

Abstract
Background
Phenylketonuria results from a deficiency of the enzyme phenylalanine hydroxylase. Dietary restriction of phenylalanine keeps blood phenylalanine concentration low. Most natural foods are excluded from diet and supplements are used to supply other nutrients. Recent publications report a decrease in blood phenylalanine concentration in some patients treated with sapropterin dihydrochloride. We examined the evidence for the use of sapropterin dihydrochloride to treat phenylketonuria. This is an update of a previously published Cochrane Review.
Objectives
To assess the safety and efficacy of sapropterin dihydrochloride in lowering blood phenylalanine concentration in people with phenylketonuria.
Search methods
We identified relevant trials from the Group's Inborn Errors of Metabolism Trials Register. Date of last search: 11 August 2014.
We also searched ClinicalTrials.gov and Current controlled trials. Last search: 4 September 2014
We contacted the manufacturers of the drug (BioMarin Pharmaceutical Inc.) for information regarding any unpublished trials.
Selection criteria
Randomized controlled trials comparing sapropterin with no supplementation or placebo in people with phenylketonuria due to phenylalanine hydroxylase deficiency.
Data collection and analysis
Two authors independently assessed trials and extracted outcome data.
Main results
Two placebo‐controlled trials were included. One trial administered 10 mg/kg/day sapropterin in 89 children and adults with phenylketonuria whose diets were not restricted and who had previously responded to saproterin.This trial measured change in blood phenylalanine concentration. The second trial screened 90 children (4 to 12 years) with phenylketonuria whose diet was restricted, for responsiveness to sapropterin. Forty‐six responders entered the placebo‐controlled part of the trial and received 20 mg/kg/day sapropterin. This trial measured change in both phenylalanine concentration and protein tolerance. Both trials reported adverse events. The trials showed an overall low risk of bias; but both are Biomarin‐sponsored. One trial showed a significant lowering in blood phenylalanine concentration in the sapropterin group (10 mg/kg/day), mean difference ‐238.80 μmol/L (95% confidence interval ‐343.09 to ‐134.51); a second trial (20 mg/kg/day sapropterin) showed a non‐significant difference, mean difference ‐51.90 μmol/L (95% confidence interval ‐197.27 to 93.47). The second trial also reported a significant increase in phenylalanine tolerance, mean difference18.00 mg/kg/day (95% confidence interval 12.28 to 23.72) in the 20 mg/kg/day sapropterin group.
Authors' conclusions
There is evidence of short‐term benefit from using sapropterin in some people with sapropterin‐responsive forms of phenylketonuria; blood phenylalanine concentration is lowered and protein tolerance increased. There are no serious adverse events associated with using sapropterin in the short term.
There is no evidence on the long‐term effects of sapropterin and no clear evidence of effectiveness in severe phenylketonuria.
Keywords: Adult; Child; Child, Preschool; Humans; Biopterin; Biopterin/administration & dosage; Biopterin/adverse effects; Biopterin/analogs & derivatives; Phenylalanine; Phenylalanine/blood; Phenylketonurias; Phenylketonurias/blood; Phenylketonurias/drug therapy; Randomized Controlled Trials as Topic
Plain language summary
The use of sapropterin to lower phenylalanine concentration in blood in people with phenylketonuria.
Phenylketonuria occurs due to an inherited deficiency of the enzyme phenylalanine hydroxylase. If untreated it causes an excessive accumulation of the amino acid phenylalanine in the body which prevents normal brain development. The established treatment for phenylketonuria consists of dietary restriction of natural protein but with prescribed phenylalanine‐free amino acid, mineral and vitamin supplements. With this treatment the long‐term outcome for people with phenylketonuria is excellent but the diet is onerous. Sapropterin dihydrochloride, the cofactor for phenylalanine hydroxylase, could lower phenylalanine concentration significantly in phenylketonuria and might allow a relaxation of dietary restrictions. The review identified two trials of sapropterin dihydrochloride; one in children and adults with no restricted diet and one in just children whose diet was restricted. The trials used different doses of sapropterin dihydrochloride (10 mg/kg/day and 20 mg/kg/day). We could not combine any data due to different formats of presentation. We found evidence to show that some people with mild or moderate phenylketonuria can benefit from the use of sapropterin dihydrochloride in the short term; the concentration of blood phenylalanine was lowered after treatment in both trials. The trial with the higher dose also measured the outcome change in protein tolerance. It reported an increase in protein tolerance in response to sapropterin. There were no adverse effects associated with the use of sapropterin dihydrochloride in the short term. We found no evidence on the effects of long‐term treatment. We could not draw any conclusions on its benefits in severe phenylketonuria.
Background
Please refer to the glossary for definition of technical terms (Appendix 1).
Description of the condition
Phenylketonuria (PKU) is an autosomal recessive mendelian disorder (OMIM 261600) characterized by an increase of phenylalanine in blood and body fluids (Scriver 2001).
Phenylketonuria manifests itself as increased phenylalanine concentration in blood (hyperphenylalaninemia (HPA)). Ninety‐eight per cent of HPA is due to mutations in the gene coding for phenylalanine hydroxylase (PAH) enzyme (E.C.1.14.16.1). Two percent of HPA is due to a defect in tetrahydrobiopterin (BH4) metabolism which is an essential co‐factor for the activity of PAH (Baulny 2007).
The incidence of PKU due to mutations in the PAH gene is about 1 in 10,000 in individuals of European and Oriental Asian origin (Scriver 2007).
On the basis of blood phenylalanine concentration while on a normal protein intake, HPA due to PAH deficiency can be classified into classic PKU (phenylalanine > 1200 μmol/L); mild PKU (phenylalanine 600 to 1200 μmol/L); and mild HPA (phenylalanine < 600 μmol/L but more than the upper reference limit) (Williams 2008).
In untreated PKU, the infant appears normal for the first few months of life, but later on shows features of progressive encephalopathy. Other features include growth failure, microcephaly, seizures, intellectual impairment, eczema, lightly pigmented skin and musty odour (Scriver 2001; Baulny 2007; Williams 2008).
Metabolic derangement in PKU
Phenylalanine is an essential amino acid provided by protein in the diet. A small amount of this is used for protein synthesis and the rest is hydroxylated to tyrosine and further metabolized. The phenylalanine hydroxylation requires PAH, dihydropteridine reductase (DHPR) and BH4. When conversion to tyrosine is blocked, phenylalanine accumulates in the body fluids (Scriver 2001). Above a threshold level (600 μmol) the high phenylalanine level is thought to result in neurotoxicity and lead to mental retardation (Scriver 2001). Threshold for damage is generally considered to be between 360 to 600 μmol/L.
Description of the intervention
The aim of the treatment for PKU is to maintain plasma phenylalanine within the recommended range for the age, which prevents neurological damage. The mainstay of treatment for PKU is a phenylalanine‐restricted diet. The treatment is to be instituted by 20 days of age, should be aggressive with regular monitoring of blood phenylalanine levels. Ideally the strict dietary restriction should continue into adult life (MRC (UK) 1993; Cockburn 1996; NIH Consensus Development Panel 2001).
The phenylalanine‐restricted diet is designed in a way that allows a decrease in blood phenylalanine concentration and provides sufficient tyrosine (now an essential amino acid) and other nutrients required for optimal growth and development of the child (Baulny 2007). This involves a measured allowance of natural protein in the diet to provide the phenylalanine requirement for growth and health as guided by routine and regular blood phenylalanine monitoring. All high protein foodstuffs such as meat are excluded. Foods such as some fruits and vegetables which contain less natural protein are allowed in measured amounts while others which have negligible amounts of natural protein are allowed 'freely'. Commercially available supplements of amino acids that lack phenylalanine are to be taken on a daily basis (Baulny 2007; Williams 2008).
A strict compliance with such a diet has been shown to be compatible with better cognitive and motor function, behavioral temperament and executive function. An early termination of treatment affects IQ scores and there may be abnormal neurological features later in life (Scriver 2001). Non‐compliance in teenagers and adults showed subtle cognitive impairments relative to controls (Channon 2007) and was associated with an increase in the rate of eczema, asthma, mental disorders, headache, hyperactivity and hypoactivity (Koch 2002).
The diet itself is very restrictive in nature, the supplements used have an unpleasant taste and odour and there is always the risk of nutritional deficiencies. Some progress has been achieved in recent years in these aspects with newer and better formulations (Giovanni 2007). In spite of these advances, there is still potential for severe compromise of quality of life (Wappner 1999; Scriver 2007). As a result of this, by late adolescence and adulthood at least 75% of people with PKU are non‐compliant (Koch 2002). Therefore, other methods of treatment have been actively sought.
BH4 supplementation
Sapropterin dihydrochloride is an orally active synthetic form of BH4. There are several reports in the literature of people with PKU who responded to pharmacological doses of BH4 loading with a reduction in blood phenylalanine levels. All these individuals had mutations in the PAH gene. Defects in BH4 synthesis or regeneration (primary BH4 defect) had been ruled out (Kure 1999; Lindner 2001; Nuoffer 2001; Spaapen 2001; Trefz 2001; Matalon 2002; Muntau 2002; Steinfeld 2002; Shintaku 2004).
Individuals responsive to BH4 are identified initially by performing a BH4 loading test. A positive response to BH4 is arbitrarily considered as a decrease of 30% or more of blood phenylalanine concentration 24 hours after administration of BH4 (Michals‐Matalon 2007). The protocol for the BH4 loading test is variable (Blau 2004; Shintaku 2004).
How the intervention might work
Prevalence of BH4 responsiveness has been variable in different studies (Bernegger 2002; Muntau 2002; Fiege 2005; Fiori 2005; Matalon 2005; Boveda 2007; Fiege 2007). There was variation in the intensity of response which was independent of the severity of the PKU, dose of BH4 used in the loading test, duration of the test and genotype. Even people with the same genotype showed differences in the intensity of response (Lindner 2001; Steinfeld 2003; Leuzzi 2006). There are a few reports of long‐term use of BH4 that show that BH4 allows relaxation of the dietary restrictions in people with PKU without any adverse effects (Cerone 2004; Shintaku 2004; Steinfeld 2004; Belanger‐Quintana 2005; Fiori 2005; Burlina 2009). Some authors have reported beneficial effects in a few people with severe classic PKU (Hennermann 2005; Matalon 2005) .Most of the individuals who respond have mild to moderate PKU. All the people with PKU who respond positively to BH4 have at least some residual PAH enzyme activity. Sapropterin, by enhancing the activity of residual PAH enzyme, may increase tolerance to phenylalanine and allow a less restrictive diet (Blau 2004; Erlandsen 2004).
Why it is important to do this review
Following the restrictive diet for PKU is beneficial but compliance after childhood is difficult to achieve as it can severely compromise the quality of life.
Sapropterin dihydrochloride administration can potentially allow a relaxation of diet or even permit discontinuation of dietary treatment in some individuals. Even in those who respond suboptimally to its administration may allow a partial relaxation of dietary restrictions making it easier to cope with a life‐long regimen. However, most of the individuals who respond to BH4 have mild to moderate PKU and some of them may not need aggressive treatment. It is therefore very important to assess critically the safety and efficacy of sapropterin dihydrochloride in a clinical setting. This review is an update of a previously published review (Somaraju 2010; Somaraju 2012).
Objectives
To assess the effectiveness and safety of sapropterin dihydrochloride in decreasing blood phenylalanine levels in people with PKU.
Methods
Criteria for considering studies for this review
Types of studies
Randomized controlled trials, published and unpublished.
Types of participants
Children and adults with PKU due to PAH deficiency, who are responsive to sapropterin dihydrochloride. Individuals with PKU due to primary defect in BH4 metabolism will be excluded.
Types of interventions
Oral supplementation of sapropterin (in any dose, frequency or duration) compared with no supplementation or placebo. This intervention can be used either in combination with, or instead of, a phenylalanine‐restricted diet.
Types of outcome measures
Primary outcomes
Change in blood phenylalanine concentration
Secondary outcomes
Adverse events which may be associated with sapropterin
Validated quality of life measures (e.g. Profile of Quality of Life in Chronically Ill (PLC))
Validated measures of Intelligence and neuro‐psychometric performance (e.g. Wechsler Intelligence Scales)
Measures of nutritional status and growth
Change in protein (phenylalanine) tolerance (assessed by giving a standard amount of protein or phenylalanine and measuring the level of blood phenylalanine; increase in tolerance is defined when the protein or phenylalanine intake does not increase the blood phenylalanine level)
Search methods for identification of studies
Electronic searches
We identified relevant trials from the Group's Inborn Errors of Metabolism Trials Register using the terms: kuvan OR ohenoptin OR sapropterin. The Inborn Errors of Metabolism Trials Register is compiled from electronic searches of the Cochrane Central Register of Controlled Trials (CENTRAL) (updated each new issue of The Cochrane Library), weekly searches of MEDLINE and the prospective handsearching of one journal ‐ Journal of Inherited Metabolic Disease. Unpublished work was identified by searching through the abstract books of the Society for the Study of Inborn Errors of Metabolism conference and the SHS Inborn Error Review Series. For full details of all searching activities for the register, please see the relevant section of the Cystic Fibrosis and Genetic Disorders Group Module.
Date of the latest search of the Group's Inborn Errors of Metabolism Register: 11 August 2014.
Additionally we undertook searches of the following registers on 4 September 2014 (see Appendices):
ClinicalTrials.gov
Current controlled trials
Searching other resources
We contacted the manufacturers of the drug (BioMarin Pharmaceutical Inc. and Merck KgaA) for information regarding any unpublished trials.
Data collection and analysis
Selection of studies
Two authors, US and MM, assessed the trials independently for inclusion in the review. We planned to resolve any disagreements that may arise through discussion. There were no disagreements between the authors.
Data extraction and management
We independently extracted the data from eligible trials using a trial selection and data extraction form modified for this review.
We planned to group outcome data into those measured at two, four and six weeks, monthly up to one year, and every three months thereafter. If data were reported at other time periods we planned to include them also. We also planned to contact authors for possible measurements of outcome data at other time periods and if available include these also in the analysis.
The data was reported at three weeks and six weeks for change in phenylalanine concentration and at ten weeks for change in protein (phenylalanine) tolerance. We contacted the authors for the outcome data measured at other time points but have not received any reply.
Assessment of risk of bias in included studies
We assessed the risk of bias of the included trials using the domain‐based evaluation as described in the Cochrane Handbook for Systematic Reviews of Intervention (Higgins 2011)
We assessed the following domains as having either a low, unclear or high risk of bias:
randomisation;
concealment of allocation;
blinding of participants, personnel and outcome assessors;
incomplete outcome data;
selective outcome reporting.
We also assessed the trials for other potential sources of bias.
Measures of treatment effect
For binary outcomes we measured the treatment effect as risk ratios (RR) with 95% confidence intervals (95% CIs). For continuous outcomes with outcome measurements on the same scale, we presented the results as mean differences (MDs) with 95% CIs. Where the continuous outcomes were measured using different scales, we used the standardised mean difference (SMD).
Unit of analysis issues
We also planned to include results from eligible cross‐over trials using methods recommended by Elbourne (Elbourne 2002). In order to allow an intention‐to‐treat analysis we planned to seek data on the number of participants by allocated treatment group, irrespective of compliance and whether or not the participant was later thought to be ineligible or otherwise excluded from treatment. However there were no eligible cross‐over trials to be included in the review.
Dealing with missing data
In order to do a more complete review we contacted the drug manufacturers (Biomarin Pharmaceutical Inc.) of the included trials for data; as yet we have not received any reply.
For the outcome 'Change in blood phenylalanine concentration', although data was measured at more frequent intervals in one trial it was only reported at six weeks as end‐point data (Levy 2007). In the second trial, we could not use the data reported at weekly intervals for three weeks, as they only gave the details in sapropterin group (Trefz 2009). Also, both the trials measured nutritional status but this outcome was not reported by either.
Assessment of heterogeneity
We planned to quantify the impact of statistical heterogeneity in the meta‐analysis using a measure (I2) of the degree of inconsistency in the trials' results. This measure describes the percentage of total variation across trials that is due to heterogeneity rather than chance. The values of I2 lie between 0% and 100%, and a simplified categorization of heterogeneity that we plan to use is as follows (Higgins 2003):
0% to 40% : might not be important;
30% to 60% : may represent moderate heterogeneity;
50% to 90% : may represent substantial heterogeneity;
75% to 100% : considerable heterogeneity.
Since the two included trials reported data at different time points, we could not combine any data.
Assessment of reporting biases
If we are able to include 10 or more trials in a future update, we plan to use a funnel plot to assess whether the review is subject to publication bias. If asymmetry is detected we also planned to assess other possible causes such as selection bias, reporting bias, true heterogeneity and artefact. Since the review included only two trials we could not assess whether the review was subject to publication bias.
In order to assess outcome reporting bias, we compared the protocols of the trials (available via ClinicalTrials.gov) to the published reports.
Data synthesis
As we did not identify any significant heterogeneity (we were not able to combine any data), we have analysed data using a fixed‐effect analysis.
Subgroup analysis and investigation of heterogeneity
In future, if we find sources of heterogeneity and if we are able to include 10 or more trials, we plan to conduct meta‐analysis by subgroups, and stratify participants according to:
Severity of PKU at baseline (classic PKU: phenylalanine > 1200 μmol/L ; mild PKU: phenylalanine 600 to 1200 μmol/L; and mild HPA: phenylalanine < 600 μmol/L but more than the upper reference limit);
Dosage of sapropterin dihydrochloride used (10 mg/kg or 20 mg/kg).
Sensitivity analysis
We planned to perform sensitivity analyses to determine the impact of risk of bias in trials on outcome, including and excluding trials with a high risk of bias regarding methods of treatment allocation. If there were any eligible crossover trials, we planned to conduct sensitivity analyses including and excluding these.
Results
Description of studies
Results of the search
In the original review, searches of the Group's Trials Register identified one trial (Levy 2007). Searches of the ClinicalTrials.gov registry identified a further trial (Trefz 2009), for which a full published report was supplied by the BioMarin Pharmaceutical Inc. Both the trials were eligible for inclusion in the review.
For the 2012 update of this review, no trials were identified from the search of the Group's Trials Register. However, the search of ClinicalTrials.gov and Current controlled trials identified four new and potentially relevant trials (Gropmann 2011; Nwose 2008; NCT01376908; NCT01114737). All four trials were ongoing. One was clearly not eligible for inclusion (Nwose 2008) and the remaining three were potentially eligible for inclusion in a future version and their progress has been monitored (Gropmann 2011; NCT01376908; NCT01114737).
For the 2014 update of the review, a search of the Cochrane Cystic Fibrosis and Genetic Disorders Group's Inborn Errors of Metabolism Trials Register identified two trials (Gramer 2009; Utz 2012). Both the trials were not eligible for inclusion. The search of ClinicalTrials.gov identified one new ongoing trial which is potentially eligible for inclusion in future (NCT01977820). Two of the trials identified in the previous search were completed (NCT01376908; NCT01114737). For both of the trials, according to the sponsors Merck KgaA (NCT01376908) and Biomarin Pharmaceutical Inc. (NCT01114737), full manuscripts will be published in a few months. These two trials are listed in the 'Studies awaiting classification' section and their progress will be monitored for possible inclusion in future updates of the review. One trial identified in the previous 2012 search was terminated due to poor recruitment and is listed in the 'Excluded studies' section (Gropmann 2011). The search of 'Current controlled trials' did not identify any trials.
Included studies
Trial Design
Both trials were multicenter, randomized, double‐blind, placebo‐controlled trials (Levy 2007; Trefz 2009). One trial was six weeks in duration (Levy 2007) and the second lasted 10 weeks (Trefz 2009).
Both trials consisted of two parts, the first part being a 'run‐in' lasting eight days in each case where participants received sapropterin at a dose of 10 mg/kg/d (Levy 2007) and 20 mg/kg/d (Trefz 2009). The Levy trial included those participants who experienced a reduction of 30% or more in blood phenylalanine concentration (Levy 2007). In Trefz trial, those participants who had at least a 30% reduction in blood phenylalanine and had a blood phenylalanine ≤ 300 μmol/L were included in Part 2 (Trefz 2009). The Levy trial screened participants six weeks prior to start of trial and randomized 89 participants who were responsive to sapropterin in Part 2 of the trial (Levy 2007). Trefz screened 90 individuals and randomized 46 participants who were sensitive into the sapropterin and control groups in a 3:1 ratio (Trefz 2009).
Participants
There were 89 participants in one trial (who had previously been involved in a Phase 1 screening study) (Levy 2007) and in the second trial, of the 90 screened for responsiveness to sapropterin, 46 were eligible for inclusion (Trefz 2009). One trial enrolled adults and children eight years or older, with PKU due to PAH deficiency (Levy 2007). The Trefz trial only enrolled children aged between 8 and 12 years old (Trefz 2009).
The Levy trial included participants with a blood phenylalanine of over 450 μmol/L (Levy 2007), where as the Trefz trial included participants with a phenylalanine tolerance of less than 1000 mg/d, and blood phenylalanine under 480 μmol/L (Trefz 2009).
In the Levy trial, all the participants had abandoned or relaxed a strict low phenylalanine diet (Levy 2007), but in the Trefz trial they were described as being under a phe‐restricted diet (Trefz 2009).
Interventions
In both included trials, the participants were randomized into sapropterin and control groups in the second part of the trial. Levy employed a dose of sapropterin 10 mg/kg/day and Trefz used a dose of sapropterin 20 mg/kg/day (Trefz 2009); both were compared to placebo. In the Trefz trial, participants received phenylalanine supplements at two weekly intervals, starting from week three onwards until the end of the trial period (Trefz 2009).
Outcomes
The Levy trial reported 'change in blood phenylalanine concentration' (Levy 2007) and the Trefz trial reported both change in blood phenylalanine concentration and in protein (phenylalanine) tolerance (Trefz 2009). Both trials reported adverse events (Levy 2007; Trefz 2009).
Excluded studies
Four trials were excluded (Gramer 2009; Gropmann 2011; Nwose 2008; Utz 2012). One trial did not include participants and outcome measures relevant to the review (Nwose 2008); one study was not an RCT (Gramer 2009); one trial (a pharmacogenetic test of responsiveness to sapropterin dihydrochloride) was excluded as the outcomes were not relevant to the review (Utz 2012); and a final trial, which was likely to be eligible for inclusion, was terminated prematurely due to poor recruitment and was excluded (Gropmann 2011).
Risk of bias in included studies
Allocation
For both the included trials, randomisation lists using block sizes of four were generated by a computer program and an interactive voice response system was used to allocate participants into sapropterin and control groups. The block sizes were not revealed to the trial sponsors and trial investigators until the trials were completed. So we judged the domains of allocation and concealment of allocation as having a low risk of bias for both the trials.
Blinding
Both the trials were described as double‐blinded and gave details of blinding (sapropterin and placebo tablets were identical in taste and appearance). The participants, personnel and outcome assessors were blinded. Therefore we judged there to be a low risk of bias for blinding for both the included trials.
Incomplete outcome data
In the Levy trial there was one withdrawal from the trial in the sapropterin group before closing due to an inability to comply with the trial course. The data were not included in the analysis as the participant did not receive even one dose of trial drug. There was another withdrawal from the control group due to non‐compliance with specified dosing, but the data was included in the analysis.
In the Trefz trial, one participant randomized to the sapropterin group did not return for week 0 visit. The data were not included in the analysis.
Since the reasons for withdrawal were clearly described and as withdrawals were unlikely to be related to outcomes, we judged that both trials had a low risk of bias due to incomplete outcome data.
Selective reporting
We compared the protocols of both the trials, available at the ClinicalTrials.gov web site, and also the 'Methods' section with the results reported in the final paper. All the detailed outcomes in the protocol were reported in the published reports; however, in the Levy trial they measured data at several time points (weeks zero, one, two, four, and six) but only reported them at six weeks as end point data. For the Trefz trial, we could not include the data reported at weekly intervals as they did not include details of the control group. Hence we judged there to be a high risk of bias for this domain for both trials.
Other potential sources of bias
BioMarin Pharmaceutical Inc. sponsored both of the trials included in this review.
Effects of interventions
As the two trials included in the review reported data at different time points, we could not combine the data for analysis. We contacted the lead authors of the trials for additional data. For the Levy trial we were advised to contact the Biomarin pharmaceutical Inc. as they owned the data. We have contacted the BioMarin Pharmaceutical Inc. but have not received any reply (December 2009). We are also waiting for a reply from the author of the Trefz trial (December 2009). Sensitivity analyses, subgroup analyses or assessment of heterogeneity could not be undertaken in this version of the review as the data could not be combined.
Primary outcome
1. Change in blood phenylalanine concentration
This outcome was reported by both the included trials. Data could not be combined as they were at different time points. The Trefz trial measured this outcome weekly for three weeks. There was a non‐significant decrease in phenylalanine concentration from baseline in sapropterin group when compared with control group at three weeks, mean difference (MD) ‐51.90 μmol/L (95% CI ‐197.27 to 93.47) (Analysis 1.1) (Trefz 2009). Levy measured this outcome at weeks one, two, four and six, but only reported at six weeks. There was a significant decrease in blood phenylalanine concentration from baseline in sapropterin group when compared with control group MD ‐238.80 μmol/L (95% CI ‐343.09 to ‐134.51) (Analysis 1.1) (Levy 2007).
1.1. Analysis.
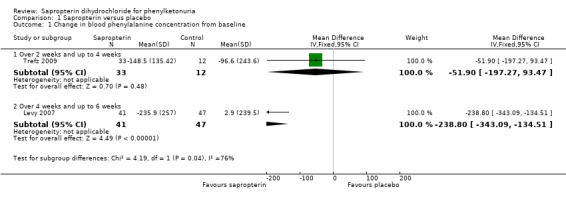
Comparison 1 Sapropterin versus placebo, Outcome 1 Change in blood phenylalanine concentration from baseline.
Both the trials also reported a significant difference in the absolute values of blood phenylalanine concentrations between the treatment groups. The MD in blood phenylalanine concentration between sapropterin and control groups at three weeks was ‐135.20 μmol/L (95% CI ‐187.92 to ‐82.48) (Analysis 1.2) (Trefz 2009). The MD in blood phenylalanine concentration between the treatment groups was ‐245.00 μmol/L (95% CI ‐349.47 to ‐140.53) at six weeks (Analysis 1.2) (Levy 2007).
1.2. Analysis.
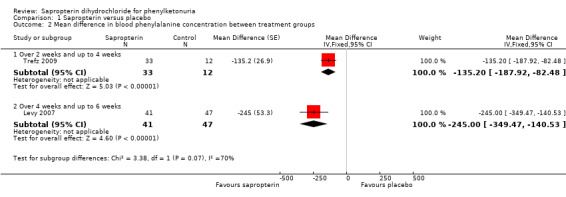
Comparison 1 Sapropterin versus placebo, Outcome 2 Mean difference in blood phenylalanine concentration between treatment groups.
Secondary outcomes
1. Adverse events which may be associated with sapropterin
We were able to combine data for several adverse events reported in both the trials like upper respiratory tract infection, headache, vomiting, abdominal pain, diarrhoea, pyrexia and bone pain (Analysis 1.3) (Levy 2007; Trefz 2009). There was no significant difference between the groups for these events. No serious adverse events were reported by either trial.
1.3. Analysis.
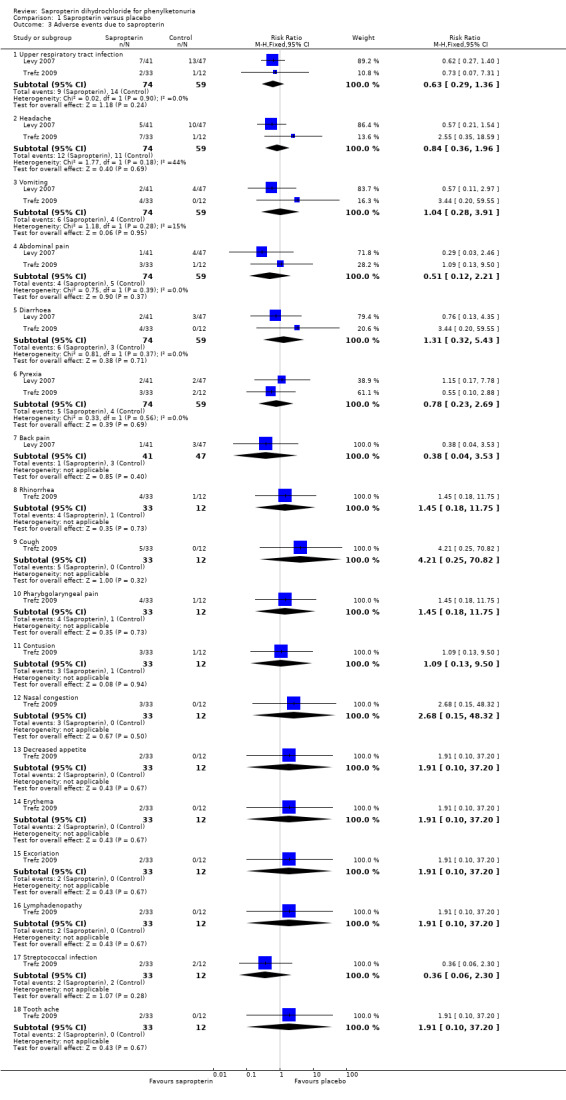
Comparison 1 Sapropterin versus placebo, Outcome 3 Adverse events due to sapropterin.
2. Validated quality of life measures
This outcome was not measured in either trial included in the review (Levy 2007; Trefz 2009).
3. Validated measures of Intelligence and neuro‐psychometric performance
This outcome was not measured in either trial included in the review (Levy 2007; Trefz 2009).
4. Measures of nutritional status and growth
This outcome was not reported in either trial included in the review (Levy 2007; Trefz 2009).
5. Change in protein (phenylalanine) tolerance
This outcome was measured by the Trefz trial (Trefz 2009). There was a significant increase in phenylalanine tolerance in the sapropterin group. The MD for the total phenylalanine supplement tolerated between the groups was 18.00 mg/Kg/day (95% CI 12.28 to 23.72) (Analysis 1.4) (Trefz 2009). The total phenylalanine intake in sapropterin group changed significantly from MD 0.50 mg/kg/day (95% CI 5.67 to 4.67) at baseline to 20.30 mg/kg/day (95% CI 9.29 to 31.31) at the end of 10 weeks (Analysis 1.5).The MD between the groups for change in phenylalanine tolerance was 17.70 mg/kg/day (95% CI 8.88 to 26.52) (Analysis 1.6) (Trefz 2009).
1.4. Analysis.
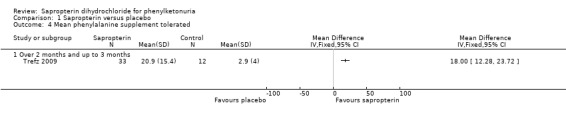
Comparison 1 Sapropterin versus placebo, Outcome 4 Mean phenylalanine supplement tolerated.
1.5. Analysis.
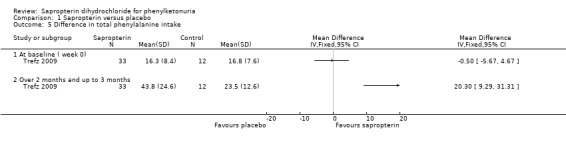
Comparison 1 Sapropterin versus placebo, Outcome 5 Difference in total phenylalanine intake.
1.6. Analysis.

Comparison 1 Sapropterin versus placebo, Outcome 6 Change in phenylalanine tolerance.
Discussion
It was reported by several authors that sapropterin dihydrochloride could reduce the blood phenylalanine concentration in some individuals with PKU due to PAH deficiency (Bernegger 2002; Kure 1999) It was proposed that using sapropterin would reduce the dietary restrictions in this group of people (Burton 2007; Steinfeld 2004). In this review we attempted to study the effectiveness of sapropterin to lower the blood phenylalanine concentration, safety of its use and other potential benefits such as improvement in nutritional status, cognitive performance, quality of life and protein tolerance. We identified two randomized controlled trials, one measuring the change in blood phenylalanine concentration (Levy 2007), and the other the change in blood phenylalanine concentration and in phenylalanine tolerance (Trefz 2009). Both trials also reported on adverse events (Levy 2007; Trefz 2009).
Summary of main results
The review identifies that there is benefit to be derived from using sapropterin in those individuals with PKU due to PAH deficiency who are responsive to sapropterin. Sapropterin significantly lowered the blood phenylalanine concentration with or without dietary restriction (Levy 2007; Trefz 2009) and improved phenylalanine tolerance (Trefz 2009). There are no serious adverse events associated with the treatment in the short term.
Overall completeness and applicability of evidence
The two trials included in the review provided data for the assessment of effectiveness and safety of sapropterin in the short term (up to 10 weeks). There were no data on the effect of treatment on nutritional status, intelligence, and quality of life. Conclusions on long‐term benefits cannot be drawn due to the short‐term nature of the included trials. The relative effectiveness of treatment with sapropterin in individuals with difference in severity of disease could not be assessed as the included trials did not provide sufficient data about the disease status in participants.
Quality of the evidence
This review includes only two trials of short duration which precludes any conclusions on the long‐term effects of sapropterin. The lack of information about the severity of disease in included participants limits the generalised application of findings to all individuals with PKU who are responsive to sapropterin, irrespective of the disease status.
Potential biases in the review process
Biomarin Pharmaceuticals Inc. sponsored both the trials included in the review. The two review authors' independent assessment of included trials and data extraction minimised potential for additional bias. Neither of the review authors have any conflict of interest.
Agreements and disagreements with other studies or reviews
The findings of the review are in agreement with earlier non‐randomized trials that sapropterin would lower blood phenylalanine concentration and improve protein tolerance in individuals with PKU due to PAH deficiency in mild to moderate disease (Lindner 2001; Steinfeld 2003; Leuzzi 2006). We found no evidence to conclude on the long‐term benefits (Cerone 2004; Shintaku 2004; Steinfeld 2004; Belanger‐Quintana 2005; Fiori 2005; Burlina 2009) or on the benefits in people with severe PKU (Hennermann 2005; Matalon 2005).
Authors' conclusions
Implications for practice.
There is evidence to suggest that treatment with sapropterin lowers blood phenylalanine concentration and improves protein tolerance in individuals with PKU who are responsive to sapropterin. However, there is lack of data on the effectiveness of the treatment on intelligence, growth and quality of life and in people with severe PKU. Treatment with sapropterin is not associated with any serious adverse events in the short term.
Implications for research.
There is a need for randomized controlled trials to study the long‐term effectiveness and safety of sapropterin and its effect on outcomes like intelligence, nutritional status and growth and quality of life. There is also a necessity to investigate whether treatment with sapropterin has any role in the management of people with severe PKU.
What's new
| Date | Event | Description |
|---|---|---|
| 5 March 2015 | New search has been performed | A search of the Cochrane Cystic Fibrosis and Genetic Disorders Group's Inborn Errors of Metabolism Trials Register identified two trials (Gramer 2009; Utz 2012). Both the trials were not eligible for inclusion. The search of ClinicalTrials.gov identified one new ongoing trial which is potentially eligible for inclusion in future (NCT01977820). Two of the trials identified in the previous search were completed (NCT01376908; NCT01114737). For both of the trials, according to the sponsors Merck KgaA (NCT01376908) and Biomarin Pharmaceutical Inc. (NCT01114737), full manuscripts will be published later in 2015. These two trials are listed in the 'Studies awaiting classification' section and their progress will be monitored for possible inclusion in future updates of the review. One trial identified in the previous 2012 search was terminated due to poor recruitment and is listed in the 'Excluded studies' section (Gropmann 2011). The search of 'Current controlled trials' did not identify any trials. |
| 5 March 2015 | New citation required but conclusions have not changed | Minor changes were made throughout the review. One new, potentially relevant, ongoing trial is listed in the 'Ongoing studies' section of the review (NCT01977820). Two trials, currently listed in 'Studies awaiting classification' section are expected be completed and published later in 2015. These will be assessed for inclusion in a future update of the review (NCT01376908; NCT01114737). |
History
Protocol first published: Issue 4, 2009 Review first published: Issue 6, 2010
| Date | Event | Description |
|---|---|---|
| 24 March 2014 | Amended | Contact details updated. |
| 19 October 2012 | New citation required but conclusions have not changed | Minor amendments have been made throughout the text for this updated version of the review. |
| 29 June 2012 | New search has been performed | A search of the Cochrane Cystic Fibrosis and Genetic Disorders Group's Haemoglobinopathies Trials Register did not identify any trials potentially eligible for inclusion in the review. A search of ClinicalTrials.gov and Current controlled trials identified two new trials, one which may be eligible for inclusion at a future date but is currently ongoing (Prasad 2010a) and one which is not eligible for inclusion and now listed in Excluded studies (Nwose 2008). |
Acknowledgements
We would like to thank Nikki Jahnke for her support during the writing of this review.
Appendices
Appendix 1. Glossary
| Technical term | Explanation |
| confidence interval | A measure of the uncertainty around the main finding of a statistical analysis. Estimates of unknown quantities, such as the odds ratio comparing an experimental intervention with a control, are usually presented as a point estimate and a 95% confidence interval. This means that if someone were to keep repeating a study in other samples from the same population, 95% of the confidence intervals from those studies would contain the true value of the unknown quantity. Alternatives to 95%, such as 90% and 99% confidence intervals, are sometimes used. Wider intervals indicate lower precision; narrow intervals, greater precision. |
| correlation coefficient | A correlation coefficient can range from ‐1 for perfect negative correlation, to +1 for perfect positive correlation (with perfect meaning that all the points lie on a straight line). A correlation coefficient of 0 means that there is no linear relationship between the variables. |
| cross‐over trial | A type of clinical trial comparing two or more interventions, in which the participants , upon completion of the course of one treatment, are switched to another. |
| encephalopathy | A disease of the brain; especially one involving alterations of brain structure |
| heterogeneity | Used in a general sense to describe the variation in, or diversity of participants, interventions and measurement of outcomes across a set of studies, or the variation in internal validity of those studies. |
| intention‐to‐treat analysis | A strategy for analysing data from a randomised controlled trial. All participants are included in the arm to which they were allocated, whether or not they received (or completed) the intervention given to that arm. Intention‐to‐treat analysis prevents bias caused by the loss of participants, which may disrupt the baseline equivalence established by randomisation and which may reflect non‐adherence to the protocol. The term is often misused in trial publications when some participants were excluded. |
| meta‐analysis | The use of statistical techniques in a systematic review to integrate the results of included studies. Sometimes misused as a synonym for systematic reviews, where the review includes a meta‐analysis. |
| microcephaly | An abnormally small head of a newborn, a congenitally small brain. |
| neuro‐psychometric testing | Detailed testing of memory and other aspects of intellectual functioning such as planning, speed of thinking, abstract thinking, calculation, language (including speech and reading), visio‐spatial function and attention and concentration. It is often used to assess if there is evidence of intellectual or memory decline. When performed serially (eg at annual intervals) it can be used to detect evidence of change in intellectual functioning with time. |
| neurotoxicity | Toxicity to nervous tissue (both brain and peripheral nerves). |
| random‐effects model | A statistical model in which both within‐study sampling error (variance) and between‐studies variation are included in the assessment of the uncertainty (confidence interval) of the results of a meta‐analysis. When there is heterogeneity among the results of the included studies beyond chance, random‐effects models will give wider confidence intervals than fixed‐effect models. |
| standard deviation | A measure of the spread or dispersion of a set of observations, calculated as the average difference from the mean value in the sample. |
Appendix 2. Search strategy: ClinicalTrials.gov (Date searched: 01 September 2009 for all years)
| Search term |
| (kuvan OR phenoptin OR Sapropterin) AND (phenylketonuria OR PKU) |
Appendix 3. Search strategy: Current Controlled Trials (Date searched: 01 September 2009 for all years)
| Register | Search term |
| metaRegister (active register) | (kuvan OR phenoptin OR Sapropterin) AND (phenylketonuria OR PKU) |
Data and analyses
Comparison 1. Sapropterin versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Change in blood phenylalanine concentration from baseline | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 1.1 Over 2 weeks and up to 4 weeks | 1 | 45 | Mean Difference (IV, Fixed, 95% CI) | ‐51.90 [‐197.27, 93.47] |
| 1.2 Over 4 weeks and up to 6 weeks | 1 | 88 | Mean Difference (IV, Fixed, 95% CI) | ‐238.8 [‐343.09, ‐134.51] |
| 2 Mean difference in blood phenylalanine concentration between treatment groups | 2 | Mean Difference (Fixed, 95% CI) | Subtotals only | |
| 2.1 Over 2 weeks and up to 4 weeks | 1 | 45 | Mean Difference (Fixed, 95% CI) | ‐135.2 [‐187.92, ‐82.48] |
| 2.2 Over 4 weeks and up to 6 weeks | 1 | 88 | Mean Difference (Fixed, 95% CI) | ‐245.0 [‐349.47, ‐140.53] |
| 3 Adverse events due to sapropterin | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3.1 Upper respiratory tract infection | 2 | 133 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.63 [0.29, 1.36] |
| 3.2 Headache | 2 | 133 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.84 [0.36, 1.96] |
| 3.3 Vomiting | 2 | 133 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.04 [0.28, 3.91] |
| 3.4 Abdominal pain | 2 | 133 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.51 [0.12, 2.21] |
| 3.5 Diarrhoea | 2 | 133 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.31 [0.32, 5.43] |
| 3.6 Pyrexia | 2 | 133 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.78 [0.23, 2.69] |
| 3.7 Back pain | 1 | 88 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.38 [0.04, 3.53] |
| 3.8 Rhinorrhea | 1 | 45 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.45 [0.18, 11.75] |
| 3.9 Cough | 1 | 45 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.21 [0.25, 70.82] |
| 3.10 Pharybgolaryngeal pain | 1 | 45 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.45 [0.18, 11.75] |
| 3.11 Contusion | 1 | 45 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.09 [0.13, 9.50] |
| 3.12 Nasal congestion | 1 | 45 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.68 [0.15, 48.32] |
| 3.13 Decreased appetite | 1 | 45 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.91 [0.10, 37.20] |
| 3.14 Erythema | 1 | 45 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.91 [0.10, 37.20] |
| 3.15 Excoriation | 1 | 45 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.91 [0.10, 37.20] |
| 3.16 Lymphadenopathy | 1 | 45 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.91 [0.10, 37.20] |
| 3.17 Streptococcal infection | 1 | 45 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.36 [0.06, 2.30] |
| 3.18 Tooth ache | 1 | 45 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.91 [0.10, 37.20] |
| 4 Mean phenylalanine supplement tolerated | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 4.1 Over 2 months and up to 3 months | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5 Difference in total phenylalanine intake | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 5.1 At baseline ( week 0) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.2 Over 2 months and up to 3 months | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 6 Change in phenylalanine tolerance | 1 | Mean Difference (Fixed, 95% CI) | Totals not selected | |
| 6.1 Over 2 months and up to 3 months | 1 | Mean Difference (Fixed, 95% CI) | 0.0 [0.0, 0.0] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Levy 2007.
| Methods | Randomized, double‐blind placebo‐controlled trial. Multicentre, North America and Europe. |
|
| Participants | 89 children and adults with PKU, over 8 years of age, with blood phe ≥450μmol/L; individuals who had a reduction of 30% or more in blood phe concentration after 8 days of treatment with sapropterin at a dose of 10 mg/kg in a previous screening test (PKU 001) were eligible for the trial. Participants had been involved in a phase 1 screening study. |
|
| Interventions | Sapropterin 10 mg/kg/day versus placebo; treatment was for a period of 6 weeks. | |
| Outcomes | Change in blood phe concentration. | |
| Notes | Data published only as a six‐week trial; further details requested from the author. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Treatment allocations were made centrally from randomisation lists generated by a computer program. Each randomisation list started with a block of two, followed by blocks of four. |
| Allocation concealment (selection bias) | Low risk | Described that a central interactive voice response system was used; investigators, participants and sponsors were kept unaware of treatment allocation until database was locked; block size was not divulged to sponsors or investigators until the trial was completed. |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Stated as double blind; sapropterin and placebo tablets were identical in taste and appearance. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Two withdrawals (one from each group) described. One withdrawal from the trial in the sapropterin group before closing due to an inability to comply with the trial course. The data were not included in the analysis as the participant did not receive even one dose of trial drug. There was another withdrawal from the control group due to non‐compliance with specified dosing, but the data was included in the analysis. |
| Selective reporting (reporting bias) | High risk | Comparison of the trial protocol available at the ClinicalTrials.gov web site and also the 'Methods' section with the results reported in the final paper showed all the detailed outcomes in the protocol were reported in the published reports. However, in the Levy trial they measured data at several time points (weeks 0, 1, 2, 4,and 6) but only reported them at 6 weeks. |
| Other bias | Unclear risk | Sponsored by BioMarin Pharmaceutical Inc. |
Trefz 2009.
| Methods | Randomized, double‐blind placebo‐controlled trial. Multicentre, North America and Europe. |
|
| Participants | 90 children with PKU between 4 to 12 years of age, under phe‐restricted diet with a phe tolerance ≤ 1000 mg/d, and blood phe ≤ 480 μmol/L; exclusion criteria: history of organ transplantation, usage of investigational agent within 30 days before screening, serum alanine aminotransferase levels more than twice upper limit of normal, concurrent disease, using drugs that inhibit folate synthesis, primary BH4 deficiency. 46 children eligible for inclusion (see 'Notes' below). |
|
| Interventions | Sapropterin 20 mg/kg/d versus placebo; treatment was for a period of 10 weeks. | |
| Outcomes | Change in blood phe concentration. Change in phe tolerance. |
|
| Notes | Trial conducted in two parts. Eligible participants (N = 90) entered part 1, received oral sapropterin 20 mg/kg/d for eight days. Those who had a ≥30% reduction in blood phe and had a blood phe ≤ 300 μmol/L were included in part 2 (N = 46) which was for 10 weeks. The phe supplement was added at the beginning of week 3. Only part 2 of the trial is eligible to be included in the analysis. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Described that randomisation was performed by a computer program and interactive voice response system using block sizes of four. |
| Allocation concealment (selection bias) | Low risk | Described that a central interactive voice response system was used; stated that block sizes were not divulged to investigators or trial sponsor until the trial was completed. |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Stated as double‐blind. Sapropterin and placebo tablets had similar taste and appearance. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | One participant randomized to the sapropterin group did not return to for week 0 visit. The data was not included in the analysis. |
| Selective reporting (reporting bias) | High risk | Comparison of the trial protocol available at the ClinicalTrials.gov web site and also the 'Methods' section with the results reported in the final paper showed all the detailed outcomes in the protocol were reported in the published reports. But the data could not be included in meta‐analysis as the details in control group were not given. |
| Other bias | Unclear risk | Sponsored by BioMarin Pharmaceutical Inc. |
BH4: tetrahydrobiopterin Phe: phenylalanine PKU: phenylketonuria
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Gramer 2009 | Not an RCT. |
| Gropmann 2011 | Trial was terminated prematurely due to poor recruitment. |
| Nwose 2008 | Participants and outcome measures are not relevant to the review. |
| Utz 2012 | Outcomes not relevant to the review. A pharmacogenetic test of responsiveness to sapropterin dihydrochloride. |
RCT: randomized controlled trial
Characteristics of studies awaiting assessment [ordered by study ID]
NCT01114737.
| Methods | Allocation: randomized Endpoint classification: safety and efficacy trial Intervention model: parallel assignment Masking: double blind (participant, caregiver, investigator, outcome assessor) Primary purpose: treatment |
| Participants | People with PKU. 8 years to 65 years |
| Interventions | Oral sapropterin dihydrochloride (20 mg/kg/day) versus placebo |
| Outcomes |
Primary outcome measures Evaluate the therapeutic effects of sapropterin dihydrochloride on the symptoms of ADHD and on global function compared to placebo, in subjects with a blood phe level reduction after treatment ADHD change will be measured as a change in ADHD from baseline to week 13 using the Attention‐Deficit Hyperactivity Disorder Rating Scale and Adult ADHD Self‐Report Scale (ADHD RS/ASRS) measurement Global function will be measured as a change in global function using the Clinical Global Impression‐Improvement (CGI‐I) scale rating compared from baseline to week 13. Secondary outcome measures Evaluate the therapeutic effects of sapropterin dihydrochloride on the symptoms of anxiety and depression compared to placebo, in subjects with a blood phe level reduction after treatment Evaluate the durability of any therapeutic effects of sapropterin dihydrochloride on neuropsychiatric symptoms and global function of subjects who have a blood phe level reduction after treatment. Determine if sapropterin dihydrochloride has a therapeutic effect on neuropsychiatric symptoms in PKU people who do not have a blood phe reduction after treatment Assess the safety of sapropterin dihydrochloride when administered as therapy to these participants |
| Notes | Estimated completion date: January 2013. Estimated enrolment: 200 participants ClinicalTrials.gov Identifier: NCT01114737 |
NCT01376908.
| Methods | Allocation: randomized Endpoint classification: safety and efficacy study Intervention model: parallel assignment Masking: open label Primary purpose: treatment |
| Participants | Children less than 4 years old with PKU |
| Interventions | Kuvan, phe‐restricted diet |
| Outcomes | Dietary phe tolerance after 26 weeks, Levels of blood phe, Change from baseline in dietary phe tolerance after 26 weeks, number of participants with adverse events after 26 weeks and 3 years |
| Notes | Participants randomized into phe‐restricted diet only and Kuvan + phe‐restricted diet groups. Trial period is 26 weeks, with an extension period of 3 years. |
Phe: phenylalanine PKU: phenylketonuria
Characteristics of ongoing studies [ordered by study ID]
NCT01977820.
| Trial name or title | Sapropterin on Cognitive Abilities in Young Adults With Phenylketonuria |
| Methods | Allocation: randomized Endpoint classification: safety/efficacy study Intervention model: parallel assignment Primary purpose: treatment Masking: double blind (participant, investigator) |
| Participants | Adults with PKU |
| Interventions | Sapropterin vs placebo |
| Outcomes | Number of participants with adverse event |
| Starting date | February 2014 |
| Contact information | Merck KgaA |
| Notes | Estimated completion date: January 2016 |
Contributions of authors
US conceived and developed the protocol with inputs from MM.
Both US and MM extracted the data, US wrote the review with inputs from MM.
US acts as guarantor of the review.
Sources of support
Internal sources
No sources of support supplied
External sources
-
National Institute for Health Research, UK.
This systematic review was supported by the National Institute for Health Research, via Cochrane Infrastructure funding to the Cochrane Cystic Fibrosis and Genetic Disorders Group.
Declarations of interest
None known.
New search for studies and content updated (no change to conclusions)
References
References to studies included in this review
Levy 2007 {published and unpublished data}
- Lee P, Treacy EP, Crombez E, Wasserstein M, Waber L, Wolff J, et al. Safety and efficacy of 22 weeks of treatment with sapropterin dihydrochloride in patients with phenylketonuria. American Journal of Medical Genetics. Part A 2008; Vol. 146A, issue 22:2851‐9. [DOI] [PubMed]
- Levy H, Milanowski A, Chakrapani A, Cleary M, Trefz F, Whitley C, et al. A phase 3 study of the efficacy of sapropterin in reducing phe levels in subjects with phenylketonuria [abstract]. Journal of Inherited Metabolic Disease 2006;29(Suppl 1):13. [Google Scholar]
- Levy HL, Milanowski A, Chakrapani A, Cleary M, Lee P, Trefz FK, et al. Efficacy of sapropterin dihydrochloride (tetrahydrobiopterin, 6R‐BH4) for reduction of phenylalanine concentration in patients with phenylketonuria: a phase III randomised placebo‐controlled study. Lancet 2007;370(9586):504‐10. [DOI] [PubMed] [Google Scholar]
Trefz 2009 {published and unpublished data}
- Trefz FK, Burton BK, Longo N, Casanova MM‐P, Gruskin DJ, Dorenbaum A, et al. Efficacy of sapropterin dihydrochloride in increasing phenylalanine tolerance in children with phenylketonuria: a phase III, randomized, double‐blind, placebo‐controlled study. Journal of Pediatrics 2009;154(5):700‐7. [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Gramer 2009 {published data only}
- Gramer G, Garbade SF, Blau N, Lindner M. Pharmacokinetics of tetrahydrobiopterin following oral loadings with three single dosages in patients with phenylketonuria. Journal of Inherited Metabolic Disease 2009;32(1):52‐7. [DOI] [PubMed] [Google Scholar]
Gropmann 2011 {unpublished data only}
- Gropmann AL. Neuroimaging and Neurocognitive Assessment and Response to Sapropterin Dihydrochloride Treatment in Phenylketonuria. www.ClinicalTrials.gov [accessed 19 October 2012]. [NCT01412437]
Nwose 2008 {unpublished data only}
- Nwose D. A Phase 1 Study to Evaluate Effects of Sapropterin Dihydrochloride on QTc Intervals in Healthy Adult Subjects. www.ClinicalTrials.gov [accessed 19 October 2012].
Utz 2012 {published data only}
- Utz JR, Lorentz CP, Markowitz D, Rudser KD, Diethelm‐Okita B, Erickson D, Whitley CB. START, a double blind, placebo‐controlled pharmacogenetic test of responsiveness to sapropterin dihydrochloride in phenylketonuria patients. Molecular genetics and metabolism 2012;105(2):193‐7. [DOI] [PubMed] [Google Scholar]
References to studies awaiting assessment
NCT01114737 {unpublished data only}
- NCT01114737. Safety and therapeutic effects of sapropterin dihydrochloride on neuropsychiatric symptoms in phenylketonuria (PKU) patients. www.ClinicalTrials.gov (accessed 19 October 2012).
NCT01376908 {unpublished data only}
- NCT01376908. Kuvan® in Phenylketonuria Patients Less Than 4 Years Old (SPARK). www.ClinicalTrials.gov (accessed 19 October 2012).
References to ongoing studies
NCT01977820 {unpublished data only}
- NCT01977820. Sapropterin on cognitive abilities in young adults with phenylketonuria. https://clinicaltrials.gov/ct2/show/NCT01977820 (accessed 07 January 2015).
Additional references
Baulny 2007
- Baulny HO, Abadie V, Feille F, Parscau L. Management of phenylketonuria and hyperphenylalaninemia. Journal of Nutrition 2007;137(6 Suppl 1):1561S‐3S. [DOI] [PubMed] [Google Scholar]
Belanger‐Quintana 2005
- Belanger‐Quintana A, Garcia MJ, Castro M, Desviat LR, Perez B, Mejia B, et al. Spanish BH4‐responsive phenylalanine hydroxylase‐deficient patients: evolution of seven patients on long‐term treatment with tetrahydrobiopterin. Molecular Genetics and Metabolism 2005;86(Suppl 1):S61‐6. [DOI] [PubMed] [Google Scholar]
Bernegger 2002
- Bernegger C, Blau N. High frequency of tetrahydrobiopterin‐responsiveness among hyperphenylalaninemias: a study of 1919 patients observed from 1988 to 2002. Molecular Genetics and Metabolism 2002;77(4):304‐13. [DOI] [PubMed] [Google Scholar]
Blau 2004
- Blau N, Erlandsen H. The metabolic and molecular bases of tetrahydrobiopterin‐responsive phenylalanine hydroxylase deficiency. Molecular Genetics and Metabolism 2004;82(2):101‐11. [DOI] [PubMed] [Google Scholar]
Boveda 2007
- Boveda MD, Couce ML, Castineiras DE, Cocho JA, Perez B, Ugarte M, et al. The tetrahydrobiopterin loading test in 36 patients with hyperphenylalaninemia: evaluation of response and subsequent treatment. Journal of Inherited Metabolic Disease 2007;30(5):812. [DOI] [PubMed] [Google Scholar]
Burlina 2009
- Burlina A, Blau N. Effect of BH(4) supplementation on phenylalanine tolerance. Journal of Inherited Metabolic Disease 2009;32(1):40‐5. [DOI] [PubMed] [Google Scholar]
Burton 2007
- Burton BK, Grange DK, Milanowski G, Feillet F, Crombwz EA, Abadie V, et al. The response of patients with phenylketonuria and elevated serum phenylalanine to treatment with oral sapropterin dihydrochloride (6R‐tetrahydrobiopterin): a phase II, multicentre, open‐label, screening study. Journal of Inherited Metabolic Disease 2007;30:700‐7. [DOI] [PubMed] [Google Scholar]
Cerone 2004
- Cerone R, Schiaffino MC, Fantasia AR, Perfumo M, Birk Moller L, Blau N. Long‐term follow‐up of a patient with mild tetrahydrobiopterin‐responsive phenylketonuria. Molecular Genetics and Metabolism 2004;81(2):137‐9. [DOI] [PubMed] [Google Scholar]
Channon 2007
- Channon S, Goodman G, Zlotowitz S, Mockler C, Lee PJ. Effects of dietary management of phenylketonuria on long‐term cognitive outcome. Archives of Disease in Childhood 2007;92(3):213‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Cockburn 1996
- Cockburn F, Clark BJ. Recommendations for protein and amino acid intake in phenylketonuric patients. European Journal of Pediatrics 1996;155(Suppl 1):125‐9. [DOI] [PubMed] [Google Scholar]
Elbourne 2002
- Elbourne DR, Altman DG, Higgins JP, Curtin F, Worthington HV, Vail A. Meta‐analyses involving cross‐over trials: methodological issues. International Journal of Epidemiology 2002;31(1):140‐9. [DOI] [PubMed] [Google Scholar]
Erlandsen 2004
- Erlandsen H, Pey AL, Gamez A, Perez B, Desviat LR, Aguado C, et al. Correction of kinetic and stability defects by tetrahydrobiopterin in phenylketonuria patients with certain phenylalanine hydroxylase mutations. Proceedings of National Academy of Science USA 2004;101(48):16903‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Fiege 2005
- Fiege B, Bonafe L, Ballhausen D, Baumgartner M, Thony B, Meili D, et al. Extended tetrahydrobiopterin loading test in the diagnosis of cofactor‐responsive phenylketonuria: a pilot study. Molecular Genetics and Metabolism 2005;86(Suppl 1):S91‐5. [DOI] [PubMed] [Google Scholar]
Fiege 2007
- Fiege B, Blau N. Assessment of tetrahydrobiopterin responsiveness in phenylketonuria. Journal of Pediatrics 2007;150(6):627‐30. [DOI] [PubMed] [Google Scholar]
Fiori 2005
- Fiori L, Fiege B, Riva E, Giovanni M. Incidence of BH4‐responsiveness in phenylalanine‐hydroxylase‐deficient Italian patients. Molecular Genetics and Metabolism 2005;86(Suppl 1):S67‐74. [DOI] [PubMed] [Google Scholar]
Giovanni 2007
- Giovanni M, Verduci E, Salvatici E, Fiori L. Phenylketonuria: Dietary and therapeutic challenges. Journal of Inherited Metabolic Disease 2007;30(2):145‐52. [DOI] [PubMed] [Google Scholar]
Hennermann 2005
- Hennermann JB, Buhrer C, Blau N, Vetter B, Monch E. Long‐term treatment with tetrahydrobiopterin increases phenylalanine tolerance in children with severe phenotype of phenylketonuria. Molecular Genetics and Metabolism 2005;86(Suppl 1):S86‐90. [DOI] [PubMed] [Google Scholar]
Higgins 2003
- Higgins JPT, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analyses. BMJ 2003;327(7414):557‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration 2011. Available from www.cochrane‐handbook.org.
Koch 2002
- Koch R, Burton B, Hoganson G, Peterson R, Rhead W, Rouse B, et al. Phenylketonuria in adulthood: a collaborative study. Journal of Inherited Metabolic Disease 2002;25(5):333‐46. [DOI] [PubMed] [Google Scholar]
Kure 1999
- Kure S, Hou DC, Ohura T, Iwamoto H, Suzuki S, Sugiyama N, et al. Tetrahydrobiopterin‐responsive phenylalanine hydroxylase deficiency. Journal of Pediatrics 1999;135(3):375‐8. [DOI] [PubMed] [Google Scholar]
Leuzzi 2006
- Leuzzi V, Carducci C, Carducci C, Chiarotti F, Artiola C, Giovanniello T, et al. The spectrum of phenylalanine variations under tetrahydrobiopterin load in subjects affected by phenylalanine hydroxylase deficiency. Journal of Inherited Metabolic Disease 2006;29(1):38‐46. [DOI] [PubMed] [Google Scholar]
Lindner 2001
- Lindner M, Haas D, Zschocke J, Burgard P. Tetrahydrobiopterin responsiveness in phenylketonuria differs between patients with the same genotype. Molecular Genetics and Metabolism 2001;73(1):104‐6. [DOI] [PubMed] [Google Scholar]
Matalon 2002
- Matalon R, Koch R, Michals‐Matalon K, Mosley K, Stevens R. Tetrahydrobiopterin‐responsive phenylalanine hydroxylase mutations. Journal of Inherited Metabolic Disease 2002;25(Suppl 1):23. [Google Scholar]
Matalon 2005
- Matalon R, Michals‐Matalon K, Koch R, Grady J, Tyring S, Stevens RC. Response of patients with phenylketonuria in the US to tetrahydrobiopterin. Molecular Genetics and Metabolism 2005;86(Suppl 1):S17‐21. [DOI] [PubMed] [Google Scholar]
Michals‐Matalon 2007
- Michals‐Matalon K, Bhatia G, Guttler F, Tyring SK, Matalon R. Response of phenylketonuria to tetrahydrobiopterin. Journal of Nutrition 2007;137(6 Suppl 1):1564S‐7S. [DOI] [PubMed] [Google Scholar]
MRC (UK) 1993
- Medical Research Council (UK). Phenylketonuria due to phenylalanine hydroxylase deficiency: an unfolding story. BMJ 1993;306(6870):115‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Muntau 2002
- Muntau AC, Roschinger W, Habich M, Demmelmain H, Hoffmann B, Sommerhoff CP, et al. Tetrahydrobiopterin as an alternative treatment for mild phenylketonuria. New England Journal of Medicine 2002;347(26):2122‐32. [DOI] [PubMed] [Google Scholar]
NIH Consensus Development Panel 2001
- National Institutes of Health Consensus Development Panel. National Institutes of Health Consensus Development Conference statement: phenylketonuria: screening and management. Pediatrics 2000;108(4):972‐82. [DOI] [PubMed] [Google Scholar]
Nuoffer 2001
- Nuoffer JM, Thony B, Romstad A, Blau N. A patient with phenylketonuria successfully treated with tetrahydrobiopterin. Journal of Inherited Metabolic Disease 2001;24(Suppl 1):29. [Google Scholar]
Scriver 2001
- Scriver CR, Seymour K. Hyperphenylalaninemia : Phenylalanine Hydroxylase Deficiency. In: Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Kinzler KW, Vogelstein B editor(s). The Metabolic and Molecular Bases of Disease. 8th Edition. Vol. 2, New York: McGraw‐Hill, 2001:1667‐724. [Google Scholar]
Scriver 2007
- Scriver CR. The PAH gene, phenylketonuria, and a paradigm shift. Human Mutation 2007;28(9):831‐45. [DOI] [PubMed] [Google Scholar]
Shintaku 2004
- Shintaku H, Kure S, Ohura T, Okano Y, Ohwada M, Sugiyama N, et al. Long‐term treatment and diagnosis of tetrahydrobiopterin‐responsive hyperphenylalaninemia with a mutant phenylalanine hydroxylase gene. Pediatric Research 2004;55(3):425‐30. [DOI] [PubMed] [Google Scholar]
Spaapen 2001
- Spaapen LJM, Bakker JA, Velter C, Loots W, Rubio ME, Forget PP, et al. Tetrahydrobiopterin‐responsive phenylalanine hydroxylase deficiency in Ditch neonates. Journal of Inherited Metabolic Disease 2001;24(3):325‐58. [DOI] [PubMed] [Google Scholar]
Steinfeld 2002
- Steinfeld R, Kohlschutter A, Zschocke J, Lindner M, Ullrich K, Lukacs Z. Tetrahydrobipterin monotherapy for phenylketonuria patients with common mild mutations. European Journal of Pediatrics 2002;161(7):403‐5. [DOI] [PubMed] [Google Scholar]
Steinfeld 2003
- Steinfeld R, Kohlschutter A, Ullrich K, Lukacs Z. A hypothesis on the biochemical mechanism of BH(4) ‐responsiveness in phenylalanine hydroxylase deficiency. Amino Acids 2003;25(1):63‐8. [DOI] [PubMed] [Google Scholar]
Steinfeld 2004
- Steinfeld R, Kohlschutter A, Ullrich K, Lukacs Z. Efficiency of long‐term tetrahydrobiopterin monotherapy in phenylketonuria. Journal of Inherited Metabolic Disease 2004;27(4):449‐53. [DOI] [PubMed] [Google Scholar]
Trefz 2001
- Trefz FK, Aulehla‐Scholz C, Blau N. Successful treatment of phenylketonuria with tetrahydrobiopterin. European Journal of Pediatrics 2001;160(5):315. [DOI] [PubMed] [Google Scholar]
Wappner 1999
- Wappner R, Cho S, Kronmal RA, Schuett V, Seashore MR. Management of phenylketonuria for optimal outcome: a review of guidelines for phenylketonuria management and a report of surveys of parents, patients, and clinic directors. Pediatrics 1999;104(6):e68. [DOI] [PubMed] [Google Scholar]
Williams 2008
- Williams RA, Mamotte CDS, Burnett JR. Phenylketonuria: an inborn error of phenylalanine metabolism. Clinical Biochemist Reviews 2008;29(1):31‐41. [PMC free article] [PubMed] [Google Scholar]
References to other published versions of this review
Somaraju 2010
- Somaraju UR, Merrin M. Sapropterin dihydrochloride for phenylketonuria. Cochrane Database of Systematic Reviews 2010, Issue 6. [DOI: 10.1002/14651858.CD008005.pub2] [DOI] [PubMed] [Google Scholar]
Somaraju 2012
- Somaraju UR, Merrin M. Sapropterin dihydrochloride for phenylketonuria. Cochrane Database of Systematic Reviews 2012, Issue 12. [DOI: 10.1002/14651858.CD008005.pub3] [DOI] [PubMed] [Google Scholar]