

Abstract
Background
Despite the high incidence of muscle weakness in individuals with amyotrophic lateral sclerosis (ALS) or motor neuron disease (MND), the effects of exercise in this population are not well understood. This is an update of a review first published in 2008.
Objectives
To systematically review randomised and quasi‐randomised studies of exercise for people with ALS or MND.
Search methods
We searched The Cochrane Neuromuscular Disease Group Specialized Register (2 July 2012), CENTRAL (2012, Issue 6 in The Cochrane Library), MEDLINE (January 1966 to June 2012), EMBASE (January 1980 to June 2012), AMED (January 1985 to June 2012), CINAHL Plus (January 1938 to June 2012), LILACS (January 1982 to June 2012), Ovid HealthSTAR (January 1975 to December 2012). We also searched ProQuest Dissertations & Theses A&I (2007 to 2012), inspected the reference lists of all papers selected for review and contacted authors with expertise in the field.
Selection criteria
We included randomised or quasi‐randomised controlled trials of people with a diagnosis of definite, probable, probable with laboratory support, or possible ALS, as defined by the El Escorial criteria. We included progressive resistance or strengthening exercise, and endurance or aerobic exercise. The control condition was no exercise or standard rehabilitation management. Our primary outcome measure was improvement in functional ability, decrease in disability or reduction in rate of decline as measured by a validated outcome tool at three months. Our secondary outcome measures were improvement in psychological status or quality of life, decrease in fatigue, increase in, or reduction in rate of decline of muscle strength (strengthening or resistance studies), increase in, or reduction in rate of decline of aerobic endurance (aerobic or endurance studies) at three months and frequency of adverse effects. We did not exclude studies on the basis of measurement of outcomes.
Data collection and analysis
Two review authors independently assessed trial quality and extracted the data. We collected adverse event data from included trials. The review authors contacted the authors of the included studies to obtain information not available in the published articles.
Main results
We identified two randomised controlled trials that met our inclusion criteria, and we found no new trials when we updated the searches in 2012. The first, a study with overall unclear risk of bias, examined the effects of a twice‐daily exercise program of moderate load endurance exercise versus "usual activities" in 25 people with ALS. The second, a study with overall low risk of bias, examined the effects of thrice weekly moderate load and moderate intensity resistance exercises compared to usual care (stretching exercises) in 27 people with ALS. After three months, when the results of the two trials were combined (43 participants), there was a significant mean improvement in the Amyotrophic Lateral Sclerosis Functional Rating Scale (ALSFRS) measure of function in favour of the exercise groups (mean difference 3.21, 95% confidence interval 0.46 to 5.96). No statistically significant differences in quality of life, fatigue or muscle strength were found. In both trials adverse effects, investigators reported no adverse effects such as increased muscle cramping, muscle soreness or fatigue
Authors' conclusions
The included studies were too small to determine to what extent strengthening exercises for people with ALS are beneficial, or whether exercise is harmful. There is a complete lack of randomised or quasi‐randomised clinical trials examining aerobic exercise in this population. More research is needed.
Keywords: Humans, Amyotrophic Lateral Sclerosis, Amyotrophic Lateral Sclerosis/psychology, Amyotrophic Lateral Sclerosis/therapy, Exercise Therapy, Exercise Therapy/methods, Exercise Tolerance, Motor Neuron Disease, Motor Neuron Disease/psychology, Motor Neuron Disease/therapy, Muscle Weakness, Muscle Weakness/therapy, Physical Endurance, Quality of Life, Randomized Controlled Trials as Topic
Plain language summary
Therapeutic exercise for people with amyotrophic lateral sclerosis or motor neuron disease
Muscle weakness is very common in people with amyotrophic lateral sclerosis (ALS), which is also known as motor neuron disease (MND). A weak muscle can be damaged if overworked because it is already functioning close to its maximal limits. Because of this, some experts have discouraged exercise programs for people with ALS. However, if a person with ALS is not active, deconditioning (loss of muscle performance) and weakness from lack of use occurs, on top of the deconditioning and weakness caused by the disease itself. If the reduced level of activity persists, many organ systems can be affected and a person with ALS can develop further deconditioning and muscle weakness, and muscle and joint tightness may occur leading to contractures (abnormal distortion and shortening of muscles) and pain. These all make daily activities harder to do. This review found only two randomised studies of exercise in people with ALS. The trials compared an exercise program with usual care (stretching exercises). Combining the results from the two trials (43 participants), exercise produced a greater average improvement in function (measured using an ALS‐specific measurement scale) than usual care. There were no other differences between the two groups. There were no reported adverse events due to exercise. The studies were too small to determine to what extent exercise for people with ALS is beneficial or whether exercise is harmful. We found no new trials when we updated the searches in 2012. More research is needed.
Summary of findings
Summary of findings for the main comparison. Exercise for people with amyotrophic lateral sclerosis or motor neuron disease.
| Exercise for people with amyotrophic lateral sclerosis or motor neuron disease | ||||||
| Patient or population: people with amyotrophic lateral sclerosis or motor neuron disease Settings: Intervention: exercise | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Control | Exercise | |||||
| ALS Functional Rating Scale (ALSFRS) score at 3 months Scale from: 0 to 40 (higher is better). | The mean ALSFRS score at 3 months ranged across control groups from 14 to 35 | The mean ALSFRS score at 3 months in the intervention groups was 3.21 higher (0.46 to 5.96 higher) | ‐ | 43 (2 studies) | ⊕⊕⊝⊝ low1,2 | |
| Short‐Form‐36 Health Survey (SF‐36) score at 3 months Scale from: 0 to 100 (higher is better). | The mean SF‐36 score at 3 months in the control groups was 80 | The mean SF‐36 score at 3 months in the intervention groups was 2.70 higher (3.1 lower to 8.5 higher) | ‐ | 18 (1 study) | ⊕⊕⊕⊝ moderate3 | |
| Fatigue Severity Scale score at 3 months Scale from: 0 to 63 (lower is better). | The mean Fatigue Severity Scale score at 3 months in the control groups was 35 to 59 | The mean Fatigue Severity Scale score at 3 months in the intervention groups was 6.25 lower (13.82 lower to 1.31 higher) | ‐ | 43 (2 studies) | ⊕⊕⊝⊝ low1,2 | |
| Manual Muscle Testing score at 3 months Right and left shoulder abduction, elbow flexion and extension, finger abduction and extension, hip flexion, knee flexion and extension, foot dorsiflexion and plantarflexion assessed and graded 0 to 5 Medical Research Council scale. Twenty individual muscle grades summed. Scale from: 0 to 100 (higher is better). | The mean Manual Muscle Testing score at 3 months in the control groups was 87.3 | The mean Manual Muscle Testing score at 3 months in the intervention groups was 10.9 lower (23.56 lower to 1.76 higher) | ‐ | 18 (1 study) | ⊕⊕⊕⊝ moderate3 | |
| Upper extremity maximum voluntary isometric contraction score at 3 months Quantitative Muscle Assessment (QMA) system. Data were normalized, summed and averaged to yield an U/E megascore (higher is better). | The mean upper extremity maximum voluntary isometric contraction score at 3 months in the control groups was ‐9.47 | The mean upper extremity maximum voluntary isometric contraction score at 3 months in the intervention groups was 1.48 lower (4.78 lower to 1.82 higher) | ‐ | 22 (1 study) | ⊕⊕⊕⊝ moderate4 | |
| Lower extremity maximum voluntary isometric contraction score at 3 months Quantitative Muscle Assessment (QMA) system. Data were normalized, summed and averaged to yield a L/E megascore (higher is better). | The mean lower extremity maximum voluntary isometric contraction score at 3 months in the control groups was ‐23.5 | The mean lower extremity maximum voluntary isometric contraction score at 3 months in the intervention groups was 2.51 higher (2.05 lower to 7.07 higher) | ‐ | 20 (1 study) | ⊕⊕⊕⊝ moderate4 | |
| Adverse effects related to the intervention | See comment | See comment | Not estimable | 43 (2 studies) | See comment | No adverse effects reported |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; RR: risk ratio; ALS: amyotrophic lateral sclerosis | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Two small studies. No allocation concealment in either study. No blinding of assessors and no intention‐to‐treat analysis in one of the studies. Intevention group loss to follow‐up = 22.2%, control group loss to follow‐up = 12%. 2 Different exercise interventions used. 3 Small study. No allocation concealment. No blinding of assessors. Intervention group loss to follow‐up = 28.6%, control group loss to follow‐up = 27.3%. 4 Small study. Loss to follow‐up in intervention group = 15.4% (0% control group).
Background
Motor neuron diseases (MND) include a heterogeneous spectrum of inherited and sporadic clinical disorders of the upper motor neurons (UMN), lower motor neurons (LMN) or a combination of both (Table 2). MND are characterized by progressive degeneration and loss of motor neurons in the spinal cord, brain stem, or motor cortex, and manifest clinically as muscle weakness, atrophy, and corticospinal tract signs and symptoms in various combinations (Rowland 1982; Swash 2000a).
1. Motor neuron diseases (adapted from: Swash 2000a).
| Motor neuron disease |
| Sporadic ALS (Charcot) Progressive bulbar palsy Progressive muscular atrophy Primary lateral sclerosis Familial ALS Guamanian (Western Pacific) ALS Monomelic amyotrophy (Hirayama's disease) Spinal muscular atrophy type 4 Kennedy's disease |
ALS: amyotrophic lateral sclerosis
Amyotrophic lateral sclerosis (ALS) is the most common, disabling, and fatal motor neuron disease among adults. Except in a very few geographic areas, worldwide annual incidence rates have been reported to be 0.4 to 2.4 cases per 100,000, with the incidence increasing with each decade of life, until at least the seventh decade. Prevalence rates of ALS are 4 to 10 cases per 100,000 (Gubbay 1985; Haverkamp 1995; Norris 1993; Pradas 1993; Ringel 1993), and in 5% to 10% of individuals the disease is inherited as an autosomal dominant trait, familial ALS (FALS) (Mulder 1986; Strong 1991). Rare cases of juvenile onset ALS are inherited in an autosomal recessive or dominant pattern (Hamida 2000). The most frequent presenting impairment is focal, asymmetrical weakness beginning in the upper or lower extremity or weakness in the bulbar muscles (Gubbay 1985; Norris 1993). Muscle weakness is considered the cardinal sign of ALS. Initial muscle weakness usually occurs in isolated muscles, most often distally, and is followed by progressive weakness and functional limitations (Mitsumoto 1998; Swash 2000b).
Despite the high incidence of muscle weakness in individuals with ALS or MND, the effects of exercise are not well understood. In individuals with ALS or MND, the safe range for therapeutic exercise may be narrowed. It is postulated that a weak muscle is more susceptible to overwork damage, because it is already functioning close to its maximal limits (Coble 1985). In the past, some experts have discouraged exercise programs because of fear of overuse weakness, and have suggested that no exercise other than everyday activities is indicated (Sinaki 1978). The possibility of inducing overwork damage in individuals with ALS or MND through excessive exercise or strengthening exercises is a concern. For example, it has been reported that highly repetitive or heavy resistance exercise can cause prolonged loss of muscle strength in weakened or denervated muscle (Bennett 1958; Johnson 1971), and epidemiologic data showing a higher incidence of ALS in people performing intense physical activity at work or for leisure before disease onset have led some clinicians to caution against exercise for people with ALS (Kurtzke 1991; Strickland 1996). However, studies of individuals with other neuromuscular diseases have found that exercise programs are beneficial and did not produce overuse weakness (Aitkens 1993; Einarsson 1991; Florence 1984; Kilmer 1994; Lindeman 1995; McCartney 1988; Milner‐Brown 1988; Vignos 1983).
Three animal studies have reported that endurance exercise training at moderate intensities slowed disease progression in superoxide dismutase 1 (SOD1) transgenic mice (Kaspar 2005; Kirkinezos 2003; Veldink 2003). However, a fourth study found that high endurance exercise training had detrimental effects in male mice (Mahoney 2004). Kirkinezos and colleagues demonstrated that treadmill running five days a week at 13 m/min led to a significant increase in the life span of G93A‐SOD1 male mice. There was a trend toward increased female mice life span, but the increase was not significant (Kirkinezos 2003). Veldink and colleagues found that treadmill running at 16 m/min significantly delayed onset and prolonged survival in transgenic low‐copy hSOD1 female but not male mice (Veldink 2003). The benefits of exercise were most pronounced in the study by Kaspar and colleagues: SOD1 transgenic mice who were exposed to an exercise wheel six hours per day beginning in the presymptomatic state survived 33% longer (163 days versus 122 days) than those not exposed to a running wheel (Kaspar 2005). Conversely, high‐intensity treadmill training at 22 m/min was found to not affect onset of disease in G93A‐SOD1 male and female mice. However, the exercise was found to hasten death in male, but not female mice (Mahoney 2004). More recently, two types of exercise using the SOD1 mouse model were compared: swimming‐based, high‐frequency and high‐amplitude exercise versus running. The swimming group maintained constant body weight longer during disease progression, exhibited sustained motor function, and had a 20% increase in life span (Deforges 2009).
A marked reduction in activity level secondary to ALS or MND can lead to cardiovascular deconditioning and disuse weakness, superimposed on the weakness caused by the disease itself. Reduced physical activity, particularly if prolonged, produces muscle atrophy, reduced strength of tendons and ligaments, and osteoporosis. Strength loss through inactivity and disuse may significantly debilitate individuals with ALS or MND, making them susceptible to deconditioning, and muscle and joint tightness which lead to contractures and pain. Exercise programs might have positive physiological and psychological effects for people with ALS or MND, especially when implemented before significant muscular atrophy occurs. As new drugs to slow the progression of ALS or MND become available, people may live longer. Interventions such as exercise, that minimize impairments, maximize function and improve quality of life, will become increasingly important. This review was first published in 2008 and this update was completed in 2012.
Objectives
To systematically review all RCTs and quasi‐RCTs that examine the effects of exercise for people with ALS or MND.
Methods
Criteria for considering studies for this review
Types of studies
We searched for all RCTs and quasi‐RCTs involving exercise, exercise therapy or physical rehabilitation for people with ALS or MND.
Types of participants
We included participants with a diagnosis of definite, probable, probable with laboratory support, or possible ALS, as defined by the El Escorial criteria (Brooks 2000).
Types of interventions
We included any of the following forms of exercise:
progressive resistance or strengthening exercise ‐ static or dynamic; and,
endurance or aerobic exercise.
The control condition was either no exercise or standard rehabilitation management (for example, range of motion exercise or stretching exercise).
Types of outcome measures
Primary outcomes
Our primary outcome measures included:
Improvement in functional ability, decrease in disability or reduction in rate of decline as measured by a validated outcome tool at three months; for example ALS Functional Rating Scale (ALSFRS) (CNTF 1996), ALSFRS‐Revised (Cedarbaum 1999), Schwab & England Rating Scale (Schwab 1969), the Appel ALS Scale (Appel 1987), the ALS Severity Scale (Hillel 1989), the Norris Scale (Norris 1974).
If a study included more than one of the primary outcomes listed above, ALSFRS or ALSFRS‐R was the first choice for primary outcome; Schwab & England Rating Scale was the second choice; ALS Severity Scale was the third choice; Appel ALS Scale was the fourth choice; and the Norris Scale was the fifth choice. If the primary outcome measures had differed across studies, change in primary measures would have been converted to a percentage of before treatment value for comparison, when possible.
Secondary outcomes
Secondary outcome measures included:
Improvement in psychological status or quality of life as measured by a validated outcome tool at three months; e.g. SF‐36 (Ware 1993), Sickness Impact Profile (Bergner 1981) or ALSAQ‐40 (Jenkinson 1999b; Jenkinson 1999a).
Decrease in fatigue as measured by a fatigue measurement tool at three months; for example, Fatigue Severity Scale (Krupp 1989) or Fatigue Visual Analogue Scale (VAS).
Increase in, or reduction in rate of decline of muscle strength, as measured by the Medical Research Council (MRC) scale, hand‐held or isokinetic dynamometry, or maximum voluntary isometric contraction (MVIC) at three months, for strengthening or progressive resistance studies.
Increase in, or reduction in rate of decline of aerobic endurance, as measured by cardiac output (CO), oxygen uptake (VO2), maximal and submaximal heart rate (HR) at three months, for aerobic or endurance studies.
The frequency of adverse effects related to the intervention throughout the study period. Serious adverse events were defined as those events that were life‐threatening or required prolonged hospitalisation. Specific adverse events included: arrhythmias; myocardial infarction; hypo‐ or hypertension; syncope; dizziness; musculoskeletal trauma; soft‐tissue injury; progression of ALS‐related weakness beyond expected as a result of typical disease progression; fatigue; increase in cramps and fasciculations.
Search methods for identification of studies
Electronic searches
We searched The Cochrane Neuromuscular Disease Group Specialized Register (2 July 2012), CENTRAL (2012, Issue 6 in the Cochrane Library), MEDLINE (January 1966 to June 2012), EMBASE (January 1980 to June 2012), AMED (January 1985 to June 2012), CINAHL Plus (January 1937 to June 2012), LILACS (January 1982 to June 2012), and OVID HealthSTAR (January 1975 to December 2012).
The detailed search strategies are in the appendices: Appendix 1 (CENTRAL), Appendix 2 (MEDLINE), Appendix 3 (EMBASE), Appendix 4 (AMED), Appendix 5 (CINAGHL Plus), Appendix 6 (LILACS), and OVID HealthSTAR (Appendix 7).
Searching other resources
We also searched ProQuest Dissertations & Theses A&I (Appendix 8) to identify any unpublished theses, inspected the reference lists of all papers selected and conference proceedings for relevant studies and contacted their authors, and experts in the field to identify additional published or unpublished data. We did not include any restrictions with respect to language or date of publication, except for ProQuest Dissertations & Theses A&I) which we searched from 2007 to December 2012.
Data collection and analysis
Selection of studies
The two review authors (VDBH and JMF) screened titles and abstracts of all publications obtained by the search strategy. We obtained and assessed the full texts of all potentially relevant studies and assessed them for inclusion and exclusion criteria. We identified excluded studies with reasons for exclusion. The two review authors initially discussed any disagreement about inclusion and there was no need to refer to a third author (in the original review) or the Cochrane Neuromuscular Disease Group (for this update) for resolution of differences.
Data extraction and management
We contacted the primary author of potentially eligible studies when necessary to resolve ambiguities in the reported methodologies or results, and to seek additional pertinent information not described in the paper. The two review authors extracted data independently. One review author entered data into the Cochrane software Review Manager (RevMan), and a second review author checked the data entered.
Assessment of risk of bias in included studies
The two review authors evaluated the quality of the included studies independently using the Cochrane 'Risk of bias' tool (Higgins 2011). The review authors were not blinded to trial author, institution or journal of publication of results. The two assessors then discussed their conclusions together. We graded each trial with respect to its risk of bias, with particular attention to the following: the randomisation procedure, allocation concealment, blinding, incomplete outcome data, selective reporting and other sources of bias (explicit inclusion or exclusion criteria, documentation of intervention, outcome criteria, and how the study dealt with baseline differences of experimental groups). Aspects of risk of bias were graded as at: "High risk of bias", "Low risk of bias" or "Unclear risk of bias". If agreement between the two review authors had been poor, we would have reassessed the studies and attempted to reach agreement by consensus. There were no disagreements between the two review authors.
Measures of treatment effect
The difference of primary interest was the absolute change in primary and secondary outcome measures. When sufficient data were available, we conducted statistical analysis as described in The Cochrane Handbook for Systematic Reviews of Interventions (current version Higgins 2011) using RevMan (currently RevMan 5.2) (RevMan 2012). For continuous outcomes, we expressed results as mean difference (MD) and 95% confidence intervals (CI). If there were dichotomous outcomes, risk ratio (RR) and 95% CI would have been used.
Data synthesis
We would have stratified the studies in sub‐categories according to:
bulbar versus non‐bulbar ALS; and
early stage versus middle stage versus late stage ALS, as determined by the functional ability scores (primary outcome).
We would have made comparisons separately according to type of control group (no treatment, other treatment). We would have attempted pooling of trials if at least two of the trials had comparable exercise protocols with the same conditions and comparable outcome measurements. We did expect heterogeneity among studies, given the likelihood of various exercise protocols and would have performed separate analyses for each intervention type (for example, strengthening or aerobic exercise). If the results were heterogeneous, we would have attempted to determine potential sources of heterogeneity with various subgroup and sensitivity analyses, on the basis of risk of bias.
Results
Description of studies
The number of papers found by the new, current strategies are MEDLINE 39; EMBASE 39; AMED 3; CINAHL Plus 72; Cochrane Neuromuscular Disease Group Specialized Register 93; CENTRAL 23; OVID HealthStar 37; and ProQuest Dissertations & Theses A&I 153.
We immediately excluded all but two papers because they did not report a RCT or quasi‐RCT (that is to say, they were a case report, or a descriptive paper about exercise or management of people with ALS or MND, et cetera). The two included RCTs are described in Characteristics of included studies and the findings are summarized in Table 1.
The first included study (Drory 2001) evaluated the benefits of a 15 min, twice‐daily exercise program in people with early and middle stage ALS. The exercise program was described as individualized (based on general health, neurologic status and fitness level), moderate load, endurance type exercises for the limbs and trunk, but no specific details about the exercise protocol were provided. All participants met the diagnosis of definite or probable ALS, as defined by the El Escorial criteria. Fourteen people were randomised into the exercise group and were compared to 11 people who were instructed to continue with usual activities of daily living. The individualized exercise program was prescribed by a physical therapist, demonstrated, written down and reviewed at each clinic visit. Changes in the exercise protocol were made at each clinic visit based on changes in status. Every 14 days, people in both groups were contacted by telephone. The purpose of the telephone call for the exercise group was to check adherence to prevent drop‐out. The participants in the control group were asked how they were, if they had any problems with swallowing or breathing, et cetera.
The second included study (Dal Bello‐Haas 2007) compared a thrice weekly resistance exercise program to daily stretching stretching exercises in people with early stage ALS. All participants met the definite, probable or probable with laboratory support ALS El Escorial criteria, and 13 were randomised into a resistance group who completed daily upper and lower extremity stretching exercises and resistance exercises three times per week. The resistance exercise program consisted of individualized (based on limitations and tolerance), progressive, moderate load and moderate intensity upper and lower extremity exercises. Fourteen were randomized into a usual care group and were instructed to complete daily stretching exercises only. For both groups, exercises were prescribed by a physical therapist unblinded to group assignment, demonstrated to the participants, written down and reviewed and revised as needed at each monthly clinic visit. Participants were asked to complete exercise logs and to note whether they exercised, what exercises they completed and if they noticed any adverse effects. Participants were also telephoned every two weeks, interviewed at the monthly clinic visit by the unblinded physical therapist and were asked about compliance and adverse effects.
In both studies, functional ability was measured with the ALSFRS (CNTF 1996); the Short Form‐36 Health Survey (SF‐36) (Ware 1993) was used to measure quality of life; and fatigue was measured using the Fatigue Severity Scale (Krupp 1989). In the Drory 2001 study, muscle strength was measured using manual muscle strength testing (MMT), using the zero to five MRC scale. Five muscle groups in each limb were tested (shoulder abduction, elbow flexion and extension, finger abduction and extension, hip flexion, knee flexion and extension, dorsiflexion and plantarflexion) and MMT scores were added. In addition, spasticity and pain were measured using validated measures, but are not reported. Changes in muscle strength were monitored as a safety outcome using upper and lower extremity MVIC in the Dal Bello‐Haas 2007 study. Data were normalized, summed and averaged to yield upper and lower extremity megascores (Andres 1986).
Risk of bias in included studies
Using the Cochrane 'Risk of bias' criteria, we determined that the Drory 2001 study was at unclear risk of bias overall, and Dal Bello‐Haas 2007 was at low risk of bias (see Figure 1 and Characteristics of included studies). In both studies, there were no baseline differences between groups in age, duration of the disease, bulbar onset or function in both studies, although in the study by Dal Bello‐Haas 2007 there was a trend toward worse physical role SF‐36 subscale scores in the usual care group. The institution statistician performed the randomisation using computer software in the Drory 2001 study. In the Dal Bello‐Haas 2007 study, participants selected an opaque envelope that contained group assignment. In both studies, allocation concealment was not done, and it would be difficult to blind participants to the exercise intervention. The outcomes assessor was not blinded to group assignment in Drory 2001, but was blinded in the study by Dal Bello‐Haas 2007.
1.
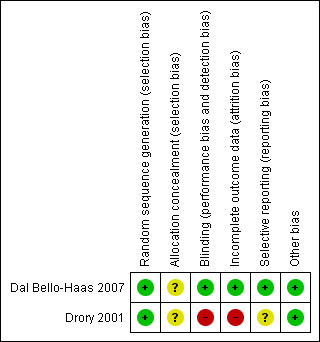
Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Effects of interventions
See: Table 1
The primary outcome measure was improvement in functional ability, decrease in disability or reduction in rate of decline as measured by a validated outcome tool at three months. The ALSFRS was published as an outcome measure in both studies, and was the primary outcome measure for the Dal Bello‐Haas 2007 study. When the results of both studies were pooled, the MD was 3.21 (95% CI 0.46 to 5.96) in favour of exercise (see Analysis 1.1).
1.1. Analysis.
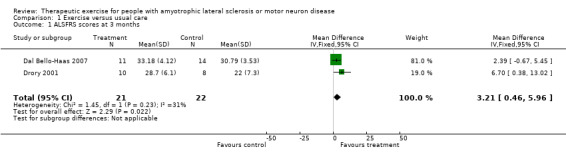
Comparison 1 Exercise versus usual care, Outcome 1 ALSFRS scores at 3 months.
Secondary outcome measures included:
Improvement in psychological status or quality of life as measured by a validated outcome tool at three months. The SF‐36 was used as an outcome measure for both studies. Drory 2001 examined total SF‐36 scores, while Dal Bello‐Haas 2007 published individual SF‐36 subscale scores. MDs in total SF‐36 scores at three months were 2.70 (95% CI ‐3.10 to 8.50); this result was not statistically significant using the Mann Whitney test (P value = 0.36) (see Analysis 1.2). Although the MD in the Physical Function subscale score favoured the treatment group, none of the SF‐36 subscale scores demonstrated significant changes in MD between groups (see Analysis 1.3 to Analysis 1.12).
Decrease in fatigue as measured by a fatigue measurement tool at three months. The Fatigue Severity Scale was used as an outcome measure for both studies. The MD was ‐6.25 (95% CI ‐13.82 to 1.31) and favoured the treatment group, a non‐significant difference (see Analysis 1.13).
Increase in, or reduction in rate of decline of muscle strength, as measured by the MRC scale, hand‐held or isokinetic dynamometry, or maximum voluntary isometric contraction at three months. Drory 2001 used MMT scores (MRC scale) as an outcome. The MD in total MMT scores between groups at three months was ‐10.90 (95% CI ‐23.56 to 1.76), a non‐significant difference (see Analysis 1.14). The MD in upper and lower extremity MVIC scores were not statistically significant (Analysis 1.15 and Analysis 1.16).
Increase in, or reduction in rate of decline of aerobic endurance, as measured by cardiac output (CO), oxygen uptake (VO2), maximal and submaximal heart rate (HR) at three months, for aerobic or endurance studies. Aerobic endurance was not examined in either study.
The frequency of adverse effects related to the intervention throughout the study period. There were no reported adverse events related to the exercise protocols.
1.2. Analysis.
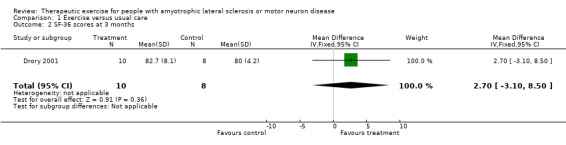
Comparison 1 Exercise versus usual care, Outcome 2 SF‐36 scores at 3 months.
1.3. Analysis.
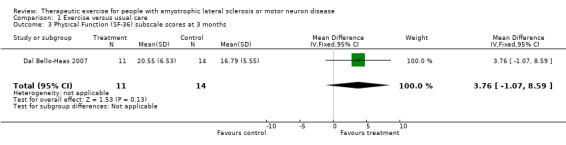
Comparison 1 Exercise versus usual care, Outcome 3 Physical Function (SF‐36) subscale scores at 3 months.
1.12. Analysis.

Comparison 1 Exercise versus usual care, Outcome 12 Mental Health summary scores.
1.13. Analysis.
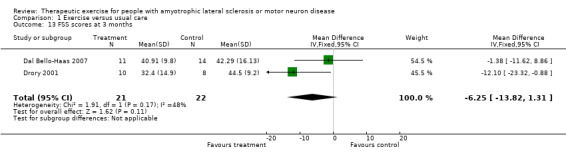
Comparison 1 Exercise versus usual care, Outcome 13 FSS scores at 3 months.
1.14. Analysis.

Comparison 1 Exercise versus usual care, Outcome 14 MMT scores at 3 months.
1.15. Analysis.
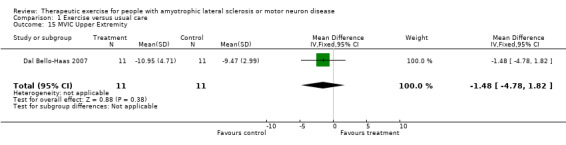
Comparison 1 Exercise versus usual care, Outcome 15 MVIC Upper Extremity.
1.16. Analysis.
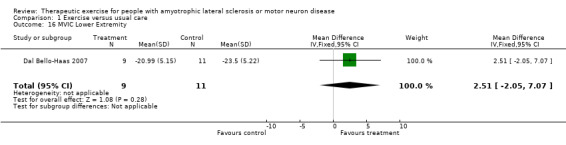
Comparison 1 Exercise versus usual care, Outcome 16 MVIC Lower Extremity.
The drop‐out rate at three months was 28.6% in the exercise group and 27.3% in the control group in the Drory 2001, and 15.4% in the resistance group and 0% in the control group in the Dal Bello‐Haas 2007 study. High drop‐out rates are not unusual in ALS clinical studies, particularly those requiring longer follow‐up. Reasons for drop‐out included difficulty attending the clinic for follow‐up due to respiratory and mobility problems, rapid disease deterioration resulting in mechanical ventilation or death, long distances to travel to the clinic for follow‐up, fatigue, and lack of interest in continuing in the study (Drory 2001); and, subsequent enrollment into a clinical drug trial, fractured wrist secondary to an auto car accident, depression, perceived disease progression or perceived lack of benefit, non‐adherence, and bypass surgery (Dal Bello‐Haas 2007).
Discussion
Only two identified studies on the effects of exercise in people with ALS or MND met the inclusion criteria. The risk of bias of one study was judged as unclear overall (no allocation concealment, the evaluator assessing the outcome measures was not blinded) and the other as at low risk of bias. The MD in the primary outcome, function as measured by the ALSFRS, was statistically significant, while MDs in secondary outcome measures between groups were non‐significant. Both studies followed participants beyond three months, Drory 2001 for 12 months and Dal Bello‐Haas 2007 for six months. Drory 2001 reported no differences between groups at six months, although there was a trend toward less deterioration in most outcome measures. Dal Bello‐Haas 2007 reported higher ALSFRS and SF‐36 physical function subscale scores in the treatment group at six months. Rate of drop‐outs continued to increase over time in both studies. In the Drory 2001 study, so few participants remained in the study at nine and 12 months that analyses could not be completed.
In both trials adverse effects, such as increased muscle cramping, muscle soreness or fatigue, were not reported by the investigators. In the Dal Bello‐Haas 2007, one participant discontinued the study because of bypass surgery. The participant had a history of cardiac problems (hypercholesterolemia and hypertension controlled with medications, previous myocardial infarction), and was informed of a blockage but surgery was not performed at that time. The participant was being followed by a cardiologist and was given initial clearance by the cardiologist to participate in the ALS exercise trial. Although not statistically significant, MDs in MMT scores favoured the control group in the Drory 2001 study, but the lower extremity MVIC scores favoured the treatment group in the Dal Bello‐Haas 2007 study. In Drory 2001, from baseline to month three, participants in the exercise group had a mean change of ‐0.33 in FSS scores compared to a mean change of +18.0 in the control group, indicating less fatigue in the exercise group. Similarily, from baseline to month three, participants in the exercise group had a mean change of ‐0.25 in muscle pain scores compared to a mean change of +1.8 in the control group, indicating less muscle pain. Thus, whether the MMT findings (Drory 2001) represent overwork weakness cannot be determined.
The dearth of research highlights the need for further investigation in this area. Only two trials examining the effect of exercise in people with ALS met the inclusion criteria and were included. Several factors regarding the disease, the population and type of intervention may be responsible for this finding. First, despite the relatively high prevalence of muscle weakness in people with ALS, the rapidly progressive nature of the disease makes recruitment to trials with large numbers difficult. Currently, there is no cure for ALS and about 50% of people die within two to three years after diagnosis (Norris 1993; Ringel 1993). It is logical to expect that people with ALS would prefer to choose to be randomised into a disease‐modifying pharmaceutical trial. Thus, the willingness of participants to be randomly allocated into an exercise trial, as opposed to a trial that might slow disease progression, is a particularly important issue. Second, ALS is highly variable from person to person. The extent and areas of disease involvement, the stage and severity of illness, the rate of disease progression, and the severity of respiratory and bulbar manifestations can all affect enrollment and continued participation with the intervention. Third, the motivation and commitment of individual participants to undertake the exercise component alone or with the assistance of a caregiver, in the face of continued physical and emotional loss and uncertainty may deter some from agreeing to participate, and contribute to high drop‐out rates.
Current clinical management for people with ALS is predominantly individualized rehabilitation that may or may not include strengthening or aerobic exercise prescription. The continued lack of robust evidence regarding the efficacy and benefits of exercise in people with ALS may influence the availability, accessibility and quality of rehabilitation services provided for this group. Exercise, when prescribed appropriately, may be physically and psychologically important for people with ALS, especially in the earlier stages of the disease and before significant muscular atrophy or deconditioning occurs. Although exercise may not improve the strength of muscles already weakened by ALS, strengthening exercises with low to moderate weights, and aerobic exercises such as swimming, walking, and bicycling, at submaximal levels may be important components of an overall management plan.
We did find some non‐randomised literature. Two early case studies reported positive effects of specific strengthening and endurance exercises in individuals with ALS (Bohannon 1983; Sanjak 1987), and Pinto and colleagues reported that people who participated in endurance exercises while on BiPAP had significantly greater Functional Impact Measure scores, slower Spinal Norris Score decline and less FVC decline compared to those who did not exercise (Pinto 1999). Animal model studies suggest that although high intensity endurance exercise may be detrimental, low or moderate intensity endurance exercise may be of some benefit (Kirkinezos 2003; Mahoney 2004; Veldink 2003), and a Cochrane review has found that in people with neuromuscular disorders, specifically myotonic dystrophy and facioscapulohumeral muscular dystrophy, moderate‐intensity strength training appeared not to be harmful (van der Kooi 2011).
Despite the lack of sufficient and adequate research evidence, some discourage strengthening or aerobic exercise programs for people with ALS because of fear of overuse weakness and believe that no exercise other than everyday activities is indicated. In people with ALS, the safe range for therapeutic exercise narrows. The degree to which this safe range narrows is dependent on the extent of disease involvement and the rate of disease progression. A weak or denervated muscle is more susceptible to overwork damage because it is already functioning close to its maximal limits. Activities of daily living alone may provide a training stimulus to weak muscles, and additional exercise that would strengthen healthy muscles may actually cause overwork damage. The remaining motor units will respond to training, and these motor units must work harder to handle a given amount of exercise stress. Thus, the possibility of inducing overwork damage in individuals with ALS through excessive exercise is a concern. On the other hand, a marked reduction in activity level secondary to ALS can lead to cardiovascular deconditioning and disuse weakness beyond the amount caused by the disease itself. Reduced physical activity, particularly if prolonged, reduces function of the neuromuscular system, in addition to the skeletal and other organ systems. With insufficient activity, disuse atrophy develops because the strength of muscle contractions are less than the total tension a muscle is capable of producing, and as contractile proteins are lost, muscle weakness progresses. Strength loss through inactivity and disuse can significantly debilitate individuals with ALS, making them highly susceptible to cardiovascular deconditioning, and muscle and joint tightness that lead to contractures and pain. Thus, special attention is needed when designing a strengthening or aerobic exercise program for a person with ALS: the balance between prevention of overuse fatigue and weakness and disuse atrophy must be taken into consideration.
The lack of substantial evidence regarding the role of exercise in people with ALS supports the need to develop future, high quality, sufficiently powered trials. At present, many questions related to the role of exercise and ALS remain unanswered, including: (1) which people would benefit most from strengthening or aerobic exercise; (2) when is the most appropriate time to initiate exercise in people with ALS; (3) what degree of disease involvement or disease progression would supercede any beneficial effects of exercise; (4) what is the most appropriate type of exercise for people with ALS; (5) what are the most appropriate exercise parameters and protocols; (6) what occurs at the physiologic level when people with ALS exercise. In addition, until relevant research evidence about the effects of exercise is available, the impact of exercise on the overall economic burden of care cannot be considered. Cost‐benefit analyses are only relevant if the benefit of exercise has been demonstrated.
Considering the nature of ALS and the controversy surrounding exercise and ALS, it is critical that future research designs include:
Compatible groups: specific diagnostic criteria should be provided for all participants included in trials. The range of disease severity, and severity of impairments and function should be noted to allow readers to assess the generalizability of the results to their patient or patient group. Group participants should be stratified or matched for disease severity.
Adequate allocation concealment.
Blinding of the outcome assessor.
A clear description of the exercise intervention, including the mode of exercise, intensity, progression, frequency, duration per exercise session, duration of the entire programme, muscle groups exercised, supervision of exercise protocol, and compliance assessment.
Level of activity at baseline.
Monitoring of adverse events and reporting of reasons for drop‐out.
Standardized outcome measures: well validated outcome measures that are able to assess positive and negative effects of exercise should be utilized, including measures of muscle function or aerobic capacity, function, fatigue and quality of life.
Authors' conclusions
Implications for practice.
Two studies were included but were too small to determine to what extent strengthening exercises for people with ALS are beneficial. Currently, there is a complete lack of randomised or quasi‐randomised clinical trials examining aerobic exercise in people with ALS.
Implications for research.
More research is needed to elucidate whether strengthening or aerobic exercise is beneficial or harmful in people with ALS. Well‐controlled studies are needed to determine the ideal exercise prescription for people with ALS, in terms of both which exercise protocols are most beneficial or cause undue risks, and whether there is a sub‐set of people with ALS who respond more positively to exercise, both physically and psychologically.
What's new
| Date | Event | Description |
|---|---|---|
| 30 December 2012 | New citation required but conclusions have not changed | Update with new searches fully incorporated. A 'Summary of findings' table has been added. |
| 2 July 2012 | New search has been performed | The review has been updated with a new search, but no new relevant studies were found. |
History
Protocol first published: Issue 2, 2005 Review first published: Issue 2, 2008
| Date | Event | Description |
|---|---|---|
| 20 February 2008 | New citation required and conclusions have changed | Substantive amendment |
Acknowledgements
This update is dedicated to our dear friend and colleague, Lisa S Krivickas, M.D, who co‐authored the original version of this review.
The editorial base of the Cochrane Neuromuscular Disease Group is supported by the Motor Neurone Disease Assocation and the MRC Centre for Neuromuscular Diseases.
Appendices
Appendix 1. CENTRAL search strategy
#1 MeSH descriptor Motor Neuron Disease explode all trees #2 (moto* neuron* disease* or moto?neuron* disease) #3 "Amyotrophic Lateral Sclerosis" #4 ("Lou Gehrig*" and (disease* or syndrome*)) #5 (#1 OR #2 OR #3 OR #4) #6 MeSH descriptor Exercise Therapy explode all trees #7 "exercise therapy" or "exercise training" or "exercise program" #8 "strength training" or "aerobic training" or "aerobic exercise" or "resistive exercise" or "muscle exercise" #9 "training program" or "endurance training" #10 rehabilitation and exercise #11 "physical therapy" or physiotherapy #12 (#6 OR #7 OR #8 OR #9 OR #10 OR #11) #13 (#5 AND #12)
Appendix 2. MEDLINE (OvidSP) search strategy
Database: Ovid MEDLINE(R) <1946 to June Week 3 2012> Search Strategy: ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐ 1 randomized controlled trial.pt. (330201) 2 controlled clinical trial.pt. (84375) 3 randomized.ab. (233876) 4 placebo.ab. (132230) 5 drug therapy.fs. (1543331) 6 randomly.ab. (168558) 7 trial.ab. (242070) 8 groups.ab. (1106725) 9 or/1‐8 (2867649) 10 exp animals/ not humans.sh. (3736636) 11 9 not 10 (2435187) 12 exp Motor Neuron Disease/ (17466) 13 (moto$1 neuron$1 disease$1 or moto?neuron$1 disease).mp. (5793) 14 ((Lou Gehrig$1 adj5 syndrome$1) or (Lou Gehrig$1 adj5 disease)).mp. (64) 15 charcot disease.tw. (11) 16 Amyotrophic Lateral Sclerosis.mp. (14118) 17 or/12‐16 (21019) 18 exp Exercise Therapy/ (25513) 19 (exercise therap$ or exercise training or exercise program$).mp. (32128) 20 (strength training or aerobic training or aerobic exercise$ or resistive exercise$ or muscle exercise$).mp. (7262) 21 (training program$ or endurance training).mp. (23660) 22 (rehabilitation and exercise$).mp. (11013) 23 Physical Therapy Modalities/ (25908) 24 (physiotherapy or physical therap$).mp. (37928) 25 or/18‐24 (97126) 26 9 and 17 and 25 (38)
Appendix 3. EMBASE (OvidSP) search strategy
Database: Embase <1980 to 2012 Week 26> Search Strategy: ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐ 1 crossover‐procedure.sh. (34246) 2 double‐blind procedure.sh. (109462) 3 single‐blind procedure.sh. (16047) 4 randomized controlled trial.sh. (324293) 5 (random$ or crossover$ or cross over$ or placebo$ or (doubl$ adj blind$) or allocat$).tw,ot. (877566) 6 trial.ti. (132039) 7 or/1‐6 (1004389) 8 (animal/ or nonhuman/ or animal experiment/) and human/ (1187619) 9 animal/ or nonanimal/ or animal experiment/ (3281792) 10 9 not 8 (2719455) 11 7 not 10 (920638) 12 limit 11 to embase (713539) 13 crossover‐procedure/ (34246) 14 double‐blind procedure/ (109462) 15 randomized controlled trial/ (324293) 16 single‐blind procedure/ (16047) 17 (random$ or factorial$ or crossover$ or cross over$ or cross‐over$ or placebo$ or (doubl$ adj blind$) or (singl$ adj blind$) or assign$ or allocat$ or volunteer$).tw. (1132880) 18 or/13‐17 (1210587) 19 human/ (13505526) 20 18 and 19 (900503) 21 nonhuman/ or human/ (16641800) 22 18 not 21 (202983) 23 20 or 22 (1103486) 24 motor neuron disease/ or amyotrophic lateral sclerosis/ (22904) 25 (moto$1 neuron$1 disease$1 or moto?neuron$1 disease$1).mp. (8496) 26 ((Lou Gehrig$1 adj5 syndrome$1) or (Lou Gehrig$1 adj5 disease)).mp. (107) 27 charcot disease.tw. (18) 28 amyotrophic lateral sclerosis.tw. (14103) 29 or/24‐28 (25487) 30 exp kinesiotherapy/ (40077) 31 (exercise therap$ or exercise training or exercise program$).mp. (20199) 32 (strength training or aerobic training or aerobic exercise$ or resistive exercise$ or muscle exercise$).mp. (15812) 33 (training program$ or endurance training).mp. (31548) 34 (rehabilitation and exercise$).mp. (17724) 35 exp physiotherapy/ (45386) 36 (physiotherapy or physical therap$).mp. (57446) 37 or/30‐36 (150581) 38 23 and 29 and 37 (41) 39 remove duplicates from 38 (39)
Appendix 4. AMED (OvidSP) search strategy
Database: AMED (Allied and Complementary Medicine) <1985 to June 2012> Search Strategy: ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐ 1 Randomized controlled trials/ (1531) 2 Random allocation/ (302) 3 Double blind method/ (437) 4 Single‐Blind Method/ (27) 5 exp Clinical Trials/ (3193) 6 (clin$ adj25 trial$).tw. (5421) 7 ((singl$ or doubl$ or treb$ or trip$) adj25 (blind$ or mask$ or dummy)).tw. (2228) 8 placebos/ (518) 9 placebo$.tw. (2496) 10 random$.tw. (12741) 11 research design/ (1676) 12 Prospective Studies/ (463) 13 meta analysis/ (108) 14 (meta?analys$ or systematic review$).tw. (1843) 15 control$.tw. (27490) 16 (multicenter or multicentre).tw. (725) 17 ((study or studies or design$) adj25 (factorial or prospective or intervention or crossover or cross‐over or quasi‐experiment$)).tw. (9706) 18 or/1‐17 (42382) 19 motor neuron disease/ (90) 20 (moto$1 neuron$1 disease$1 or moto?neuron$1 disease).mp. (163) 21 ((Lou Gehrig$1 adj5 syndrome$1) or (Lou Gehrig$1 adj5 disease)).mp. (2) 22 charcot disease.tw. (1) 23 Amyotrophic Lateral Sclerosis/ (172) 24 amyotrophic lateral sclerosis.tw. (245) 25 or/19‐24 (384) 26 Exercise Therapy/ (5009) 27 (exercise therap$ or exercise training or exercise program$).mp. (6355) 28 (strength training or aerobic training or aerobic exercise$ or resistive exercise$ or muscle exercise$).mp. (1333) 29 (training program$ or endurance training).mp. (1979) 30 (rehabilitation and exercise$).mp. (5275) 31 Physiotherapy/ (9156) 32 (physiotherapy or physical therap$).mp. (16150) 33 or/26‐32 (24943) 34 18 and 25 and 33 (3)
Appendix 5. CINAHL Plus (EBSCOhost) search strategy
Monday, July 02, 2012 9:36:37 AM S32 S18 and S23 and S31 72 S31 S24 or S25 or S26 or S27 or S28 or S29 or S30 82125 S30 physiotherap* or physical therap* 40312 S29 (MH "Physical Therapy") 19592 S28 rehabilitation and exercise* 11990 S27 resistive exercise* or muscle exercise* or endurance training 3667 S26 strength training or aerobic training or aerobic exercise* or training program* 16908 S25 exercise therap* or exercise training or exercise program* 17946 S24 (MH "Therapeutic Exercise+") 27429 S23 S19 or S20 or S21 or S22 4778 S22 ("Amyotrophic Lateral Sclerosis") 1950 S21 Lou Gehrig* and ( disease* or syndrome* ) 31 S20 (moto* neuron* disease* or moto?neuron* disease) 852 S19 MH Motor Neuron Diseases+ 4516 S18 S1 or S2 or S3 or S4 or S5 or S6 or S7 or S8 or S9 or S10 or S11 or S12 or S13 or S14 or S15 or S16 or S17 544729 S17 ABAB design* 76 S16 TI random* or AB random* 110869 S15 ( TI (cross?over or placebo* or control* or factorial or sham? or dummy) ) or ( AB (cross?over or placebo* or control* or factorial or sham? or dummy) ) 229065 S14 ( TI (clin* or intervention* or compar* or experiment* or preventive or therapeutic) or AB (clin* or intervention* or compar* or experiment* or preventive or therapeutic) ) and ( TI (trial*) or AB (trial*) ) 77341 S13 ( TI (meta?analys* or systematic review*) ) or ( AB (meta?analys* or systematic review*) ) 22527 S12 ( TI (single* or doubl* or tripl* or trebl*) or AB (single* or doubl* or tripl* or trebl*) ) and ( TI (blind* or mask*) or AB (blind* or mask*) ) 18089 S11 PT ("clinical trial" or "systematic review") 102544 S10 (MH "Factorial Design") 823 S9 (MH "Concurrent Prospective Studies") or (MH "Prospective Studies") 180529 S8 (MH "Meta Analysis") 14237 S7 (MH "Solomon Four‐Group Design") or (MH "Static Group Comparison") 30 S6 (MH "Quasi‐Experimental Studies") 5465 S5 (MH "Placebos") 7589 S4 (MH "Double‐Blind Studies") or (MH "Triple‐Blind Studies") 24428 S3 (MH "Clinical Trials+") 143550 S2 (MH "Crossover Design") 9355 S1 (MH "Random Assignment") or (MH "Random Sample") or (MH "Simple Random Sample") or (MH "Stratified Random Sample") or (MH "Systematic Random Sample") 56940 Bottom of Form
Appendix 6. LILACS search strategy
(Mh motor neuron disease OR Mh amyotrophic lateral sclerosis) [Words] and exp E02.779.483 or exercise or training or physical therapy or rehabilitation [Words] and ((Pt randomized controlled trial OR Pt controlled clinical trial OR Mh randomized controlled trials OR Mh random allocation OR Mh double‐blind method OR Mh single‐blind method) AND NOT (Ct animal AND NOT (Ct human and Ct animal)) OR (Pt clinical trial OR Ex E05.318.760.535$ OR (Tw clin$ AND (Tw trial$ OR Tw ensa$ OR Tw estud$ OR Tw experim$ OR Tw investiga$)) OR ((Tw singl$ OR Tw simple$ OR Tw doubl$ OR Tw doble$ OR Tw duplo$ OR Tw trebl$ OR Tw trip$) AND (Tw blind$ OR Tw cego$ OR Tw ciego$ OR Tw mask$ OR Tw mascar$)) OR Mh placebos OR Tw placebo$ OR (Tw random$ OR Tw randon$ OR Tw casual$ OR Tw acaso$ OR Tw azar OR Tw aleator$) OR Mh research design) AND NOT (Ct animal AND NOT (Ct human and Ct animal)) OR (Ct comparative study OR Ex E05.337$ OR Mh follow‐up studies OR Mh prospective studies OR Tw control$ OR Tw prospectiv$ OR Tw volunt$ OR Tw volunteer$) AND NOT (Ct animal AND NOT (Ct human and Ct animal))) [Words]
Appendix 7. OVID Healthstar search strategy
December 30, 2012 Amyotrophic Lateral Sclerosis/rh [Rehabilitation]limit 1 to (humans and yr="2007 ‐ 2012")
Appendix 8. ProQuest Dissertations & Theses A&I search strategy
December 30, 2012
all(amyotrophic lateral sclerosis) OR (motor neuron disorder) OR (motor neurone disease) OR all(lou gehrig) OR all(charcot)
Date: From January 01 2007 to December 31 2012
Manuscript type: Master's theses, Doctoral dissertations
Language: English
Results narrowed by Index term (keyword)
Data and analyses
Comparison 1. Exercise versus usual care.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 ALSFRS scores at 3 months | 2 | 43 | Mean Difference (IV, Fixed, 95% CI) | 3.21 [0.46, 5.96] |
| 2 SF‐36 scores at 3 months | 1 | 18 | Mean Difference (IV, Fixed, 95% CI) | 2.70 [‐3.10, 8.50] |
| 3 Physical Function (SF‐36) subscale scores at 3 months | 1 | 25 | Mean Difference (IV, Fixed, 95% CI) | 3.76 [‐1.07, 8.59] |
| 4 Physical Role (SF‐36) subscale scores at 3 months | 1 | 25 | Mean Difference (IV, Fixed, 95% CI) | 0.52 [‐0.72, 1.76] |
| 5 Bodily Pain (SF‐36) subscale scores at 3 months | 1 | 25 | Mean Difference (IV, Fixed, 95% CI) | ‐0.09 [‐1.40, 1.22] |
| 6 General Health (SF‐36) subscale scores at 3 months | 1 | 24 | Mean Difference (IV, Fixed, 95% CI) | 1.02 [‐2.82, 4.86] |
| 7 Vitality (SF‐36) subscale scores at 3 months | 1 | 25 | Mean Difference (IV, Fixed, 95% CI) | ‐1.93 [‐4.55, 0.69] |
| 8 Social Function (SF‐36) subscale scores at 3 months | 1 | 25 | Mean Difference (IV, Fixed, 95% CI) | ‐0.20 [‐1.69, 1.29] |
| 9 Emotional Role (SF‐36) subscale scores at 3 months | 1 | 25 | Mean Difference (IV, Fixed, 95% CI) | 0.69 [‐0.16, 1.54] |
| 10 Mental Health (SF‐36) subscale scores at 3 months | 1 | 25 | Mean Difference (IV, Fixed, 95% CI) | 0.66 [‐0.86, 2.18] |
| 11 Physical Health summary scores | 1 | 25 | Mean Difference (IV, Fixed, 95% CI) | 5.0 [‐4.46, 14.46] |
| 12 Mental Health summary scores | 1 | 25 | Mean Difference (IV, Fixed, 95% CI) | ‐0.79 [‐5.90, 4.32] |
| 13 FSS scores at 3 months | 2 | 43 | Mean Difference (IV, Fixed, 95% CI) | ‐6.25 [‐13.82, 1.31] |
| 14 MMT scores at 3 months | 1 | 18 | Mean Difference (IV, Fixed, 95% CI) | ‐10.90 [‐23.56, 1.76] |
| 15 MVIC Upper Extremity | 1 | 22 | Mean Difference (IV, Fixed, 95% CI) | ‐1.48 [‐4.78, 1.82] |
| 16 MVIC Lower Extremity | 1 | 20 | Mean Difference (IV, Fixed, 95% CI) | 2.51 [‐2.05, 7.07] |
1.4. Analysis.
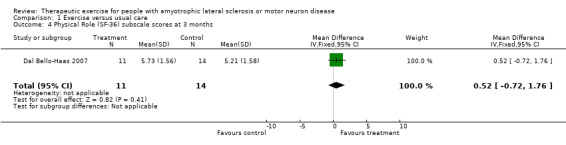
Comparison 1 Exercise versus usual care, Outcome 4 Physical Role (SF‐36) subscale scores at 3 months.
1.5. Analysis.
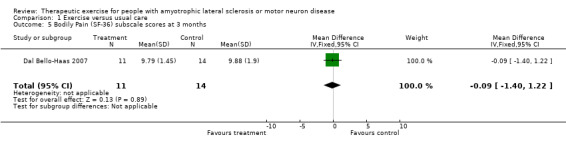
Comparison 1 Exercise versus usual care, Outcome 5 Bodily Pain (SF‐36) subscale scores at 3 months.
1.6. Analysis.

Comparison 1 Exercise versus usual care, Outcome 6 General Health (SF‐36) subscale scores at 3 months.
1.7. Analysis.
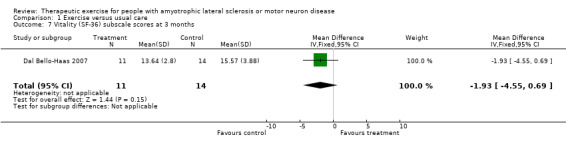
Comparison 1 Exercise versus usual care, Outcome 7 Vitality (SF‐36) subscale scores at 3 months.
1.8. Analysis.

Comparison 1 Exercise versus usual care, Outcome 8 Social Function (SF‐36) subscale scores at 3 months.
1.9. Analysis.
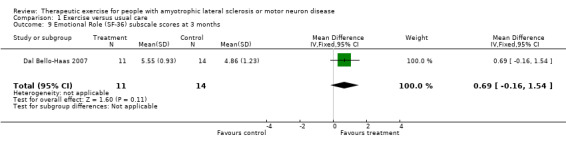
Comparison 1 Exercise versus usual care, Outcome 9 Emotional Role (SF‐36) subscale scores at 3 months.
1.10. Analysis.
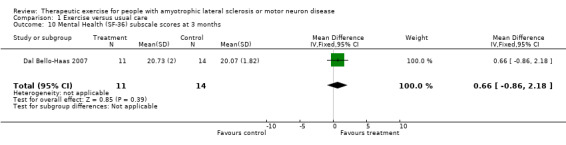
Comparison 1 Exercise versus usual care, Outcome 10 Mental Health (SF‐36) subscale scores at 3 months.
1.11. Analysis.
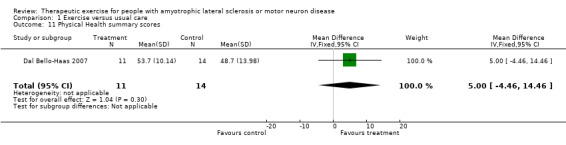
Comparison 1 Exercise versus usual care, Outcome 11 Physical Health summary scores.
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Dal Bello‐Haas 2007.
| Methods | 6 month parallel group, randomized trial. | |
| Participants | 27 people with definite or probable, probable with laboratory‐supported ALS (El Escorial criteria), aged 41 to 80 years. Early stage ALS (Stage 1 or 2, Sinaki & Mulder staging). FVC > 90% predicted and ALSFRS > 30. Able to ambulate with or without assistive device. Able to breathe without any form of mechanical ventilation. Able to understand and comply with instructions. | |
| Interventions | Exercise group: thrice weekly individualized, progressive, moderate intensity, moderate load resistance exercises and daily stretching exercises performed at home. Control group: daily stretching exercises performed at home. | |
| Outcomes | Outcomes measured monthly for 6 months. Function, using ALSFRS*. Quality of life, using SF‐36*. Fatigue using FSS*. Muscle strength, using MVIC*. | |
| Notes | Multi‐site trial. Treatment group drop‐out rates: 15.4.6% at 3 months, 38.4% at 6 months. Control group drop‐out rates: 0% at 3 months, 28.6% at 6 months. Continued high drop‐out rates continued after 6 months. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "twenty‐seven eligible, consecutive subjects were randomly assigned" (p. 2004) Comment: probably done |
| Allocation concealment (selection bias) | Unclear risk | Not reported |
| Blinding (performance bias and detection bias) All outcomes | Low risk | "all monthly evaluations were performed by a second physical therapist ...who was blinded to group assignment" (p. 2004) Comment: probably done "participants were prescribed an exercise program by a research physical therapist who was unblinded to group assignment" (p. 2004) Participants were not blinded to group assignment Comment: blinding of participants and prescribing physical therapist not feasible |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Enrolled subjects with FVC ≥90% of predicted and ALSFRS ≥ 30; high drop‐out rates: 28.6% usual care group and 38.4% resistance exercise group at 6 months; trend toward worse Physical Role SF‐36 subscale scores in the usual care group at baseline "dropout rates and reasons for dropout did not differ significantly between groups" (p. 2004) "we completed a stringent intention‐to‐treat analysis and imputed usual care group means at month 6 for missing data points for both the resistance exercise and the usual care group subjects" (p. 2004) Comment: probably done |
| Selective reporting (reporting bias) | Low risk | All data published |
| Other bias | Low risk | None noted |
Drory 2001.
| Methods | 12 month parallel group, quasi‐randomised trial. | |
| Participants | 25 people with definite or probable ALS (El Escorial criteria), aged 41 to 80 years. Mild to moderate stages of ALS. Able to ambulate with or without assistive device. Able to breathe without any form of mechanical ventilation. Able to understand and comply with instructions. | |
| Interventions | Exercise group: 15 min of twice daily moderate load, endurance type exercises for limbs and trunk; performed at home. Control group: usual activities of daily living. | |
| Outcomes | Outcomes measured at baseline 3, 6, 9 and 12 months. Muscle strength, using MMT (MRC scale)*. Function, using ALSFRS*. Quality of life, using SF‐36*. Fatigue using FSS*. Pain using VAS. Spasticity using Ashworth Scale. | |
| Notes | Single site trial, conducted at Tel Aviv Sourasky Medical Centre. Treatment group drop‐out rates: 28.6% at 3 months, 42.9% at 6 months. Control group drop‐out rates: 27.3% at 3 months, 45.5% at 6 months. Continued high drop‐out rates continued after 6 months. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "patients were assigned randomly to two groups . . . " (p.134) |
| Allocation concealment (selection bias) | Unclear risk | Not reported |
| Blinding (performance bias and detection bias) All outcomes | High risk | Not stated; authors reported assessors not blinded Participants were not blinded to group assignment Comment: blinding of participants and prescribing physical therapist not feasible |
| Incomplete outcome data (attrition bias) All outcomes | High risk | High attrition rates ‐ treatment group, 14 enrolled, 10 evaluated at 3 months, 8 at 6 months, 5 at 9 months, 3 at 12 months; control group ‐ 11 enrolled, eight evaluated at 3 months, six evaluated at 6 months, three evaluated at 9 months and 2 evaluated at 12 months Comment: attrition rates not accounted for statistically at 3 and 6 months |
| Selective reporting (reporting bias) | Unclear risk | Outcome data presented graphically |
| Other bias | Low risk | None noted |
* Scores at 3 months analysed in review
Differences between protocol and review
This review has a published protocol (Dal Bello‐Haas 2005). We updated the methods for 'Risk of bias' assessment according to the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011) and added a 'Risk of bias summary' and 'Summary of findings' figure.
Contributions of authors
For the original version of the review, VDBH and JMF assessed studies for inclusion and exclusion criteria, assessed the quality of included studies, and extracted data independently. VDBH contacted primary authors of potentially eligible studies and experts in the field, entered the data, and developed and wrote the initial and final drafts of the protocol and review texts. JMF and LSK checked the data entered and comments from JMF and LSK were incorporated into the final version of the review. For the 2012 update, VDBH checked the text and updated it, VDBH and JF independently checked searches for RCTs, VDBH and JF carried out risk of bias assessments. VDBH and JF approved the final text.
Declarations of interest
VDBH and JMF are co‐investigators of one of the included randomised trials. VDBH received a grant from the Amyotrophic Lateral Sclerosis Association to conduct the study.
JMF has an interest in the design & implementation of outcome measures for clinical trials in neuromuscular disorders. She has been involved in this arena through her appointment in the Department of Neurology at Washington University School of Medicine in St. Louis and in the position of a consultant for several biotech, biomedical & pharmaceutical companies.
New search for studies and content updated (no change to conclusions)
References
References to studies included in this review
Dal Bello‐Haas 2007 {published and unpublished data}
- Dal Bello‐Haas VP, Florence JM, Kloos AD, Scheirbecker J, Lopate G, Hayes SM, et al. A randomized controlled trial of resistance exercise in individuals with ALS. Neurology 2007;68(23):2003‐7. [PUBMED: 17548549] [DOI] [PubMed] [Google Scholar]
Drory 2001 {published and unpublished data}
- Drory VE, Goltsman E, Reznik JG, Mosek A, Korczyn AD. The value of muscle exercise in patients with amyotrophic lateral sclerosis. Journal of Neurological Sciences 2001;191(1‐2):133‐7. [PUBMED: 11677004] [DOI] [PubMed] [Google Scholar]
Additional references
Aitkens 1993
- Aitkens SG, McCrory MA, Kilmer DD, Bernauer EM. Moderate resistance exercise program: its effect in slowly progressive neuromuscular disease. Archives of Physical Medicine & Rehabilitation 1993;74(7):711‐5. [DOI] [PubMed] [Google Scholar]
Andres 1986
- Andres PL, Hedlund W, Finison L, Conlon T, Felmus M, Munsat TL, et al. Quantitative motor assessment in amyotrophic lateral sclerosis. Neurology 1986;36(7):937‐41. [DOI] [PubMed] [Google Scholar]
Appel 1987
- Appel V, Stewart SS, Smith G, Appel SH. A rating scale for amyotrophic lateral sclerosis: description and preliminary experience. Annals of Neurology 1987;22(3):328‐33. [DOI] [PubMed] [Google Scholar]
Bennett 1958
- Bennett RL, Knowlton GC. Overwork weakness in partially denervated skeletal muscle. Clinical Orthopaedics and Related Research 1958;12:22‐9. [PubMed] [Google Scholar]
Bergner 1981
- Bergner M, Bobbitt RA, Carter WB, Gilson BS. The Sickness Impact Profile: development and final revision of a health status measure. Medical Care 1981;19(8):787‐805. [DOI] [PubMed] [Google Scholar]
Bohannon 1983
- Bohannon RW. Results of resistance exercise on a patient with amyotrophic lateral sclerosis. Physical Therapy 1983;63(6):965‐8. [DOI] [PubMed] [Google Scholar]
Brooks 2000
- Brooks BR, Miller RG, Swash M, Munsat TL, World Federation of Neurology Research Group on Motor Neuron Diseases. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotrophic Lateral Sclerosis and Other Motor Neuron Disorders 2000;1(5):293‐9. [DOI] [PubMed] [Google Scholar]
Cedarbaum 1999
- Cedarbaum JM, Stambler N, Malta E, Fuller C, Hilt D, Thurmond B. The ALSFRS‐R: A revised ALS functional rating scale that incorporates assessments of respiratory function. BDNF ALS Study Group (Phase III). Journal of the Neurological Sciences 1999;169(1‐2):13‐21. [DOI] [PubMed] [Google Scholar]
CNTF 1996
- The ALS CNTF Treatment Study (ACTS) Phase I‐II Study Group. The amyotrophic lateral sclerosis functional rating scale: Assessment of activities of daily living in patients with amyotrophic lateral sclerosis. Archives of Neurology 1996;53(2):141‐7. [PubMed] [Google Scholar]
Coble 1985
- Coble NO, Maloney FP. Interdisciplinary rehabilitation of multiple sclerosis and neuromuscular disorders. In: Maloney FP, Burks JS, Ringel SP editor(s). Effects of Exercise in Neuromuscular Disease. New York: JB Lippincott, 1985:228‐38. [Google Scholar]
Deforges 2009
- Deforges S, Branchu J, Biondi O, Grondard C, Pariset C, Lécolle S, et al. Motoneuron survival is promoted by specific exercise in a mouse model of amyotrophic lateral sclerosis. Journal of Physiology 2009;587(Pt 14):3561‐751. [DOI] [PMC free article] [PubMed] [Google Scholar]
Einarsson 1991
- Einarsson G. Muscle conditioning in late poliomyelitis. Archives of Physical Medicine and Rehabilitation 1991;72(1):11‐4. [PubMed] [Google Scholar]
Florence 1984
- Florence JM, Hagberg JM. Effects of training on the exercise response of neuromuscular disease patients. Medicine and Science in Sports and Exercise 1984;16(5):460‐5. [DOI] [PubMed] [Google Scholar]
Gubbay 1985
- Gubbay SS, Kahana E, Zilber N, Cooper G, Pintov S, Leibowitz Y. Amyotrophic lateral sclerosis: A study of its presentation and prognosis. Journal of Neurology 1985;232(5):295‐300. [DOI] [PubMed] [Google Scholar]
Hamida 2000
- Hamida MB, Hentati F. Juvenile amyotrophic lateral sclerosis. In: Brown R Jr, Meininger V, Swash M editor(s). Amyotrophic Lateral Sclerosis. London: Martin Dunitz Ltd, 2000:59‐79. [Google Scholar]
Haverkamp 1995
- Haverkamp LJ, Appel V, Appel SH. Natural history of amyotrophic lateral sclerosis in database population. Validation of a scoring system and a model for survival prediction. Brain 1995;118(Pt 3):707‐19. [DOI] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Altman DG, Sterne JAC (editors). Chapter 8: Assessing risk of bias in included studies. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions. Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011 Available from www.cochrane‐handbook.org.
Hillel 1989
- Hillel AD, Miller RM, Yorkston K, McDonald E, Norris FH, Konikow N. Amyotrophic Lateral Sclerosis Severity Scale. Neuroepidemiology 1989;8(3):142‐50. [DOI] [PubMed] [Google Scholar]
Jenkinson 1999a
- Jenkinson C, Fitzpatrick R, Brennan M, Bromberg M, Swash M. Evidence for the validity and reliability of the ALS assessment questionnaire: the ALSAQ‐40. Amyotrophic Lateral Sclerosis and Other Motor Neuron Disorders 1999;1(1):33–40. [DOI] [PubMed] [Google Scholar]
Jenkinson 1999b
- Jenkinson C, Fitzpatrick R, Brennan C, Bromberg M, Swash M. Development and validation of a short measure of health status for individuals with amyotrophic lateral sclerosis/motor neurone disease: the ALSAQ‐40. Journal of Neurology 1999;246(Suppl 3):16–21. [DOI] [PubMed] [Google Scholar]
Johnson 1971
- Johnson EW, Braddom R. Overwork weakness in facioscapulohumeral muscular dystrophy. Archives of Physical Medicine and Rehabilitation 1971;52(7):333‐6. [PubMed] [Google Scholar]
Kaspar 2005
- Kaspar BK, Frost LM, Christian L, Umapathi P, Gafe FH. Synergy of insulin‐like growth factor‐1 and exercise in amyotrophic lateral sclerosis. Annals of Neurology 2005;57(5):649‐55. [DOI] [PubMed] [Google Scholar]
Kilmer 1994
- Kilmer DD, McCrory MA, Wright NC, Aitkens SG, Bernauer EM. The effect of a high resistance exercise program in slowly progressive neuromuscular disease. Archives of Physical Medicine and Rehabilitation 1994;75(5):560‐3. [PubMed] [Google Scholar]
Kirkinezos 2003
- Kirkinezos IG, Hernandez D, Bradley WG, Moraes CT. Regular exercise is beneficial to a mouse model of amyotrophic lateral sclerosis. Annals of Neurology 2003;53(6):804‐7. [DOI] [PubMed] [Google Scholar]
Krupp 1989
- Krupp LB, LaRocca NG, Muir‐Nash J, Steinberg AD. The fatigue severity scale. Application to patients with multiple sclerosis and systemic lupus erythematosus. Archives of Neurology 1989;46(10):1121‐23. [DOI] [PubMed] [Google Scholar]
Kurtzke 1991
- Kurtzke JF. Risk factors in amyotrophic lateral sclerosis. Advances in Neurology 1991;56:245‐70. [PubMed] [Google Scholar]
Lindeman 1995
- Lindeman E, Leffers P, Spaans F, Drukker J, Reuben J, Kerckhoffs M. Strength training in patients with myotonic dystrophy and hereditary motor and sensory neuropathy: a randomized clinical trial. Archives of Physical Medicine and Rehabilitation 1995;76(7):612‐20. [DOI] [PubMed] [Google Scholar]
Mahoney 2004
- Mahoney DJ, Rodriguez C, Devries M, Yasuda N, Tarnopolsky M. Effects of high‐intensity endurance exercise training in the G93A mouse model of amyotrophic lateral sclerosis. Muscle & Nerve 2004;29(5):656‐62. [DOI] [PubMed] [Google Scholar]
McCartney 1988
- McCartney N, Moroz D, Garner SH, McComas AJ. The effects of strength training in patients with selected neuromuscular disorders. Medicine and Science in Sports and Exercise 1988;20(4):362‐8. [DOI] [PubMed] [Google Scholar]
Milner‐Brown 1988
- Milner‐Brown HS, Miller RG. Muscle strengthening through high resistance weight training in patients with neuromuscular disorders. Archives of Physical Medicine and Rehabilitation 1988;69(1):14‐9. [PubMed] [Google Scholar]
Mitsumoto 1998
- Mitsumoto H, Chad DA, Pioro EK. Clinical features: Signs and symptoms. In: Mitsumoto H, Chad DA, Pioro EK editor(s). Amyotrophic Lateral Sclerosis. Philadelphia: F.A. Davis Company, 1998:47‐64. [Google Scholar]
Mulder 1986
- Mulder DW, Kurland LT, Offord KP, Beard CM. Familial adult motor neuron disease: amyotrophic lateral sclerosis. Neurology 1986;36(4):511‐7. [DOI] [PubMed] [Google Scholar]
Norris 1974
- Norris FH Jr, Calanchini PR, Fallat RF, Panchari S, Jewett B. The administration of guanidine in amyotrophic lateral sclerosis. Neurology 1974;24(8):721‐8. [DOI] [PubMed] [Google Scholar]
Norris 1993
- Norris F, Shepherd R, Denys E, Mukai E, Elias L, Holden D. Onset, natural history and outcome in idiopathic adult motor neuron disease. Journal of the Neurological Sciences 1993;118(1):48‐55. [DOI] [PubMed] [Google Scholar]
Pinto 1999
- Pinto AC, Alves M, Nogueira A, Evangelista T, Carvalho J, Coelho A, et al. Can amyotrophic lateral sclerosis patients with respiratory insufficiency exercise?. Journal of the Neurological Sciences 1999;169(1‐2):69‐75. [DOI] [PubMed] [Google Scholar]
Pradas 1993
- Pradas J, Finison L, Andres PL, Thornell B, Hollander D, Munsat TL. The natural history of amyotrophic lateral sclerosis and the use of natural history controls in therapeutic trials. Neurology 1993;43(4):751‐5. [DOI] [PubMed] [Google Scholar]
RevMan 2012 [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.2. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2012.
Ringel 1993
- Ringel SP, Murphy JR, Alderson MK, Bryan W, England JD, Miller RG, et al. The natural history of amyotrophic lateral sclerosis. Neurology 1993;43(7):1316‐22. [DOI] [PubMed] [Google Scholar]
Rowland 1982
- Rowland LP. Diverse forms of motor neuron disease. Advances in Neurology 1982;36:1‐13. [PubMed] [Google Scholar]
Sanjak 1987
- Sanjak M, Reddan W, Brooks BR. Role of muscular exercise in amyotrophic lateral sclerosis. Neurology Clinics 1987;5(2):251‐68, vi. [PubMed] [Google Scholar]
Schwab 1969
- Schwab R, England A. Projection technique for evaluating surgery in Parkinson's disease. In: Gillingham J, Donaldson I editor(s). Third Symposium on Parkinson's Disease. Edinburgh: Livingstone, 1969. [Google Scholar]
Sinaki 1978
- Sinaki M, Mulder DW. Rehabilitation techniques for patients with amyotrophic lateral sclerosis. Mayo Clinic Proceedings 1978;53(3):173‐8. [PubMed] [Google Scholar]
Strickland 1996
- Strickland D, Smith SA, Dolliff G, Goldman L, Roelofs R. Physical activity, trauma, and ALS: a case‐control study. Acta Neurologica Scandinavica 1996;94(1):45‐50. [DOI] [PubMed] [Google Scholar]
Strong 1991
- Strong MJ, Hudson AJ, Alvord WG. Familial amyotrophic lateral sclerosis, 1850‐1989: A statistical analysis of the world literature. Canadian Journal of Neurological Sciences 1991;18(1):45‐58. [DOI] [PubMed] [Google Scholar]
Swash 2000a
- Swash M, Desai J. Motor neuron disease: classification and nomenclature. Amyotrophic Lateral Sclerosis and Other Motor Neuron Disorders 2000;1(2):105‐12. [DOI] [PubMed] [Google Scholar]
Swash 2000b
- Swash M. Clinical features and diagnosis of amyotrophic lateral sclerosis. In: Brown R Jr, Meininger V, Swash M editor(s). Amyotrophic Lateral Sclerosis. London: Martin Dunitz Ltd, 2000:3‐30. [Google Scholar]
van der Kooi 2011
- Voet NBM, Kooi EL, Riphagen II, Lindeman E, Engelen BGM, Geurts ACH. Strength training and aerobic exercise training for muscle disease. Cochrane Database of Systematic Reviews 2010, Issue 1. [DOI: 10.1002/14651858.CD003907.pub3] [DOI] [PubMed] [Google Scholar]
Veldink 2003
- Veldink JH, Bar PR, Joosten EAJ, Otten M, Wokke JHJ, Berg LH. Sexual differences in onset of disease and response to exercise in a transgenic model of ALS. Neuromuscular Disease 2003;13(9):737‐43. [DOI] [PubMed] [Google Scholar]
Vignos 1983
- Vignos PJ Jr. Physical models of rehabilitation in neuromuscular disease. Muscle & Nerve 1983;6(5):323‐38. [DOI] [PubMed] [Google Scholar]
Ware 1993
- Ware JE, Snow KK, Kosinski M, Gandek B. SF‐36 Health Survey: Manual and Interpretation Guide. Boston: Health Institute, New England Medical Center, 1993. [Google Scholar]
References to other published versions of this review
Dal Bello‐Haas 2005
- DalBello‐Haas V, Florence J, Krivickas L. Therapeutic exercise for people with amyotrophic lateral sclerosis/motor neuron disease. Cochrane Database of Systematic Reviews 2005, Issue 2. [DOI: 10.1002/14651858.CD005229] [DOI] [PubMed] [Google Scholar]
Dal Bello‐Haas 2008
- Dal Bello‐Haas V, Florence JM, Krivickas LS. Therapeutic exercise for people with amyotrophic lateral sclerosis or motor neuron disease. Cochrane Database of Systematic Reviews 2008, Issue 2. [DOI: 10.1002/14651858.CD005229.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]