

Abstract
This is an invited editorial on the editorial on the original manuscript HEP4‐19‐0099.R1 “Liver‐targeted TLR7 agonist combined with Entecavir promotes a functional cure in the woodchuck model of chronic HBV.”
Abbreviations
- anti‐HBs
antibody to hepatitis B surface antigen
- CHB
chronic hepatitis B
- HBV
hepatitis B virus
- IFN
interferon
- IL
interleukin
- NK
natural killer
- TLR
toll‐like receptor
- WHeAg
woodchuck hepatitis e antigen
- WHV
woodchuck hepatitis virus
Although nucleos(t)ide analogue therapy is efficient in reducing hepatitis B virus (HBV) replication and liver inflammation in patients with chronic hepatitis B (CHB), the therapy rarely achieves functional cure of HBV, a virological situation characterized by negativity of HBV virological parameters and antibody to hepatitis B surface antigen (anti‐HBs) seroconversion. This is why new therapies that not only inhibit viral replication but stimulate the defective antiviral immunity of CHB patients are being developed.
Toll‐like receptor (TLR) (TLR‐7, TLR‐8, TLR‐9) and retinoic acid–inducible gene 1 (RIG‐I) agonists are the major representatives of such class of immune therapy. They are designed to activate host antiviral immunity in a non–antigen‐specific way. Their rationale resides in the fact that even though HBV replication is not activating innate immunity in hepatocytes, it is responsive to the antiviral effect of interferons (IFNs) and other cytokines. Thus, activating type I IFN responses directly in hepatocytes (by RIG‐I agonists) or intrahepatic immune cells (by TLR‐7, TLR‐8, TLR‐9 agonists) should inhibit HBV replication. Furthermore, the localized activation of IFN type I responses may trigger a cascade of immunological events (e.g., release of cytokines, alteration of antigen presentation) that lead to natural killer (NK) cell activation and possibly recovery of HBV‐specific T cells and B cells (Fig. 1).1
Figure 1.
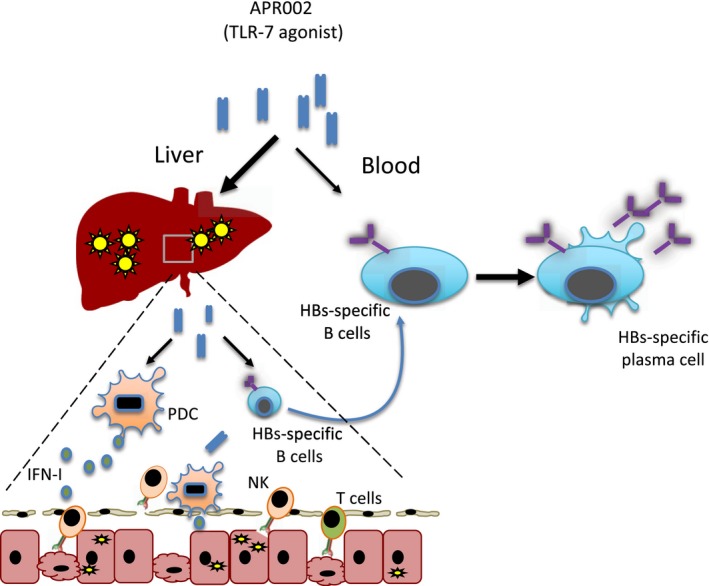
TLR‐7 agonist's immunological mechanisms. TLR‐7 agonist (as APR002) acts by targeting intrahepatic plasmacytoid dendritic cells (PDCs) and B cells. TLR‐7 agonist triggers type I IFN production in PDC. Type I IFNs have a direct antiviral effect on HBV‐infected hepatocytes but also activate intrahepatic NK or T cells. TLR‐7 agonist on B cells can induce their functional maturation to plasma cells (both in the liver and peripheral blood).
In this issue of Hepatology Communications, Korolowizc et al. published a study describing the safety and therapeutic efficacy of a new TLR‐7‐activating compound (APR002) used alone or in association with entecavir in the woodchuck model of woodchuck hepatitis virus (WHV) infection. Therapy with TLR‐7 agonists has already been reported in chimpanzees,2 woodchucks,3 and humans.4, 5 The difference with this new compound (APR002) in comparison with others (i.e., GS‐9620) is the fact that APR002 has been designed to have preferentially intrahepatic delivery. By minimizing systemic exposure, the authors argued that it should reduce the poor tolerability observed in animal models treated previously with TLR‐7 agonist. The hepato‐selective design of APR002 is principally mediated by active uptake by the organic‐anion‐transporting polypeptide 1B1/3 transporters, highly expressed on the sinusoidal membrane of hepatocytes. Korolowizc et al. first showed in mice that APR002 is indeed preferentially localized in the liver with a serum to liver ratio of approximately 30, an undeniable improvement in comparison to the ratio of approximately 6 for GS‐9620. They also reported in mice that APR002 induces lower release of proinflammatory cytokines (interleukin [IL]‐6 and tumor necrosis factor α) in comparison to GS‐9620 and then demonstrated APR002’s ability to activate IFN‐α genes in the liver of normal uninfected woodchucks. APR002’s therapeutic efficacy was then tested: Four comparable cohorts of woodchucks chronically infected with WHV (5 woodchucks per group) were treated either with entecavir alone, APR002 monotherapy, or a complex combination of entecavir and different doses of APR002 (see table 1 of the article for posology).
Treatment with APR002 was well tolerated. The weekly oral dosing of APR002 for 12 weeks did not induce systemic immunotoxicity or changes in clinical chemistry and hematological parameters. Increases of liver enzymes were observed primarily but not exclusively in the APR002‐treated animals and reversed in all animals at the end of the study. However, elevated liver enzymes were also observed in a single animal treated with APR002, only it was found dead. The mortality was attributed to preexisting kidney disfunction.
As expected, APR002 treatment (alone or in combination) induced a clear activation of innate immunity in the liver and blood of infected animals (tested by measuring the activation of IFN‐stimulated genes) and clear reduction of WHV parameters (WHV DNA and WH surface antigen [WHsAg] and WH e antigen [WHeAg]) that was less pronounced than in the animals treated with entecavir only. Remarkably, only in the animals treated with combination therapy (groups 3 and 4) did the authors observe a sustained negativity of WHV antigen and seroconversion to anti‐HBs (3 animals), which occurred approximately 8 to 10 weeks after therapy.
This small but very well conducted study provides important new information that might help fine‐tune the treatment with these immunotherapeutic agents. First, the preferential hepato‐selectivity and the absence of signs of systemic toxicity and alteration of hematological parameters, linked with an efficient activation of intrahepatic innate immunity, suggest that APR002 might have a better safety profile than GS‐9620. Perhaps future clinical trials with APR002 in CHB patients will be designed using a drug posology that is similar to what was used in animals that seroconverted. It is important to remember that the lack of therapeutic efficacy observed in the human trials of GS‐9620 could have been principally caused by the low dose used to minimize possible side effects (a dose approximately 35 to 100 times lower than the maximum ones used in woodchucks and chimpanzees was administered to patients4, 5).
However, it appears inevitable that immunotherapies, designed to trigger intrahepatic immunity, activate liver inflammation. Despite the lack of systemic toxicity, APR002 treatment induced in many animals a transient alteration of transaminase, and this clinical perturbation should perhaps be considered a direct sign of drug activity and not an undesirable side effect. Nevertheless, this remains a problem in the design of human trials because it is difficult to fully predict the severity of hepatic flares after activation of intrahepatic innate immunity. Their severity is likely multifactorial, related to environmental, clinical, and genetic factors. A very recent publication on a case of fulminant hepatic failure in a child infected with hepatitis A virus has identified a deletion in IL‐18‐binding protein (IL‐18BP) as the cause of excessive activation of NK cells with uncontrolled killing of hepatocytes.6 If this case illustrates the importance of genetic factors in the development of severe hepatic failure after intrahepatic innate immune activation, it also shows that IL‐18BP might be the perfect antidote to control hepatic flares caused by innate immune activation, and therefore a possible new alley for future experimental trials on immunotherapy for CHB.
One other interesting observation of the Korolowizc et al. study is the fact that the virological and immunological features of the APR002‐treated woodchucks that achieved the equivalence of functional HBV cure (anti‐WHs+) indicate that the therapeutic efficacy of APR002 is linked with immunological modulation (particularly on B cells) rather than increased antiviral potency. The analysis of virological markers (WHV DNA, WHsAg, and WHeAg) in animals treated with entecavir alone or with the APR002 and entecavir combination showed that the inhibition of WHV virological markers was more marked in the entecavir‐alone group. The authors suggested that a low level of hepatic metabolism induced by APR002 reduced entecavir’s antiviral potency, because the drug needs to be transformed into its active form. Regardless of the causes of such virological findings, the fact is that the anti‐WHs seroconversion detected in animals treated with combination therapy was therefore not directly related to an increased level of viral inhibition (and type I IFN gene activation in the liver).
Therefore, even though a threshold of viral inhibition appears indispensable for subsequent seroconversion (not a single animal treated with APR002 monotherapy developed anti‐WHs antibodies), the success of the therapy in these animals is likely dependent on the ability of TLR‐7 agonists to induce functional maturation of antiviral B‐cell responses.7
The study of humoral immunity in patients with CHB has been neglected for a long time, but the development of new methods has recently permitted the study of HBV‐specific B cells at a single‐cell level8, 9 and led to the demonstration that hepatitis B surface antigen–specific B cells in CHB patients are not fully deleted but rather present functional maturation defects.
It appears that the correct use of immunotherapies (such as TLR‐7 agonist) targeting B‐cell maturation cannot be separated from more precise understanding of the requirement of HBs‐specific B‐cell restoration in the treated patients. The recent analysis of T‐cell and NK‐cell immunological parameters in GS‐9620‐treated CHB patients5 has shown that, despite the lack of therapeutic efficacy, the treatment induced a boost of HBV‐specific T‐cell responses. These data open the question of why not a single CHB patient experienced anti‐HBs seroconversion despite a partial recovery of T‐cell immunity. The features of HBV‐specific B cells were not analyzed, but the lack of patients experiencing anti‐HBs seroconversion clearly argues that a functional reconstitution of the B‐cell compartment did not occur.
As we discussed previously, the therapeutic discrepancy between TLR‐7 agonist efficacy in animal models and patients may be explained by the different quantity and frequency of drug administration tested.
However, in our opinion, one other variable that should be considered is that while TLR‐7 agonist therapy has been tested in classic adult anti‐HBe+ CHB patients, the features of the animal models (WHeAg+, barely elevated or normality of alanine aminotransferase infected for 1‐2 years and not for decades) treated with TLR‐7 agonists appear to be more similar to the HBeAg+ chronic infection (or immunotolerant subjects). We do not know whether HBV‐specific B cells might be less or differently altered, as we have seen for T cells,10 in different phases of CHB infection.
In conclusion, this carefully designed study not only reports the development of a new and potentially safer compound but stimulates immunological analysis that can help fine‐tune the treatment with TLR‐7 agonists and achieve functional HBV cure in selected CHB patients.
Supported by the National Medical Research Council (MOH‐STaR17nov‐0001 and NMRC/TCR/014‐NUHS/2015).
Potential conflict of interest: Dr. Bertoletti consults for, advises, and received grants from Gilead and Spring Bank.
See Article on Page https://doi.org/10.1002/hep4.1397
References
Author names in bold designate shared co‐first authorship.
- 1. Maini MK, Gehring AJ. The role of innate immunity in the immunopathology and treatment of HBV infection. J Hepatol 2016;64:S60‐S70. [DOI] [PubMed] [Google Scholar]
- 2. Lanford RE, Guerra B, Chavez D, Giavedoni L, Hodara VL, Brasky KM, et al. GS‐9620, an oral agonist of toll‐like receptor‐7, induces prolonged suppression of hepatitis B virus in chronically infected chimpanzees. Gastroenterology 2013;144:1508‐1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Menne S, Tumas DB, Liu KH, Thampi L, AlDeghaither D, Baldwin BH, et al. Sustained efficacy and seroconversion with the toll‐like receptor 7 agonist GS‐9620 in the woodchuck model of chronic hepatitis B. J Hepatol 2015;62:1237‐1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Janssen HLA, Brunetto MR, Kim YJ, Ferrari C, Massetto B, Nguyen A‐H, et al. Safety, efficacy and pharmacodynamics of vesatolimod (GS‐9620) in virally suppressed patients with chronic hepatitis B. J Hepatol 2018;68:431‐440. [DOI] [PubMed] [Google Scholar]
- 5. Boni C, Vecchi A, Rossi M, Laccabue D, Giuberti T, Alfieri A, et al. TLR7 agonist increases responses of hepatitis B virus‐specific T cells and natural killer cells in patients with chronic hepatitis B treated with nucleos(t)ide analogues. Gastroenterology 2018;154:1764‐1777. [DOI] [PubMed] [Google Scholar]
- 6. Belkaya S, Michailidis E, Korol CB, Kabbani M, Cobat A, Bastard P, et al. Inherited IL‐18BP deficiency in human fulminant viral hepatitis. J Exp Med 2019;158:jem.20190669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rubtsova K, Rubtsov AV, van Dyk LF, Kappler JW, Marrack P. T‐box transcription factor T‐bet, a key player in a unique type of B‐cell activation essential for effective viral clearance. Proc Natl Acad Sci U S A 2013;110:E3216‐E3224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Salimzadeh L, Le Bert N, Dutertre C‐A, Gill US, Newell EW, Frey C, et al. PD‐1 blockade partially recovers dysfunctional virus‐specific B cells in chronic hepatitis B infection. J Clin Invest 2018;128:4573‐4587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Burton AR, Pallett LJ, McCoy LE, Suveizdyte K, Amin OE, Swadling L, et al. Circulating and intrahepatic antiviral B cells are defective in hepatitis B. J Clin Invest 2018;128:4588‐4603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kennedy PTF, Sandalova E, Jo J, Gill U, Ushiro‐Lumb I, Tan AT, et al. Preserved T‐cell function in children and young adults with immune‐tolerant chronic hepatitis B. Gastroenterology 2012;143:637‐645. [DOI] [PubMed] [Google Scholar]