zhou M., Wu X., Eastham-Anderson J., Moore H., Roose-Girma M., Chinn Y., Hang J. Q., Warming S., et al. (2015) Therapeutic antibodies reveal Notch control of transdifferentiation in the adult lung. Nature 528, 127–131 10.1038/nature15715 [DOI] [PubMed] [Google Scholar]
- 48. Vollersen N., Hermans-Borgmeyer I., Cornils K., Fehse B., Rolvien T., Triviai I., Jeschke A., Oheim R., Amling M., Schinke T., and Yorgan T. A. (2018) High bone turnover in mice carrying a pathogenic Notch2 mutation causing Hajdu-Cheney syndrome. J. Bone Miner. Res. 33, 70–83 10.1002/jbmr.3283 [DOI] [PubMed] [Google Scholar]
- 49. Zhao B., Grimes S. N., Li S., Hu X., and Ivashkiv L. B. (2012) TNF-induced osteoclastogenesis and inflammatory bone resorption are inhibited by transcription factor RBP-J. J. Exp. Med. 209, 319–334 10.1084/jem.20111566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zanotti S., Smerdel-Ramoya A., and Canalis E. (2011) Hairy and enhancer of split (HES)1 is a determinant of bone mass. J. Biol. Chem. 286, 2648–2657 10.1074/jbc.M110.183038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Shang Y., Smith S., and Hu X. (2016) Role of Notch signaling in regulating innate immunity and inflammation in health and disease. Protein Cell 7, 159–174 10.1007/s13238-016-0250-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Rechsteiner M., and Rogers S. W. (1996) PEST sequences and regulation by proteolysis. Trends Biochem. Sci. 21, 267–271 10.1016/S0968-0004(96)10031-1 [DOI] [PubMed] [Google Scholar]
- 53. Cenciarelli C., Marei H. E., Zonfrillo M., Casalbore P., Felsani A., Giannetti S., Trevisi G., Althani A., and Mangiola A. (2017) The interference of Notch1 target Hes1 affects cell growth, differentiation and invasiveness of glioblastoma stem cells through modulation of multiple oncogenic targets. Oncotarget 8, 17873–17886 10.18632/oncotarget.15013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Palomero T., Sulis M. L., Cortina M., Real P. J., Barnes K., Ciofani M., Caparros E., Buteau J., Brown K., Perkins S. L., Bhagat G., Agarwal A. M., Basso G., Castillo M., Nagase S., et al. (2007) Mutational loss of PTEN induces resistance to NOTCH1 inhibition in T-cell leukemia. Nat. Med. 13, 1203–1210 10.1038/nm1636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wong G. W., Knowles G. C., Mak T. W., Ferrando A. A., and Zúñiga-Pflücker J. C. (2012) HES1 opposes a PTEN-dependent check on survival, differentiation, and proliferation of TCRbeta-selected mouse thymocytes. Blood 120, 1439–1448 10.1182/blood-2011-12-395319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Xie S., Chen M., Yan B., He X., Chen X., and Li D. (2014) Identification of a role for the PI3K/AKT/mTOR signaling pathway in innate immune cells. PLoS ONE 9, e94496 10.1371/journal.pone.0094496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lee S. E., Woo K. M., Kim S. Y., Kim H. M., Kwack K., Lee Z. H., and Kim H. H. (2002) The phosphatidylinositol 3-kinase, p38, and extracellular signal-regulated kinase pathways are involved in osteoclast differentiation. Bone 30, 71–77 10.1016/S8756-3282(01)00657-3 [DOI] [PubMed] [Google Scholar]
- 58. Ottaviani S., Tahiri K., Frazier A., Hassaine Z. N., Dumontier M. F., Baschong W., Rannou F., Corvol M. T., Savouret J. F., and Richette P. (2010) Hes1, a new target for interleukin 1β in chondrocytes. Ann. Rheum. Dis. 69, 1488–1494 10.1136/ard.2009.120816 [DOI] [PubMed] [Google Scholar]
- 59. Tanaka S. (2017) RANKL-independent osteoclastogenesis: a long-standing controversy. J. Bone Miner. Res. 32, 431–433 10.1002/jbmr.3092 [DOI] [PubMed] [Google Scholar]
- 60. Imayoshi I., Shimogori T., Ohtsuka T., and Kageyama R. (2008) Hes genes and neurogenin regulate non-neural versus neural fate specification in the dorsal telencephalic midline. Development 135, 2531–2541 10.1242/dev.021535 [DOI] [PubMed] [Google Scholar]
- 61. Yeon Won H., Hwan Mun S., Shin B., and Lee S. K. (2016) Contradictory role of CD97 in basal and tumor necrosis factor-induced osteoclastogenesis in vivo. Arthritis Rheumatol. 68, 1301–1313 10.1002/art.39538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Dempster D. W., Compston J. E., Drezner M. K., Glorieux F. H., Kanis J. A., Malluche H., Meunier P. J., Ott S. M., Recker R. R., and Parfitt A. M. (2013) Standardized nomenclature, symbols, and units for bone histomorphometry: a 2012 update of the report of the ASBMR Histomorphometry Nomenclature Committee. J. Bone Miner. Res. 28, 2–17 10.1002/jbmr.1805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lee S. H., Rho J., Jeong D., Sul J. Y., Kim T., Kim N., Kang J. S., Miyamoto T., Suda T., Lee S. K., Pignolo R. J., Koczon-Jaremko B., Lorenzo J., and Choi Y. (2006) v-ATPase V0 subunit d2-deficient mice exhibit impaired osteoclast fusion and increased bone formation. Nat. Med. 12, 1403–1409 10.1038/nm1514 [DOI] [PubMed] [Google Scholar]
- 64. Kouadjo K. E., Nishida Y., Cadrin-Girard J. F., Yoshioka M., and St-Amand J. (2007) Housekeeping and tissue-specific genes in mouse tissues. BMC Genomics 8, 127 10.1186/1471-2164-8-127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zanotti S., Smerdel-Ramoya A., and Canalis E. (2013) Nuclear factor of activated T-cells (Nfat)c2 inhibits Notch signaling in osteoblasts. J. Biol. Chem. 288, 624–632 10.1074/jbc.M112.340455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Sisto M., Lisi S., D'Amore M., and Lofrumento D. D. (2014) Rituximab-mediated Raf kinase inhibitor protein induction modulates NF-kappaB in Sjogren syndrome. Immunology 143, 42–51 10.1111/imm.12288 [DOI] [PMC free article] [PubMed] [Google Scholar]