

Abstract
Cancer cells undergo metabolic reprogramming to support cell proliferation, growth, and dissemination. Alterations in lipid metabolism, and specifically the uptake and synthesis of fatty acids (FAs), comprise one well-documented aspect of this reprogramming. Recent studies have revealed an expanded range of roles played by FA in promoting the aggressiveness of cancer while simultaneously identifying new potential targets for cancer therapy. This article provides a brief review of these advances in our understanding of FA metabolism in cancer, highlighting both recent discoveries and the inherent challenges caused by the metabolic plasticity of cancer cells in targeting lipid metabolism for cancer therapy.
Keywords: cancer, fatty acid metabolism, lipid metabolism, metabolic plasticity
Introduction
Lipids are a diverse group of hydrophobic biomolecules that include sterols, acylglycerols, phospholipids, and sphingolipids. Most lipids are synthesized from fatty acids (FAs), a family of molecules consisting of a terminal carboxyl group and a hydrocarbon chain of various carbon lengths and levels of saturation. In the past three decades, FAs have attracted increased research attention, and even some controversy, because of studies linking dietary fat in the Western diet to various human malignancies, including cancers of the colon, breast, and prostate.1–3 Meanwhile, other findings have linked endogenous FA synthesis to human cancer. In the 1980s, Ookhtens et al. used in vivo14C labelling of the central carbon metabolism in Ehrlich ascites tumors to show that almost all esterified FAs in the tumors are derived from de novo synthesis, although they noted that tumor tissues also acquire small amounts of FAs from circulating blood.4 In the mid-1990s, Kuhajda et al. revealed that OA-519, a highly expressed tumor-specific antigen in human breast cancer, encodes fatty acid synthase (FASN),5 and since this landmark discovery, numerous studies have confirmed that many human cancers display aberrant activation of de novo FA synthesis. It now appears that FA metabolism, including FA uptake, synthesis, modification, and degradation, are all altered in cancer, which enables cancer cells to proliferate, grow, and disseminate to distant organs.6 While several excellent reviews have covered the general role of lipid metabolism in human cancer,6–12 here, we will consider the expanded range of roles played by FA in promoting the aggressiveness of cancer, in particular metastasis, and discuss the challenges inherent in targeting lipid metabolism for cancer therapy.
FA metabolism in cancer
Cell proliferation requires that new biological membranes be assembled from lipids. FAs, the building blocks of lipids, can be acquired exogenously (through diet) or endogenously synthesized (Fig. 1). In well-nourished individuals, de novo FA synthesis only minimally contributes to the lipid content of most adult tissues, as normal cells preferentially use exogenous FAs to satisfy their lipid requirements.13 The uptake of FAs from the circulation is facilitated by protein-mediated transport. Several proteins have been implicated in FA uptake, including fatty acid translocase (FAT)/CD36, fatty acid transport proteins (FATPs)/SLC27A, LDLR, and fatty acid binding proteins (FABPs).14
Figure 1 .
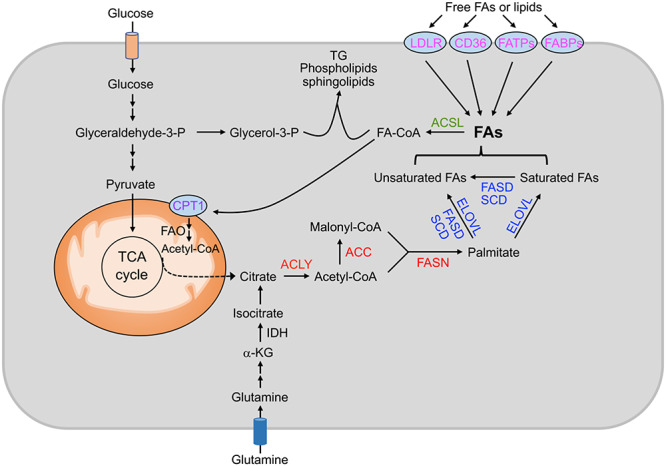
Fatty acid (FA) metabolism and possible strategies for targeting lipid metabolism in cancer: red highlights protein targets involved in de novo FA synthesis; blue highlights protein targets involved in FA modification; magenta highlights protein targets involved in FA uptake; green highlights protein targets involved in FA activation; purple highlights protein targets involved in FA degradation. ACC, acetyl-CoA carboxylases; ACLY, ATP citrate lyase; ACSL, long-chain acetyl-CoA synthetase; CPT1, carnitine palmitoyl transferase 1; ELOVL, fatty acid elongase; FABPs, fatty acid binding proteins; FADS, fatty acid desaturase; FAO: fatty acid β-oxidation; FASN, fatty acid synthase; FATPs, transport proteins. Glyceraldehyde-3-p, glyceraldehyde-3-phosphate; glycerol-3-p, glycerol-3-phosphate; IDH, isocitrate dehydrogenase; α-KG, α-ketoglutarate; TCA, tricarboxylic acid; TG, triacylglycerols; SCD, stearoyl-CoA desaturase.
While normal cells primarily depend on the uptake of FAs, cancer cells reactivate de novo FA synthesis, irrespective of circulating lipid levels,4,5,15 which indicates the crucial role played by FA synthesis in tumorigenesis. FA synthesis is an anabolic process that converts nutrient-derived carbons into FAs. These carbons are mainly provided by citrate, which is normally generated from glucose-derived pyruvate flux into the tricarboxylic acid (TCA) cycle. Notably, cancer cells display metabolic plasticity in producing citrate for FA synthesis. During hypoxia or in cancer cells with malfunctioning mitochondria, the TCA cycle is inhibited, and citrate is generated from reductive carboxylation of glutamine-derived α-ketoglutarate by the NADPH-dependent isocitrate dehydrogenase.16–18 Through the action of ATP-citrate lyase (ACLY), citrate is cleaved into oxaloacetate and acetyl-coenzyme A (acetyl-CoA), which is the primary substrate for FA synthesis. Next, acetyl-CoA carboxylase (ACC), the rate-limiting enzyme, converts acetyl-CoA into malonyl-CoA. Malonyl-CoA is then committed to FA synthesis and is involved in elongation of FA through FASN until palmitate is formed. Additional modification of FAs can be performed by elongases and desaturases to produce FAs at various carbon lengths and levels of saturation. The free FAs are then esterified via the glycerol phosphate pathway to become the hydrophobic tails of phospholipids and sphingolipids, which, together with cholesterol, form key constituents of all biological membranes.19 FAs can also be used to generate triacylglycerols (TGs) and cholesterol esters (CEs) for energy storage in the form of lipid droplets, which, when necessary, can provide fuel for cellular bioenergetics through FA β-oxidation (FAO).20 It is worth noting that while the activation of de novo FA synthesis in cancer cells is now widely recognized, the role of exogenous FAs in tumorigenesis has long been underappreciated. Recent studies have demonstrated that many cancer cells also scavenge FAs from their environment, and that de novo synthesis and exogenous FAs are equally important in driving cancer progression,21–24 which has substantial implications for cancer treatment (see discussion below).
Expanded role of FA metabolism in cancer
Although aberrant FA metabolism helps explain how cancer cells satisfy their lipid requirements, it is not entirely clear what advantages cancer cells obtain by acquiring more lipids. Recent studies, however, suggest that alterations in FA metabolism, including FA uptake, synthesis, modification and degradation, may play distinctive roles in different metabolic niches and during different stages of tumor development. Here we summarize current knowledge regarding the newly discovered roles played by FA metabolism in tumorigenesis beyond its canonical roles in membrane biosynthesis, energy storage, signal transduction, and post-translational modifications of proteins, which have been reviewed in depth elsewhere.6–12
Protecting cells from lipotoxicity
Viable cells must maintain a proper ratio of saturated to unsaturated FAs in their biological membranes. It has been shown that the accumulation of excess saturated FAs in membranes triggers mitochondrial dysfunction, enhanced reactive oxygen species (ROS), and endoplasmic reticulum (ER) stress.25,26 To prevent these outcomes, FA desaturation is carried out by stearoyl-CoA desaturases (SCDs) in an oxygen-dependent manner. Of the two SCD isoforms (SCD1 and SCD5) found in humans, SCD1 is the principle enzyme responsible for desaturation, and is highly expressed in a wide range of tissues. SCD1 converts saturated FAs into monounsaturated FAs to increase the levels of unsaturated FAs. Overexpression of SCD1 has been found in tumors of various cancer types, including prostate, breast, liver, kidney,27–30 and particularly lung cancer, where it correlates with aggressiveness and reduced patient survival.31 As a consequence, inhibition of SCD1 can increase the ratio of saturated to unsaturated FAs, leading to ER stress and reduced proliferation in cancer cells.26,27,30,32,33 The growth of solid tumors is often characterized by limited concentrations of oxygen, which markedly inhibits the activity of SCD1.34 To cope with this hypoxic stress, cancer cells scavenge unsaturated FAs from the environment, or release unsaturated FAs from lipid droplets to maintain lipid homostasis,34,35 suggesting that buffering cancer cells from lipotoxicity constitutes a critical function of FA metabolism. Interestingly, Ras-transformed cells also display hypoxic metabolic phenotypes with increased reliance on FA uptake even under normoxic conditions,34 which may render Ras-driven tumors sensitive to therapies targeting the uptake of FAs.
Impacting cell migration and drug resistance by altering membrane fluidity
The lipid composition of a membrane affects its fluidity: saturated FAs confer a more rigid and organized membrane and decrease membrane fluidity, while unsaturated FAs have at least one cis double bond that distorts the hydrophobic chain, leading to loose membrane packing and a consequent increase in membrane fluidity.36,37 Membrane fluidity is a key physical property dictating cell adhesion, migration, and metastatic potential,38,39 and recent studies have shown that treatment of breast cancer cells with chemical inhibitors blocking the metastasis signature genes decreases membrane fluidity and inhibits cell migration and lung metastasis. Importantly, addition of oleic acid, an unsaturated FA, to these cancer cells restores previous levels of membrane fluidity and distant metastasis.39 Furthermore, lung cancer patients with high plasma membrane fluidity generally face a worse prognosis than those with less fluid membranes.40 Intriguingly, cancer cells with reactivated FA synthesis display high levels of lipid saturation and low membrane fluidity, and are resistant to chemotherapeutics because of reduced drug uptake, highlighting the positive correlation between membrane fluidity and membrane permeability, and suggesting a possible connection between FA synthesis and drug resistance.41 Together, these reports indicate how cellular lipids, the end products of lipogenesis, and their composition are altered to change membrane fluidity in support of various aspects of cancer.
Meeting high energy demands of metastatic cells through FAO
Cells that undergo loss of attachment (LOA) to the extracellular matrix display inhibition of glucose uptake and catabolism, which results in loss of cellular ATP.42,43 During the invasion-metastasis cascade, metastatic cells exit their primary sites of growth and translocate systemically in the circulation. Therefore, successful intravasation and extravasation during metastasis require that cancer cells generate or obtain more ATP through some other means. Among metabolic pathways, FAO is the most energy-efficient way to generate ATPs to satisfy the energy needs of cancer cells and is perhaps particularly crucial for disseminated tumor cells, which must withstand the selective pressure of metastatic colonization. Indeed, to survive the colonization process, metastatic ovarian cancer cells catabolize lipids acquired from omental adipocytes to meet their high demand for ATP through FAO.44 Additionally, they also express high levels of monoacylglycerol lipase (MAGL), an enzyme which releases free FAs from lipids. Blockade of MAGL impairs tumor growth and metastasis, which are both rescued by free FAs present in a high fat diet (HFD),45 further underscoring the importance of FAO in promoting the aggressiveness of cancer. In addition, a recent study found that a population of metastasis-initiating cells (MICs) express high levels of the FA receptor CD36 and lipid metabolism genes, and are unique in their ability to initiate metastasis.21 CD36 takes lipids up from the extracellular environment, allowing MICs to meet their high demand for ATP through FAO. This is essential for MICs to anchor and survive at metastatic sites. Importantly, administration of a HFD or palmitic acid has been shown to boost the metastatic potential of cancer cells in a CD36-dependent manner. Consistent with these findings, CD36 or its associated gene signature significantly correlates with poor clinical outcomes in patients with cancers of various histological origins.21 The functional significance of FAO in metastatic progress was also recently observed in tumor metastasis to lymph nodes, where researchers found that lymph node metastasis requires that tumor cells undergo a metabolic shift toward FAO, with the transcription coactivator yes-associated protein (YAP) driving the upregulation of FAO-related metabolic genes.46 These studies strongly suggest that inhibition of the FAO metabolic pathway may merit exploration as a potential target for mitigating distant metastasis, which is the leading cause of cancer-related deaths.
Targeting FA metabolism for cancer therapy
As FAs are essential for every aspect of all cellular processes, limiting their availability to cancer cells holds great promising as a therapeutic approach. We will therefore detail below several possible strategies for limiting FA availability to cancer cells (Fig. 1).
Targeting de novo FA synthesis
Considerable efforts have been made to target de novo FA synthesis for cancer treatment as this pathway is of minor importance in normal tissue but is reactivated in cancers of a range of histological origins.9 The expression of the enzymes involved in FA synthesis is mainly regulated by a master transcriptional regulator, the sterol regulatory element binding protein 1 (SREBP-1).47 SREBP-1 has two splice variants: SREBP-1a and SREBP-1c. These activate the FA biosynthetic genes, including ACLY, ACC, FASN, and SCD-1. Therefore, targeting SREBP-1 could inhibit expression levels of lipogenic genes and potentially represent an efficient means of blocking tumor growth. However, targeting transcription factors such as SREBPs remains a daunting task, because they are widely considered undruggable. A potential strategy might be to develop inhibitors targeting the association between SREBP and the SREBP-cleavage activating protein (SCAP), an escort protein that plays an essential role in SREBP activation by mediating ER-to-Golgi transport of SREBP. One example of such an inhibitor is fatostatin, which has been shown to inhibit the growth and metastasis of prostate cancer in preclinical studies.22,48,49 One effect of the inhibition of SREBPs is the loss of SCD-1 expression and consequent FA desaturation, which causes ER stress because of abnormal accumulation of saturated FAs.32,33 Another strategy is to target upstream regulators of SREBPs, such as liver X receptors, which are required for insulin-dependent SREBP-1c transcriptional regulation.50 However, the impacts on cell growth of preventing LXR activation may not be entirely a result of downregulation of SREBP expression alone.
The other way to block FA synthesis is to directly target the enzymes involved in FA synthesis. FASN is the most studied lipogenic enzyme, and its increased expression strongly correlates with poor prognosis in many cancers.7,9 As a result, the FASN inhibitors C75, cerulenin, orlistat, and IPI-9119 have been the subjects of extensive preclinical studies.51–54 However, some FASN inhibitors have produced severe side effects in mice, partly as a result of the essential role of FASN in adult neuronal stem cell function, which has raised concerns regarding their use in the clinic.55,56 And, surprisingly, stable FASN knockdown has been found to enhance cell migration and distant lung metastasis in a xenograft mouse model.57 Although it may seem counterintuitive that suppression of de novo FA synthesis could promote metastasis, it is possible that inhibition of FA biosynthesis during the invasion-metastasis cascade could benefit migratory cells by diverting nutrients and bioenergetics away from anabolic processes and reserving them for cell invasion, suggesting that it is beneficial to target FASN in a tumor-stage dependent manner.
Targeting other enzymes within the FA synthesis pathway has also been evaluated, including ACLY and ACC, and has been shown to inhibit cancer cell growth and proliferation.58–60 Interestingly, through regulation of acetyl-CoA, nuclear ACLY is required for histone acetylation, and may therefore affect cancer cell proliferation on many levels.61 It is worth noting that FAs can not only be synthesized endogenously, but can also be acquired exogenously. Cancer cells take advantage of this metabolic plasticity, which may contribute to rapid drug resistance. Indeed, recent studies have shown that lipogenic inhibitors decrease prostate cancer cell viability only in the absence of exogenous lipid sources such as lipoprotein,62 highlighting the importance of the development of combinatorial treatments.
Targeting FA modification
Newly synthesized palmitate can be elongated or desaturated to form a complex collection of FAs. The family of elongases comprises seven members (ELOVL1–7), while the family of desaturases includes only two members (SCD1 and SCD5). Inhibition of ELOVL6 in squamous cell carcinoma cells has been shown to significantly attenuate tumor growth in vivo.63 Meanwhile, inhibition of SCD1 blocks cancer cell growth and proliferation by selectively depleting monounsaturated and polyunsaturated FAs.64–66 Mechanistically, blocking SCD-1 enhances the saturation levels of cardiolipin, a mitochondrial lipid that regulates mitochondria-dependent cell death. As a result, the mitochondria in SCD-1 compromised cells undergo ultrastructural changes, release cytochrome c, and quickly induce cell death. It should be noted that Ras-transformed cells rely on FA uptake to acquire unsaturated FAs and display de novo resistance to SCD1 inhibition.34 Furthermore, some liver and lung carcinomas display metabolic plasticity and employ alternative means to generate unsaturated FAs, that is converting palmitate into sapienate using the FADS2 enzyme, which renders them resistant to SCD1 inhibition.67
Targeting FA uptake
In addition to de novo synthesis, cancer cells satisfy their lipid requirements through the uptake of exogeneous FAs using various membrane proteins, such as CD36, FATPs, LDLR, and FABPs. Because most lipid chaperones are membrane receptor proteins, targeting FA uptake pathways represents a promising strategy for cancer therapy. Indeed, the CD36 antibody has shown significant anti-tumor or anti-metastatic efficacy in preclinical studies.21,23 Additionally, pharmacological blockade of FATPs with small-molecule inhibitors has been found to abrogate lipid transport into melanoma cells and reduce melanoma growth and invasion.68 FABP4, a mediator of lipid trafficking in adipocytes, is highly expressed in adipocytes and in metastatic ovarian cancer cells at the adipocyte‑cancer cell interface, but not in primary ovarian tumors. Either pharmacological inhibition or genetic ablation of FABP4 has been found to drastically inhibit lipid accumulation in ovarian cancer cells and adipocyte-mediated omental metastases.44 These findings suggest that FA uptake and transport will prove another important target pathway for anticancer therapy.
Targeting FA activation
FAs must be activated into FA-CoAs by a member of the long-chain acyl-CoA synthetase (ACSL) family before they can be incorporated into either TGs or phospholipids, or broken down into acetyl-CoA to aid ATP generation. In mammals, there are five ACSL members, namely ACSL1, ACSL3, ACSL4, ACSL5, and ACSL6. Targeting these activating enzymes has the advantage of blocking use of FAs regardless of whether they are synthesized de novo or acquired exogenously. However, various roles played by different members of the ACSL family in cancer can be complex. For example, ACSL1 may play a potentially oncogenic role in liver and breast cancer,69,70 but a potentially tumor-suppressive role in lung squamous cell carcinoma.71 While ACSL4 is upregulated in colon, breast, liver, and prostate cancer,72–75 it is downregulated in gastric cancer.76 Therefore, when designing cancer treatment through targeting ACSLs, it is important to consider tissue specificities and the distinctive roles of each ACSL isoform in cancer. Notably, ACSL4 is also essential for the induction of ferroptosis,77,78 a form of regulated cell death induced by the build-up of toxic lipid peroxides. Induction of ferroptic cell death may represent a therapeutic strategy against various types of cancer with high levels of ACSL4.
Targeting FA storage and mobilization
When cellular lipids are in excess, they are diverted to storage as TGs and cholesterol esters in the lipid droplets (LDs), which are specialized organelles with key functions in lipid storage and energy homeostasis.20 LD accumulation has been reported in many cancers and is enhanced in cancer cells exposed to hypoxia or nutrient starvation.79–82 Recent studies have shown that LDs protect cancer cells against nutrient and oxidative stress, contributing to cancer cell survival and growth,80,82 suggesting that LD biogenesis could be an attractive target for strategies designed to diminish the resistance of cancer cells to stress. The inhibition of LD biogenesis may provide a therapeutic advantage by not only increasing lipotoxicity but also concomitantly blocking lipid acquisition and associated survival mechanisms. Emerging studies imply that targeting the molecules involved in LD biogenesis, such as lysophosphatidylcholine acyltransferase 2 (LPCAT2) and FABPs,80,83 holds promise for inhibiting LD formation and tumor growth in vivo.
When needed, the stored lipid esters are mobilized by the action of lipases in a three-step process, catalyzed by adipose triglyceride lipase (ATGL), hormone sensitive lipase (HSL), and monoacylglycerol lipase (MAGL), which releases one FA in each step from the glycerol backbone.84 Although these three lipases have important functions in the adipose tissue, their roles in cancer have not been extensively studied. MAGL is upregulated in several cancers and promotes aggressiveness of cancer through upregulation of endocannabinoid and FA signaling pathways. Knockdown or pharmacological inhibition of MAGL by JZL184 inhibits free FA levels and tumorigenesis of melanoma and ovarian cancer cells.45 The role of ATGL in cancer is relatively complex. ATGL is overexpressed in high-grade breast tumors and inhibition of ATGL by knockdown or a chemical inhibitor such as atglistatin suppresses the growth of several types of cancer cells.85,86 However, recent immunohistochemical analysis of ATGL expression in human malignancies has found that ATGL protein levels are significantly reduced in non-small cell lung cancers, pancreatic adenocarcinoma, and leiomyosarcoma compared to adjacent normal tissues.87 Additionally, mice lacking ATGL spontaneously develop lung tumors.87 These contradictory results may suggest that the role of ATGL in cancer is dependent on specific tissue types.
Targeting FA degradation
FAs provide twice as much ATP as carbohydrates, and as a result are an important energy source for cell growth, survival, and metastasis when nutrients are scarce.21,22,44,68,88 Activated FAs are broken down through FAO in mitochondria to generate acetyl-CoA, NADH, and FADH2. Acetyl-CoA feeds into the TCA cycle, while NADH and FADH2 enter the electron transport chain to produce ATP. The enzyme carnitine palmitoyl transferase 1 (CPT1) provides the first and rate-limiting step of FA transport into mitochondria for oxidation to carbon dioxide. Knockdown or inhibition of CPT1 by inhibitors (such as etomoxir and ST1326) suppresses cancer cell growth.89,90 Interestingly, c-myc-driven cancer appears to be very sensitive to CPT1 inhibition.91 These data suggest that CPT1 could represent a potential new target in anticancer treatment.
Conclusions and perspectives
Cancer cells rely on FAs for all biological activities. Our review provides an update on the expanded role of FA metabolism in cancer as well as emerging opportunities to target this process. FA metabolism can be perceived as a network of pathways notable for its plasticity. This metabolic plasticity has key implications for targeted therapies and subsequent drug resistance. The capability of cancer cells to switch between FA synthesis, FA uptake, and degradation ensures their fitness when exposed to the temporary fluctuations in the availability of nutrients and oxygen. Although chemical inhibitors for specific metabolic pathways already exist, successful therapies targeting FA metabolism may depend on an understanding of the specific metabolic plasticity associated with a particular type of cancer, and may require targeting multiple sources of FAs simultaneously, and perhaps in combination with stringent dietary regimens.
Acknowledgments
We extend a sincere apology to those whose work was not discussed or cited in this review because of space limitations. We thank Thomas Garvey for editing the manuscript. This work was supported by the Startup funds at Duke University School of Medicine to M.C. and the National Institutes of Health (Grants No. 5R01CA205001-03 and 5R01CA200853-03) to J.H.
Conflict of interest
JH reports advisory roles for Kingmed Diagnostics, Gemflower Healthcare, Optrascan and MoreHealth.
References
- 1. Fleshner N, Bagnell PS, Klotz L, et al. Dietary fat and prostate cancer. J Urol 2004;171:S19–24. doi: 10.1097/01.ju.0000107838.33623.19. [DOI] [PubMed] [Google Scholar]
- 2. Reddy BS. Dietary fat and colon cancer: Animal model studies. Lipids 1992;27:807–13. [DOI] [PubMed] [Google Scholar]
- 3. Carroll KK. Dietary fat and breast cancer. Lipids 1992;27:793–7. [DOI] [PubMed] [Google Scholar]
- 4. Ookhtens M, Kannan R, Lyon I, et al. Liver and adipose tissue contributions to newly formed fatty acids in an ascites tumor. Am J Physiol 1984;247:R146–53. doi: 10.1152/ajpregu.1984.247.1.R146. [DOI] [PubMed] [Google Scholar]
- 5. Kuhajda FP, Jenner K, Wood FD, et al. Fatty acid synthesis: A potential selective target for antineoplastic therapy. Proc Natl Acad Sci U S A 1994;91:6379–83. doi: 10.1073/pnas.91.14.6379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rohrig F, Schulze A. The multifaceted roles of fatty acid synthesis in cancer. Nat Rev Cancer 2016;16:732–49. doi: 10.1038/nrc.2016.89. [DOI] [PubMed] [Google Scholar]
- 7. Kuhajda FP. Fatty-acid synthase and human cancer: New perspectives on its role in tumor biology. Nutrition 2000;16:202–8. [DOI] [PubMed] [Google Scholar]
- 8. Swinnen JV, Brusselmans K, Verhoeven G. Increased lipogenesis in cancer cells: New players, novel targets. Curr Opin Clin Nutr Metab Care 2006;9:358–65. doi: 10.1097/01.mco.0000232894.28674.30. [DOI] [PubMed] [Google Scholar]
- 9. Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer 2007;7:763–77. doi: 10.1038/nrc2222. [DOI] [PubMed] [Google Scholar]
- 10. Santos CR, Schulze A. Lipid metabolism in cancer. FEBS J 2012;279:2610–23. doi: 10.1111/j.1742-4658.2012.08644.x. [DOI] [PubMed] [Google Scholar]
- 11. Baenke F, Peck B, Miess H, et al. Hooked on fat: The role of lipid synthesis in cancer metabolism and tumour development. Dis Model Mech 2013;6:1353–63. doi: 10.1242/dmm.011338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Currie E, Schulze A, Zechner R, et al. Cellular fatty acid metabolism and cancer. Cell Metab 2013;18:153–61. doi: 10.1016/j.cmet.2013.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Weiss L, Hoffmann GE, Schreiber R, et al. Fatty-acid biosynthesis in man, a pathway of minor importance. Purification, optimal assay conditions, and organ distribution of fatty-acid synthase. Biol Chem Hoppe Seyler 1986;367:905–12. [DOI] [PubMed] [Google Scholar]
- 14. Glatz JF, Luiken JJ, Bonen A. Membrane fatty acid transporters as regulators of lipid metabolism: Implications for metabolic disease. Physiol Rev 2010;90:367–417. doi: 10.1152/physrev.00003.2009. [DOI] [PubMed] [Google Scholar]
- 15. Sabine JR, Abraham S, Chaikoff IL. Control of lipid metabolism in hepatomas: Insensitivity of rate of fatty acid and cholesterol synthesis by mouse hepatoma BW7756 to fasting and to feedback control. Cancer Res 1967;27:793–9. [PubMed] [Google Scholar]
- 16. Metallo CM, Gameiro PA, Bell EL, et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 2011;481:380–4. doi: 10.1038/nature10602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mullen AR, Wheaton WW, Jin ES, et al. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature 2011;481:385–8. doi: 10.1038/nature10642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wise DR, Ward PS, Shay JE, et al. Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of alpha-ketoglutarate to citrate to support cell growth and viability. Proc Natl Acad Sci U S A 2011;108:19611–6. doi: 10.1073/pnas.1117773108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yen CL, Stone SJ, Koliwad S, et al. Thematic review series: Glycerolipids. DGAT enzymes and triacylglycerol biosynthesis. J Lipid Res 2008;49:2283–301. doi: 10.1194/jlr.R800018-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Farese RV Jr, Walther TC. Lipid droplets finally get a little R-E-S-P-E-C-T. Cell 2009;139:855–60. doi: 10.1016/j.cell.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pascual G, Avgustinova A, Mejetta S, et al. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature 2017;541:41–5. doi: 10.1038/nature20791. [DOI] [PubMed] [Google Scholar]
- 22. Chen M, Zhang J, Sampieri K, et al. An aberrant SREBP-dependent lipogenic program promotes metastatic prostate cancer. Nat Genet 2018;50:206–18. doi: 10.1038/s41588-017-0027-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Watt MJ, Clark AK, Selth LA, et al. Suppressing fatty acid uptake has therapeutic effects in preclinical models of prostate cancer. Sci Transl Med 2019;11. doi: 10.1126/scitranslmed.aau5758. [DOI] [PubMed] [Google Scholar]
- 24. Yue S, Li J, Lee SY, et al. Cholesteryl ester accumulation induced by PTEN loss and PI3K/AKT activation underlies human prostate cancer aggressiveness. Cell Metab 2014;19:393–406. doi: 10.1016/j.cmet.2014.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ackerman D, Simon MC. Hypoxia, lipids, and cancer: Surviving the harsh tumor microenvironment. Trends Cell Biol 2014;24:472–8. doi: 10.1016/j.tcb.2014.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ariyama H, Kono N, Matsuda S, et al. Decrease in membrane phospholipid unsaturation induces unfolded protein response. J Biol Chem 2010;285:22027–35. doi: 10.1074/jbc.M110.126870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fritz V, Benfodda Z, Rodier G, et al. Abrogation of de novo lipogenesis by stearoyl-CoA desaturase 1 inhibition interferes with oncogenic signaling and blocks prostate cancer progression in mice. Mol Cancer Ther 2010;9:1740–54. doi: 10.1158/1535-7163.Mct-09-1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Holder AM, Gonzalez-Angulo AM, Chen H, et al. High stearoyl-CoA desaturase 1 expression is associated with shorter survival in breast cancer patients. Breast Cancer Res Treat 2013;137:319–27. doi: 10.1007/s10549-012-2354-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Huang GM, Jiang QH, Cai C, et al. SCD1 negatively regulates autophagy-induced cell death in human hepatocellular carcinoma through inactivation of the AMPK signaling pathway. Cancer Lett 2015;358:180–90. doi: 10.1016/j.canlet.2014.12.036. [DOI] [PubMed] [Google Scholar]
- 30. von Roemeling CA, Marlow LA, Wei JJ, et al. Stearoyl-CoA desaturase 1 is a novel molecular therapeutic target for clear cell renal cell carcinoma. Clin Cancer Res 2013;19:2368–80. doi: 10.1158/1078-0432.Ccr-12-3249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Huang J, Fan XX, He J, et al. SCD1 is associated with tumor promotion, late stage and poor survival in lung adenocarcinoma. Oncotarget 2016;7:39970–9. doi: 10.18632/oncotarget.9461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Griffiths B, Lewis CA, Bensaad K, et al. Sterol regulatory element binding protein-dependent regulation of lipid synthesis supports cell survival and tumor growth. Cancer Metab 2013;1:3. doi: 10.1186/2049-3002-1-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Williams KJ, Argus JP, Zhu Y, et al. An essential requirement for the SCAP/SREBP signaling axis to protect cancer cells from lipotoxicity. Cancer Res 2013;73:2850–62. doi: 10.1158/0008-5472.Can-13-0382-t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kamphorst JJ, Cross JR, Fan J, et al. Hypoxic and Ras-transformed cells support growth by scavenging unsaturated fatty acids from lysophospholipids. Proc Natl Acad Sci U S A 2013;110:8882–7. doi: 10.1073/pnas.1307237110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ackerman D, Tumanov S, Qiu B, et al. Triglycerides promote lipid homeostasis during hypoxic stress by balancing fatty acid saturation. Cell Rep 2018;24:2596–605.e2595. doi: 10.1016/j.celrep.2018.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zalba S, Ten Hagen TL. Cell membrane modulation as adjuvant in cancer therapy. Cancer Treat Rev 2017;52:48–57. doi: 10.1016/j.ctrv.2016.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Golden GM, McKie JE, Potts RO. Role of stratum corneum lipid fluidity in transdermal drug flux. J Pharm Sci 1987;76:25–8. [DOI] [PubMed] [Google Scholar]
- 38. Schaeffer BE, Curtis AS. Effects on cell adhesion and membrane fluidity of changes in plasmalemmal lipids in mouse L929 cells. J Cell Sci 1977;26:47–55. [DOI] [PubMed] [Google Scholar]
- 39. Zhao W, Prijic S, Urban BC, et al. Candidate antimetastasis drugs suppress the metastatic capacity of breast cancer cells by reducing membrane fluidity. Cancer Res 2016;76:2037–49. doi: 10.1158/0008-5472.Can-15-1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sok M, Sentjurc M, Schara M, et al. Cell membrane fluidity and prognosis of lung cancer. Ann Thorac Surg 2002;73:1567–71. doi: 10.1016/s0003-4975(02)03458-6. [DOI] [PubMed] [Google Scholar]
- 41. Rysman E, Brusselmans K, Scheys K, et al. De novo lipogenesis protects cancer cells from free radicals and chemotherapeutics by promoting membrane lipid saturation. Cancer Res 2010;70:8117–26. doi: 10.1158/0008-5472.Can-09-3871. [DOI] [PubMed] [Google Scholar]
- 42. Schafer ZT, Grassian AR, Song L, et al. Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature 2009;461:109–13. doi: 10.1038/nature08268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Carracedo A, Weiss D, Leliaert AK, et al. A metabolic prosurvival role for PML in breast cancer. J Clin Invest 2012122:3088–100. doi: 10.1172/jci62129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Nieman KM, Kenny HA, Penicka CV, et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat Med 2011;17:1498–503. doi: 10.1038/nm.2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Nomura DK, Long JZ, Niessen S, et al. Monoacylglycerol lipase regulates a fatty acid network that promotes cancer pathogenesis. Cell 2010;140:49–61. doi: 10.1016/j.cell.2009.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lee CK, Jeong SH, Jang C, et al. Tumor metastasis to lymph nodes requires YAP-dependent metabolic adaptation. Science 2019;363:644–9. doi: 10.1126/science.aav0173. [DOI] [PubMed] [Google Scholar]
- 47. Horton JD, Shah NA, Warrington JA, et al. Combined analysis of oligonucleotide microarray data from transgenic and knockout mice identifies direct SREBP target genes. Proc Natl Acad Sci U S A 2003;100:12027–32, 10.1073/pnas.1534923100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kamisuki S, Mao Q, Abu-Elheiga L, et al. A small molecule that blocks fat synthesis by inhibiting the activation of SREBP. Chem Biol 2009;16:882–92. doi: 10.1016/j.chembiol.2009.07.007. [DOI] [PubMed] [Google Scholar]
- 49. Li X, Chen YT, Hu P, Huang WC. Fatostatin displays high antitumor activity in prostate cancer by blocking SREBP-regulated metabolic pathways and androgen receptor signaling. Mol Cancer Ther 2014;13:855–66. doi: 10.1158/1535-7163.Mct-13-0797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chen W, Chen G, Head DL, et al. Enzymatic reduction of oxysterols impairs LXR signaling in cultured cells and the livers of mice. Cell Metab 2007;5:73–9. doi: 10.1016/j.cmet.2006.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Carvalho MA, Zecchin KG, Seguin F, et al. Fatty acid synthase inhibition with Orlistat promotes apoptosis and reduces cell growth and lymph node metastasis in a mouse melanoma model. Int J Cancer 2008;123:2557–65. doi: 10.1002/ijc.23835. [DOI] [PubMed] [Google Scholar]
- 52. Pizer ES, Thupari J, Han WF, et al. Malonyl-coenzyme-a is a potential mediator of cytotoxicity induced by fatty-acid synthase inhibition in human breast cancer cells and xenografts. Cancer Res 2000;60:213–8. [PubMed] [Google Scholar]
- 53. Pizer ES, Wood FD, Heine HS, et al. Inhibition of fatty acid synthesis delays disease progression in a xenograft model of ovarian cancer. Cancer Res 1996;56:1189–93. [PubMed] [Google Scholar]
- 54. Zadra G, Ribeiro CF, Chetta P, et al. Inhibition of de novo lipogenesis targets androgen receptor signaling in castration-resistant prostate cancer. Proc Natl Acad Sci U S A 2019;116:631–40. doi: 10.1073/pnas.1808834116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Knobloch M, Braun SM, Zurkirchen L, et al. Metabolic control of adult neural stem cell activity by Fasn-dependent lipogenesis. Nature 2013;493:226–30. doi: 10.1038/nature11689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Loftus TM, Jaworsky DE, Frehywot GL, et al. Reduced food intake and body weight in mice treated with fatty acid synthase inhibitors. Science 2000;288:2379–81. doi: 10.1126/science.288.5475.2379. [DOI] [PubMed] [Google Scholar]
- 57. Jiang L, Xiao L, Sugiura H, et al. Metabolic reprogramming during TGFbeta1-induced epithelial-to-mesenchymal transition. Oncogene 2015;34:3908–16. doi: 10.1038/onc.2014.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hatzivassiliou G, Zhao F, Bauer DE, et al. ATP citrate lyase inhibition can suppress tumor cell growth. Cancer Cell 2005;8:311–21. doi: 10.1016/j.ccr.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 59. Migita T, Narita T, Nomura K, et al. ATP citrate lyase: Activation and therapeutic implications in non-small cell lung cancer. Cancer Res 2008;68:8547–54. doi: 10.1158/0008-5472.Can-08-1235. [DOI] [PubMed] [Google Scholar]
- 60. Beckers A, Organe S, Timmermans L, et al. Chemical inhibition of acetyl-CoA carboxylase induces growth arrest and cytotoxicity selectively in cancer cells. Cancer Res 2007;67:8180–7. doi: 10.1158/0008-5472.Can-07-0389. [DOI] [PubMed] [Google Scholar]
- 61. Wellen KE, Hatzivassiliou G, Sachdeva UM, et al. ATP-citrate lyase links cellular metabolism to histone acetylation. Science 2009;324:1076–80. doi: 10.1126/science.1164097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ros S, Santos CR, Moco S, et al. Functional metabolic screen identifies 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 4 as an important regulator of prostate cancer cell survival. Cancer Discov 2012;2:328–43. doi: 10.1158/2159-8290.Cd-11-0234. [DOI] [PubMed] [Google Scholar]
- 63. Marien E, Meister M, Muley T, et al. Phospholipid profiling identifies acyl chain elongation as a ubiquitous trait and potential target for the treatment of lung squamous cell carcinoma. Oncotarget 2016;7:12582–97. doi: 10.18632/oncotarget.7179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Mason P, Liang B, Li L, et al. SCD1 inhibition causes cancer cell death by depleting mono-unsaturated fatty acids. PLoS One 2012;7e33823. doi: 10.1371/journal.pone.0033823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Peck B, Schug ZT, Zhang Q, et al. Inhibition of fatty acid desaturation is detrimental to cancer cell survival in metabolically compromised environments. Cancer Metab 2016;4:6. doi: 10.1186/s40170-016-0146-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Potze L, Di Franco S, Grandela C, et al. Betulinic acid induces a novel cell death pathway that depends on cardiolipin modification. Oncogene 2016;35:427–37. doi: 10.1038/onc.2015.102. [DOI] [PubMed] [Google Scholar]
- 67. Vriens K, Christen S, Parik S, et al. Evidence for an alternative fatty acid desaturation pathway increasing cancer plasticity. Nature 2019;566:403–6. doi: 10.1038/s41586-019-0904-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Zhang M, Di Martino JS, Bowman RL, et al. Adipocyte-derived lipids mediate melanoma progression via FATP proteins. Cancer Discov 2018;8:1006–25. doi: 10.1158/2159-8290.Cd-17-1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Cui M, Xiao Z, Wang Y, et al. Long noncoding RNA HULC modulates abnormal lipid metabolism in hepatoma cells through an miR-9-mediated RXRA signaling pathway. Cancer Res 2015;75:846–57. doi: 10.1158/0008-5472.Can-14-1192. [DOI] [PubMed] [Google Scholar]
- 70. Wang Y, Cai X, Zhang S, et al. HBXIP up-regulates ACSL1 through activating transcriptional factor Sp1 in breast cancer. Biochem Biophys Res Commun 2017;484:565–71. doi: 10.1016/j.bbrc.2017.01.126. [DOI] [PubMed] [Google Scholar]
- 71. Huang W, Jin Y, Yuan Y, et al. Validation and target gene screening of hsa-miR-205 in lung squamous cell carcinoma. Chin Med J (Engl) 2014;127:272–8. [PubMed] [Google Scholar]
- 72. Cao Y, Dave KB, Doan TP, Prescott SM. Fatty acid CoA ligase 4 is up-regulated in colon adenocarcinoma. Cancer Res 2001;61:8429–34. [PubMed] [Google Scholar]
- 73. Sung YK, Hwang SY, Park MK, et al. Fatty acid-CoA ligase 4 is overexpressed in human hepatocellular carcinoma. Cancer Sci 2003;94:421–4. doi: 10.1111/j.1349-7006.2003.tb01458.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Wu X, Deng F, Li Y, et al. ACSL4 promotes prostate cancer growth, invasion and hormonal resistance. Oncotarget 2015;6:44849‑63. doi: 10.18632/oncotarget.6438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Wu X, Li Y, Wang J, et al. Long chain fatty acyl-CoA synthetase 4 is a biomarker for and mediator of hormone resistance in human breast cancer. PLoS One 2013;8:e77060. doi: 10.1371/journal.pone.0077060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Ye X, Zhang Y, Wang X, et al. Tumor-suppressive functions of long-chain acyl-CoA synthetase 4 in gastric cancer. IUBMB Life 2016;68:320–7. doi: 10.1002/iub.1486. [DOI] [PubMed] [Google Scholar]
- 77. Dixon SJ, Winter GE, Musavi LS, et al. Human haploid cell genetics reveals roles for lipid metabolism genes in nonapoptotic cell death. ACS Chem Biol 2015;10:1604–9. doi: 10.1021/acschembio.5b00245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Doll S, Proneth B, Tyurina YY, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol 2017;13:91–8. doi: 10.1038/nchembio.2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Accioly MT, Pacheco P, Maya-Monteiro CM, et al. Lipid bodies are reservoirs of cyclooxygenase-2 and sites of prostaglandin-E2 synthesis in colon cancer cells. Cancer Res 2008;68:1732–40. doi: 10.1158/0008-5472.Can-07-1999. [DOI] [PubMed] [Google Scholar]
- 80. Bensaad K, Favaro E, Lewis CA, et al. Fatty acid uptake and lipid storage induced by HIF-1alpha contribute to cell growth and survival after hypoxia-reoxygenation. Cell Rep 2014;9:349–65. doi: 10.1016/j.celrep.2014.08.056. [DOI] [PubMed] [Google Scholar]
- 81. Bozza PT, Viola JP. Lipid droplets in inflammation and cancer. Prostaglandins Leukot Essent Fatty Acids 2010;82:243–50. doi: 10.1016/j.plefa.2010.02.005. [DOI] [PubMed] [Google Scholar]
- 82. Cabodevilla AG, Sánchez-Caballero L, Nintou E, et al. Cell survival during complete nutrient deprivation depends on lipid droplet-fueled beta-oxidation of fatty acids. J Biol Chem 2013;288:27777–88. doi: 10.1074/jbc.M113.466656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Cotte AK, Aires V, Fredon M, et al. Lysophosphatidylcholine acyltransferase 2-mediated lipid droplet production supports colorectal cancer chemoresistance. Nat Commun 2018;9:322. doi: 10.1038/s41467-017-02732-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Lass A, Zimmermann R, Oberer M, Zechner R. Lipolysis - a highly regulated multi-enzyme complex mediates the catabolism of cellular fat stores. Prog Lipid Res 2011;50:14–27. doi: 10.1016/j.plipres.2010.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Wang YY, Attané C, Milhas D, et al. Mammary adipocytes stimulate breast cancer invasion through metabolic remodeling of tumor cells. JCI Insight 2017;2:e87489. doi: 10.1172/jci.insight.87489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Zagani R, El-Assaad W, Gamache I, Teodoro JG. Inhibition of adipose triglyceride lipase (ATGL) by the putative tumor suppressor G0S2 or a small molecule inhibitor attenuates the growth of cancer cells. Oncotarget 2015;6:28282–95. doi: 10.18632/oncotarget.5061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Al-Zoughbi W, Pichler M, Gorkiewicz G, et al. Loss of adipose triglyceride lipase is associated with human cancer and induces mouse pulmonary neoplasia. Oncotarget 2016;7:33832–40. doi: 10.18632/oncotarget.9418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Beyaz S, Mana MD, Roper J, et al. High-fat diet enhances stemness and tumorigenicity of intestinal progenitors. Nature 2016;531:53–8. doi: 10.1038/nature17173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Schlaepfer IR, Rider L, Rodrigues LU, et al. Lipid catabolism via CPT1 as a therapeutic target for prostate cancer. Mol Cancer Ther 2014;13:2361–71. doi: 10.1158/1535-7163.Mct-14-0183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Ricciardi MR, Mirabilii S, Allegretti M, et al. Targeting the leukemia cell metabolism by the CPT1a inhibition: Functional preclinical effects in leukemias. Blood 2015;126:1925–9. doi: 10.1182/blood-2014-12-617498. [DOI] [PubMed] [Google Scholar]
- 91. Pacilli A, Calienni M, Margarucci S, et al. Carnitine-acyltransferase system inhibition, cancer cell death, and prevention of myc-induced lymphomagenesis. J Natl Cancer Inst 2013;105:489–98. doi: 10.1093/jnci/djt030. [DOI] [PubMed] [Google Scholar]