

Abstract
Apixaban is an oral, direct factor Xa inhibitor that inhibits both free and clot-bound factor Xa, and has been approved for clinical use in several thromboembolic disorders, including reduction of stroke risk in non-valvular atrial fibrillation, thromboprophylaxis following hip or knee replacement surgery, the treatment of deep vein thrombosis or pulmonary embolism, and prevention of recurrent deep vein thrombosis and pulmonary embolism. The absolute oral bioavailability of apixaban is ~ 50%. Food does not have a clinically meaningful impact on the bioavailability. Apixaban exposure increases dose proportionally for oral doses up to 10 mg. Apixaban is rapidly absorbed, with maximum concentration occurring 3–4 h after oral administration, and has a half-life of approximately 12 h. Elimination occurs via multiple pathways including metabolism, biliary excretion, and direct intestinal excretion, with approximately 27% of total apixaban clearance occurring via renal excretion. The pharmacokinetics of apixaban are consistent across a broad range of patients, and apixaban has limited clinically relevant interactions with most commonly prescribed medications, allowing for fixed dosages without the need for therapeutic drug monitoring. The pharmacodynamic effect of apixaban is closely correlated with apixaban plasma concentration. This review provides a summary of the pharmacokinetic, pharmacodynamic, biopharmaceutical, and drug–drug interaction profiles of apixaban. Additionally, the population-pharmacokinetic analyses of apixaban in both healthy subjects and in the target patient populations are discussed.
Key Points
| Apixaban, a direct factor Xa inhibitor, has predictable pharmacokinetic and pharmacodynamic properties that are consistent across a wide range of patients, including the elderly and those with moderate renal impairment. |
| The fast onset of action, low potential for food or drug interactions, and lack of requirement for routine monitoring during clinical use make apixaban a potentially useful option to simplify anticoagulation treatment. |
Introduction
Warfarin and other vitamin K antagonists (VKAs) are highly effective oral anticoagulants but are limited by a narrow therapeutic window, drug and food interactions, and the requirement for frequent monitoring [1, 2]. Direct oral anticoagulants including dabigatran (a direct thrombin inhibitor) and the direct factor Xa (FXa) inhibitors including rivaroxaban, apixaban, edoxaban, and betrixaban, have been developed to overcome some of the limitations associated with VKAs allowing fixed dosages without routine therapeutic monitoring.
Apixaban is a direct FXa inhibitor that has been approved in many countries for several indications [3, 4]. The results of the key phase III clinical trials supporting its approval demonstrated that apixaban is an important alternative to existing anticoagulant therapies, such as VKAs or aspirin or low-molecular-weight heparin (LMWH), with an improved benefit–risk profile. In patients with nonvalvular atrial fibrillation (NVAF), apixaban 5 mg twice daily (BID) significantly reduced the risk of stroke or systemic embolism by 21%, major bleeding by 31%, and death by 11% compared with warfarin [5]. Similarly, in patients with NVAF for whom VKA therapy had failed or was considered unsuitable, apixaban reduced the risk of stroke or emic embolism by > 50% compared with aspirin without a significant increase in the risk of major bleeding [6]. Apixaban 2.5 mg BID demonstrated superior efficacy to enoxaparin 40 mg once daily (QD) and numerically similar efficacy to enoxaparin 30 mg BID (non-inferiority criteria were not met) without increasing major bleeding events for prophylaxis against venous thromboembolism (VTE) in patients undergoing knee or hip replacement surgery [7–9]. Furthermore, compared with enoxaparin 1 mg/kg BID followed by VKA, apixaban (10 mg BID for 7 days followed by 5 mg BID for 6 months) demonstrated non-inferior efficacy for the treatment of VTE, but with a significantly lower risk of major bleeding (a 69% reduction) [10]. After completion of initial treatment for VTE, extended anticoagulation with the approved dose of apixaban 2.5 mg BID significantly reduced the risk of recurrent VTE compared with placebo, without increasing major bleeding [11]. This review summarizes the pharmacokinetic (PK)/pharmacodynamic (PD), biopharmaceutical, and drug–drug interaction profiles of apixaban as well as the potential clinical implications in patients based on a large global clinical development program.
Chemical and Physicochemical Properties
Apixaban, 1-(4-methoxyphenyl)-7-oxo-6-(4-(2-oxopiperidin-1-yl)phenyl)-4,5,6,7-tetrahydro-1H-pyrazolo[3,4-c]pyridine-3-carboxamide (Fig. 1), is a structurally novel neutral bi-cyclic pyrazole with a molecular weight of 459.5 g/mol, aqueous solubility of 40–50 μg/mL, and Caco-2 permeability value of 0.9 × 10−6 cm/s [12].
Fig. 1.
Chemical structure of apixaban
Mode of Action
Apixaban is a potent, direct, oral, reversible, and highly selective inhibitor of FXa (inhibitory constant = 0.08 nM [0.037 ng/mL] at 25 °C) [12, 13] that does not require antithrombin III for antithrombotic activity [14]. Apixaban inhibits free and clot-bound FXa, as well as prothrombinase activity, which inhibits clot growth [15]. By inhibiting FXa, apixaban decreases thrombin generation and thrombus development. It has no direct effect on platelet aggregation, but indirectly inhibits platelet aggregation induced by thrombin [16]. In a rabbit arteriovenous shunt thrombosis model, apixaban inhibited thrombus formation in a dose-dependent manner (half maximal inhibitory concentration = 329 nM) [12].
Pharmacokinetic Properties
A summary of the absorption, distribution, metabolism, and elimination of apixaban is shown in Fig. 2 [4, 17–19]. The PK properties of apixaban following single- or multiple-dose administration are summarized in Tables 1, 2. Apixaban concentration was determined using a validated liquid chromatography-tandem mass spectroscopy method [20]. Blood samples were citrated and were stored frozen at − 20 °C until they were analyzed. Intra- and inter-assay precision values for replicate quality-control samples were ≤ 5.36%. The lower limit of quantification for apixaban is 1 ng/mL with a dynamic range of 1.00–1000 ng/mL.
Fig. 2.

Absorption, distribution, metabolism, and elimination of apixaban [3, 4, 17–19]. *Data from subjects following oral administration without bile collection. CYP cytochrome P450, Vss volume of distribution at steady state
Table 1.
Apixaban pharmacokinetic parameters following a single-dose administration [17]
Reproduced with premission from Frost et al. Br J Clin Pharmacol. 2013;75:476–87
| Dose (mg) | n |
Cmax (ng/mL) GM (CV %) |
AUC0–∞ (ng·h/mL), GM (CV %) |
AUC0–t (ng·h/mL) GM (CV %) |
Tmax (h) Median (min, max) |
t1/2 (h) Mean (SD) |
|---|---|---|---|---|---|---|
| Single-ascending-dose study | ||||||
| Oral solution | ||||||
| 0.5 | 6 | 9.1 (20) | 61.9 (16) | 52.7 (23) | 1.5 (1.0, 4.0) | 3.6 (1.1) |
| 1.0 | 6 | 23.5 (35) | 174.4 (31) | 162.6 (33) | 1.8 (1.0, 3.0) | 4.3 (1.6) |
| 2.5 | 6 | 52.5 (35) | 437.5 (41) | 421.1 (42) | 1.5 (1.0, 3.0) | 6.8 (2.0) |
| Oral tablet | ||||||
| 5 | 6 | 104.7 (25) | 1016.6 (37) | 976.6 (36) | 3.3 (2.5, 4.0) | 15.2 (8.5) |
| 10 | 6 | 176.3 (42) | 1303.6 (40) | 1266.5 (38) | 3.0 (2.0, 4.0) | 11.1 (5.8) |
| 25 | 6 | 365.1 (17) | 4010.0 (19) | 3868.9 (22) | 3.0 (2.5, 4.0) | 26.8a (33.7) |
| 50 | 7 | 685.2 (22) | 7556.5 (25) | 7096.7 (23) | 2.5 (2.0, 4.0) | 19.7 (15.3) |
| Food effect study, oral tablet | ||||||
| 10 fasted | 21 | 150.8 (28) | 1789.0 (31) | 1762.2 (32) | 3.0 (1.5, 6.0) | 11.5 (4.3) |
| 10 fed | 21 | 165.0 (18) | 1867.8 (30) | 1811.5 (30) | 4.0 (1.0, 9.0) | 11.3 (2.9) |
AUC0–∞ area under the plasma concentration–time curve from time zero extrapolated to infinity, AUC0–t area under the plasma concentration–time curve from time zero to time of the last observed concentration, Cmax maximum plasma concentration, CV % percentage coefficient of variance, GM geometric mean, h hours, SD standard deviation, t1/2 plasma terminal half-life, Tmax time to Cmax
aThe summary statistics for t1⁄2 in the 25-mg panel were calculated using data from all six subjects, including one subject in whom the calculated t1⁄2 exceeded 95 h
Table 2.
Apixaban pharmacokinetic parameters following a single- and multiple-dose (days 1 and 7) administration [21]
Reproduced with permission from Frost et al. Br J Clin Pharmacol. 2013;76:776–86
| Apixaban dose (mg) and regimen |
Cmax (ng/mL), GM (CV %) |
Cmina (ng/mL), GM (CV %) |
AUCtaub (ng·h/mL), GM (CV %) |
Tmax (h), median (min, max) |
AI, GM (CV %) |
t1/2 (h), mean (SD) |
|---|---|---|---|---|---|---|
| Day 1 | ||||||
| 2.5 BID (n = 6) | 51.0 (27) | 14.2 (53) | 353.3 (25) | 3.5 (2.0, 12.0) | – | – |
| 5 BID (n = 6) | 81.9 (18) | 25.3 (20) | 600.6 (20) | 3.5 (3.0, 6.0) | – | – |
| 10 BID (n = 6) | 226.2 (38) | 72.7 (27) | 1608.3 (30) | 4.0 (2.0, 4.0) | – | – |
| 25 BID (n = 6) | 425.3 (24) | 129.0 (33) | 3108.6 (25) | 3.5 (2.0, 4.0) | – | – |
| 10 QD (n = 6) | 178.4 (19) | 14.5 (27) | 1589.6 (20) | 4.0 (3.0, 4.0) | – | – |
| 25 QD (n = 6) | 310.0 (12) | 36.5 (31) | 2868.1 (7) | 4.0 (2.0, 6.0) | – | – |
| Day 7 | ||||||
| 2.5 BID (n = 5) | 62.3 (37) | 21.0 (17) | 462.8 (35) | 3.0 (3.0, 9.0) | 1.3 (18) | 8.1 (1.8) |
| 5 BID (n = 6) | 128.5 (10) | 49.6 (20) | 1051.9 (9) | 4.0 (2.0, 4.0) | 1.8 (22) | 11.7 (3.3) |
| 10 BID (n = 6) | 329.8 (45) | 103.8 (57) | 2424.9 (47) | 3.0 (2.0, 4.0) | 1.5 (33) | 10.9 (2.9) |
| 25 BID (n = 6) | 716.6 (21) | 281.1 (38) | 5850.3 (16) | 3.5 (1.0, 4.0) | 1.9 (17) | 15.2 (7.2) |
| 10 QD (n = 6) | 201.4 (15) | 26.8 (43) | 2015.7 (16) | 3.5 (3.0, 4.0) | 1.3 (23) | 14.9 (7.2) |
| 25 QD (n = 6) | 428.9 (20) | 55.3 (33) | 4248.3 (19) | 3.0 (2.0, 4.0) | 1.5 (17) | 15.3 (4.3) |
AI accumulation index, AUCtau area under the plasma concentration–time curve from time zero to the time of last measureable concentration, BID twice daily, Cmax maximum plasma concentration, Cmin minimum plasma concentration, CV % percentage coefficient of variance, GM geometric mean, h hours, QD once daily, SD standard deviation, t1/2 plasma terminal half-life, Tmax time to Cmax
aCmin defined as apixaban concentration 12 or 24 h after morning dosing for BID or QD regimens, respectively
btau = 12 h for BID panels and 24 h for QD panels
Absorption, Bioavailability, and Biopharmaceutical Profile
The maximum plasma concentration (Cmax) of apixaban occurs 3–4 h after oral administration [17, 21]. The absorption of apixaban appears to occur primarily in the small intestine and decreases progressively throughout the gastrointestinal tract [22]. Compared with oral administration, the bioavailability of 2.5 mg of apixaban solution was approximately 60% and 84% lower when released in the distal small bowel and ascending colon, respectively [22]. For oral doses up to 10 mg, the absolute bioavailability of apixaban is ~ 50% [23, 24], resulting from the incomplete absorption [18] and first-pass metabolism in the gut and liver [25, 26].
Food does not have a clinically significant effect on the bioavailability of apixaban. Apixaban exposure following administration of 10 mg of apixaban with food (high-fat, high-calorie meal) was similar to apixaban exposure when administered in the fasting state, with geometric mean ratios (fed/fasted) for Cmax and area under the concentration–time curve from time zero to infinity (AUC0–∞) [90% confidence interval (CI)] of 1.10 (1.004, 1.197) and 1.04 (1.004, 1.086) [17]. In another study using the apixaban commercial 5-mg tablet, administration with a high-fat, high-calorie meal reduced apixaban Cmax and AUC0–∞ by 14.9% and 20.1%, respectively [27]. The exposure reduction was considered not clinically significant.
Several alternatives for administering apixaban were also studied. When 10 mg of apixaban (2 × 5-mg tablets) was crushed and suspended in 30 mL of water, Cmax and AUC0–∞ met bioequivalence criteria compared with administration of whole tablets [27]. When apixaban was crushed and mixed with 30 g of applesauce, Cmax and AUC0–∞ decreased by 21.1% and 16.4%, respectively. Administration of an apixaban 5-mg crushed tablet via nasogastric tube (NGT) resulted in exposure (Cmax and AUC0–∞) equivalent to that obtained after administration of 5 mg of apixaban as a solution (12.5 mL × 0.4 mg/mL) via an oral syringe [28]. An apixaban solution formulation is being investigated for use in patients unable to swallow oral dosage forms [28, 29], such as certain hospitalized patients and children in ongoing pediatric studies. Oral administration of apixaban solution resulted in exposure (AUC0–∞) equivalent to that obtained following administration of apixaban tablets [28]. The bioavailability of apixaban solution administered via NGT and flushed with 5% dextrose in water or infant formula (Similac®; Abbott Laboratories, Abbott Park, IL, USA) was found to be generally comparable: bioequivalent with 5% dextrose in water and 19% lower Cmax and 8% lower AUC0–∞ with infant formula to that of apixaban solution administered orally. These results suggest both 5% dextrose in water and infant formula could be used to flush apixaban solution through an NGT [28]. In addition, administration of apixaban solution via NGT in the presence of a liquid meal challenge with BoostPlus® (Nestlé HealthCare Nutrition, Fremont, MI, USA) resulted in a modest reduction in apixaban exposure (Cmax and AUC0–∞ values 32% and 19% lower, respectively), compared with the corresponding values observed after administration of apixaban solution via an oral syringe [28]. These results support several alternatives for administering apixaban that maintain exposure similar to that of intact oral tablets [28].
Protein Binding and Distribution
The in vitro plasma protein binding of apixaban in humans is approximately 87% and it is predominantly bound to albumin [30]. When studied in subjects administered apixaban, protein binding was ~ 93% in healthy subjects and comparable in subjects with end-stage renal disease (ESRD; creatinine clearance [CrCl] < 15 mL/min) and in subjects with mild-to-moderate hepatic impairment, suggesting that protein binding was not altered by ESRD or mild-to-moderate hepatic impairment [31, 32].
The volume of distribution is approximately 21 L, suggesting distribution mainly into extracellular fluid, which comprises vascular and interstitial fluid [3, 4, 23, 24]. The blood to plasma ratio of apixaban is 0.9:1 in humans, suggesting that apixaban is uniformly distributed between plasma and red blood cells [16]. It is unknown whether apixaban or its metabolites are excreted in human breast milk. A tissue distribution study in rats showed that apixaban was excreted in milk (~ 10% of the maternal dose) [33]. The milk to plasma AUC0–∞ ratio in rats was 30:1, indicating that apixaban can accumulate in milk.
Metabolism and Elimination
Total plasma clearance of apixaban is ~ 3.3 L/h and renal clearance is ~ 0.9 L/h (~ 27% of total clearance), as determined by two intravenous (IV) studies [3, 4, 23, 24]. The apparent elimination half-life (t1/2) is ~ 12 h [17, 21, 34–36]. Elimination involves multiple pathways, including metabolism as well as biliary and renal elimination of the unchanged parent compound and direct intestinal excretion [18, 37]. Apixaban is not a high-extraction-ratio drug.
The metabolic pathways for apixaban include O-demethylation, hydroxylation, and sulfation of hydroxylated O-demethyl apixaban [18], with metabolism primarily occurring via cytochrome P450 (CYP) 3A4/5, with minor contributions from CYP1A2, CYP2C8, CYP2C9, CYP2C19, and CYP2J2 [25]. After an oral dose of apixaban, unchanged apixaban is the major drug-related component in human plasma with no active circulating metabolites present [18]. When a 20-mg radiolabeled dose was administered orally, 56.0% of the dose was recovered in feces and 24.5% was eliminated in urine and unchanged apixaban was the major component in both feces (60.7%) and urine (87.7%). When bile was collected following administration of the 20-mg dose, the recovery was 46.7% in feces and 28.8% in urine. The radioactivity identified in biliary excreta during a 5-h collection window was approximately 5% of total radioactivity recovery, suggesting that biliary excretion is a minor pathway of elimination and that fecal recovery consisted of both absorbed and unabsorbed drug because the fecal recovery of total radioactivity was larger than that seen in bile.
In experiments conducted in bile duct cannulated rats and dogs, 20–50% of an IV dose of apixaban was excreted fecally, which identified direct intestinal excretion as a contributor to apixaban elimination [26]. The direct intestinal excretion appears to play a role in humans as well. In an open-label, randomized, crossover study in which subjects were administered activated charcoal either 2 or 6 h after a single oral dose of apixaban 20 mg, the results showed that apixaban terminal t1/2 was reduced from 13.4 h to approximately 5 h while there was little effect on peak apixaban plasma concentrations [37]. This increased elimination of apixaban by activated charcoal may be due to adsorption of unabsorbed apixaban and interruption of apixaban reabsorption after biliary and/or direct intestinal excretion.
The total and renal elimination of apixaban were characterized in two IV studies including 50 healthy subjects administered a single IV dose of apixaban (0.5–5 mg) [23, 24]. In these studies, apixaban renal clearance was 27% of total clearance on average.
Dose Proportionality and Time Dependency
Apixaban exposure increases proportionally with doses up to 10 mg, but at doses ≥ 25 mg, less-than-proportional increases are observed, likely due to dissolution-limited bioavailability [17, 34, 38]. The dose-proportionality analysis showed that apixaban exposure increased proportionally to dose across the approved dose range (2.5–10 mg) [34]. Exposure was less than proportional for doses greater than 10 mg; this appeared to be driven by the less-than-proportional increases in exposure following administration of the 25- and 50-mg doses. Dose proportionality was further assessed across 2.5–10 mg using single-dose PK data from 17 phase I studies (data on file). This analysis showed that both Cmax and AUC0–∞ showed a dose-proportional increase across the 2.5- to 10-mg dose range.
There is no time dependency in apixaban pharmacokinetics and single-dose data predict multiple-dose pharmacokinetics. Following BID doses of apixaban, steady-state concentrations were reached by day 3, consistent with the apparent elimination t1/2 of 12 h, as illustrated in Fig. 3 [21]. Apixaban showed a modest accumulation (less than two fold) with BID dosing consistent with the apparent elimination t1/2.
Fig. 3.

Mean (+ standard deviation) plasma apixaban concentration vs. time profiles on days 1 and 7 [21].
Reproduced with permission from Frost et al. Br J Clin Pharmacol. 2013;76:776–86
Pharmacokinetic Effects of Intrinsic Factors in Special Populations
The effects of intrinsic factors on the pharmacokinetics of apixaban from phase I studies in special populations are described below, and selected results are summarized in Fig. 4.
Fig. 4.
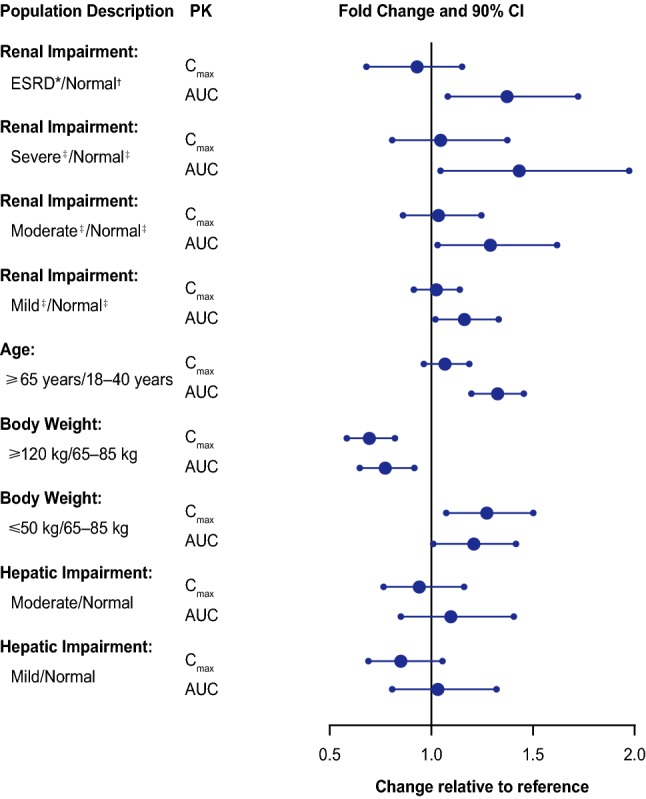
Effect of intrinsic factors on the pharmacokinetics of apixaban [4]. *Subjects with end-stage renal disease (ESRD) treated with intermittent hemodialysis; reported pharmacokinetic (PK) findings are following a single dose of apixaban post-hemodialysis. †Creatinine clearance (CrCl) > 80 mL/min. ‡Severe, moderate, mild, and normal reflect CrCl of 15 mL/min, 40 mL/min, 65 mL/min, and 100 mL/min based on regression analyses. AUC area under the plasma concentration–time curve, CI confidence interval, Cmax maximum plasma concentration. Reproduced with permission from Eliquis (apixaban) US prescribing information
Age
A study in healthy male and female subjects, aged either 18–40 years (young) or 65–79 years (elderly), found that the Cmax of apixaban was similar in both age groups but the AUC0–∞ was 32% higher in the elderly subjects [39]. The study showed a direct relationship between apixaban clearance and creatinine clearance, suggesting that renal function may have contributed to the differences in apixaban exposures between groups. Population-PK analyses using intensive PK data collected in phase I studies and sparse PK data from phase II and III studies for each approved indication showed that age alone had a small impact on apixaban exposure [40–42]. In NVAF, for example, 80- and 40-year-old subjects are predicted to have 5% higher and 11.5% lower daily AUC values at steady state, respectively, compared with a reference 65-year-old subject. In addition, results from sub-analyses of phase III trials with apixaban found that age did not affect the benefit–risk profile of apixaban [10, 11, 43–45], and therefore no dose adjustment of apixaban is required based on age alone [3, 4].
Sex
In the study of healthy subjects that evaluated age, Cmax and AUC0–∞ of apixaban were approximately 18% and 15% higher in female than in male subjects. This difference in exposure is considered modest and unlikely to be clinically significant [39]. Population-PK analyses also showed a < 20% increase in apixaban daily exposure in female versus male subjects for approved indications [40–42]. Further, no meaningful differences were seen between male and female subjects in the primary efficacy or safety outcomes in the key phase III trials for apixaban [5, 6, 10, 11, 44]. No dose adjustment of apixaban is required based on sex [3, 4].
Body Weight
In healthy subjects, those with low body weight (≤ 50 kg) had approximately 27% and 20% higher apixaban Cmax and AUC0–∞, respectively, compared with the reference body weight group (65–85 kg) [46]. Conversely, those with a high body weight (≥ 120 kg) had approximately 31% and 23% lower apixaban Cmax and AUC0–∞, respectively, compared with the reference group [46]. Apixaban renal clearance was similar across weight groups [46]. In addition, population-PK analyses showed that body weight explained the between-subject variability for apparent volume of distribution and the effect was less than proportional [40–42]. Results from subanalyses of phase III trials with apixaban found that body weight did not affect the benefit–risk profile of apixaban [10, 11, 47] and no dose adjustment is required based on body weight alone [3, 4].
Race
In healthy subjects, pharmacokinetics in Asian (Japanese and Chinese) subjects were similar to pharmacokinetics in non-Asian subjects [34–36]. Population-PK analyses showed that Asian subjects with NVAF or VTE had a 13.5% and 20.2% increase in apixaban AUC, respectively; however, this difference was not considered clinically meaningful [41, 42]. Polymorphisms of CYP3A5 and P-glycoprotein (P-gp) have been suggested as contributing factors to ethnic differences in apixaban pharmacokinetics [48, 49]. Results from subanalyses of phase III trials with apixaban found that race did not affect the benefit–risk profile of apixaban [50]. No dose adjustment is required based on race or ethnicity [3, 4].
Renal Impairment
Consistent with the limited contribution of renal clearance to the overall clearance of apixaban (~ 27%), the impact of renal impairment on apixaban exposure was modest. Renal impairment showed no effect on apixaban Cmax. The regression analysis of AUC0–∞ vs. CrCl showed that in subjects with mild (CrCl of 65 mL/min), moderate (40 mL/min), and severe (15 mL/min) renal impairment, apixaban AUC0–∞ increased by 16%, 29%, and 44%, respectively, compared with healthy subjects with normal renal function (CrCl of 100 mL/min) [51]. Population-PK analyses showed consistent results [40–42]. For example, NVAF subjects with mild, moderate, and severe renal impairment are estimated to have approximately 9%, 28%, and 55% higher daily AUC values at steady state, respectively, compared with subjects with normal renal function. In subjects with ESRD maintained on dialysis, apixaban AUC0–∞ following administration after completion of a dialysis session was 36% higher than that in healthy subjects with normal renal function. Hemodialysis was associated with a reduction in apixaban exposure of approximately 14% [31].
The limited renal contribution to apixaban elimination and the modest increase in apixaban exposure in subjects with ESRD undergoing hemodialysis suggest that, from a PK perspective, apixaban could be used without dose modification in these patients. However, it is important to note that subjects with severe renal impairment (CrCl < 30 mL/min in VTE prevention orthopedic studies or CrCl < 25 mL/min in NVAF and VTE treatment studies) or ESRD maintained on hemodialysis were excluded in clinical efficacy and safety studies of apixaban. Limited clinical outcomes data indicate that the benefit–risk profile of apixaban appears to be maintained in the presence of severe renal impairment: those randomized to apixaban experienced lower bleeding rates compared with warfarin among 269 subjects with NVAF who had CrCl 25 to < 30 mL/min in the phase III ARISTOTLE study, consistent with the results in the overall population [52]. Further evaluation of the benefit–risk profile of apixaban in patients with NVAF and ESRD is ongoing (NCT02942407, NCT02933697). The prescribing guidance depends on the approved indication and the region. For example in the USA, no dose adjustment is recommended for apixaban due to renal function alone based on the phase I results in subjects undergoing hemodialysis [4, 31]. In patients with NVAF, a reduced dose of apixaban (2.5 mg BID) should be administered if patients have at least two of the following characteristics: age ≥ 80 years, body weight ≤ 60 kg, or serum creatinine ≥ 1.5 mg/dL, consistent with the apixaban dose-reduction criteria used in NVAF phase III clinical trials [5, 6], as these are considered inherent risk factors for bleeding in the NVAF population [53]. In Europe, a lower dose of apixaban (2.5 mg BID) should be taken by patients with NVAF with severe renal impairment (CrCl 15–29 mL/min) and apixaban is not recommended in patients with CrCl < 15 mL/min, or in patients undergoing dialysis [3].
Hepatic Impairment
Mild (Child–Pugh Class A) and moderate (Child–Pugh Class B) hepatic impairment had no appreciable effect on the pharmacokinetics of apixaban. Point estimates (90% CI) vs. healthy subjects for AUC0–∞ were 1.03 (0.798, 1.32) and 1.09 (0.849, 1.41), respectively, for subjects with mild or moderate hepatic impairment [32]. Protein binding in subjects with mild or moderate hepatic impairment (data on file: unbound fraction of 7.1% and 6.8%, respectively) was comparable to that in healthy subjects (data on file: 7.9%). No dose adjustment is required in patients with mild or moderate hepatic impairment [4]. Tirona et al. evaluated the impact of nonalcoholic fatty liver disease on apixaban pharmacokinetics and reported no difference between healthy subjects and subjects with nonalcoholic fatty liver disease [54]. The pharmacokinetics of apixaban in subjects with severe (Child–Pugh Class C) hepatic impairment was not evaluated. As patients with severe hepatic impairment may have intrinsic coagulation abnormalities and there is little clinical experience with apixaban in these patients, use of apixaban is not recommended in patients with severe hepatic impairment [3, 4].
Pediatric Population
Limited PK data are available from a multiple-dose PK study of apixaban using an oral solution (0.4 mg/mL) in pediatric patients at risk of VTE and who had an indwelling central venous catheter [55]. Of the eight patients enrolled, six patients aged 12 to < 18 years received apixaban 0.66 mg/m2 BID for 10 days, and two patients aged 6–11 years received apixaban 0.60 mg/m2 BID for 10 days. Apixaban pharmacokinetics was characterized by a two-compartment population-PK model, with first-order absorption and elimination. Allometric scaling of body weight was used to adjust for differences in oral clearance and volume of distribution. The oral clearance of apixaban was 4.86 L/h in the adolescent subjects, consistent with that in adults (data on file, [40–42]). A single-dose apixaban PK/PD study and multiple efficacy and safety studies in pediatric patients are ongoing (NCT01707394, NCT02464969, NCT02981472, NCT02369653).
Drug Interactions
Effects of Apixaban on the Pharmacokinetics of Other Drugs
In vitro assessment showed that apixaban did not induce or inhibit major CYP enzymes or interfere with transport of P-gp substrates [25]. When apixaban was evaluated for the potential to inhibit CYP3A4, CYP2C9, CYP2C19, and CYP2D6 in human liver microsomes, half-maximal inhibitory concentration values were > 45 μM [25]. For induction effects of apixaban, there was little or no effect on the activities of CYP1A2, CYP2B6, and CYP3A4/5 in the primary cultures of human hepatocytes, nor any significant increase in messenger RNA levels. Therefore, drug–drug interactions in which apixaban alters the pharmacokinetics of other drugs are not expected. Apixaban did not inhibit digoxin transport in Caco-2 cells [26]. The lack of effect on P-gp was confirmed by a phase I study of co-administration of apixaban (20 mg QD) with digoxin (0.25 mg QD), a P-gp substrate, which showed no impact on digoxin Cmax or AUC [56].
Effects of Other Drugs on the Pharmacokinetics of Apixaban
Apixaban is a substrate for CYP enzymes, primarily CYP3A4/5, and for efflux transporters P-gp and breast cancer resistance protein (BCRP) [25, 26]. Both P-gp and BCRP are human ATP-binding cassette transporters with a significant overlap in substrate specificity and BCRP-specific inhibitors are limited [57]. Modulation of CYP3A4 and P-gp by other agents is the most likely mechanism for PK interactions involving apixaban, and therefore this was the focus of phase I drug–drug interaction studies as summarized in Fig. 5.
Fig. 5.

Effect of co-administered drugs on the pharmacokinetics of apixaban [4]. AUC area under the plasma concentration–time curve from time zero extrapolated to infinity, CI confidence interval, Cmax maximum plasma concentration, CYP3A4 cytochrome P450 3A4, P-gp P-glycoprotein, PK pharmacokinetics. Reproduced with permission from Eliquis (apixaban) US prescribing information
The largest effect on apixaban exposure was observed in the presence of ketoconazole, a representative strong inhibitor of CYP3A4 and P-gp, and rifampin, a representative strong inducer of both CYP3A4 and P-gp [23, 58]. Following co-administration with ketoconazole, apixaban Cmax and AUC0–∞ were approximately 1.6- and two-fold higher, respectively, than values observed following administration of apixaban alone [58]. Following co-administration with rifampin, apixaban Cmax and AUC0–∞ were 42% and 54% lower, respectively, than values following administration of apixaban alone [23]. Other strong inhibitors of CYP3A4 and P-gp (e.g., itraconazole, ritonavir) would be expected to increase blood apixaban concentrations, and simultaneous use with other strong inducers of CYP3A4 and P-gp (e.g., carbamazepine, phenytoin, St. John’s wort) would be expected to reduce blood apixaban concentrations [23, 58]. The use of strong CYP3A4 inhibitors and inducers was prohibited in apixaban phase III clinical trials. Given the limited clinical experience, the prescribing guidance for strong inhibitors or inducers of CYP3A4 and P-gp depends on the approved indication and the region. Concomitant administration of apixaban with strong inhibitors of both CYP3A4 and P-gp is generally not recommended, with some regions, including the USA, recommending to reduce the apixaban dose by 50% for patients who would otherwise be receiving apixaban doses of 5 mg or 10 mg BID [4]. Co-administration of apixaban with strong inducers of both CYP3A4 and P-gp is generally not recommended, especially for patients receiving treatment for VTE owing to the potential decrease in efficacy associated with decreased apixaban exposure. Some geographic regions, including Europe, recommend that co-administration with such strong inducers should be used with caution for VTE prevention in orthopedic patients and in patients with NVAF [3].
Co-administration with less potent CYP3A4 or P-gp inhibitors resulted in a more modest effect on apixaban exposure. Following administration with naproxen, a P-gp inhibitor with no activity toward CYP3A4 or BCRP, at an intestinal concentration of 6–10 mM [26], apixaban Cmax and AUC0–∞ were 61% and 54% higher, respectively, than values following administration of apixaban alone [59]. Similarly, following co-administration with diltiazem, a moderate inhibitor of CYP3A4 and a weak inhibitor of P-gp, apixaban Cmax and AUC0–∞ were 31% and 40% higher, respectively, than values observed following administration of apixaban alone [58]. Clarithromycin, an inhibitor of P-gp and a strong inhibitor of CYP3A4, led to a 1.3- and 1.6-fold increase in mean apixaban Cmax and AUC0–∞, respectively [3]. No dose adjustment of apixaban is required when administered with agents that are not considered strong inhibitors of both CYP3A4 and P-gp, such as naproxen, diltiazem, and clarithromycin [3, 4].
Agents with no known effect on either CYP3A4 or P-gp (e.g., atenolol or famotidine) had little (< 20%) or no effect on apixaban exposure [4, 56, 60]. Following co-administration of atenolol and apixaban, mean apixaban Cmax and AUC0–∞were 18% and 15% lower, respectively, than when apixaban was administered alone [56]. The administration of apixaban with famotidine, a commonly prescribed histamine H2-receptor antagonist, had no effect on apixaban Cmax or AUC0–∞. These results demonstrate that elevated gastric pH is unlikely to affect the pharmacokinetics of apixaban [60]. In addition, as famotidine is a potent inhibitor of the uptake transporter protein human organic cation transporter-3, and a moderate inhibitor of human organic cation transporter-1 and human organic cation transporter-2, the results indicate that apixaban is not a substrate for these transporters [60].
Pharmacodynamic Properties
The PD effects of apixaban observed in clinical studies were consistent with its mechanism of action: direct reversible inhibition of FXa. The relationships between apixaban plasma concentrations and clotting time measures as well as anti-FXa activity are illustrated in Fig. 6. Apixaban prolongs traditional clotting tests such as prothrombin time (PT), international normalized ratio (INR), and activated partial thromboplastin time (aPTT) [17, 21, 35]. Clotting times showed dose-related increases tracking the plasma concentration–time profile; however, changes are small, subject to a high degree of variability, and not useful in monitoring the anticoagulant effect of apixaban. The modified PT assay was developed as an exploratory PD measure of apixaban to improve the dynamic range of the PT assay [61]. As expected, the modified PT results in healthy subjects showed a more sensitive assessment of apixaban activity than INR or aPTT [17].
Fig. 6.

Scatter plots of a international normalized ratio (INR), b activated partial thromboplastin time (aPTT), c modified prothrombin time (mPT), and d, e anti-Xa activity vs. apixaban plasma concentration [35].
Reproduced with permission from Yamahira et al. Int J Clin Pharmacol Ther. 2014;52:564–73
Ex vivo thrombin generation mediated by tissue factor in platelet-poor plasma was studied in healthy subjects [34]. Single doses of apixaban (2.5–50 mg) produced transient dose-related changes in parameters of the thrombin generation curve. Lag time and time to peak increased by ~ 65% 3 h after administration of apixaban 2.5 mg, while the peak value (maximum thrombin concentration) and endogenous thrombin potential decreased by approximately 40% and 12.5%, respectively. The effect of apixaban on thrombin generation parameters was evident through ≥ 12 h after dosing. In addition, effects of four-factor prothrombin complex concentrates (PCC) on the pharmacodynamics of apixaban were studied in healthy subjects following a single infusion of either a heparin-free four-factor PCC or a heparin-containing four-factor PCC [62]. Following administration of apixaban 10 mg BID, endogenous thrombin potential returned to pre-apixaban levels 4 h after the initiation of a 30-min PCC infusion, compared with 45 h following placebo. Mean endogenous thrombin potential continued to increase and exceeded pre-apixaban values, reaching a maximum (34–51% increase over pre-apixaban levels) at 21 h after initiating PCC.
Anti-FXa activity, determined with a single-step chromogenic Rotachrom® Heparin assay (Stago, Parsippany, NJ, USA), exhibits a close direct linear relationship with apixaban plasma concentration, reaching maximum values at the time of apixaban peak plasma concentrations as expected as a bioassay for apixaban exposure [31, 35, 46]. The relationship between apixaban plasma concentration and anti-FXa activity was linear over a wide range of apixaban doses. The dose- and concentration-related changes observed following apixaban administration were more pronounced, and less variable, with anti-FXa activity compared with clotting tests. Anti-FXa activity in LMWH units was collected in phase III clinical studies of apixaban using the Rotachrom® Heparin chromogenic assay and a PK–anti-FXa activity analysis was performed for each indication [40–42]. The anti-FXa activity consistently exhibited a close linear relationship with apixaban plasma concentration across indications in target populations [40–42]. The predicted anti-FXa activity levels for each indication at each dose level are summarized in Table 3 [3, 40–42]. Other measures of hemostasis, including template bleeding time and agonist-induced platelet aggregation (agonists: adenosine 5′-diphosphate, arachidonic acid, or collagen), do not reflect apixaban activity.
Table 3.
Predicted apixaban steady-state maximum plasma concentration (Cmax) and minimum plasma concentration (Cmin) and factor Xa (anti-FXa) activity in approved indications [40–42]
Reproduced with permission from the European Union summary of product characteristics: Eliquis (apixaban tablets); and from Byon et al. CPT Pharmacometrics Syst Pharmacol. 2017;6(5):340–9
| Apixaban dose and regimen | Cmax (ng/mL) | Cmin (ng/mL) | Anti-FXa activity maximum (IU/mL) | Anti-FXa activity minimum (IU/mL) |
|---|---|---|---|---|
| Median (5th, 95th percentile) | ||||
| Prevention of VTE: elective hip or knee replacement surgery | ||||
| 2.5 mg BID | 77 (41, 146) | 51 (23, 109) | 1.3 (0.67, 2.4) | 0.84 (0.37, 1.8) |
| Prevention of stroke and systemic embolism: NVAF | ||||
| 2.5 mg BIDa | 123 (69, 221) | 79 (34, 162) | 1.8 (1.0, 3.3) | 1.2 (0.51, 2.4) |
| 5 mg BID | 171 (91, 321) | 103 (41, 230) | 2.6 (1.4, 4.8) | 1.5 (0.61, 3.4) |
| Treatment of VTE and prevetion of recurrent VTE | ||||
| 2.5 mg BID | 67 (30, 153) | 32 (11, 90) | 1.1 (0.47, 2.4) | 0.51 (0.17, 1.4) |
| 5 mg BID | 132 (59, 302) | 63 (22, 177) | 2.1 (0.93, 4.8) | 1.0 (0.35, 2.8) |
| 10 mg BID | 251 (111, 572) | 120 (41, 335) | 4.0 (1.8, 9.1) | 1.9 (0.65, 5.3) |
BID twice daily, NVAF nonvalvular atrial fibrillation, VTE venous thromboembolism
aDose-adjusted population based on two of three dose reduction criteria in the ARISTOTLE study [5]
An in vitro study in pediatric plasma spiked with apixaban demonstrated that endogenous baseline factor X levels, measured by a DiaPharma® Factor X assay (DiaPharma, West Chester, OH, USA) using Russell’s viper venom, were 68%, 54%, and 43% lower in the infant (> 1 month to ≤ 6 months), neonate (birth to ≤ 1 month), and umbilical cord groups when compared with adults, while the levels in the older pediatric age groups appeared to be comparable to those in the adults [63]. Despite the lower factor X levels in neonates and infants, the inhibition of FXa of apixaban, measured by the DiaPharma® Factor X assay in pediatric subjects, appeared to be generally consistent with the effect observed in adults, noting that apixaban at 110 ng/mL resulted in 17%, 16%, and 21% greater inhibition of FXa in the infant, neonate, and umbilical cord blood groups, respectively, relative to the effect observed in adult plasma. While other measures, such as Rotachrom anti-FXa activity, PT, and a modified PT, appeared to be consistent across age groups, this observation may be due to methodologic limitations of these tests.
Pharmacodynamic Interactions with Anticoagulants and Antiplatelet Agents
After combined administration of LMWH, enoxaparin (40-mg single dose), with apixaban (5-mg single dose), an additive effect on anti-FXa activity was observed [64]. Agents associated with serious bleeding that are not recommended for concomitant use with apixaban include unfractionated heparins and heparin derivatives (including LMWH), FXa-inhibiting oligosaccharides (e.g., fondaparinux), direct thrombin II inhibitors (e.g., desirudin), thrombolytic agents, glycoprotein IIb/IIIa receptor antagonists, thienopyridines (e.g., clopidogrel), dipyridamole, dextran, sulfinpyrazone, VKA, and other oral anticoagulants. Pharmacokinetic or PD interactions were not evident when apixaban was co-administered with aspirin 325 mg QD [65]. Co-administration of 500 mg naproxen with apixaban had no additional impact on platelet aggregation beyond that of naproxen [59]. Similarly, co-administration of apixaban with either clopidogrel (75 mg QD), prasugrel (60 mg followed by 10 mg QD), or the combination of clopidogrel 75 mg and aspirin 162 mg QD did not show a relevant increase in template bleeding time or further inhibition of platelet aggregation compared with administration of the antiplatelet agents without apixaban [65, 66]. Increases in clotting tests (PT, INR, and aPTT) were consistent with the effects of apixaban alone (data on file). Despite these findings, apixaban should be used with caution when co-administered with nonsteroidal anti-inflammatory drugs (including aspirin) and other antiplatelet agents because these medicinal products typically increase the bleeding risk and some individuals may have a more pronounced PD response when antiplatelet agents are co-administered with apixaban.
Effect on Electrocardiography
In a thorough QT study, apixaban had no effect on the QTc interval, with a peak placebo-adjusted, time-matched, Fridericia-corrected change from baseline QTc (ΔΔQTc) interval of 1.51 ms (one-sided upper 95% CI 3.71 ms) following administration of a supratherapeutic dose of 50 mg QD for 3 days [38].
Pharmacokinetics in Patient Populations
A population-PK analysis was performed using intensive PK data collected in phase I studies and sparse PK data from phase II and III studies for each approved indication [40–42]. The base structural model was consistently a two-compartment model with first-order absorption and first-order elimination. The effect of renal function on total clearance of apixaban was incorporated in the base model with empirical separation of total clearance into renal and non-renal components. As apixaban exposure was less than proportional with doses greater than 10 mg, this was also consistently incorporated in the base model as a relationship between apixaban relative bioavailability and dose.
Prevention of Venous Thromboembolism After Hip or Knee Replacement Surgery
A population-PK analysis was performed using data from the phase I studies and ~ 1000 subjects receiving apixaban after a total knee or hip replacement in phase II/III clinical trials [40]. The results showed that apixaban clearance decreased with increasing age and was lower in female subjects compared with male subjects (< 25% impact on apixaban AUC exposure). Subjects with mild, moderate, and severe renal impairment were predicted to have median AUC exposures at steady state ~ 15%, 38%, and 58% higher, respectively, than for subjects with normal renal function. Apixaban clearance was slightly lower immediately after surgery (< 25%), returning close to the pretreatment value by the fourth day after surgery. The reduced blood flow to the liver, kidney, and gastrointestinal tract might result in this change in apixaban clearance in the days after surgery.
Treatment of Deep Vein Thrombosis and Pulmonary Embolism
A population-PK analysis was performed using data from the phase I studies and ~ 700 subjects receiving apixaban for VTE treatment or prevention of recurrent VTE in phase II/III clinical trials [41]. The results showed that age, sex, and Asian race had a < 25% impact on apixaban AUC exposure while subjects with severe renal impairment were predicted to have a 56% higher exposure than subjects with normal renal function. There was no difference in apixaban pharmacokinetics between healthy subjects and subjects receiving apixaban for VTE treatment or prevention of recurrent VTE: for a typical subject (60-year-old non-Asian male individual weighing 85 kg with CrCl of 100 mL/min), the apparent renal and non-renal clearance of apixaban were estimated to be 1.83 L/h and 2.52 L/h, respectively.
Reducing Stroke Risk in Patients with Nonvalvular Atrial Fibrillation
A population-PK analysis was performed to describe apixaban pharmcokinetics in subjects with NVAF including data from the phase I studies and ~ 3000 NVAF subjects in phase II/III clinical trials [42]. Predictive covariates of apixaban pharmacokinetics included age, sex, Asian race, renal function, and patient status (NVAF subjects vs. healthy subjects), although individual covariate effects generally resulted in a < 25% change in apixaban exposure, except for severe renal impairment, which resulted in an apixaban exposure increase of 55%. The effect of Japanese race was assessed as an ad-hoc analysis and found to be small, consistent with the effect of Asian race. Subjects with NVAF were estimated to have a slightly higher apixaban AUC exposure compared with phase I subjects (< 20%). In subjects whose apixaban dose was reduced to 2.5 mg BID because they met at least two of the three criteria (age ≥ 80 years, body weight ≤ 60 kg, or serum creatinine ≥ 1.5 mg/dL), apixaban median AUC was ~ 27% lower than that in subjects receiving 5 mg BID, and there was a large overlap between the two dose groups.
Exposure–Response Analyses with Bleeding or Efficacy Endpoints
In VTE prevention in orthopedic surgery subjects, a Cox proportional hazards model showed a statistically significant relationship between individual daily AUC at steady state and any bleeding endpoint [40]; a two-fold increase in apixaban daily AUC is expected to increase bleeding frequencies from 6.18% to 7.25% and from 9.32% to 10.9% in subjects following knee or hip replacement surgery, respectively. There was no statistically significant relationship between apixaban exposure and VTE outcomes using logistic regression [67]. The predicted daily AUC at steady state, Cmax, or minimum plasma concentration and their corresponding anti-FXa activity values showed that there was considerable overlap in the individual predicted values for those with or without bleeding (data on file). Similar observations were made for apixaban exposure and bleeding endpoints in subjects with NVAF (data on file).
For the treatment of VTE or prevention of recurrent VTE, logistic regression analyses were performed between individual daily AUC at steady state and bleeding endpoint (composite of adjudicated major or adjudicated clinically relevant nonmajor bleeding) or efficacy endpoint (adjudicated symptomatic VTE or VTE-related death) [41]. The results found no statistically significant relationship for either bleeding or efficacy endpoints and showed that the range of individual predicted values of daily AUC at steady state, Cmax, minimum plasma concentration, and the corresponding anti-FXa activity values for subjects with efficacy or bleeding events was entirely contained within the range of values from subjects without events [41]. Considering these data, there is no defined therapeutic exposure range or discernable threshold of apixaban concentration that would predict safety or efficacy outcomes for individual subjects.
Variability and Monitoring of Apixaban Exposure
After incorporating important covariates, apixaban pharmacokinetics exhibited moderate between-subject variability: ~ 40% and ~ 25% for oral plasma clearance and central volume of distribution, respectively, in the final population-PK analyses. The residual unexplained variability was ~ 30%.
While treatment with apixaban does not require routine monitoring of exposure, knowledge of apixaban exposure may help to inform clinical decisions, e.g., overdose and emergency surgery. The final population-PK and PK–anti-FXa activity models developed for each indication were used for simulations to predict the steady-state apixaban and anti-FXa activity levels in target patients. These exposure levels predicted from the population-PK and PK–anti-FXa activity analyses for each indication and at each dose level are summarized in Table 3 [3, 42]. While anti-FXa activity was reported in LMWH units in multiple apixaban clinical studies, the current assay uses an apixaban-specific calibrator and anti-FXa activity values are reported in apixaban concentration units, enabling a timely evaluation of apixaban exposure [31, 68]. Apixaban clinical studies that used both LMWH and apixaban calibrators and controls demonstrate a very high correlation between methods [35, 39]. Considering that apixaban exposure and anti-FXa activity are expected to fluctuate within a dosing interval, in contrast to INR with warfarin, it is critical to examine adherence and to accurately collect previous dosing history and time of blood sample when interpreting exposure levels, especially in routine care settings [69]. It should also be noted that factors such as impaired hepatic function and administration of exogenous PCC known to impact other clotting tests (INR, PT) do not impact apixaban anti-FXa activity determined with a one-step chromogenic assay regardless of the calibrators and controls used (i.e., results reported in LMWH units or ng/mL).
Antidote
The administration of activated charcoal may be useful in the management of apixaban overdose or accidental ingestion. When administered 2 or 6 h after a single oral dose of apixaban 20 mg, activated charcoal reduced apixaban AUC by approximately 50% and 27%, respectively [37]. Hemodialysis has a small impact on apixaban exposure (a reduction in apixaban exposure of approximately 14%) and thus is not recommended as an effective means of managing apixaban overdose [3, 4]. Based on the four-factor PCC study results mentioned above, PCC, activated PCC, or recombinant factor VIIa may be considered for treatment of overdose. It is important to note that four-factor PCC did not affect apixaban pharmacokinetics or anti-FXa activity [62].
Andexanet alfa, a recombinant coagulation FXa, inactivated-zhzo is approved in the USA as an antidote for apixaban and rivaroxaban when reversal of anticoagulation is needed because of life-threatening or uncontrolled bleeding [70]. Andexanet alfa acts as an FXa decoy that binds to FXa inhibitors in the blood, preventing them from binding to and inhibiting native FXa [71]. The native FXa is then available to participate in the coagulation process and restore hemostasis [72]. ANNEXA-A was a phase III, randomized, placebo-controlled study to evaluate the reversal of anticoagulation of apixaban (5 mg BID) using andexanet in healthy older subjects (50–75 years of age) [71]. Subjects were randomized in a 3:1 ratio to receive andexanet (IV bolus alone or IV bolus followed by a 2-h infusion) or placebo. Anti-FXa activity, measured using a chromogenic assay, was reduced by 94% following the andexanet bolus compared with 21% in those receiving placebo and was maintained when the IV bolus was followed by infusion. The preliminary results from ANNEXA-4, the ongoing, prospective, open-label, single-arm study of andexanet alfa in patients with acute major bleeding, showed that subjects who were taking apixaban had the median anti-FXa activity reduced by 93% with an andexanet bolus, which remained similar during the 2-h infusion. Twelve hours after the andexanet infusion, effective hemostasis was reported in 75% of the subjects [73].
Conclusions
Apixaban, a direct FXa inhibitor, has predictable PK and PD properties that are consistent across the range of different patient populations studied, including the elderly and those with renal impairment. The fast onset of action, low potential for food or drug interactions, and lack of requirement for routine monitoring during clinical use make apixaban a suitable option to simplify anticoagulation treatment.
Acknowledgements
The authors thank Yan Song for her contributions to the development of this manuscript.
Compliance with Ethical Standards
Funding
This study was funded by Bristol-Myers Squibb and Pfizer Inc. Rob Coover, MPH, and Claire Line, PhD, employees of Caudex, provided editorial assistance, and this assistance was funded by Bristol-Myers Squibb and Pfizer Inc.
Conflict of interest
Wonkyung Byon and Rebecca A. Boyd are current or former employees of Pfizer, Inc., and own stock/stock options. Samira Garonzik is an employee of Bristol-Myers Squibb. Charles E. Frost is an employee of, owns stock/stock options in, has received travel support and payment for lectures from, and is an inventor on patents with Bristol-Myers Squibb.
Ethics approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards.
Consent to participate
Informed consent was obtained from all individual participants included in the studies.
References
- 1.Bristol-Myers Squibb. Coumadin (wafarin sodium) prescribing information. 2017. http://packageinserts.bms.com/pi/pi_coumadin.pdf. Accessed 2 Feb 2018.
- 2.Lip GYH, Banerjee A, Boriani G, Chiang CE, Fargo R, Freedman B, et al. Antithrombotic therapy for atrial fibrillation: CHEST guideline and expert panel report. Chest. 2018;154(5):1121–1201. doi: 10.1016/j.chest.2018.07.040. [DOI] [PubMed] [Google Scholar]
- 3.Bristol-Myers Squibb, Pfizer EEIG. EU summary of product characteristics: Eliquis (apixaban tablets). 2018. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002148/WC500107728.pdf. Accessed 8 Apr 2019.
- 4.Bristol-Myers Squibb Company PI. Eliquis (apixaban) prescribing information. 2018. http://packageinserts.bms.com/pi/pi_eliquis.pdf. Accessed 14 Jun 2018.
- 5.Granger CB, Alexander JH, McMurray JJ, Lopes RD, Hylek E, Hanna M, et al. Apixaban versus warfarin in patients with atrial fibrillation. N Engl J Med. 2011;365(11):981–992. doi: 10.1056/NEJMoa1107039. [DOI] [PubMed] [Google Scholar]
- 6.Connolly SJ, Eikelboom J, Joyner C, Diener HC, Hart R, Golitsyn S, et al. Apixaban in patients with atrial fibrillation. N Engl J Med. 2011;364(9):806–817. doi: 10.1056/NEJMoa1007432. [DOI] [PubMed] [Google Scholar]
- 7.Lassen MR, Raskob GE, Gallus A, Pineo G, Chen D, Hornick P. Apixaban versus enoxaparin for thromboprophylaxis after knee replacement (ADVANCE-2): a randomised double-blind trial. Lancet. 2010;375(9717):807–815. doi: 10.1016/S0140-6736(09)62125-5. [DOI] [PubMed] [Google Scholar]
- 8.Lassen MR, Gallus A, Raskob GE, Pineo G, Chen D, Ramirez LM. Apixaban versus enoxaparin for thromboprophylaxis after hip replacement. N Engl J Med. 2010;363(26):2487–2498. doi: 10.1056/NEJMoa1006885. [DOI] [PubMed] [Google Scholar]
- 9.Lassen MR, Raskob GE, Gallus A, Pineo G, Chen D, Portman RJ. Apixaban or enoxaparin for thromboprophylaxis after knee replacement. N Engl J Med. 2009;361(6):594–604. doi: 10.1056/NEJMoa0810773. [DOI] [PubMed] [Google Scholar]
- 10.Agnelli G, Buller HR, Cohen A, Curto M, Gallus AS, Johnson M, et al. Oral apixaban for the treatment of acute venous thromboembolism. N Engl J Med. 2013;369(9):799–808. doi: 10.1056/NEJMoa1302507. [DOI] [PubMed] [Google Scholar]
- 11.Agnelli G, Buller HR, Cohen A, Curto M, Gallus AS, Johnson M, et al. Apixaban for extended treatment of venous thromboembolism. N Engl J Med. 2013;368(8):699–708. doi: 10.1056/NEJMoa1207541. [DOI] [PubMed] [Google Scholar]
- 12.Pinto DJ, Orwat MJ, Koch S, Rossi KA, Alexander RS, Smallwood A, et al. Discovery of 1-(4-methoxyphenyl)-7-oxo-6-(4-(2-oxopiperidin-1-yl)phenyl)-4,5,6,7-tetrahydro-1H-pyrazolo[3,4-c]pyridine-3-carboxamide (apixaban, BMS-562247), a highly potent, selective, efficacious, and orally bioavailable inhibitor of blood coagulation factor Xa. J Med Chem. 2007;50(22):5339–5356. doi: 10.1021/jm070245n. [DOI] [PubMed] [Google Scholar]
- 13.Luettgen JM, Knabb RM, He K, Pinto DJ, Rendina AR. Apixaban inhibition of factor Xa: microscopic rate constants and inhibition mechanism in purified protein systems and in human plasma. J Enzyme Inhib Med Chem. 2011;26(4):514–526. doi: 10.3109/14756366.2010.535793. [DOI] [PubMed] [Google Scholar]
- 14.Ansell J. Factor Xa or thrombin: is factor Xa a better target? J Thromb Haemost. 2007;5(Suppl. 1):60–64. doi: 10.1111/j.1538-7836.2007.02473.x. [DOI] [PubMed] [Google Scholar]
- 15.Jiang X, Crain EJ, Luettgen JM, Schumacher WA, Wong PC. Apixaban, an oral direct factor Xa inhibitor, inhibits human clot-bound factor Xa activity in vitro. Thromb Haemost. 2009;101(4):780–782. doi: 10.1160/TH08-07-0486. [DOI] [PubMed] [Google Scholar]
- 16.Wong PC, Pinto DJ, Zhang D. Preclinical discovery of apixaban, a direct and orally bioavailable factor Xa inhibitor. J Thromb Thrombolysis. 2011;31(4):478–492. doi: 10.1007/s11239-011-0551-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Frost C, Wang J, Nepal S, Schuster A, Barrett YC, Mosqueda-Garcia R, et al. Apixaban, an oral, direct factor Xa inhibitor: single-dose safety, pharmacokinetics, pharmacodynamics and food effect in healthy subjects. Br J Clin Pharmacol. 2013;75(2):476–487. doi: 10.1111/j.1365-2125.2012.04369.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Raghavan N, Frost CE, Yu Z, He K, Zhang H, Humphreys WG, et al. Apixaban metabolism and pharmacokinetics after oral administration to humans. Drug Metab Dispos. 2009;37(1):74–81. doi: 10.1124/dmd.108.023143. [DOI] [PubMed] [Google Scholar]
- 19.Zhang D, He K, Raghavan N, Wang L, Mitroka J, Maxwell BD, et al. Comparative metabolism of 14C-labeled apixaban in mice, rats, rabbits, dogs, and humans. Drug Metab Dispos. 2009;37(8):1738–1748. doi: 10.1124/dmd.108.025981. [DOI] [PubMed] [Google Scholar]
- 20.Pursley J, Shen JX, Schuster A, Dang OT, Lehman J, Buonarati MH, et al. LC-MS/MS determination of apixaban (BMS-562247) and its major metabolite in human plasma: an application of polarity switching and monolithic HPLC column. Bioanalysis. 2014;6(15):2071–2082. doi: 10.4155/bio.14.66. [DOI] [PubMed] [Google Scholar]
- 21.Frost C, Nepal S, Wang J, Schuster A, Byon W, Boyd RA, et al. Safety, pharmacokinetics and pharmacodynamics of multiple oral doses of apixaban, a factor Xa inhibitor, in healthy subjects. Br J Clin Pharmacol. 2013;76(5):776–786. doi: 10.1111/bcp.12106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Byon W, Nepal S, Schuster AE, Shenker A, Frost CE. Regional gastrointestinal absorption of apixaban in healthy subjects. J Clin Pharmacol. 2018;58(7):965–971. doi: 10.1002/jcph.1097. [DOI] [PubMed] [Google Scholar]
- 23.Vakkalagadda B, Frost C, Byon W, Boyd RA, Wang J, Zhang D, et al. Effect of rifampin on the pharmacokinetics of apixaban, an oral direct inhibitor of factor Xa. Am J Cardiovasc Drugs. 2016;16(2):119–127. doi: 10.1007/s40256-015-0157-9. [DOI] [PubMed] [Google Scholar]
- 24.Frost C, Yu Z, Nepal S, Bragat A, Moore K, Shenker A, et al. Apixaban, a direct factor Xa inhibitor: single-dose pharmacokinetics and pharmacodynamics of an intravenous formulation [abstract 148] J Clin Pharmacol. 2008;48:1132. [Google Scholar]
- 25.Wang L, Zhang D, Raghavan N, Yao M, Ma L, Frost CE, et al. In vitro assessment of metabolic drug-drug interaction potential of apixaban through cytochrome P450 phenotyping, inhibition, and induction studies. Drug Metab Dispos. 2010;38(3):448–458. doi: 10.1124/dmd.109.029694. [DOI] [PubMed] [Google Scholar]
- 26.Zhang D, He K, Herbst J, Kolb J, Shou W, Wang L, et al. Characterization of efflux transporters involved in distribution and disposition of apixaban. Drug Metab Dispos. 2013;41(4):827–835. doi: 10.1124/dmd.112.050260. [DOI] [PubMed] [Google Scholar]
- 27.Song Y, Chang M, Suzuki A, Frost RJ, Kelly A, LaCreta F, et al. Evaluation of crushed tablet for oral administration and the effect of food on apixaban pharmacokinetics in healthy adults. Clin Ther. 2016;38(7):1674–1685. doi: 10.1016/j.clinthera.2016.05.004. [DOI] [PubMed] [Google Scholar]
- 28.Song Y, Wang X, Perlstein I, Wang J, Badawy S, Frost C, et al. Relative bioavailability of apixaban solution or crushed tablet formulations administered by mouth or nasogastric tube in healthy subjects. Clin Ther. 2015;37(8):1703–1712. doi: 10.1016/j.clinthera.2015.05.497. [DOI] [PubMed] [Google Scholar]
- 29.Song Y, Worthington J, Wang X. Development of apixaban oral solution formulation: excipients and palatability. Clin Pharmacol Ther. 2013;93(Suppl. 1):S120–S121. [Google Scholar]
- 30.He K, Luettgen JM, Zhang D, He B, Grace JE, Jr, Xin B, et al. Preclinical pharmacokinetics and pharmacodynamics of apixaban, a potent and selective factor Xa inhibitor. Eur J Drug Metab Pharmacokinet. 2011;36(3):129–139. doi: 10.1007/s13318-011-0037-x. [DOI] [PubMed] [Google Scholar]
- 31.Wang X, Tirucherai G, Marbury TC, Wang J, Chang M, Zhang D, et al. Pharmacokinetics, pharmacodynamics, and safety of apixaban in subjects with end-stage renal disease on hemodialysis. J Clin Pharmacol. 2016;56(5):628–636. doi: 10.1002/jcph.628. [DOI] [PubMed] [Google Scholar]
- 32.Frost C, Yu Z, Wang J, Li T, Ziegler C, Schuster A, et al. Single-dose safety and pharmacokinetics of apixaban in subjects with mild or moderate hepatic impairment. Clin Pharmacol Ther. 2009;85(Suppl. 1):S34.
- 33.Wang L, He K, Maxwell B, Grossman SJ, Tremaine LM, Humphreys WG, et al. Tissue distribution and elimination of [14C]apixaban in rats. Drug Metab Dispos. 2011;39(2):256–264. doi: 10.1124/dmd.110.036442. [DOI] [PubMed] [Google Scholar]
- 34.Frost C, Shenker A, Jhee S, Yu Z, Wang J, Bragat A, et al. Evaluation of the single-dose pharmacokinetics and pharmacodynamics of apixaban in healthy Japanese and Caucasian subjects. Clin Pharmacol. 2018;10:153–163. doi: 10.2147/CPAA.S169505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yamahira N, Frost C, Fukase H, Yu Z, Wang J, Pursley J, et al. Safety, tolerability, pharmacokinetics, and pharmacodynamics of multiple doses of apixaban in healthy Japanese male subjects. Int J Clin Pharmacol Ther. 2014;52(7):564–573. doi: 10.5414/CP201967. [DOI] [PubMed] [Google Scholar]
- 36.Cui Y, Song Y, Wang J, Yu Z, Schuster A, Barrett YC, et al. Single- and multiple-dose pharmacokinetics, pharmacodynamics, and safety of apixaban in healthy Chinese subjects. Clin Pharmacol. 2013;5:177–184. doi: 10.2147/CPAA.S51981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang X, Mondal S, Wang J, Tirucherai G, Zhang D, Boyd RA, et al. Effect of activated charcoal on apixaban pharmacokinetics in healthy subjects. Am J Cardiovasc Drugs. 2014;14(2):147–154. doi: 10.1007/s40256-013-0055-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Frost C, Nepal S, Byon W, Moore K, Reeves RA, Boyd R, et al. Randomized, blinded, placebo- and positive-controlled crossover study to determine the effect of multiple doses of apixaban on the QTc interval. J Clin Pharmacol. 2015;55(5):549–555. doi: 10.1002/jcph.447. [DOI] [PubMed] [Google Scholar]
- 39.Frost CE, Song Y, Shenker A, Wang J, Barrett YC, Schuster A, et al. Effects of age and sex on the single-dose pharmacokinetics and pharmacodynamics of apixaban. Clin Pharmacokinet. 2015;54(6):651–662. doi: 10.1007/s40262-014-0228-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Leil TA, Frost C, Wang X, Pfister M, LaCreta F. Model-based exposure-response analysis of apixaban to quantify bleeding risk in special populations of subjects undergoing orthopedic surgery. CPT Pharmacometrics Syst Pharmacol. 2014;3:e136. doi: 10.1038/psp.2014.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Byon W, Sweeney K, Frost C, Boyd R. Population pharmacokinetics, pharmacodynamics, and exploratory exposure-response analyses of apixaban in subjects treated for venous thromboembolism. CPT Pharmacometrics Syst Pharmacol. 2017;6(5):340–349. doi: 10.1002/psp4.12184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cirincione B, Kowalski K, Nielsen J, Roy A, Thanneer N, Byon W, et al. Population pharmacokinetics of apixaban in subjects with non-valvular atrial fibrillation. CPT Pharmacometrics Syst Pharmacol. 2018;7(11):728–738. doi: 10.1002/psp4.12347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Halvorsen S, Atar D, Yang H, De Caterina R, Erol C, Garcia D, et al. Efficacy and safety of apixaban compared with warfarin according to age for stroke prevention in atrial fibrillation: observations from the ARISTOTLE trial. Eur Heart J. 2014;35(28):1864–1872. doi: 10.1093/eurheartj/ehu046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pineo GF, Gallus AS, Raskob GE, Chen D, Ramirez LM, Ramacciotti E, et al. Apixaban after hip or knee arthroplasty versus enoxaparin: efficacy and safety in key clinical subgroups. J Thromb Haemost. 2013;11(3):444–451. doi: 10.1111/jth.12109. [DOI] [PubMed] [Google Scholar]
- 45.Ng KH, Shestakovska O, Connolly SJ, Eikelboom JW, Avezum A, Diaz R, et al. Efficacy and safety of apixaban compared with aspirin in the elderly: a subgroup analysis from the AVERROES trial. Age Ageing. 2016;45(1):77–83. doi: 10.1093/ageing/afv156. [DOI] [PubMed] [Google Scholar]
- 46.Upreti VV, Wang J, Barrett YC, Byon W, Boyd RA, Pursley J, et al. Effect of extremes of body weight on the pharmacokinetics, pharmacodynamics, safety and tolerability of apixaban in healthy subjects. Br J Clin Pharmacol. 2013;76(6):908–916. doi: 10.1111/bcp.12114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sandhu RK, Ezekowitz J, Andersson U, Alexander JH, Granger CB, Halvorsen S, et al. The ‘obesity paradox’ in atrial fibrillation: observations from the ARISTOTLE (Apixaban for Reduction in Stroke and Other Thromboembolic Events in Atrial Fibrillation) trial. Eur Heart J. 2016;37(38):2869–2878. doi: 10.1093/eurheartj/ehw124. [DOI] [PubMed] [Google Scholar]
- 48.Ueshima S, Hira D, Fujii R, Kimura Y, Tomitsuka C, Yamane T, et al. Impact of ABCB1, ABCG2, and CYP3A5 polymorphisms on plasma trough concentrations of apixaban in Japanese patients with atrial fibrillation. Pharmacogenet Genomics. 2017;27(9):329–336. doi: 10.1097/FPC.0000000000000294. [DOI] [PubMed] [Google Scholar]
- 49.Ueshima S, Hira D, Kimura Y, Fujii R, Tomitsuka C, Yamane T, et al. Population pharmacokinetics and pharmacogenomics of apixaban in Japanese adult patients with atrial fibrillation. Br J Clin Pharmacol. 2018;84(6):1301–1312. doi: 10.1111/bcp.13561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Goto S, Zhu J, Liu L, Oh BH, Wojdyla DM, Aylward P, et al. Efficacy and safety of apixaban compared with warfarin for stroke prevention in patients with atrial fibrillation from East Asia: a subanalysis of the Apixaban for Reduction in Stroke and Other Thromboembolic Events in Atrial Fibrillation (ARISTOTLE) Trial. Am Heart J. 2014;168(3):303–309. doi: 10.1016/j.ahj.2014.06.005. [DOI] [PubMed] [Google Scholar]
- 51.Chang M, Yu Z, Shenker A, Wang J, Pursley J, Byon W, et al. Effect of renal impairment on the pharmacokinetics, pharmacodynamics, and safety of apixaban. J Clin Pharmacol. 2016;56(5):637–645. doi: 10.1002/jcph.633. [DOI] [PubMed] [Google Scholar]
- 52.Stanifer J, Chertow G, Hohnloser S, Wojdyla D, Garoznik S, Byon W, et al. Apixaban versus warfarin in patients with atrial fibrillation and stage 4 chronic kidney disease. J Am Soc Nephrol. 2017;28(Suppl. 1):218. https://www.asn-online.org/education/kidneyweek/2017/program-abstract.aspx?controlId=2787114. Accessed 6 May 2019.
- 53.Lip GY, Frison L, Halperin JL, Lane DA. Comparative validation of a novel risk score for predicting bleeding risk in anticoagulated patients with atrial fibrillation: the HAS-BLED (Hypertension, Abnormal Renal/Liver Function, Stroke, Bleeding History or Predisposition, Labile INR, Elderly, Drugs/Alcohol Concomitantly) score. J Am Coll Cardiol. 2011;57(2):173–180. doi: 10.1016/j.jacc.2010.09.024. [DOI] [PubMed] [Google Scholar]
- 54.Tirona RG, Kassam Z, Strapp R, Ramu M, Zhu C, Liu M, et al. Apixaban and rosuvastatin pharmacokinetics in nonalcoholic fatty liver disease. Drug Metab Dispos. 2018;46(5):485–492. doi: 10.1124/dmd.117.079624. [DOI] [PubMed] [Google Scholar]
- 55.Perlstein I, Suryawanshi S, Elefant E, Wang Z, Cohen L, Abu Tarif M, et al. Multiple-dose study to evaluate apixaban pharmacokinetics, pharmacodynamics, safety, and tolerability in pediatric subjects with an indwelling central venous catheter. Clin Pharmacol Ther. 2014;95(Suppl.):S61–2.
- 56.Frost C, Song Y, Yu Z, Wang J, Lee LS, Schuster A, et al. The effect of apixaban on the pharmacokinetics of digoxin and atenolol in healthy subjects. Clin Pharmacol. 2017;9:19–28. doi: 10.2147/CPAA.S115687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mao Q, Unadkat JD. Role of the breast cancer resistance protein (BCRP/ABCG2) in drug transport: an update. AAPS J. 2015;17(1):65–82. doi: 10.1208/s12248-014-9668-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Frost CE, Byon W, Song Y, Wang J, Schuster AE, Boyd RA, et al. Effect of ketoconazole and diltiazem on the pharmacokinetics of apixaban, an oral direct factor Xa inhibitor. Br J Clin Pharmacol. 2015;79(5):838–846. doi: 10.1111/bcp.12541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Frost C, Shenker A, Gandhi MD, Pursley J, Barrett YC, Wang J, et al. Evaluation of the effect of naproxen on the pharmacokinetics and pharmacodynamics of apixaban. Br J Clin Pharmacol. 2014;78(4):877–885. doi: 10.1111/bcp.12393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Upreti VV, Song Y, Wang J, Byon W, Boyd RA, Pursley JM, et al. Effect of famotidine on the pharmacokinetics of apixaban, an oral direct factor Xa inhibitor. Clin Pharmacol. 2013;5(1):59–66. doi: 10.2147/CPAA.S41999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Barrett YC, Wang Z, Knabb RM. A novel prothrombin time assay for assessing the anticoagulant activity of oral factor Xa inhibitors. Clin Appl Thromb Hemost. 2013;19(5):522–528. doi: 10.1177/1076029612441859. [DOI] [PubMed] [Google Scholar]
- 62.Song Y, Wang Z, Perlstein I, Wang J, LaCreta F, Frost RJA, et al. Reversal of apixaban anticoagulation by 4-factor prothrombin complex concentrates in healthy subjects: a randomized 3-period crossover study. J Thromb Haemost. 2017;15(11):2125–2137. doi: 10.1111/jth.13815. [DOI] [PubMed] [Google Scholar]
- 63.Yetman RJ, Barrett YC, Wang Z, Adamczyk R, Wang J, Ramacciotti E, et al. Apixaban pharmacodynamic activity in umbilical cord, paediatric, and adult plasma. Thromb Haemost. 2017;117(8):1518–1527. doi: 10.1160/TH16-06-0423. [DOI] [PubMed] [Google Scholar]
- 64.Barrett YC, Wang J, Song Y, Pursley J, Wastall P, Wright R, et al. A randomised assessment of the pharmacokinetic, pharmacodynamic and safety interaction between apixaban and enoxaparin in healthy subjects. Thromb Haemost. 2012;107(5):916–924. doi: 10.1160/TH11-09-0634. [DOI] [PubMed] [Google Scholar]
- 65.Frost C, Yu Z, Wastall P, Nepal S, Moore K, Shenker A, et al. Tolerability, pharmacokinetics, and pharmacodynamics of apixaban, aspirin, and clopidogrel following coadministration in healthy subjects. Clin Pharmacol Drug Dev. 2014;3(Suppl. 1):29–30. [Google Scholar]
- 66.AbuTariff M, Bui A, Pursley J, He J, La Creta F, Frost C. Lack of pharmacokinetic interaction between apixaban and prasugrel in healthy subjects. Clin Pharmacol Drug Dev. 2014;3(Suppl. 1):30–31. [Google Scholar]
- 67.Leil TA, Feng Y, Zhang L, Paccaly A, Mohan P, Pfister M. Quantification of apixaban’s therapeutic utility in prevention of venous thromboembolism: selection of phase III trial dose. Clin Pharmacol Ther. 2010;88(3):375–382. doi: 10.1038/clpt.2010.106. [DOI] [PubMed] [Google Scholar]
- 68.Barrett YC, Wang Z, Frost C, Shenker A. Clinical laboratory measurement of direct factor Xa inhibitors: anti-Xa assay is preferable to prothrombin time assay. Thromb Haemost. 2010;104(6):1263–1271. doi: 10.1160/TH10-05-0328. [DOI] [PubMed] [Google Scholar]
- 69.Gulilat M, Tang A, Gryn SE, Leong-Sit P, Skanes AC, Alfonsi JE, et al. Interpatient variation in rivaroxaban and apixaban plasma concentrations in routine care. Can J Cardiol. 2017;33(8):1036–1043. doi: 10.1016/j.cjca.2017.04.008. [DOI] [PubMed] [Google Scholar]
- 70.Portola Pharmaceuticals. Andexxa [prescribing information]. 2018. https://www.andexxa.com/prescribing-information/. Accessed 8 Apr 2019.
- 71.Siegal DM, Curnutte JT, Connolly SJ, Lu G, Conley PB, Wiens BL, et al. Andexanet alfa for the reversal of factor Xa inhibitor activity. N Engl J Med. 2015;373(25):2413–2424. doi: 10.1056/NEJMoa1510991. [DOI] [PubMed] [Google Scholar]
- 72.Crowther M, Kitt M, Lorenz T, Mathur V, Lu G, Hutchaleelaha A, et al. A phase 2 randomized, double-blind, placebo-controlled trial of PRT064445, a novel, universal antidote for direct and indirect factor Xa inhibitors [abstract] J Thromb Haemost. 2013;11(Suppl. 2):30. [Google Scholar]
- 73.Connolly SJ, Milling TJ, Jr, Eikelboom JW, Gibson CM, Curnutte JT, Gold A, et al. Andexanet alfa for acute major bleeding associated with factor Xa inhibitors. N Engl J Med. 2016;375(12):1131–1141. doi: 10.1056/NEJMoa1607887. [DOI] [PMC free article] [PubMed] [Google Scholar]