

Abstract
Recent reports have suggested a reproducible association between the rs11121615 SNP, located within an intron of the castor zinc finger 1 (CASZ1) gene, and varicose veins. This study aimed to determine if this variant is also differentially associated with the various clinical classifications of chronic venous disease (CVD). The rs11121615 SNP was genotyped in two independent cohorts from New Zealand (n = 1876 controls /1606 CVD cases) and the Netherlands (n = 1626/2966). Participants were clinically assessed using well-established CVD criteria. The association between the rs11121615 C-allele and varicose veins was validated in both cohorts. This was strongest in those with higher clinical severity classes and was not significant in those with non-varicose vein CVD. Functional analysis of the rs11121615 variant demonstrated that the risk allele was associated with increased enhancer activity. This study demonstrates that the CASZ1 gene associated C-allele of rs11121615 has a significant, reproducible, association with CVD (CEAP C ≥ 2 meta-odds ratio 1.31, 95% CI 1.27–1.34, P = 1 × 10−98, PHet = 0.25), but not with non-varicose vein (CEAP C1, telangiectasia or reticular veins) forms of venous disease. The effect size of this association therefore appears to be susceptible to influence by phenotypic heterogeneity, particularly if a cohort includes a large number of cases with lower severity CVD.
Subject terms: Functional genomics, Genetic association study
Introduction
Chronic venous disease is common1 and is associated with a significant socioeconomic burden2, accounting for an estimated 1–2% of total health budget costs in Western Europe and the United States of America3. Family-based studies suggest that CVD has a strong genetic risk component4–6. Although a large number of candidate gene-based studies have been previously undertaken, these have failed to produce robust genetic associations7. In recent years several genome-wide association studies (GWAS) have been conducted with a specific focus on varicose veins of the lower limbs. The first of these was conducted by 23andMe, examining European participants with self-declared varicose veins. This work was presented in poster form at the 64th Annual Meeting of the American Society of Human Genetics in 20148. This was followed by a GWAS examining more clinically verified cohorts of German participants9. Interestingly, there appeared to be little overlap between the results of these two GWAS. Very recently Shadrina and colleagues performed a validation of the top hits from both of these two GWAS, using two independent cohorts from Russia and the United Kingdom (UK Biobank)10. By far the strongest association, both in terms of effect size and statistical power, was with rs11121615, initially identified in the 23andMe study. This single nucleotide polymorphism (SNP) is located within an intron of the castor zinc finger 1 (CASZ1) gene however, to date, the functional consequence of this variant is unclear.
We therefore undertook a validation study, in two independent cohorts from New Zealand (NZ) and the Netherlands (NL), to compare the association of this risk variant with different clinical classifications of venous disease. In addition, we performed functional analyses to determine the potential biological mechanism(s) through which the risk allele may be able to influence disease pathobiology.
Results
The demographics of the NZ and NL cohorts are shown in Table 1. The proportion of participants with CEAP C1 disease reflected the differing designs of the two studies (NZ: selected cases referred for clinical venous assessment, NL: a population-based cohort study). Accordingly, over half (57.5%) of the NL case cohort were in the CEAP C1 group while only 6% of the NZ cases had this clinical classification.
Table 1.
Demographics of the New Zealand and Netherlands cohorts divided by chronic venous disease clinical classification.
| Controls, CEAP C0 | Self-declared VV | CEAP C1 | CEAP C2 | CEAP C3 | CEAP C4 | CEAP C5-6 | Varicose veins CEAP ≥ C2 | |
|---|---|---|---|---|---|---|---|---|
| New Zealand, n | 1876 | 564 | 97 | 535 | 60 | 202 | 148 | 1509 |
| Age | 68.8 (7.6) | 67.7 (9.5) | 71.2 (10.5) | 60.4 (14.7) | 63.3 (12.0) | 60.8 (11.8) | 63.0 (12.6) | 64.3 (12.3) |
| sex (% male) | 64.6 | 50.0 | 56.4 | 38.7 | 46.6 | 55.3 | 56.7 | 47.7 |
| The Netherlands, n | 1626 | — | 1704 | 925 | 268 | 62 | 7 | 1262 |
| age | 68.4 (9.6) | — | 69.4 (9.8) | 71.9 (9.7) | 75.3 (9.1) | 76.0 (10.9) | 67.5 (14.1) | 72.8 (9.8) |
| sex (% male) | 53.4 | — | 34.2 | 39.7 | 38.4 | 40.3 | 57.4 | 39.5 |
Participants were characterised using the clinical classification matrix of the CEAP chronic venous disease criteria, except a group of self-declared NZ participants, who were visually inspected to confirm varicose veins and exclude those with CEAP C1.
CEAP clinical classifications; (C0) No evidence of venous disease, (C1) telangiectasia or reticular veins, (C2) uncomplicated varicose veins, (C3) edema, (C4) Skin changes ascribed to venous disease (pigmentation or eczema, lipodermatosclerosis), (C5) healed or (C6) active venous ulcer. Age is represented as mean (standard deviation).
The association between rs11121615 and chronic venous disease
The rs11121615 SNP was in Hardy-Weinberg equilibrium in both the NZ (cases P = 0.86 and controls P = 0.10) and NL (cases P = 0.22 and controls = 0.07) cohorts.
The rs11121615 minor allele frequencies (MAF) for both cohorts are summarized in Figs 1 and 2. The frequency of the minor C-allele was similar in the control cohorts from both NZ and NL (MAF 0.30 and 0.32 respectively, P = 0.15). There was no statistical difference in the allele frequencies between controls and case participants with CEAP C1 venous disease in either study.
Figure 1.
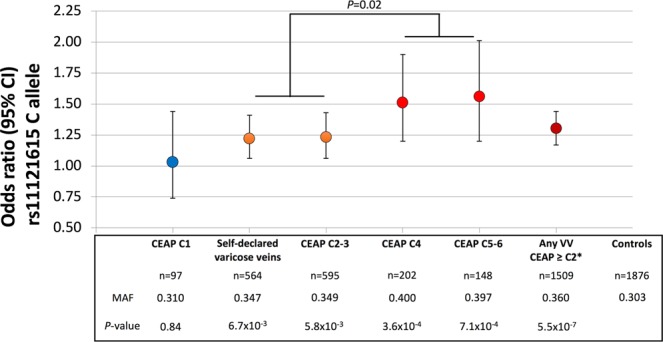
rs11121615 C-allele (CASZ1) association with chronic venous disease in the New Zealand cohort. *The ‘Any VV’ group includes those with clinically classified (CEAP ≥ C2) and the self-declared (but visually verified) varicose veins.
Figure 2.
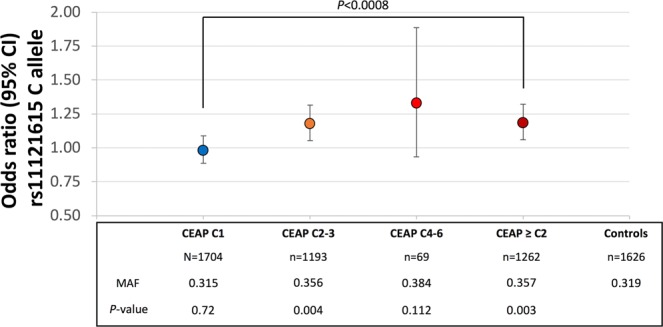
rs11121615 C-allele (CASZ1) association with chronic venous disease in the Netherlands study.
In both cohorts, those with varicose veins (CEAP ≥ C2 or self-declared but visually verified varicose veins) had a higher C-allele frequency than either controls or participants with CEAP C1. In the NL cohort, CEAP C1 participants had a significantly (P < 0.0008) lower C-allele frequency than those with a CEAP ≥ C2 (Fig. 2). Given that CEAP C1 classified venous alterations are not considered to be varicose veins11, we then excluded such individuals from the subsequent association analysis. The unadjusted odds ratio for the C-allele being associated with varicose veins (CEAP ≥ C2 or visually verified varicose veins) were 1.30 (95% CI 1.17–1.44, P = 5.5 × 10−7) and 1.18 (1.06–1.32, P = 2.5 × 10−3) in the NZ and NL cohorts (Figs 1 and 2 respectively and Supplementary Table 2). These associations remained significant after including age and sex as covariates (1.27, 1.14–1.42, P = 9.9 × 10−6 and 1.17, 1.04–1.31, P = 0.01 respectively).
A meta-analysis was performed using the major T-allele as the reference and minor C-allele as the risk variant. The odds ratios from all contributing cohorts were adjusted for age and sex. The NZ and NL C-allele associations in those with varicose veins and more advanced CVD (by excluding those with CEAP C1) were analyzed in a fixed effect model meta-analysis with the previously reported 23andMe, Russian and United Kingdom associations. The resulting odds ratio was highly statistically significant (Fig. 3, odds ratio 1.20, 95% CI 1.18–1.22, P = 1.5 × 10−90). Despite consistent directions of effect across all cohorts there was significant heterogeneity in the fixed effect model (I2 95.8, PHet < 1 × 10−8). This was driven primarily by the inclusion of the 23andMe cohort, with the meta odds ratio rising to 1.31 (95% CI 1.27–1.34, P = 9.0 × 10−99) and no significant heterogeneity in effect (I2 27.6, PHet = 0.25) when this cohort was excluded. In a random effects model the rs11121615 C-allele had an odds ratio for association with varicose veins of 1.22 (95%CI 1.09–1.36, P = 5.2 × 10−4) which increased to 1.28 (95%CI 1.22–1.35, P = 4.5 × 10−22) when the 23andMe cohort was excluded.
Figure 3.

rs11121615 C-allele (CASZ1) meta-analysis association with chronic venous disease. Both the NZ and NL cohorts consisted of chronic venous disease cases with CEAP ≥ C2. Case control status in the 23andMe cohort was self-declared (unverified). The Russian cases and controls were confirmed by clinical examination (CEAP ≥ C2), while the UK Biobank case status was electronically extracted from ICD10 clinical modification codes (I83; varicose veins of the lower extremity). Results from all cohorts were adjusted for age and sex.
Because of the well clinically phenotyped nature of the NZ and NL cohorts a further analysis was conducted to determine if the rs111211615 C-allele showed any further stratified association with the clinical classification of CVD. In both cohorts the highest C-allele frequency was observed in those with CEAP C4-6. In the NZ cohort there was a significant difference between those with either self-declared varicose veins or varicose veins with or without edema (CEAP C3 or C2 respectively) and those with CEAP ≥ C4 (Fig. 1, C-allele frequency 0.35 versus 0.40 respectively, P = 0.02).
Rs11121615 functional analysis
The ability of rs11121615 to alter regulatory element activity was examined using publicly available ENCODE and Roadmap Epigenomics project data. In NHEK (normal human epidermal keratinocytes), the rs11121615 DNA region had a H3K27ac and H3K4me1 peak, DNase I hypersensitivity sites and was annotated as an enhancer by ChromHMM (Fig. 4A). It had documented enhancer functions in multiple other primary cell types (epithelial, brain, digestive, muscle, heart, lung, liver and pancreas) (Supplementary Table 2). Further, rs1121615 resides in a region that is conserved in vertebrates (Fig. 4A). Rs11121615 is predicted to alter the binding motif of the mouse zinc-finger transcription factors Zfp128, Zic1 and Zic2 (Supplementary Fig. 1), indicating it might affect gene regulation by altering the binding affinity of transcription factors.
Figure 4.

rs11121615 affects enhancer activity within a topologically associated domain (TAD) containing CASZ1. (A) UCSC snapshot showing the following tracks: UCSC genes, 100 vertebrates basewise conservation by PhyloP, ENCODE layered ChIP-Seq data for H3K27ac and H3K4me1 from seven cell lines (NHEK cells in purple), ENCODE DNase I hypersensitivity data, and 15-state chromatin state Segmentation by HMM (ChromHMM) for nine cell lines. hg19 chromosome 1 coordinates are shown. (B) Allele-specific enhancer activity for rs11121615 was assessed using a luciferase reporter assay in HEK293 cells. The rs11121615 T variant (major allele) did not harbour enhancer activity whereas the C variant (minor allele) did. The average of five biological replicates+/− the standard error of the mean is shown. Statistical significance was determined by a one-way ANOVA followed by a Tukey’s multiple comparisons test (*P < 0.0001). (C) Hi-C contact map in NHEK cells (normal human epidermal keratinocyte) at chr1:10140000-11450000 (hg19), obtained from the 3D genome browser (http://www.3dgenome.org). TADs, calculated using the directional bias method13, are indicated by alternating orange and blue bars. NCBI reference genes are annotated below.
To experimentally confirm whether the rs11121615 DNA region harbors enhancer activity, and to assess whether the SNP alters its activity, a luciferase assay was performed in HEK293 cells. The major (T) allele did not harbour enhancer activity, while the minor (C) allele showed significant activity (Fig. 4B). This data suggested that the risk allele of rs11121615 increases enhancer activity, and gives insight into the mechanism through which it could influence gene expression.
Whether rs11121615 regulates the expression of CASZ1 and/or of genes further away was assessed using publicly available chromatin interaction (Hi-C) and expression quantitative trait loci (eQTL) data. Hi-C data in several cell lines indicates that rs11121615 is located within a topologically associated domain (TAD) spanning from DFFA/PEX14 at the upstream boundary to TARDBP at the downstream boundary (Fig. 4C). The TAD boundaries were similar in all cell types for which Hi-C data is available (data not shown). Genes within the domain included CASZ1 and C1orf127 (an uncharacterized protein-coding gene) (Fig. 4C). Genes near the TAD boundaries, such as DFFA (DNA fragmentation factor subunit alpha), PEX14 (a peroxisome membrane protein) and TARDBP (a DNA and RNA-binding protein that regulates transcription and splicing), are ‘housekeeping’ type of genes and have no known specific function in vascular-related cell types. Given that regulatory elements typically confine their activity within TADs, and TAD boundaries are enriched for housekeeping genes12,13, it is likely that the rs11121615-containing enhancer regulates the expression of CASZ1 and/or C1orf127, and not of DFFA/PEX14 or TARDBP.
eQTL data from GTEx (Genotype-Tissue Expression) and ASAP (Advanced Study of Aortic Pathology) were explored to identify eQTLs for rs1112161514,15. In GTEx, no significant eQTL for rs11121615 were present, while the ASAP study had two significant eQTLs for CTNNBIP1 (P = 0.00082) and TARDBP (P = 0.013) in heart tissue. These genes are not located within the TAD however, and it is therefore unclear whether rs11121615 might regulate the expression of these genes.
Discussion
This study validated by far the strongest previously reported genetic association with CVD8,10. Located within intron 1 of the CASZ1 gene, the minor C-allele of rs11121615, had a significant positive association with CVD in all five of the independent populations examined to date. Of note, however, the fixed effect model suggested significant heterogeneity of effect, with the outlier population appearing to be the 23andMe cohort.
When considering results of case control genetic association studies, it is important to consider the quality of both case and control phenotyping. The 23andMe study, which was the first to report the CASZ1 association, utilized self-declared case status, which does not inform the clinical severity of disease and is potentially prone to bias based on an individual’s perception of what constitutes a varicose vein. For example, in the 23andMe cohort the case prevalence in females was almost three times higher than males, an observation which does not match that reported in well-designed epidemiological studies1. Nor were cases and controls well matched for age in the study. Offsetting these issues was the very large sample size (over 20,000 cases) that was examined. In contrast, the UK Biobank dataset has over 10,000 varicose vein cases, with the advantage of improved phenotyping via electronic extraction of International Classification of Disease (ICD10) codes (I83; varicose veins of lower extremities), however, there was no clinical sub-classification of those with CVD. The Russian cohort reported by Shadrina and colleagues had cases confirmed by clinical examination and duplex ultrasound with all having visible varicose veins and reflux in either the great or small saphenous vein. The vast majority of this cohort (n = 590, 83.2%) had CEAP C2-3 classified disease.
In order to determine the possible impact of such self-declared case designation, we included a group of NZ participants who had self-reported as having varicose veins on a research recruitment health questionnaire (albeit with a degree of verification via visual observation of the patient’s lower limbs by a vascular sonographer). Encouragingly, these participants showed a similar varicose vein risk association for the rs11121615 C-allele to that of patients with CEAP C2-3 in both the NZ and Dutch studies, suggesting that, at least for this variant, the self-declared phenotype represents a valid association with varicose veins. Of note, however, those classified as having CEAP C1 venous disease did not show an association with the rs11121615 C-allele in either of the cohorts in this study, with allele frequencies being strikingly similar to venous disease-free controls. Given that CEAP C1 classified venous alterations are not considered to be varicose veins11, the rs11121615 association appears to represent a varicose vein specific effect. Moreover, this suggests that inclusion of CEAP C1 individuals in a case cohort, for example when case designation is collected via an unverified self-declared mechanism, would influence the resulting analysis towards the null hypothesis of no association.
We suggest that this is the possible explanation for the lower effect size observed in the 23andMe cohort. While the meta-analysis indicated a C-allele odds ratio of 1.2 when all cohorts were included, our data suggests that the association is more likely to be in the order of 1.3 for uncomplicated varicose veins (CEAP C2) and potentially as high as 1.5 for CVD with skin changes and venous ulcers (CEAP C4-6).
Rs11121615 is located in intron 1 of the CASZ1 gene, and thus does not directly alter protein function. Our bioinformatic analysis of the region harboring rs11121615 suggested potential enhancer functions in multiple cell types. Importantly, we demonstrated that the varicose vein associated C-allele of this SNP confers a significant increase in enhancer activity. Chromatin interaction analysis suggested that CASZ1 is a likely target of this enhancer regulation. CASZ1 is expressed in human endothelial cells and drives the expression of Epidermal Growth Factor Like-Domain 7 (EGFL7), which in turn drives the expression of the RhoA gene. RhoA mediates NF-κB dependent gene expression, including VEGFR2, and thereby stimulates angiogenesis and vascular assembly16. Taken together, our data supports the proposition that rs11121615 is a causal variant influencing risk of developing varicose veins via alterations in the transcription factor CASZ1, an upstream regulator of vascular structure.
The observation that the rs11121615 C-allele is more strongly associated with more severe forms of CVD, particularly venous ulceration, needs further investigation. If this proves to be the case, this variant may have utility identifying those individuals with varicose veins who are at greater risk of progression to more severe, higher classes of CVD.
Methods
Participants
All New Zealand participants were recruited from the Otago-Southland region of New Zealand’s South Island. Participants were either part of the Otago Vascular Genetics Research (OVGR) cohort17 or were patients referred for superficial venous disease assessment. OVGR participants completed a self-administered health questionnaire which included the question “Have you ever had, or been treated, for varicose veins”. All participants attended the recruitment facility in the local hospital, at which time the questionnaire was reviewed by experienced recruitment staff. Visual inspection of the limbs of those indicating that they had varicose veins was performed by an experienced vascular sonographer. Those with only telangiectasia or reticular veins were recoded as not having significant varicose veins. Patients referred for superficial venous disease assessment to the Otago Vascular Diagnostics Laboratory were also recruited. These patients underwent a formal assessment for lower limb superficial chronic venous disease, including full ultrasound examination, and were classified using the clinical classification matrix of the clinical, etiological, anatomical and pathological (CEAP) chronic venous disorders criteria11. For bilateral disease the class of the most severely affected limb was recorded. In brief, CEAP C0, no visible or palpable signs of venous disease; C1, telangiectasia or reticular veins; C2, varicose veins (VV), distinguished from reticular veins by a diameter of greater than or equal to 3 mm; C3, edema of venous origin; C4, skin hyperpigmentation or lipodermatosclerosis; C5, healed venous ulcer; C6, active venous ulceration.
All participants from the Netherlands (NL) were part of the Rotterdam Study18, and were clinically assessed using the same CEAP classification as the NZ venous disease cohort.
All participants gave written informed consent and all research was performed in accordance with relevant guidelines/ regulations. In New Zealand the study was approved by the Health and Disability Ethics Committee (New Zealand Ministry of Health), while in the Netherlands the Rotterdam study was approved by Erasmus Medical Center’s Medical Ethics Committee and by the review board of The Netherlands Ministry of Health, Welfare and Sports.
All participants had DNA extracted from an EDTA blood sample for genotyping. New Zealand samples were assessed using a Taqman assay (C_3092341_20, Thermofisher Scientific, MA, USA).
In the Rotterdam Study, DNA from whole blood was extracted following standard protocols19. Genome-wide SNP array data is available for the Rotterdam Study with details described by Hofman and colleagues19. SNP coverage was further expanded by imputation using the MACH software (http://csg.sph.umich.edu/abecasis/mach/index.html), with default parameters, and the 1000Genomes (GIANT Phase I version 3) as the reference panel. In total 30,072,738 markers were genotyped and/or imputed. From this data we extracted best guess genotypes for the rs11121615 SNP using GCTA20 with parameter defaults. The imputation quality score for this SNP was 0.76.
Genetic association statistical analysis
The SNP-disease associations in the NZ and NL cohorts were determined using PLINK (http://pngu.mgh.harvard.edu/purcell/plink/)21. The Fisher’s exact test was used to test allelic associations. Logistic regression was used to determine interactions with covariates (age and sex). Meta-analysis was performed using fixed and random-effects modelling using the Comprehensive Meta-Analysis software package, version 3 (www.meta-analysis.com/). Effects sizes were reported as odds ratios (OR) and 95% confidence intervals. Inter-study heterogeneity was measured using the I2 statistic.
Functional studies
Luciferase assay
Enhancer activity of the region spanning rs11121615 was assessed using a luciferase reporter assay in the human embryonic kidney cell line HEK293. A 2 kb region spanning rs11121615 was amplified from the 1000 Genomes sample HG00112 (heterozygous for rs11121615)22, cloned into the pCR®8/GW/TOPO entry vector and recombined into pGL4.23-GW (Addgene, MA, USA) using the Gateway® system (LR Clonase® Enzyme mix, Invitrogen, Thermofisher Scientific, MA, USA). Primers used to amplify the cloned regions can be found in Supplementary Table 4. The construct obtained was Sanger sequenced and contained the minor allele of rs11121615 (T). Site-directed mutagenesis (SDM) was carried out obtain a construct containing the major allele of rs11121615 (C). The SDM protocol used was adapted from the QuikChange II site-directed mutagenesis protocol (Agilent Technologies, CA, USA). PCR was performed using PfuUltra High-Fidelity DNA Polymerase (Agilent Technologies) using the primers listed in Supplementary Table 4. Parental plasmid DNA was digested with DpnI (Agilent Technologies) and mutagenized plasmids were then transformed into E. coli TOP10 chemically competent cells. SDM was confirmed using Sanger sequencing. All plasmids used for transfections were isolated using the NucleoBond Xtra Midi endotoxin-free kit (Macherey-Nagel GmbH, Germany).
One day prior to transfection, HEK293 cells were seeded at 8 × 103 cells per well in a 96-well plate in 200 μL DMEM supplemented with 10% fetal bovine serum (Moregate, NZ). Transfections were carried out using Lipofectamine 3000 (Invitrogen) with 100 ng of each pGL4.23 construct and 4 or 20 ng of pRL-TK (Renilla; Promega, WI, USA) per well. Luminescence was measured 48 hours post-transfection using the Dual Glo Luciferase Assay System (Promega), on a Perkin Elmer Victor X4 plate reader. Firefly luciferase was normalised to that of Renilla and expressed relative to pGL4.23 lacking a gateway cassette.
ChIP-seq, DNase I, Hi-C, eQTL data, and computational prediction of transcription factor binding motif affinity
Histone modification ChIP-seq, DNase I hypersensitivity and 15-state ChromHMM data was obtained from the ENCODE23 and Roadmap Epigenomics24 projects through the UCSC genome browser (https://genome.ucsc.edu/) and the 3DSNP database (http://cbportal.org/3dsnp/). Previously described Hi-C data in NHEK cells was obtained using the 3D genome browser (http://www.3dgenome.org). eQTL data were obtained from GTEx and ASAP. RegulomeDB (http://www.regulomedb.org) was used to predict alterations in transcription factor binding motifs.
Supplementary information
Acknowledgements
The authors wish to thank the study participants, without whom this work could not have been performed. This study was supported by the Health Research Council of New Zealand and H.S. and J.C. Anderson Charitable Trusts (GTJ). Genetic analysis was supported by infrastructure from Genomics Aotearoa (www.genomics-aotearoa.org.nz). HEK293 cells were kindly provided by Dr. Heather Cunliffe (Department of Pathology, University of Otago, Dunedin).
Author Contributions
G.J. designed the study, performed the genetic analysis and drafted the manuscript. J.M., C.L.S. and J.H. performed the functional analysis. L.P., T.N. and M.D.M. were responsible for the Netherlands data acquisition, interpretation. V.P. was performed the New Zealand study genotyping. J.K. and A.v.R. were involved with New Zealand clinical data acquisition. All authors reviewed the manuscript.
Data Availability
The datasets generated during the current study are available from the corresponding author on reasonable request.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information accompanies this paper at 10.1038/s41598-019-50586-2.
References
- 1.Evans CJ, Fowkes FG, Ruckley CV, Lee AJ. Prevalence of varicose veins and chronic venous insufficiency in men and women in the general population: Edinburgh Vein Study. J Epidemiol Community Health. 1999;53:149–153. doi: 10.1136/jech.53.3.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Onida S, Davies AH. Predicted burden of venous disease. Phlebology. 2016;31:74–79. doi: 10.1177/0268355516628359. [DOI] [PubMed] [Google Scholar]
- 3.Rabe E, Pannier F. Societal costs of chronic venous disease in CEAP C4, C5, C6 disease. Phlebology. 2010;25(1):64–67. doi: 10.1258/phleb.2010.010s09. [DOI] [PubMed] [Google Scholar]
- 4.Ahti TM, Makivaara LA, Luukkaala T, Hakama M, Laurikka JO. Effect of family history on the incidence of varicose veins: a population-based follow-up study in Finland. Angiology. 2009;60:487–491. doi: 10.1177/0003319709335510. [DOI] [PubMed] [Google Scholar]
- 5.Brinsuk M, Tank J, Luft FC, Busjahn A, Jordan J. Heritability of venous function in humans. Arterioscler Thromb Vasc Biol. 2004;24:207–211. doi: 10.1161/01.ATV.0000107080.48969.64. [DOI] [PubMed] [Google Scholar]
- 6.Cornu-Thenard A, Boivin P, Baud JM, De Vincenzi I, Carpentier PH. Importance of the familial factor in varicose disease. Clinical study of 134 families. J Dermatol Surg Oncol. 1994;20:318–326. doi: 10.1111/j.1524-4725.1994.tb01631.x. [DOI] [PubMed] [Google Scholar]
- 7.Krysa J, Jones GT, van Rij AM. Evidence for a genetic role in varicose veins and chronic venous insufficiency. Phlebology. 2012;27:329–335. doi: 10.1258/phleb.2011.011030. [DOI] [PubMed] [Google Scholar]
- 8.Bell, R. K. et al. In The 64th Annual Meeting of The American Society of Human Genetics. Paper no. 2082M, p. 2487. https://blog.23andme.com/wp-content/uploads/2014/10/Bell_ASHG2014_varicose.pdf (last accessed 4 April 2019).
- 9.Ellinghaus E, et al. Genome-wide association analysis for chronic venous disease identifies EFEMP1 and KCNH8 as susceptibility loci. Sci Rep. 2017;7:45652. doi: 10.1038/srep45652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shadrina A, et al. Polymorphisms of genes involved in inflammation and blood vessel development influence the risk of varicose veins. Clin Genet. 2018;94:191–199. doi: 10.1111/cge.13362. [DOI] [PubMed] [Google Scholar]
- 11.Rabe E, Pannier F. Clinical, aetiological, anatomical and pathological classification (CEAP): gold standard and limits. Phlebology. 2012;27(1):114–118. doi: 10.1258/phleb.2012.012s19. [DOI] [PubMed] [Google Scholar]
- 12.Symmons O, et al. Functional and topological characteristics of mammalian regulatory domains. Genome Res. 2014;24:390–400. doi: 10.1101/gr.163519.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dixon JR, et al. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012;485:376–380. doi: 10.1038/nature11082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Folkersen L, et al. Association of genetic risk variants with expression of proximal genes identifies novel susceptibility genes for cardiovascular disease. Circ Cardiovasc Genet. 2010;3:365–373. doi: 10.1161/CIRCGENETICS.110.948935. [DOI] [PubMed] [Google Scholar]
- 15.GTEx Consortium. The Genotype-Tissue Expression (GTEx) project. Nat Genet45, 580–585 (2013). [DOI] [PMC free article] [PubMed]
- 16.Charpentier MS, et al. CASZ1 promotes vascular assembly and morphogenesis through the direct regulation of an EGFL7/RhoA-mediated pathway. Dev Cell. 2013;25:132–143. doi: 10.1016/j.devcel.2013.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jones GT, et al. Comparison of three targeted approaches to screening for abdominal aortic aneurysm based on cardiovascular risk. Br J Surg. 2016;103:1139–1146. doi: 10.1002/bjs.10224. [DOI] [PubMed] [Google Scholar]
- 18.Ikram MA, et al. The Rotterdam Study: 2018 update on objectives, design and main results. Eur J Epidemiol. 2017;32:807–850. doi: 10.1007/s10654-017-0321-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hofman A, et al. The Rotterdam Study: 2016 objectives and design update. Eur J Epidemiol. 2015;30:661–708. doi: 10.1007/s10654-015-0082-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang J, Lee SH, Goddard ME, Visscher PM. GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet. 2011;88:76–82. doi: 10.1016/j.ajhg.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Purcell S, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Genomes Project Consortium. et al. A global reference for human genetic variation. Nature526, 68–74 (2015). [DOI] [PMC free article] [PubMed]
- 23.Rosenbloom KR, et al. ENCODE data in the UCSC Genome Browser: year 5 update. Nucleic Acids Res. 2013;41:D56–63. doi: 10.1093/nar/gks1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roadmap Epigenomics Consortium. et al. Integrative analysis of 111 reference human epigenomes. Nature518, 317–330 (2015). [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during the current study are available from the corresponding author on reasonable request.