

Abstract
Objective:
To evaluate serum and cerebrospinal fluid (CSF) levels of phosphorylated neurofilament heavy (pNfH), and to compare these to levels of neurofilament light (NfL), as biomarkers of pre-symptomatic ALS.
Design:
The study population includes 34 controls, 79 individuals at-risk for ALS, 22 ALS patients, and 14 phenoconverters. At-risk individuals are enrolled through Pre-Symptomatic Familial ALS (Pre-fALS), a longitudinal natural history and biomarker study of individuals who are carriers of any ALS-associated gene mutation, but who demonstrate no clinical evidence of disease at the time of enrollment. pNfH and NfL in serum and CSF were quantified using established enzyme-linked immunosorbent assays.
Results:
There is a longitudinal increase in serum pNfH in advance of the emergence of clinically manifest ALS. A similar pattern is observed for NfL, but with the absolute levels also frequently exceeding a normative threshold. Although CSF data are more sparse, similar patterns are observed for both neurofilaments, with absolute levels exceeding a normative threshold prior to phenoconversion. In serum, these changes are observed in the 6-12 months prior to disease among SOD1 A4V mutation carriers, and as far back as 2 and 3.5 years respectively in individuals with a FUS c.521del6 mutation and a C9ORF72 hexanucleotide repeat expansion.
Conclusions:
Serum and CSF pNfH increase prior to phenoconversion. In CSF, the temporal course of these changes is similar to NfL. In serum, however, pNfH is less sensitive to pre-symptomatic disease than NfL. The duration of pre-symptomatic disease, as defined by changes in neurofilaments, may vary depending on underlying genotype.
Keywords: Amyotrophic lateral sclerosis, Neurofilaments, Biomarkers, Pre-Symptomatic, Disease Prevention
Introduction
Growing interest in the tangible possibility of early therapeutic intervention is fueling research into the pre-symptomatic phase of a range of neurodegenerative disorders. Essential to these endeavors is the development of biomarkers that either define individuals as being at heightened short-term risk of developing clinically manifest disease, or that indicate the presence of subclinical disease prior to the emergence of clinically manifest disease. The field of Alzheimer’s disease (AD) research has led the way, currently using genetic risk (namely, the presenilin1 E280A mutation) as an eligibility criterion for the Alzheimer’s Prevention Initiative (API) trial of crenezumab () and using positron emission tomography (PET) evidence of amyloid deposition as an eligibility criterion for the Anti-Amyloid Treatment in Asymptomatic Alzheimer’s Disease (A4) Trial of solanezumab 1, with both trials enrolling cognitively normal subjects. In recent years, the concentrations of neurofilament proteins in serum and cerebrospinal fluid (CSF) have emerged as candidate biomarkers relevant to the study of a range of pre-symptomatic neurodegenerative diseases including familial AD 2–4, frontotemporal dementia 5–7, Huntington’s disease 8–10, and amyotrophic lateral sclerosis 11.
Building on our recent work describing the utility of serum and CSF neurofilament light (NfL) as the earliest biochemical biomarker of pre-symptomatic ALS 11, here we present phosphorylated neurofilament heavy (pNfH) data as well as NfL results on an additional 4 phenoconverters (including 2 with a C9orf72 repeat expansion). We also show how levels of these neurofilaments may shed light on the relationship between genotype and the duration of the pre-symptomatic phase of ALS and yield an opportunity for initiating a disease prevention trial in ALS. The interest in exploring pNfH in addition to NfL rests on the potential insights that the combined fluid expression of these isoforms may yield in understanding the early evolution of pathology in ALS. The recently reported increase of the NfL isoform and the more modest up-regulation of heavy and medium chain subunits in blood from patients with ALS may reflect differences in the immunological clearance of these proteins 12 and/or an energy-saving change in the stoichiometry of distressed neuronal cells, whereby increased NfL production compensates for the overall loss of neurofilament proteins13.
Methods
Study Population
Pre-Symptomatic Familial ALS (Pre-fALS) 14 is a longitudinal natural history and biomarker study of individuals recruited from across North America who are carriers of any ALS-causing gene mutation (in SOD1, C9orf72, TARDBP, FUS, VCP, etc.) but demonstrate, at the time of enrollment, no clinical or electromyographic evidence of disease. Asymptomatic carriers of pathogenic variants in these genes comprise the only population known to be at significantly greater risk for ALS (compared to the general population), and in whom a study of pre-symptomatic disease may realistically be considered. As described elsewhere 14, participants are followed longitudinally, with phenotypic (motor and cognitive), electrophysiological, quantitative motor and imaging assessments, along with collection of biological samples (urine, blood, and CSF). Cognition and behavior are assessed using a combination of the Edinburgh Cognitive Behavioral Assessment (ECAS) 15 and a full neuropsychological test battery that assesses domains relevant to frontotemporal spectrum dysfunction that occurs in association with ALS 16; alternate versions of tests are used to minimized the potential for learning effects. These in-person assessments are repeated approximately every 12-24 months, with intervening remote visits. Phenoconversion is defined as the emergence of definite symptoms or signs that clearly indicate manifest disease 17; those who phenoconvert continue to be followed after developing clinically manifest disease. We thus acquire longitudinal data prior to the appearance of symptoms, around the time that symptoms begin to emerge, and in the early stages of manifest disease. In addition, ALS patients as well as controls (who are either healthy individuals with no known family history of ALS or individuals who were found, as part of their Pre-fALS screening, not to harbor the genetic mutation known to cause ALS in their family) are evaluated with the same set of assessments as is used for Pre-fALS study visits. The serum and CSF samples included in this experiment were collected at study visits that took place between January 2008 and September 2017. This study was approved by the University of Miami Institutional Review Board, and all participants provided written informed consent. The study is registered on clinicaltrials.gov ().
Sample Collection, Processing, and Storage
For serum analysis, blood was collected in a red top BD vacutainer and allowed to clot upright at room temperature for 1-2 hours. Following centrifugation (1750g for 10 minutes at 4°C) serum was aliquoted into cryogenic sterile freestanding conical microtubes (Nalgene, Rochester, NY or Bio Plas Inc., San Rafael CA) and quickly stored at −80°C until use. CSF (free of macroscopic hemoglobin) was collected in polypropylene tubes, centrifuged (1750g for 10 minutes at 4°C), aliquoted using a sterile pipette into pre-capped polypropylene cryogenic sterile freestanding conical microtubes, frozen within ~30 minutes of collection, and stored at −80°C until use.
Neurofilament Quantification
Quantification of pNfH was performed using a CE marked ELISA (Euroimmun AG, Lübeck, Germany), which utilizes polyclonal capture and monoclonal detection antibodies to pNfH. The assay and its analytical performance have been described in detail previously 18. Each plate contained calibrators (0-10,000pg/mL) and quality controls; if required, samples were appropriately diluted to fall within the range of the standard curve. All samples were measured in duplicate at the same dilution. All pNfH assays were performed blind to group/disease state. For serum, inter-assay coefficients of variance are below 19% and the mean intra-assay coefficients of variance are below 10%. For CSF, inter-assay coefficients of variance are below 11% and the mean intra-assay coefficients of variance are below 10%. Serum and CSF NfL were assayed as previously described 11.
Statistical Analysis
Participant characteristics are summarized in Table 1. Neurofilament concentrations that were below the level of detection (<LOD) were excluded from the summary statistics in Table 2, but included in the analyses using an imputed value of 0.03 pg/ml for pNfH or 0.37 pg/ml for NfL, which was the lowest detectable value in our data. Due to the highly skewed distribution of neurofilament concentrations and the presence of outliers, analyses were performed using non-parametric tests or natural logarithm-transformed values where applicable. For ease of interpretation of log-transformed values, a small constant was added to neurofilament values before taking the natural logarithm, so that the lowest log-transformed value is 0 (rather than a negative value). For CSF NfL, we excluded one extreme outlying value (in an ALS affected individual) that was 5-fold higher than the next highest value. Summary statistics for numerical variables are provided in mean±SD (or, for variables with a highly skewed distribution, in median and range), and for categorical variables in frequency and percentage. For phenoconverters in the Pre-fALS study, date of phenoconversion is determined based on the earlier of either unequivocal symptoms reported by the participant, or subclinical signs of disease detected through detailed neuromuscular examination, EMG, or cognitive/behavioral testing that clearly indicate disease 17. For affected individuals, on the other hand, date of onset is based on participants’ recollection and self-report. While current efforts are underway to obtain more rigorously defined “normative threshold” of NfL or pNfH concentrations among pre-symptomatic individuals, given the constraints of the current data it is operationally defined here as the 95th percentile of all available data among the controls. Spearman rank correlation, Wilcoxon rank-sum test, or Kruskal-Wallis test were employed in the analysis of cross-sectional data. For the analysis of longitudinal data, linear regression and mixed model analysis (with random intercept and slope, unstructured covariance structure, and Kenward-Roger degrees of freedom) were employed, adjusting for baseline age in all models. Given the potential for <LOD neurofilament values to be highly influential (e.g. the by-person slope estimate may be skewed for individuals whose first or last neurofilament value was <LOD), longitudinal analyses were performed with and without these values. Both sets of analyses yielded similar results; results from analyses without <LOD are reported below as the estimates were less prone to high influential data points. For the comparison of neurofilament rate of increase between participant groups, the mixed models included a time x group interaction term. The level of statistical significance was set at 0.05 (two-sided); given the discovery and exploratory nature of this study, however, the focus of this manuscript is less on statistical testing but more on characterizing the neurofilament concentrations observed. All statistical analyses were performed, and graphics produced, using SAS 9.4 (SAS Institute Inc., Cary, NC, USA).
Table 1.
Study participant characteristics
| All Participants | Longitudinal Subset | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Control (N=34) |
At-risk (N=79) |
Converter (N=14) |
Affected (N=22) |
Control (N=13) |
At-risk (N=55) |
Converter (N=10) |
Affected (N=16) |
||
| # of collections | Median (Range) |
(n/a) | 2 (2-6) |
3 (2-7) |
3.5 (2-7) |
3 (2-7) |
|||
| Follow-up duration (years) | Median (Range) |
1.5 (0.6-7.8) |
3.7 (1.0-8.7) |
2.7 (0.3-4.0) |
1.0 (0.3-2.6) |
||||
| Baseline age (years) | Mean ± SD (Range) |
46.4 ± 11.4 (24.2-69.2) |
44.8 ± 12.4 (18.9-77.0) |
51.1 ± 12.2 (31.9-74.8) |
59.2 ± 7.7 (45.6-75.8) |
48.1 ± 11.9 (34.5-69.2) |
47.0 ± 11.4 (18.9-67.6) |
51.8 ± 11.7 (32.0-74.8) |
59.8 ± 6.9 (45.6-67.7) |
| Male | N (%) | 15 (44%) | 28 (35%) | 7 (50%) | 12 (55%) | 4 (31%) | 19 (35%) | 3 (30%) | 9 (56%) |
| Genotype |
SOD1 A4V SOD1 nonA4V C9ORF72 HRE Other Unknown |
(n/a) | 27 22 25 5 0 |
9 2 2 1 0 |
1 1 5 0 15 |
(n/a) | 21 17 13 4 0 |
6 2 1 1 0 |
1 0 4 0 11 |
| Site of onset | Bulbar Limbs Other Unknown |
(n/a) | 2 10 2 0 |
2 18 0 2 |
(n/a) | 1 7 2 0 |
1 13 0 2 |
||
| Baseline years since onset | Median (Range) |
−1.6 (−6.0, −0.1) |
2.2 (0.7, 7.6) |
−1.8 (−6.0, −0.1) |
2.0 (0.7, 7.6) |
||||
| Baseline years since diagnosis | Median (Range) |
−1.7 (−6.2, −0.3) |
1.0 (0.1, 3.4) |
−1.9 (−6.2, −0.3) |
0.9 (0.1, 3.4) |
||||
| Baseline ALSFRS-R | Mean ± SD (Range) |
(n/a) | 34.2 ± 7.8 (9-44) a,b |
(n/a) | 35.4 ± 8.5 (9-44) a,c |
||||
| Baseline ∆FRS | Mean ± SD (Range) |
(n/a) | 0.53 ± 0.38 (0.14-1.45) a |
(n/a) | 0.49 ± 0.34 (0.14-1.31) a |
||||
Baseline = first visit at which serum sample was available (with or without contemporaneous CSF collection)
Follow-up duration = time between the participant’s first and last serum sample included in this study
(n/a) = not applicable.
Baseline ALSFRS-R not available for N=2
Excluding N=1 with baseline ALSFRS-R=9, 35.5 ± 5.2 (27-44).
Excluding N=1 with baseline ALSFRS-R=9, 37.4 ± 4.0 (31-44).
Table 2.
Serum and CSF pNfH concentration
| Baseline Visit Only | All Available Visits (Baseline & Follow Up) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Control | At-risk | Converter b | Affected | Control | At-risk | Converter c | Affected | ||
| Serum pNfH: | Samples available: pNfH below LOD a: |
N=34 N=2 |
N=79 N=6 |
N=14 N=0 |
N=22 N=0 |
59 visits 3 visits |
224 visits 17 visits |
42 visits 2 visits |
68 visits 0 visit |
| Original scale (pg/ml) | Median (Range) |
22.9 (0.03-113.2) |
20.6 (1.2-212.7) |
12.7 (0.6-366.6) |
84.7 (2.0-1,156) |
25.7 (0.03-124.9) |
22.1 (0.03-212.7) |
43.4 (0.6-1,156) |
75.1 (1.1-1,156) |
| Log-transformed d | Mean ± SD (Range) |
2.9 ± 1.3 (0-4.7) |
3.0 ± 1.0 (0.8-5.4) |
2.8 ± 1.5 (0.4-5.9) |
4.2 ± 1.7 (1.1-7.1) |
3.1 ± 1.2 (0-4.8) |
2.9 ± 1.0 (0-5.4) |
3.9 ± 1.6 (0.4-7.1) |
4.2 ± 1.5 (0.7-7.1) |
| CSF pNfH: | Samples available e: | N=15 | N=46 | N=7 | N=9 | 21 visits | 116 visits | 20 visits | 23 visits |
| Original scale (pg/ml) | Median (Range) |
253 (24-1,321) |
252 (109-612) |
852 (109-3,900) |
1,657 (639-11,418) |
253 (24-1,321) |
278 (73.7-1,137) |
2,853 (109-12,244) |
1,782 (639-11,418) |
| Log-transformed d | Mean ± SD (Range) |
5.5 ± 0.9 (3.2-7.2) |
5.5 ± 0.4 (4.7-6.4) |
6.5 ± 1.2 (4.7-8.3) |
7.6 ± 0.9 (6.5-9.3) |
5.5 ± 0.8 (3.2-7.2) |
5.6 ± 0.5 (4.3-7.0) |
7.7 ± 1.3 (4.7-9.4) |
7.6 ± 0.8 (6.5-9.3) |
Baseline = first visit at which serum sample was available (with or without contemporaneous CSF collection)
N = number of participants. Visits = number of person-visits. LOD = limit of detection.
These observations were assigned the lowest detectable value in analyses and figures, but excluded from the calculation of summary statistics presented in this table.
All 14 converters were pre-symptomatic at baseline.
The 42 person-visits in the converter group included visits from when participants were pre-symptomatic and after phenoconversion.
Natural algorithm
None of the pNfH concentrations was below the limit of detection. N=11 participants (N=1 control, N=7 at-risk, N=3 converters) had CSF pNfH only at follow-up visits. Their data are included in the “All Available Visits” columns but not the “Baseline Visit Only” columns.
Results
Study Population
The study population (Table 1) comprises 34 controls; 79 at-risk individuals (who have remained pre-symptomatic throughout the duration of follow-up to date); 14 converters (who have undergone phenoconversion and followed longitudinally from the pre-symptomatic to the symptomatic phase); and 22 affected individuals (i.e. patients with ALS at the time of initial assessment). While the affected group includes 15 ALS patients in whom there is no identified genetic mutation, all individuals in the at-risk group are at genetic risk for disease, largely due to an SOD1 mutation (n=27 A4V, n=22 non-A4V) or a C9orf72 hexanucleotide repeat expansion (n=25). These at-risk individuals have been followed for a median (range) of 3.7 (1.0-8.7) years. Most of the converters were SOD1 (especially A4V) mutation carriers, but one carried a FUS mutation (c.521del6), and two a C9orf72 repeat expansion. Apart from one of the C9orf72 converters whose initial clinical manifestations were cognitive/behavioral, the earliest clinical features were motor in all other phenoconverters (Table e1). Clinical and pNfH data are available from all participants at time of initial sample collection (“baseline”). In addition, longitudinal data are available from a subset of 13 controls, 55 at-risk, 10 converters, and 16 affected, for a total of 393 person-visits (Table 1).
The control and at-risk groups are comparable in baseline age (mean ±SD = 48±12 and 47±11 years, respectively), with the converters slightly older (52±12 years) and the affected about 10 years older (60 ±7 years; p<0.001 when compared to controls or at-risk). As of the cut-off date for the pNfH samples included in this experiment, we had conducted a median of 3 (and up to 7) longitudinal visits on the N=94 participants in the longitudinal subset, with varying lengths of follow-up for the different groups (Table 1).
Baseline pNfH Concentration
Baseline serum pNfH levels (Table 2) are comparable between controls (median [range] = 23 [0.03-113] pg/ml), at-risk individuals (21 [1-213] pg/ml) and phenoconverters (13 [0.6-367] pg/ml), irrespective of genotype (Figure 1). While median serum levels are, as expected, higher in patients with ALS (85 [2.0-1156] pg/ml) as compared to controls (p<0.001), there is substantial overlap between these groups in their range of serum pNfH values. On the other hand, while lumbar puncture was only performed in 77 of 149 (52%) study participants at baseline, there is much less overlap in CSF pNfH values between ALS affected and either control or at-risk groups (Table 2, Figure 1). Serum and CSF pNfH levels are poorly correlated (Spearman rank correlation, r=0.09, p=0.4).
Figure 1. Baseline levels of pNfH in serum and CSF.
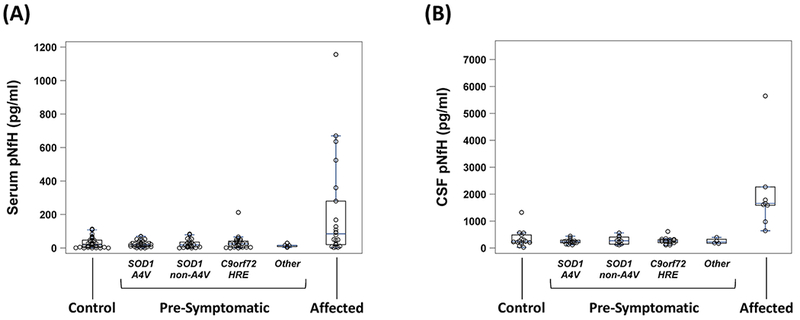
(A) Serum pNfH (pg/ml); and (B) CSF pNfH (pg/ml). Boxes show median, and 25th and 75th percentiles; whiskers extend to a maximum of 1.5 x interquartile range (IQR), or to the most extreme value if it is less than 1.5 x IQR from the 25th or 75th percentile. CSF = cerebrospinal fluid; pNfH = phosphorylated neurofilament heavy.
For the investigation of potential age effect, we focused on controls (who are free of disease), but also considered the at-risk group (most of whom are likely to be as yet minimally affected by disease, if at all) given its much larger samples size. While there is no correlation between age and baseline serum pNfH concentrations in controls (r=0.11, p=0.5), there is a strong correlation between age and CSF pNfH (r=0.71, p=0.003) (Table e2). The magnitude of this age-related CSF pNfH elevation, however, is negligible in the context of disease-related elevation among ALS patients (Figure 2). Moreover, neither serum nor CSF pNfH levels significantly differed by sex (Table e3) or the underlying genotype (at-risk group only, Table e4).
Figure 2. Scatterplot of baseline serum pNfH levels vs. age.
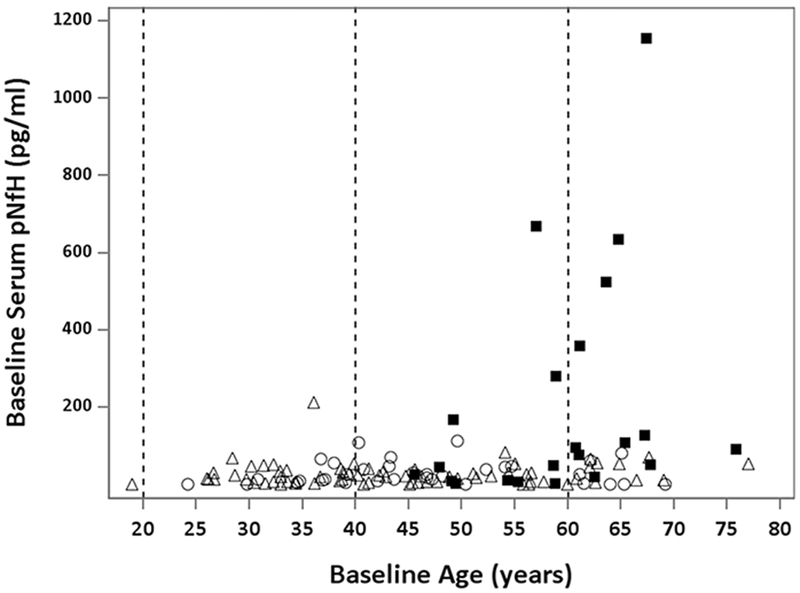
Although pNfH levels are slightly higher among older individuals in the control group (open circles) and at-risk group (open triangles), the magnitude of this increase is negligible in comparison to the levels of pNfH observed among ALS patients (closed squares); that is, older age does not explain the increase in serum pNfH among ALS patients. Vertical dashed lines demarcate age groups (< 40, 40-60 and > 60 years). ALS = amyotrophic lateral sclerosis; NfL = neurofilament light; pNfH = phosphorylated neurofilament heavy.
Longitudinal Changes in Neurofilament Concentration
Longitudinally, serum pNfH levels are relatively stable among controls, at-risk individuals who have not yet undergone phenoconversion, and ALS patients (Figure 3); the median rate of pNfH increase in these 3 groups ranges from 0.76 to 1.83 pg/ml per year, after adjusting for baseline age. In contrast, in the majority of converters with data available from multiple time points before (or shortly after) phenoconversion, we observed an increase in serum pNfH in advance of the appearance of manifest disease (Figure 4A), with an age-adjusted median rate of pNfH increase of 93.6 pg/ml per year. Moreover, in mixed model analyses using log-transformed pNfH values and adjusting for baseline age, the rate of pNfH increase is significantly higher in converters compared to controls (p=0.003, group x time interaction term), at-risk individuals (p<0.0001), and ALS affected (p=0.002).
Figure 3. Longitudinal changes in serum pNfH concentrations (pg/ml).
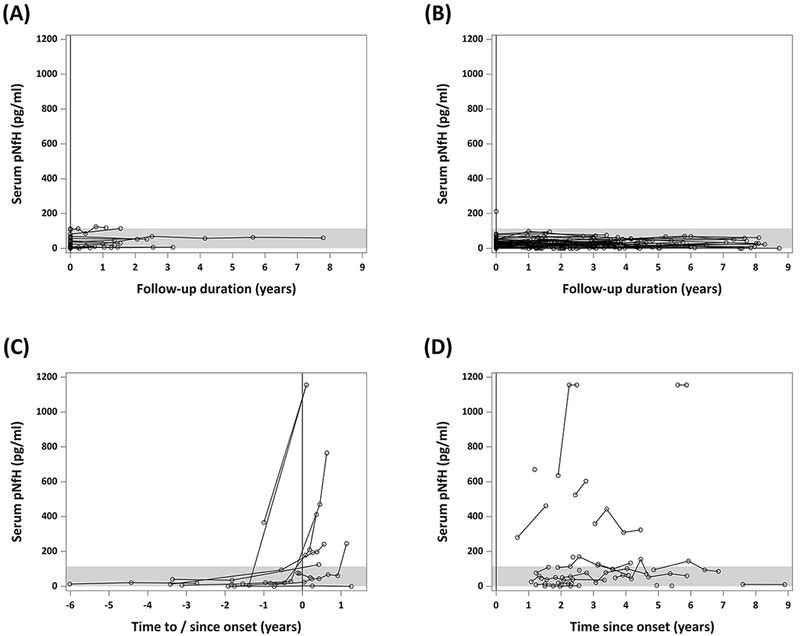
(A) Controls; (B) at-risk individuals who remain pre-symptomatic throughout follow-up; (C) phenoconverters; and (D) ALS patients. The x-axis in (A) and (B) shows years since baseline. The x-axis in (C) and (D) shows years to or since the onset of symptoms/signs, which is marked by the vertical dashed line at year = 0. ALS = amyotrophic lateral sclerosis; pNfH = phosphorylated neurofilament heavy. Shading indicates the 95th percentile of all available data among controls.
Figure 4. Longitudinal changes in serum NfL and pNfH among phenoconverters.
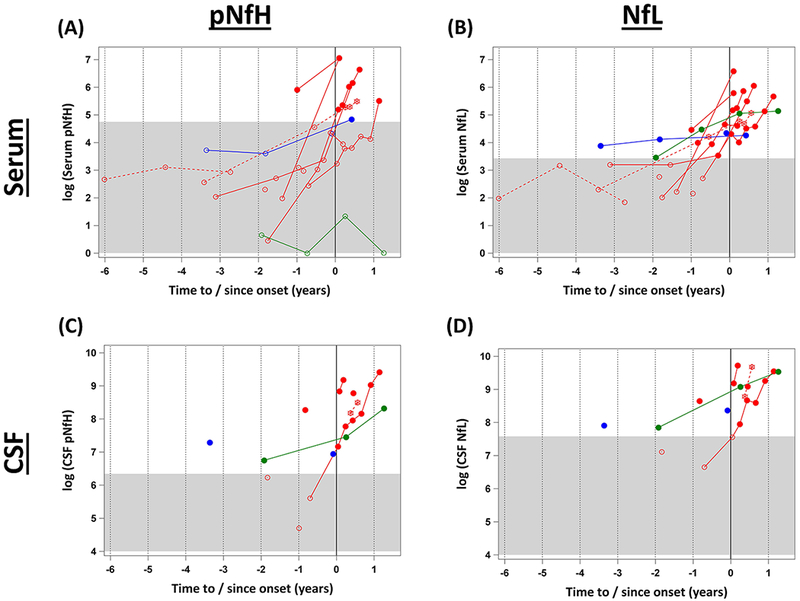
(A) Serum pNfH; (B) serum NfL; (c) CSF pNfH; (d) CSF NfL each plotted on the natural logarithm scale. The x-axis shows years to or since the onset of symptoms/signs, which is marked by the vertical dashed line at year = 0. The gray area covers the range of values within the normative threshold, defined here as the 95th percentile of serum NfL or pNfH values observed in the control group. Colors indicate genotype, with SOD1 A4V in solid red, SOD1 non-A4V in dotted red, FUS in green, and C9orf72 in blue. The closed circles mark the levels that are elevated above the normative threshold. ALS = amyotrophic lateral sclerosis; NfL = neurofilament light; pNfH = phosphorylated neurofilament heavy.
The absolute level of serum pNfH, on the other hand, exceeded the normative threshold in only one individual at about 1 year prior to phenoconversion (Figure 4A). By comparison, not only is a pre-symptomatic increase in serum NfL also observed in the majority of cases, the concentration of NfL is elevated above the normative threshold in all converters 11 (Figure 4B). CSF data are sparser. In four converters we observed an elevation in the absolute level of pNfH and NfL prior to phenoconversion; these elevations occurred, ~3.5 years, 2 years, ~9 months, and shortly before phenoconversion (Figures 4C and 4D).
Relationship Between Genotype and the Timing of Neurofilament Increase
Considering all available pNfH as well as NfL data from all 14 converters (4 with only baseline data so far, 10 with longitudinal data), we observe a relationship between genotype and the timing of initial neurofilament elevation. In both serum and CSF, the elevation is observed as far back as 6-12 months prior to phenoconversion among SOD1 A4V mutation carriers; as far back as 2 years in the single FUS c.521del6 converter; and as far back as 3.5 years in the C9orf72 HRE motor converter.
Discussion
Neurofilaments have been investigated in the pre-symptomatic phase of a range of neurodegenerative diseases (summarized in Table 3). With the exception of TRACK-HD, DIAN and our own Pre-fALS study, these studies have largely been cross-sectional, with longitudinal data available from only small numbers of participants, few (if any) phenoconverters, and therefore little opportunity to quantify neurofilament before and after phenoconversion. Moreover, these studies have focused almost exclusively on NfL. Here, we present data showing the longitudinal change in pNfH in a large number of pre-symptomatic individuals at genetic risk for ALS, with quantification of pNfH in serum and (to a lesser extent, CSF) both before and after the emergence of clinically manifest disease. We also present updated NfL data, with additional converters added to previously published data 11. The critical observations are a pre-symptomatic increase in pNfH and NfL among phenoconverters. While the temporal patterns of the increases in serum and CSF NfL and pNfH are comparable, there is one important difference. Namely, in serum, we observe longitudinal increases (i.e. positive slopes) in both NfL and pNfH prior to manifest disease, but the elevation in absolute levels to beyond our a priori conservatively defined normative threshold is consistently observed in NfL but not pNfH. Whether pNfH measured using more sensitive assays (e.g. Simoa) might yield a different conclusion is as yet unknown; it is, however, the focus of an ongoing study. In contrast to the findings in serum, CSF data, albeit sparser, show similar patterns for both NfL and pNfH. The more striking observations from our NfL data may be driven, at least in part, by the more distinct separation between the ALS affected group and the control and at-risk groups in absolute levels of serum NfL. (In CSF, on the other hand, both NfL and pNfH show comparable separation between these groups). The observation that there is significant overlap in the level of blood pNfH between controls and ALS patients is consistent with previously published data 12,18. The greater sensitivity of serum NfL (than serum pNfH) to detecting pre-symptomatic disease is perhaps also related to the strong correlation we observed between serum and CSF in NfL (r=0.79, p <0.001) but not pNfH. This in turn, may be a function of the greater methodological difficulty of accurately quantifying serum pNfH given the tendency for pNfH to be sequestrate in hetero-aggregate immune complexes 19. It may also reflect the recently reported energy-saving adaptive response to neurodegeneration in ALS, in which upregulation of NfL and downregulation of neurofilament medium (NfM) and NfH leads to shift in the highly conserved stoichiometry of neurofilament isoforms 13.
Table 3.
Published data on neurofilaments in pre-symptomatic neurodegenerative diseases
| Disease | Genotypes with Data Available | Biomarker | Matrix | Data Type | N | Compared to Controls | Pre-symptomatic Longitudinal Increase | Reference |
|---|---|---|---|---|---|---|---|---|
| HD | HTT | NfH | Plasma | Cross-sectional | 29 | No difference | -- | Wild et al 24 |
| HD | HTT | NfL | Plasma | Cross-sectional | 104 | Higher | -- | Byrne et al 8 |
| HD | HTT | NfL | Plasma | Longitudinal | 97 | -- | Yes | Byrne et al 8 |
| HD | HTT | NfL | CSF | Cross-sectional | 32 | Higher | -- | Vinther-Jensen et al 9 |
| AD | PSEN1, APP | NfL | Serum | Cross-sectional | 19 | Higher | -- | Weston et al 4 |
| AD | PESN1, APP | NfL | Serum | Longitudinal | 25 | -- | Yes | Weston et al 3 |
| AD | PSEN1, PSEN2, APP | NfL | Serum | Longitudinal | 78 | -- | Yes | Preische et al 2 |
| FTD | MAPT, GRN, C9orf72 | NfL | Serum | Cross-sectional | 44 | No difference | -- | Meeter et al 7 |
| FTD | CHMP2B | NfL | CSF | Cross-sectional | 6 | No difference (after age adjustment) | -- | Rostgaard et al 5 |
| FTD | MAPT, GRN, C9orf72 | NfL | CSF | Cross-sectional | 40 | No difference | -- | Meeter et al 7 |
| FTD | MAPT, GRN, C9orf72 | NfL | CSF | Cross-sectional | 8 | No difference | -- | Scherling et al 6 |
| FTD | GRN | NfL | CSF | Longitudinal | 2 | -- | Unclear | Meeter et al 7 |
| FTD/ALS | C9orf72 | NfL | CSF | Cross-sectional | 25 | No difference | -- | Meeter et al 25 |
| FTD/ALS | C9orf72 | NfL | CSF | Longitudinal | 2 | -- | No | Meeter et al 25 |
| ALS | C9orf72, SOD, TARDBP, FUS | NfL | CSF | Cross-sectional | 7 | No difference | -- | Weydt et al 26 |
| ALS | C9orf72, SOD, TARDBP, FUS | pNfH | CSF | Cross-sectional | 9 | No difference | -- | Weydt et al 26 |
| ALS | C9orf72, SOD, TARDBP, FUS | NfL | Blood | Cross-sectional | 11 | No difference | -- | Weydt et al 26 |
| ALS | SOD1, FUS, C9orf72 | NfL | Serum | Cross-sectional | 94 | No difference | -- | Benatar et al 11 |
| ALS | SOD1, FUS | NfL | Serum | Longitudinal | 59 | -- | Yes | Benatar et al 11 |
| ALS | SOD1, FUS, C9orf72 | NfL | CSF | Cross-sectional | 63 | No difference | -- | Benatar et al 11 |
| ALS | SOD1, FUS | NfL | CSF | Longitudinal | 32 | -- | Yes | Benatar et al 11 |
| ALS | C9orf72 | pNfH | CSF | Cross-sectional | 8 | No difference | -- | Poesen et al 27 |
HD – Huntington’s disease; AD – Alzheimer’s disease; FTD – frontotemporal dementia; NfH – Neurofilament Heavy; pNfH – Phosphorylated neurofilament heavy; NfL – Neurofilament light; CSF – cerebrospinal fluid
Importantly, having now followed a larger number of phenoconverters with different genotypes, we acquired the following insight about the relationship between the timing of neurofilament increase, post-phenoconversion survival duration, and the underlying genotype: for SOD1 A4V, an aggressive disease with median survival ~12 months, the pre-symptomatic neurofilament increase was apparent as far back as 6-12 months prior to phenoconversion. For FUS c.521del6, with a post-phenoconversion survival of 3.4 years, elevated neurofilament levels were evident at least 2 years prior to phenoconversion. For C9orf72 HRE, we have observed elevated NfL and pNfH as far back as ~3.5 years prior to phenoconversion; survival duration for this individual is unknown as (s)he has only recently developed clinically manifest disease. While the generalizability of our findings will require further observations in phenoconverters with an even broader array of genotypes, these data suggest the intriguing possibility that the duration of the pre-symptomatic phase of ALS (as defined by an increase in neurofilament levels) is proportional to that of the symptomatic phase of disease That is, the pre-symptomatic phase is longer in patients with more slowly progressive (symptomatic) disease, and shorter in patients with more aggressive manifest disease.
By providing critical insight into the pre-symptomatic phase of ALS, these neurofilament data move us closer to one of the major long-term goals of the Pre-fALS study 20 – namely, the design of a disease prevention or early treatment trial. A major challenge to the design and implementation of such a trial has, until now, been our inability to predict when someone at genetic risk for ALS will develop manifest disease 21. (Parenthetically, this is less of a problem for HD in which age and CAG repeat length may be used to predict likely age of onset 22, and in familial AD in which age of onset of parents and/or other family members may similarly be used 23.) The data presented show that a rise in serum neurofilament concentration is a prognostic biomarker predicting when manifest disease is likely to emerge. For a disease prevention trial, therefore, instead of enrolling anyone at genetic risk for ALS (without knowing when manifest disease is likely to emerge), we may now enroll the subset of this population most likely to develop manifest disease within a relatively short period of time (i.e. those in whom an increase in serum neurofilament levels is observed). Alternatively put, a pre-symptomatic increase in serum neurofilament levels may be used as a biomarker eligibility criterion for a disease prevention trial. Such an approach would not only obviate unnecessarily prolonged exposure to an experimental therapeutic (among people whom might not develop disease for many years), but would also enrich the study population for at-risk individuals most likely to meet the study endpoint of phenoconversion during a defined follow-up period. Moreover, the timing of the rise in neurofilament in relationship to phenoconversion among those with an SOD1 mutation associated with rapidly progressive disease, suggests that this may be the population in which it is most feasible to try and delay or prevent the emergence of manifest disease.
Supplementary Material
Table e1. Phenoconverters: clinical signs or symptoms indicating manifest disease
Table e2. Baseline serum and CSF pNfH levels vs. age
Table e3. Baseline serum and CSF pNfH levels, by sex
Table e4. Baseline serum and CSF pNfH levels in the at-risk group, by genotype
Acknowledgements
We extend thanks to our research team at the University of Miami for participant recruitment and evaluation (Dr. Volkan Granit, Anne-Laure Grignon, Danielle Dauphin, M. Catalina Fernandez, Danielle Sheldon, Eliana Reyes, Sumaira Hussain, Anne Cooley, Jessica Stark, Dr. Christina Bermudez, Jessica Medina, Ashley Manso) and data management (Christine Clayman); and Christine Stanislaw at Emory University for providing genetic counseling. We are also grateful to Euroimmun for in-kind support through the provision of ELISA kits. Most importantly, we are grateful to all the Pre-fALS participants as well as control and ALS affected participants for their altruism as well as their commitment and contribution to advancing ALS therapy development efforts.
Study Funding: The study was sponsored by the Muscular Dystrophy Association (Grant #4365 and #172123), the ALS Association (Grant #2015), the ALS Recovery Fund, the Kimmelman Estate, the National Institutes of Health (R01 NS105479), the Knut and Alice Wallenberg Foundation, the Swedish Research Council, and the Swedish Brain Research Foundation. Euroimmun provided pNfH assay kits as in-kind support.
Disclosures:
Drs. Benatar and Malaspina have filed a provisional patent entitled: “Determining onset of amyotrophic lateral sclerosis”. M. Benatar reports grant support from Muscular Dystrophy Association, ALS Association, ALS Recovery Fund, Kimmelman Estate, Target ALS, Eli Lilly & Company, and the National Institutes of Health (NIH) during the conduct of the study. He also reports grant support from FDA, CDC, and DOD; research support from Alexion Pharmaceuticals, UCB, Cytokinetics, Neuraltus, Biogen and Orphazyme A/S; and personal fees from NMD Pharma, Ra Pharmaceuticals, Mitsubishi-Tanabe, Avexis, UCB and Denali outside the submitted work.
J Wuu reports grant support from Muscular Dystrophy Association, ALS Association, ALS Recovery Fund, Kimmelman Estate, Eli Lilly & Company and the NIH during the conduct of the study; and grant support from FDA, CDC, and DOD, and from Target ALS Foundation outside the submitted work.
V. Lombardi reports no disclosures.
A. Jeromin has received compensation and stock options from Iron Horse Diagnostics, Inc.
R. Bowser reports grants from the ALS Association, the Muscular Dystrophy Association and Target ALS; and personal fees from Mitsubishi-Tanabe outside the submitted work. He has also received compensation and stock options from Iron Horse Diagnostics, Inc.
P. M. Andersen reports grants from the Swedish Research Council, The Swedish Brain Research Foundation, the Knut and Alice Wallenberg Foundation; research support from Orphazyme A/S, Orion Pharma and Biogen; and personal fees from Biogen, Orphazyme A/S, and Hoffman la Roche/Genentech outside the submitted work.
A. Malaspina reports grants from NIH, MND Association UK, NIHR (UK), EU2020
References
- 1.Sperling RA, Rentz DM, Johnson KA, et al. The A4 study: stopping AD before symptoms begin? Sci Transl Med 2014;6:228fs213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Preische O, Schultz SA, Apel A, et al. Serum neurofilament dynamics predicts neurodegeneration and clinical progression in presymptomatic Alzheimer’s disease. Nature medicine 2019;25:277–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weston PSJ, Poole T, O’Connor A, et al. Longitudinal measurement of serum neurofilament light in presymptomatic familial Alzheimer’s disease. Alzheimers Res Ther 2019;11:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weston PSJ, Poole T, Ryan NS, et al. Serum neurofilament light in familial Alzheimer disease: A marker of early neurodegeneration. Neurology 2017;89:2167–2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rostgaard N, Roos P, Portelius E, et al. CSF neurofilament light concentration is increased in presymptomatic CHMP2B mutation carriers. Neurology 2018;90:e157–e163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Scherling CS, Hall T, Berisha F, et al. Cerebrospinal fluid neurofilament concentration reflects disease severity in frontotemporal degeneration. Annals of neurology 2014;75:116–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Meeter LH, Dopper EG, Jiskoot LC, et al. Neurofilament light chain: a biomarker for genetic frontotemporal dementia. Annals of clinical and translational neurology 2016;3:623–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Byrne LM, Rodrigues FB, Blennow K, et al. Neurofilament light protein in blood as a potential biomarker of neurodegeneration in Huntington’s disease: a retrospective cohort analysis. Lancet Neurol 2017;16:601–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vinther-Jensen T, Bornsen L, Budtz-Jorgensen E, et al. Selected CSF biomarkers indicate no evidence of early neuroinflammation in Huntington disease. Neurology(R) neuroimmunology & neuroinflammation 2016;3:e287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wild E, Tabrizi S. Premanifest and early Huntington’s disease In: Bates G, Tabrizi S, Jones L, eds. Huntington’s Disease. Pages 86–106. New York: Oxford University Press, 2014. [Google Scholar]
- 11.Benatar M, Wuu J, Andersen PM, Lombardi V, Malaspina A. Neurofilament light: A candidate biomarker of pre-symptomatic ALS and phenoconversion. Annals of neurology 2018;84:130–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lu CH, Petzold A, Topping J, et al. Plasma neurofilament heavy chain levels and disease progression in amyotrophic lateral sclerosis: insights from a longitudinal study. J Neurol Neurosurg Psychiatry 2015;86:565–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zucchi E, Lu CH, Cho Y, et al. A motor neuron strategy to save time and energy in neurodegeneration: adaptive protein stoichiometry. J Neurochem 2018;146:631–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Benatar M, Wuu J. Pre-Symptomatic Studies in ALS: Rationale, Challenges and Approach. Neurology 2012;79:1732–1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abrahams S, Newton J, Niven E, Foley J, Bak TH. Screening for cognition and behaviour changes in ALS. Amyotrophic lateral sclerosis & frontotemporal degeneration 2013. [DOI] [PubMed] [Google Scholar]
- 16.Strong MJ, Abrahams S, Goldstein LH, et al. Amyotrophic lateral sclerosis - frontotemporal spectrum disorder (ALS-FTSD): Revised diagnostic criteria. Amyotroph Lateral Scler Frontotemporal Degener 2017;18:153–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Benatar M, Turner M, Wuu J. Defining pre-symptomatic amyotrophic lateral sclerosis. Amyotroph Lateral Scler and Frontotemporal Degeneration 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.De Schaepdryver M, Jeromin A, Gille B, et al. Comparison of elevated phosphorylated neurofilament heavy chains in serum and cerebrospinal fluid of patients with amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry 2018;89:367–373. [DOI] [PubMed] [Google Scholar]
- 19.Adiutori R, Aarum J, Zubiri I, et al. The proteome of neurofilament-containing protein aggregates in blood. Biochem Biophys Rep 2018;14:168–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Benatar M, Polak M, Kaplan S, Glass J. Preventing familial amyotrophic lateral sclerosis: Is a clinical trial feasible? Journal of the neurological sciences 2006;251:3–9. [DOI] [PubMed] [Google Scholar]
- 21.Benatar M, Polak M, Feng R. Predicting the risk of disease in familial amyotrophic lateral sclerosis. American Neurological Association 133rd Annual Meeting Salt Lake City2008. [Google Scholar]
- 22.Langbehn DR, Hayden MR, Paulsen JS. CAG-repeat length and the age of onset in Huntington disease (HD): a review and validation study of statistical approaches. American journal of medical genetics Part B, Neuropsychiatric genetics : the official publication of the International Society of Psychiatric Genetics 2010;153B:397–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ryman DC, Acosta-Baena N, Aisen PS, et al. Symptom onset in autosomal dominant Alzheimer disease: a systematic review and meta-analysis. Neurology 2014;83:253–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wild EJ, Petzold A, Keir G, Tabrizi SJ. Plasma neurofilament heavy chain levels in Huntington’s disease. Neurosci Lett 2007;417:231–233. [DOI] [PubMed] [Google Scholar]
- 25.Meeter LHH, Gendron TF, Sias AC, et al. Poly(GP), neurofilament and grey matter deficits in C9orf72 expansion carriers. Annals of clinical and translational neurology 2018;5:583–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weydt P, Oeckl P, Huss A, et al. Neurofilament levels as biomarkers in asymptomatic and symptomatic familial amyotrophic lateral sclerosis. Annals of neurology 2016;79:152–158. [DOI] [PubMed] [Google Scholar]
- 27.Poesen K, De Schaepdryver M, Stubendorff B, et al. Neurofilament markers for ALS correlate with extent of upper and lower motor neuron disease. Neurology 2017;88:2302–2309. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table e1. Phenoconverters: clinical signs or symptoms indicating manifest disease
Table e2. Baseline serum and CSF pNfH levels vs. age
Table e3. Baseline serum and CSF pNfH levels, by sex
Table e4. Baseline serum and CSF pNfH levels in the at-risk group, by genotype