

Abstract
Background
Liquid biopsy based on cell-free DNA circulating in plasma has shown solid results as a non-invasive biomarker. In the present study we evaluated the utility of circulating free DNA (cfDNA) and the sub-type tumor DNA (ctDNA) in hepatocellular cancer (HCC) patients to assess therapy response and clinical outcome.
Methods
A cohort of 13 patients recruited in the context of the SORAMIC trial with unresectable, advanced HCC and different etiological and clinicopathological characteristics was included in this exploratory study. Plasma samples were collected between liver micro-intervention and beginning of sorafenib-based systemic therapy and then in correspondence of three additional follow-ups. DNA was isolated from plasma and next generation sequencing (NGS) was performed on a panel of 597 selected cancer-relevant genes.
Results
cfDNA levels showed a significant correlation with the presence of metastases and survival. In addition cfDNA kinetic over time revealed a trend with the clinical history of the patients, supporting its use as a biomarker to monitor therapy. NGS-based analysis on ctDNA identified 28 variants, detectable in different combinations at the different time points. Among the variants, HNF1A, BAX and CYP2B6 genes showed the highest mutation frequency and a significant association with the patients’ clinicopathological characteristics, suggesting a possible role as driver genes in this specific clinical setting.
Conclusions
Taken together, the results support the prognostic value of cfDNA/ctDNA in advanced HCC patients with the potential to predict therapy response. These findings support the clinical utility of liquid biopsy in advanced HCC improving individualized therapy and possible earlier identification of treatment responders.
Keywords: Liquid biopsy, Circulating tumor DNA, Hepatocellular carcinoma, Biomarkers
Background
Hepatocellular carcinoma (HCC) is the sixth most common cancer and the second leading cause of cancer-related death globally, with a median survival of only 7 months, if untreated [1–3]. Although cirrhosis, generally resulting from chronic inflammation and oxidative stress [4], is recognized as the most frequent risk factor, patients are often diagnosed with HCC in an already advanced phase, when treatment is limited and resection or transplantation are not any longer an option. This is in part due to the limited sensitivity and specificity of the standard diagnostic modalities of imaging and biomarkers, such as serum alpha-fetoprotein (AFP) [5], in the detection of small tumors during surveillance of high risk populations. Advanced HCC often shows a poor outcome, with very limited benefit from cytotoxic agents. Targeted therapy with the multi-tyrosine kinase inhibitor sorafenib has been the mainstay of systemic treatment, nevertheless resulting in modest improvements in overall survival over placebo and often only in subgroups of patients [6]. Identification of early predictors of therapy response would be highly desirable in order to move quickly patients to more effective treatments.
Circulating cell-free DNA (cfDNA) is detectable in the plasma and serum of healthy individuals as consequence of cellular necrosis or apoptosis. Cancer patients show a higher amount of cfDNA since tumor cells divide faster than normal cells, and cfDNA is released in higher proportion [7–9]. The fraction of cfDNA derived from the tumor is indicated as circulating tumor DNA (ctDNA) [10, 11]. In the last years, analysis of both cfDNA and ctDNA has gained considerable attention as novel blood biomarkers, since quantification and kinetic analysis of cfDNA [12, 13] and molecular profiling of ctDNA [14] have demonstrated both a predictive and prognostic value. Venipuncture, with capacity for serial sampling over time, may offer advantages over standard biopsy, avoiding complications from incisions and reflecting the genetic heterogeneity of the whole tumor. In addition, due to the short half-life of circulating DNA [15], genetic analysis may offer “real time” insights into the kinetic mutations arising during therapy. The prognostic value of cfDNA/ctDNA in early HCC has been previously shown [16, 17]. The aim of the present study was to evaluate if cfDNA quantification over time may be a valid monitoring strategy to predict clinical outcome in advanced HCC patients in the context of the SORAMIC trial [18, 19], and whether molecular profiling of ctDNA may track the genetic evolution of the tumor during the combined systemic and liver-directed micro-interventional treatment.
Methods
Patients and therapy
A total of 13 male patients (median age (years ± SD): 68 ± 8.91, range 46–82) from the prospective randomized multicenter phase II SORAMIC trial (EudraCT 2009-012576-27, NCT01126645) were included in this exploratory study [18, 19]. Participants were recruited between 2011 and 2016 at different European study centers. Advanced, unresectable HCC patients received radioembolization with Yttrium-90 (90Y-RE) in combination with systemic treatment with sorafenib (n = 10) or radiofrequency ablation in combination with sorafenib (n = 2) or placebo (n = 1). Treatment efficacy was evaluated based on imaging and alfa-fetoprotein (AFP) [20, 21]. The median overall survival was 23.8 months (range 11.9 – 41.4) and only two patients showed recurrence with a progression free survival of 22.3 and 26.8 months. Blood samples were collected at four different time points: after micro-interventional therapy and before starting the systemic therapy with sorafenib (T1), approximately 8 weeks after beginning of systemic therapy (T2, median numbers of days between T1 and T2: 112), at first follow up (FU) (T3, median numbers of days between T1 and T3: 224) and at second FU (T4, median numbers of days between T1 and T4: 297). The SORAMIC trial design is given in Fig. 1, where the time points relative to blood collection are indicated. The study was approved by all the involved ethical boards and conducted in accordance with the Declaration of Helsinki [22]. Before entering the study, all patients gave their written informed consent.
Fig. 1.

SORAMIC trial design. Indicated in red are the time points chosen for discovery of the variants. SNV, single nucleotide variant; InDel, insertion and deletion; R, randomization; TX, therapy; RFA, radiofrequency ablation; 90Y-RE, 90Y-radioembolization
DNA isolation, library preparation and next-generation sequencing (NGS)
Peripheral blood (5 ml) was drawn in EDTA tubes (Becton–Dickinson, Heidelberg, Germany) and processed immediately (centrifugation 3000 rpm, 5 min, 4 °C) to collect plasma and buffy coats, which were aliquoted and stored at − 80 °C until further use. Plasma was used for extraction of cell-free DNA (cfDNA) and buffy coats were used for extraction of genomic DNA (gDNA). DNA extraction, quality control and NGS were performed at Eurofins Genomics GmbH (Konstanz, Germany). The quality and quantity of the extracted DNA was assessed via electropherograms and fluorometer concentration determinations (for details, see Additional file 1: Figure S1). The extracted DNA was used for library preparation and subsequent sequencing. A summary of the depth of coverage is shown in Additional file 2: Table S1.
The genes were selected according to their importance and in accordance with clinical guidelines such as the European Society for Medical Oncology (ESMO), the National Comprehensive Cancer Network (NCCN), and the College of American Pathologists (CAP).
NGS was performed on Illumina platform using the 150 paired-end mode. The sequencing reads were quality checked and mapped to the human genome using BWA with default parameters [23]. After target processing and alignment refinement, variants (single nucleotide variants, SNV; Insertions and deletions, InDel; and gene fusions according to Chimer DB 2.0 [24]) were discovered and annotated. To distinguish somatic from germline mutations, genomic DNA extracted from buffy coats was analyzed in parallel (Additional file 3: Table S2). The whole experimental workflow is schematically described in Fig. 2.
Fig. 2.

Schematic description of the experimental workflow. Peripheral blood was collected and processed to separate plasma from buffy coat containing white blood cells (WBC). Whole circulating DNA was extracted and used for library preparation. Next generation sequencing was performed on Illumina platform using the 150 paired-end (PE) mode. To distinguish somatic from germline mutations, genomic DNA extracted from WBC was analyzed in parallel
Statistical analysis
All statistical analyses were performed using IBM SPSS Statistics 21.0.0 (IBM Corporation, New York, N.Y.). Results for numerical data are given as median together with minimum and maximum of the sample (i.e. range). The correlation between tumor size and cfDNA concentrations at different time points were analyzed using the Pearson’s correlation coefficient and corresponding significance test. cfDNA at different time points were compared between patient groups featuring different clinicopathological characteristics using the non-parametric Mann–Whitney U test. The association between variants at different time points and patients’ clinical characteristics was evaluated using the Fisher’s exact test. Overall survival (OS) was analyzed using the Kaplan–Meier method, and survival estimates in different groups were compared using the log-rank test. Patients were clustered in two groups corresponding to high and low cfDNA concentration (high cfDNA = over the median, low cfDNA = below the median). OS was calculated from the date of primary tumor diagnosis to the date of death or the date of the last clinical follow-up. All tests were carried out two-sided. Due to the low sample size of n = 13 patients, no alpha adjustment was made. All statistical tests are interpreted at a significance level of alpha = 5% and according results are considered exploratory.
Results
Clinicopathological characteristics of the patients
Within the cohort chosen for this study, patients were divided in two subgroups: patients with an age below 65, who did not present with liver cirrhosis at time of recruitment, did not report any past alcohol abuse and, based on the SORAMIC trial [19] results, responded better to therapies; and patients over 65, who presented with liver cirrhosis at time of recruitment, reported past alcohol abuse and responded worse to therapies. None of the patients were positive for Hepatitis B, only 1 was positive for Hepatitis C, and 4 presented non-alcoholic steatohepatitis (NASH). Three patients (33.3%, for n = 4 the number of lesions was not available) presented with diffuse disease (> 20 lesions), while among the others the median number of lesions was 2 (range 0–28) and the median size of the lesions was 49 mm (range 8–160 mm). Three patients (23.0%) showed portal vein infiltration, all of them presented with a Child–Pugh A liver function, 46.1% (n = 6) of the patients were Barcelona Clinic Liver Cancer Staging (BCLC) C, while the remaining were A (n = 3, 23.0%) or B (n = 4, 30.7%). Four patients (30.7%) presented lymph node metastases and one of them (7.7%) also bone metastases. Two patients (66.6%) showed recurrence at 2 years FU. A summary of the clinicopathological characteristics of the patients is given in Table 1.
Table 1.
Clinicopathological characteristics of the patients
| ID | Age | Liver cirrhosis | HBV | HCV | Alcohol abuse | Other | Number of lesions (> 20 = DD) | Max. diameter largest lesion (mm) | PVI | CP | CP points | BCLC stage | Therapy | M | R | death | OS (months) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A | 46 | No | Neg | Neg | No | Nash | 28 | 34 | No | A | 5 | C |
RE SRFB |
No | No | 0 | 40.7 |
| B | 60 | No | Neg | Neg | No | – | 99 | 160 | No | A | 6 | C |
RE SRFB |
Yes | No | 1 | 38.6 |
| C | 61 | No | Neg | Neg | No | Nash | 99 | 8 | No | A | 5 | B |
RE SRFB |
No | No | 0 | 23.4 |
| D | 61 | No | Neg | Neg | No | Nash | na | 88 | No | A | 5 | B |
RE SRFB |
No | No | 0 | 16.1 |
| E | 59 | No | Neg | pos | No | – | na | 55 | Yes | A | 5 | C |
RE SRFB |
Yes | No | 1 | 11.7 |
| F | 68 | Yes | Neg | Neg | Yes | Nash | 3 | 91 | Yes | A | 6 | C |
RE SRFB |
Yes | No | 1 | 26.8 |
| G | 75 | Yes | Neg | Neg | Yes | – | na | na | No | A | 6 | B |
RE SRFB |
No | No | 1 | 13.7 |
| H | 68 | Yes | Neg | Neg | Yes | – | 3 | 15 | Yes | A | 5 | C |
RE SRFB |
No | No | 1 | 16.8 |
| I | 66 | Yes | Neg | Neg | Yes | – | na | 65 | No | A | 5 | C |
RE SRFB |
Yes | No | 1 | 13.0 |
| J | 69 | Yes | Neg | Neg | Yes | – | 2 | 75 | No | A | 5 | B |
RE SRFB |
No | No | 1 | 17.5 |
| K | 70 | Yes | Neg | Neg | Yes | – | 1 | 43 | No | A | 5 | A |
RFA PLC |
No | Yes | 1 | 27.1 |
| L | 82 | Yes | Neg | Neg | Yes | – | 1 | 40 | No | A | 6 | A |
RFA SRFB |
No | No | 1 | 26.3 |
| M | 73 | Yes | Neg | Neg | Yes | – | 1 | 33 | No | A | 5 | A |
RFA SRFB |
No | Yes | 1 | 32.5 |
HBV, hepatitis B virus; HCV, hepatitis C virus; DD, diffuse disease; Nash, non-alcoholic steatohepatitis; PVI, portal vein infiltration; CP, Child–Pugh, BCLC, Barcelona Clinic Liver Cancer; RE, 90Y-Radioembolization; SRFB, sorafenib; PLCB, placebo; M, metastases; R, recurrence; OS, overall survival, in months; na, not available
Correlation between cfDNA levels, patient’s clinicopathological characteristics and clinical outcome
cfDNA was isolated from plasma collected at the 4 time points (T1, T2, T3, and T4) during the SORAMIC trial. Nine of the 52 cfDNA samples (17.3%) were excluded from the subsequent analysis due to high genomic DNA contamination or because the concentration was below the threshold (Additional file 1: Figure S1). In the remaining samples, the amount of cfDNA at the different time points was ranging between 2.04 ng/ml to 160.75 ng/ml with a median of 10.2 ng/ml at T1, 15.3 ng/ml at T2, 9.3 ng/ml at T3 and 13.3 ng/ml at T4 (Table 2). We did not observe a significant correlation between cfDNA concentration at T1 and tumor volume or between cfDNA concentrations at any time points and patient’s clinical characteristics (all p > 0.1) (data not shown). On the contrary, we did observe a difference between the concentrations of cfDNA in the presence or absence of metastases, which was significant (p = 0.012) or borderline significant (p = 0.073) at T1 and T2, respectively (Fig. 3). The relative amount of cfDNA at the different time points was also evaluated with respect to overall survival (OS). Patients were grouped according to high or low cfDNA levels with respect to the median values at the different time points. We found a borderline significance at the latest time points (T3, p = 0.057; T4, p = 0.095), suggesting that patients keeping high levels of plasmatic cfDNA while receiving systemic therapy might have a worse outcome (Fig. 4). On the contrary we did not find any association between cfDNA levels at earlier time points and OS (all p > 0.1). The fact the we found a borderline significance might be due to the low number of patients included in the analysis, therefore it will be necessary to validate these preliminary results with a bigger cohort of patients.
Table 2.
Amount of cfDNA (ng/ml) and AFP (ng/ml) in plasma samples at the different time points
| Patients | T1 | T2 | T3 | T4 | Median [cfDNA] |
|---|---|---|---|---|---|
| cfDNA | cfDNA | cfDNA | cfDNA | ||
| AFP | AFP | AFP | AFP | ||
| A | na | 39.44 | 8.39 | 10.11 | 10.1 |
| 85.00 | 11.00 | 13.00 | 6.00 | ||
| B | 12.78 | 17.68 | na | 15.30 | 15.3 |
| 373.00 | 75.00 | 33.00 | 45.00 | ||
| C | 4.35 | 7.34 | 2.72 | 3.40 | 3.9 |
| 2.00 | 3.00 | 3.00 | 3.00 | ||
| D | 2.81 | 14.96 | 4.18 | 9.07 | 6.6 |
| 4.00 | 3.00 | 9.00 | 16.00 | ||
| E | 30.63 | 15.30 | 17.20 | 38.62 | 23.9 |
| 3.00 | 3.00 | 10.00 | 42.10 | ||
| F | 28.33 | 57.12 | 30.60 | 14.73 | 29.5 |
| 2.00 | 4.00 | na | 4.00 | ||
| G | 10.20 | 11.90 | 11.56 | 20.94 | 11.7 |
| 4.00 | 6.00 | 8.00 | 4.00 | ||
| H | 1.81 | 5.64 | 9.38 | 11.02 | 7.5 |
| 13.00 | 19.00 | 30.00 | 72.00 | ||
| I | 30.37 | 24.28 | 7.41 | 13.33 | 18.8 |
| 1681.00 | 1685.00 | 991.00 | 967.00 | ||
| J | 7.96 | na | 12.60 | 160.75 | 12.6 |
| 6154.00 | 2410.00 | 8732.00 | 3855.00 | ||
| K | 2.04 | 4.42 | 7.28 | 9.25 | 5.8 |
| 4.00 | 3.00 | 3.00 | 3.00 | ||
| L | 20.26 | 16.68 | 16.68 | 16.68 | 16.8 |
| 7.00 | 5.00 | 6.00 | 7.00 | ||
| M | na | na | na | 8.98 | 8.98 |
| 9.00 | 4.00 | 4.00 | 6.00 | ||
| Median [cfDNA] | 10.2 | 15.3 | 9.3 | 13.3 |
cfDNA median values are given for each patient at the four time points (last column) or for each time point for all patients (last row)
na, not available
Fig. 3.
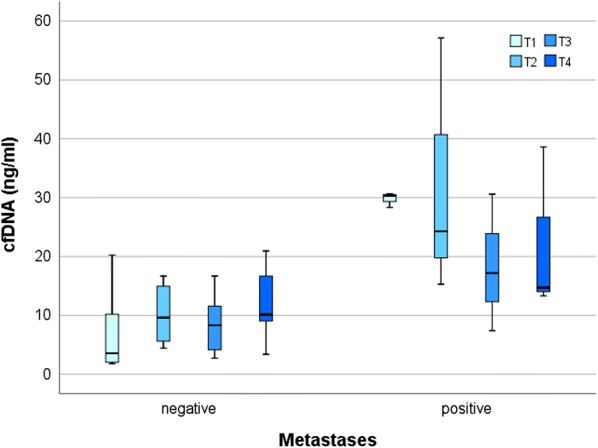
cfDNA concentrations at different time points and presence of metastases. The analysis shows that patients presenting metastases when recruited in the trial, had still a significant higher amount of plasmatic cfDNA in the timeframe between micro-interventional therapy and the beginning of sorafenib-based systemic therapy (T1, p = 0.012). Borderline significance (p = 0.073) was found after the beginning of systemic therapy (T2), while no significant difference was found at the two later time points. Comparison was performed using the Mann–Whitney U test (* ≤ 0.05)
Fig. 4.

Survival plots for HCC patients grouped according to the amount of cfDNA at the different time points. Patients’ total population was grouped according to the corresponding cfDNA concentration with respect to the median value found at each time point. Higher cfDNA concentration at later time points (T3 and T4) was showing a trend with a shorter OS (p = 0.057 and p = 0.095) while no association (p > 0.1) was found and earlier time points (T1 and T2). High and low cfDNA levels were defined as being above or below the median values at the different time points. OS was analyzed using the Kaplan–Meier method and survival estimates in the different groups were compared using the log-rank test
cfDNA in comparison to the conventional biomarker AFP
AFP is detectable in as many as 30% of HCC patients at time of diagnosis and usually remains low during the course of the disease, even with advanced HCC [20]. AFP > 400–500 ng/ml supports diagnosis of HCC [21]. At the considered time points, only 2 (15.4%) of the patients showed high AFP values (> 400 ng/ml), 4 (30.8%) showed values between 20 and 400 ng/ml and 7 (53.8%) showed values between 2 and 20 ng/ml (Table 2). It has been already shown that cfDNA has clinical diagnostic relevance, reflecting therapy response [25]. We report here in more detail three vignettes of patients with evidence of disease progression, for which cfDNA offered earlier monitoring value than conventional biomarkers such as AFP (Fig. 5). A concentration of 400 ng/ml was chosen as cut-off value. The first patient (patient B) entered in the trial with a diffuse disease in the liver and lymph nodes and bone metastases; he died after 38.6 months from enrolment in the SORAMIC trial. AFP concentration was high (373 ng/ml) before starting sorafenib treatment; however it decreased very rapidly below 50 ng/ml and remained low while assuming sorafenib, suggesting efficacy of treatment. On the contrary, we did not find a corresponding reduction in the cfDNA concentration which instead increased during time, suggesting the presence of progressive disease, as confirmed by CT imaging at follow up (Fig. 5a). The second patient (patient E) entered in the study with lymph node metastases, portal vein infiltration and disease progression on treatment with an OS of 11.7 months from the beginning of therapy. At time points considered, AFP values were measured always below 50 ng/ml; in contrast, cfDNA level, while showing a drastic decrease immediately after the beginning of the therapy, increased rapidly between 6 and 8 months from the beginning of the therapy, reflecting the rapid progression of the disease and indicating a change in the response to sorafenib (Fig. 5b). The third patient (patient K) entered in the study presenting only one hepatic lesion and no evidence of metastases. AFP was always measured below 5 ng/ml at all time points, however cfDNA showed a steadily increase, predicting the progression of the disease. The patient finally relapsed and died shortly after (Fig. 5c). In conclusion, our data, in line with other reports, showed improved diagnostic sensitivity of cfDNA over AFP [24, 26], suggesting also in advanced HCC cfDNA as a biomarker for monitoring therapy efficacy.
Fig. 5.

Comparison between cfDNA and AFP levels in three cases corresponding to patients showing different clinical characteristics. Panels a, b and c represent the described clinical cases of patients B, E and K, respectively
Somatic mutation analysis
Genomic profiling of ctDNA was undertaken at the different time points, with the screening of 597 cancer-relevant genes. Only those mutations proven to have clinical impact according to the ClinVar database (National Center for Biotechnology Information-NCBI) were further analyzed. In total 28 variants, including single nucleotide variations (SNV) or insertions and deletions (InDel) were discovered in our dataset, while no gene fusion was identified (Fig. 6). All patients showed at least one variant (median = 6 variants per patient, range 3–16), however patterns varied between patients. Variants were found in genes involved in DNA repair, multidrug resistance, cell cycle control, signal transduction, transcription control, chromatin structure, apoptosis and DNA methylation (Table 3). We detected somatic mutations in recognized HCC driver genes such as CTNNB1 and TP53, however at very low frequency. Over the 28 variants, three genes were showing the highest mutation allele frequency (MAF): HNF1A (n = 12, 92.3%), BAX (n = 9, 69.2%) and CYB2B6 (n = 6, 46.1%). We observed that in a high percentage of genes (n = 19, 68%) variants were found mainly after the beginning of sorafenib treatment, suggesting a possible clonal selection induced by the therapy itself. Nevertheless, a different kinetic was also observed. For example, in the case of HNF1A, 50% of the patients showed the variant already before starting to assume sorafenib. At T2, only one patient was still positive, suggesting a good therapy response. However at T3 the variant was showing a MAF of 82% (n = 9), a finding which could suggest a clonal expansion of cells not responsive any longer to sorafenib.
Fig. 6.

Heatmaps showing the variants discovered in the two subgroups of patients (A–E: below 65, no liver cirrhosis, no past alcohol abuse and better response to therapies; F–M: over 65, with liver cirrhosis, past alcohol abuse and worse response to therapies) at the different time points. In (a) and in (b) SNV and InDel with relative mutation frequencies are reported, respectively
Table 3.
List of variants grouped according to gene function
| Gene group | Location | AA | Codon | Patients (%) |
|---|---|---|---|---|
| DNA repair | ||||
| ATM | chr11:108121426 | – | c.1236-2A>T | 2 (15.4) |
| BLM | chr15:91304138 |
p.N515fs na |
c.1544delA c.*468delA |
1 (7.7) |
| BRCA2 | chr13:32954022 |
p.T18fs p.T3033fs |
c.52insA c.9097insA |
1 (7.7) |
| ERCC5 | chr13:103524611 |
p.K150fs p.K917fs |
c.450delA c.2751delA |
5 (38.4) |
| MSH6 | chr2:48030639 |
p.F56fs p.F1088fs – p.F958fs p.F786fs |
c.165delC c.3261delC c.*2608delC c.2871delC c.2355delC |
6 (46.1) |
| RAD50 | chr5:131931451 |
p.K722fs – – p.?661fs |
c.2165delA c.*1791delA c.*351delA c.1982delA |
4 (30.8) |
| WRN | chr8:30915970 | p.K5fs | c.15delA | 2 (15.4) |
| Multidrug resistance/xenobiotic metabolism | ||||
| ABCB1 | chr7:87160618 |
p.S893A p.S829A |
c.2677T>G c.2485T>G |
3 (23.0) |
| CYP2B6 | chr19:41515263 | p.K262R | c.785A>G | 6 (46.1) |
| CYP2C9 | chr10:96702047 | p.R144C | c.430C>T | 1 (7.7) |
| CYP2D6 | chr22:42524994 |
p.P34S p.P12S |
c.100C>T c.34C>T |
2 (15.4) |
| GSTP1 | chr11:67352689 |
– p.I105V |
c.*137A>G c.313A>G |
2 (15.4) |
| SLCO1B1 | chr12:21331549 | p.V174A | c.521T>C | 2 (15.4) |
| Cell cycle control/proliferation | ||||
| CTNNB1 | chr3:41266098 |
p.D32V p.D25V |
c.95A>T c.74A>T |
1 (7.75) |
| FGFR2 | chr10:123274794 |
p.Y261C p.Y147C p.Y287C p.Y263C p.Y286C p.Y375C p.Y376C p.Y377C p.Y260C – |
c.782A>G c.440A>G c.860A>G c.788A>G c.857A>G c.1124A>G c.1127A>G c.1130A>G c.779A>G c.*171A>G |
1 (7.75) |
| FGFR4 | chr5:176520243 |
p.G23R p.G388R |
c.67G>A c.1162G>A |
1 (7.75) |
| TP53 | chr17:7579447 |
p.G254D p.G113D p.G206D p.G152D p.G86D p.G234D |
c.374G>A c.338G>A c.617G>A c.455G>A c.257G>A c.701G>A |
1 (7.75) |
| TP53* | chr17:7579407 |
p.A86fs p.A47fs |
c.257del279 c.140del162 |
1 (7.75) |
| Signal transduction signalling | ||||
| PIK3CA | chr3:178952085 | p.H1047R | c.3140A>G | 1 (7.75) |
| PIK3CA* | chr3:178952085 | p.H1047L | c.3140A>T | 1 (7.75) |
| SOS1 | chr2:39249927 | p.S548R | c.1642A>C | 1 (7.75) |
| Chromatin structure | ||||
| ASXL1 | chr20:31022441 |
p.G641fs p.G646fs |
c.1919insG c.1934insG |
3 (23.0) |
| Liver transcription factor | ||||
| HNF1A | chr12:121432114 |
p.P291fs p.G226fs – |
c.864delG c.677delG c.*304delG |
12 (92.3) |
| Apoptosis | ||||
| BAX | chr19:49458970 |
p.E41fs p.R24fs p.E24fs |
c.121insG c.69insG c.70insG |
9 (69.2) |
| DNA methylation | ||||
| DNMT3A | chr2:25457242 |
– p.R693H p.R659H p.R882H |
c.*498G>A c.2078G>A c.1976G>A c.2645G>A |
2 (15.4) |
Chromosome location, amino acid (AA) change and codon change are indicated
(na, not available)
Association between variants, patient’s clinicopathological characteristics and overall survival
A possible association between the genetic variants and the patient’s clinicopathological characteristics was also evaluated. Due to the little size of the cohort, patients were clustered into those with variants identified only at early time points (T1 and T2) and those with variants identified only at later time points (T3 and T4). We measured a significant association between the BAX variant and portal vein invasion (p = 0.014) and between the HNF1A variant and liver cirrhosis (p = 0.032). For both genes, the significance was found only at the earlier time points. On the contrary, CYP2B showed to be significantly (p = 0.005) associated to BCLC grading only at the later time points. No further correlations were found (all p > 1.0) (Table 4).
Table 4.
Association between variant detection at different time points and patient’s clinicopathological characteristics
| Variable | Patients | CYP2B6 (T1–T2) | CYP2B6 (FU) | BAX (T1–T2) | BAX (FU) | HNF1A (T1–T2) | HNF1A (FU) |
|---|---|---|---|---|---|---|---|
| n | 8 | 8 | 4 | 6 | 5 | 12 | |
| Portal vein invasion | |||||||
| Yes | 3 | 3 | 1 | 3 | 1 | 1 | 3 |
| No | 10 | 5 | 7 | 1 | 5 | 4 | 9 |
| p | 0.231 | 0.51 | 0.014* | 1 | 1 | 1 | |
| BCLC | |||||||
| A + B | 7 | 4 | 7 | 1 | 2 | 2 | 7 |
| C | 6 | 4 | 1 | 3 | 4 | 3 | 5 |
| p | 1 | 0.005* | 0.266 | 0.286 | 0.592 | 1 | |
| Metastases | |||||||
| Yes | 4 | 3 | 1 | 2 | 3 | 2 | 3 |
| No | 9 | 5 | 7 | 2 | 3 | 3 | 9 |
| p | 0.608 | 0.217 | 0.53 | 0.266 | 1 | 0.538 | |
| Cirrhosis | |||||||
| Yes | 8 | 6 | 6 | 3 | 4 | 1 | 7 |
| No | 5 | 2 | 2 | 1 | 2 | 4 | 5 |
| p | 0.293 | 0.293 | 0.608 | 1 | 0.032* | 1 | |
| Tumor size (cm) | |||||||
| < 5 | 6 | 2 | 4 | 2 | 2 | 2 | 6 |
| > 5 | 6 | 5 | 3 | 2 | 3 | 2 | 6 |
| p | 0.242 | 1 | 1 | 1 | 1 | 1 | |
The analysis shows that there is a significant association between variants in CYP2B6, BAX and HNF1A and BCLS status (p = 0.005) portal vein invasion (p = 0.014) and absence of liver cirrhosis (p = 0.032), respectively. No association was found between any variant and presence of metastases or tumor size (all p > 1.0). Comparison was performed (for each time group separately) using the Fischer exact test
We further evaluated the prognostic value of the genetic variants with respect to OS. Patients carrying the CYP2B6 variant in the time frame T1–T2 showed a worse OS (p = 0.013). On the contrary, when the same variant was detected at later time points, no significant correlation was found (p = 0.360) (Fig. 7). No other genetic variant showed any correlation with the clinical outcome at any of the time points considered (all p > 0.1).
Fig. 7.
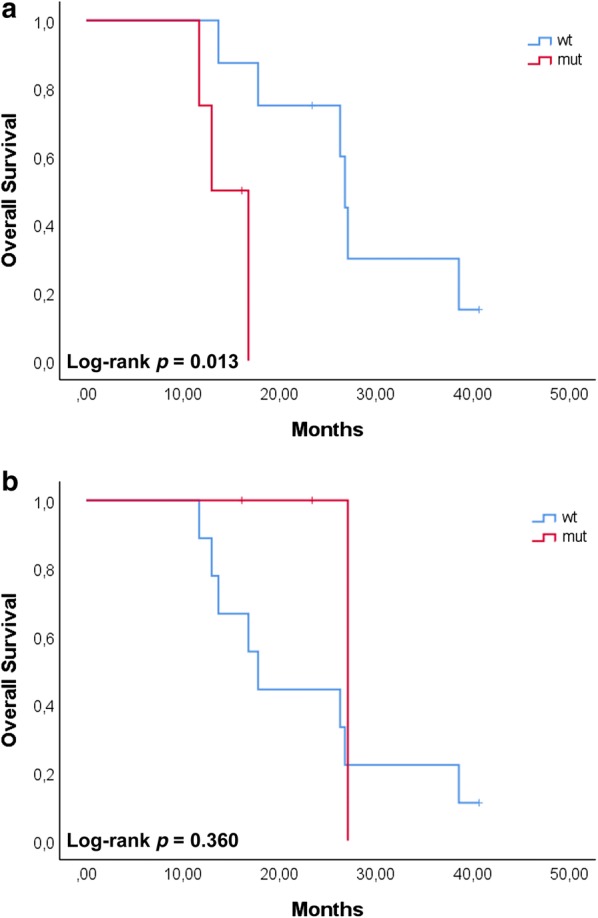
Survival plots for HCC patients carrying the CP2B6 variant. Patients were grouped according to the time points (T1–T2, a; T3–T4, b). Patients carrying the variant at T1–T2, showed a worse survival with respect to those patients carrying the variant at T3–T4. OS was analyzed using the Kaplan–Meier method and survival estimates in the different groups were compared using the log-rank test
Discussion
HCC is a heterogeneous pathology, involving multiple somatic genomic alterations responsible for different degrees of tumor differentiation and related clinical outcomes.
TP53 and CTNNB1 are the two most frequent mutations although other driver mutations have been identified [27]. Genomic analysis, based on liver biopsy, allows the molecular subtyping of HCC and opens the door to select and adapt therapies to the individual patient’s needs. Apart from being associated to a higher risk for the patient, tissue biopsy presents nevertheless some limitations: first, the genetic heterogeneity in tumor differentiation cannot be assessed by a single site sampling; second, the histopathological assessment of vascular invasion requires large biopsies; third, being highly invasive, biopsy is usually performed only once during the whole therapy. Liquid biopsy is independent of these limitations and shows predictive and prognostic value in several types of cancer including HCC [12, 13, 28]. cfDNA quantitative changes have been mainly linked to tumor’s burden and therapeutic efficacy [26], while ctDNA qualitative changes can detect genetic variations in the tumor as a whole [16, 29].
In this exploratory study we investigated the prognostic value of cfDNA/ctDNA isolated from advanced HCC patients over multiple time points in the context of the SORAMIC trial. We observed that the amount of cfDNA measured immediately after micro-intervention significantly correlated with the presence of distant metastases, supporting also in this clinical context a diagnostic value of cfDNA to unravel secondary lesions, as showed in other tumor types [25, 30, 31]. In addition, patients with post-operative high levels of cfDNA had a shorter OS. Interestingly, this holds true only at the later time points, after several weeks of sorafenib treatment, suggesting that also in advanced HCC cfDNA can support in monitoring therapy response. This finding is relevant when considering the use of other biomarkers such as AFP [32, 33] whose prognostic value is still under debate [34, 35]. Among the patients enrolled in this study, only two patients had, over the considered time points, AFP concentrations higher than the assigned cut-off value of 400 ng/ml. On the contrary, quantitative analysis of post-operative cfDNA was always feasible and showed a trend with the clinical history of the patients, supporting the use of cfDNA to monitor therapy response also in advanced HCC.
NGS analysis showed that all 13 patients presented SNV and/or InDel variants in a total of 28 genes, with 6 as the median number of variants per patient. We never found two patients with an identical profile, supporting the concept that HCC is a highly heterogeneous disease. Interestingly, only in few cases we detected the hotspot variants normally associated to HCC as those in TP53 or CTNNB1. We cannot exclude that this result is due to the low number of patients included in the study, therefore a validation study with a larger cohort will be necessary. However, these driver mutations are usually associated to specific etiological characteristics, not present in our patients’ group, such as HBV infection, dietary exposure to aflatoxin B1, or are per se very rare events [36]. The absence of these variants could be therefore associated to the clinical characteristics of the patients. In addition, in a multimodal meta-analysis run over 1494 HCC samples, it has been shown that other driver mutations, less frequent nevertheless still clinically significant, can be identified [37]. From the 28 variants, we identified in particular 3 genes displaying high MAFs. CYP2B6 (mutated in 46.1% of the patients at least at one time point) belongs to the Cytochrome P450 class of enzymes which are important catalysts among all metabolizing enzymes. The isoform CYP2B6, expressed mainly in the liver, is highly polymorphic [38] (with the haplotype CYP2B6*6 found in the Caucasian population in up to 25% of the cases) and shows clinical relevance [38, 39]. However, there are contradictory results on the effect of the haplotype *6 (h*6). On one side it has been reported that patients carrying h*6 have a reduced amount of mRNA and protein [40], with an inferior response to specific treatments. On the other side, studies demonstrated that h*6 increase the enzymatic activity of CYP2B6 [41]. In our study, we found that 46% of the patients presented a CYP2B6 variant corresponding to h*6 and that the detection of this variant at earlier time points was significantly associated with a worse prognosis and a shorter OS. Since variants in enzymes involved in the metabolism can affect the response to specific drugs [42] and CYP450 and its haplotypes are involved in the metabolism of several drugs [39, 43], we can hypothesize that those patients presenting the variant h*6 responded worse to the therapy with sorafenib and therefore had a short OS.
BAX (mutated in 69.2% of the patients at least at one time point) belongs to the Bcl-2 gene family of pro-apoptotic proteins [44]. Inactivation of BAX might be a mechanism to escape cell death, providing a survival advantage to the tumor. In colorectal cancer this particular frameshift mutation is indeed conferring an advantage to tumor progression, with p53-independent induction of apoptosis [45]. Sorafenib is a multikinase inhibitor with a strong anti-proliferative and anti-angiogenic effect on different cell types [46], displaying also a pro-apoptotic effect [47]. BAX might play a role in cellular death induced by sorafenib, and the presence of this frameshift variant might predict a low therapy efficacy due to the inactivation of the pathway inducing apoptosis. Although we did not find any significant correlation between the presence of variants of BAX and OS, we unraveled a significant correlation with portal vein invasion. Tumor cells able to overcome apoptosis display higher invasive characteristics. The fact that 3 patients carrying this mutation are positive for portal vein infiltration might support the role of BAX in tumor invasion and the possible inefficacy of sorafenib in these patients. However at the moment this is just a hypothesis and further analysis will be necessary to confirm it.
Finally, HNF1A showed the highest MAF, reaching 92.3% over the whole cohort. HNF1A codes for a transcription factor (HNF1α) which regulates liver differentiation and development [48]. This transcription factor functions as a tumor suppressor [49] and its inactivation might play an important role in tumor development [50]. A recent work of Takashima et al., demonstrated that the ectopic expression of HNF1α combined to other two transcription factors (HNF4a and FOXA3) successfully inhibited proliferation in a cellular model system [51]. If confirmed in other model system, HNF1A could be a novel target for combined therapy, especially when drug resistance is developed.
Moreover, while HNF1A inactivation is known to be frequent in hepatocellular adenoma, somatic mutations in this gene are relatively a rare event in HCC and very often bound to specific etiological characteristics such as absence of cirrhosis and viral infection [52]. In our cohort, none of the patients but one presented with viral infection and the presence of the variant showed a significant negative association with cirrhosis. Although TP53 and CTBNN1 are the two most frequently mutated genes in HCC, there are other low frequency driver genes, such as HNF1A, which nevertheless are significantly associated to patient’ survival. We can hypothesize that in the patient cohort selected for this study, HNF1A was a driver mutation. If confirmed in a larger number of patients, HNF1A could represent a novel target for personalized therapy.
A remarkable result we observed was the dynamic change of the mutation status over time. With respect to each gene taken individually, in the time frame between T1 and T4 we identified 6 categories of patients, showing: always the wildtype gene (WT); always the mutated gene (M); a single shift WT → M; a single shift M → WT; a double shift WT → M → WT; a double shift M → WT → M. With the small number of patients included in the study, for the moment we can only speculate that these changes are mirroring clonal heterogeneity found in the primary tumors [53, 54], evidenced because the analysis was done by liquid biopsy. It is reasonable to speculate that the shift WT → M might take place because the mutated clones are more resistant to therapy; therefore post-treatment samples are enriched with ctDNA carrying the mutation. These clones possibly develop spontaneously and the mutation they carry confers a selective advantage to cells. On the contrary, the shift M → WT might be observed because the mutated clones are more sensitive to therapy, which possibly could slow down their proliferation, while WT clones divide and expand faster. In the latter case we cannot exclude that ctDNA carrying the mutation is still present, however in this case the corresponding MAF is under the NGS detection threshold. Dynamic changes in clonal expansion during therapy are a known event [55], also strongly suggested by our data. Due to the limited number of samples, we cannot provide here anything more than a hypothesis. However it will be surely interesting to pursue in this direction, enlarging the number of cases and validating these preliminary results.
Conclusions
A validation study with a larger cohort and the inclusion of appropriate control groups will be necessary to confirm these preliminary results and to support their clinical value. Also a more detailed comparison between patients receiving ablation or radio-embolization should be considered to understand if different procedures possible with the micro-interventional therapy might induce the accumulation of alternative genetic mutations. However, from the results obtained from this exploratory study we can conclude that also in advanced HCC after micro-intervention cfDNA quantification is feasible, can support standard imaging analysis for the early detection of metastatic lesions and is showing a prognostic value with respect to OS. In addition, interrogating ctDNA for genetic variants can reduce understaging of clonal selection as a consequence of systemic therapy, in the potential to improve longitudinal monitoring of therapy efficacy. HCC is a highly heterogeneous tumor type; it would be therefore an advantage to tailor therapy according to the intrinsic genetic characteristics. None of the patients included in this study showed an identical genomic profile, supporting the concept that also in advanced HCC personalized therapy is necessary to improve clinical outcome. A rapid adaption of therapy in concomitance to variant appearance and therefore a more efficient treatment might be particularly important for patients already in an advanced stage of the disease and therefore with a relative short overall survival expectancy.
Supplementary information
Additional file 1: Figure S1. Size distribution of extracted cfDNA after capillary electrophoresis (CE). DNA extracted from all the plasma samples were fractionated by CE and analyzed. cfDNA displays a size of approximately 150 base pairs (bp), corresponding to one nucleosome, and multiple of this size, while gDNA has a higher molecular weight of several kilobases (kb). Samples displaying a unique peak around 150 bp were further analyzed, while samples showing a peak several kb were no further processed. Panels A and B report examples of cfDNA and gDNA as determined by CE, respectively (RFU, relative fluorescence unit; LM, lower marker; UM, upper marker).
Additional file 2: Table S1. Depth of coverage. An average coverage of at least 1000 times or more was reached for all the samples (average 1743.63, range 1182.17–2870.64), except one (sample F4) which showed a lower depth (840.41).
Additional file 3: Table S2. List of variants as detected in genomic DNA extracted from buffy coats for each patient. Chromosome location, gene name and mutation frequency are indicated. The analysis was done for all the patients except for patient E, whose corresponding cellular sample was not available. For this patient we set arbitrarily a mutation frequency threshold corresponding to the average of the other samples. Genes presenting a mutation frequency of 50 (± 5) and 100 (± 5) % were considered carrying a germline mutation and therefore were excluded from the following analysis.
Acknowledgements
This work was supported by Otto-von-Guericke University Magdeburg, Magdeburg, Germany. The authors wish to thank Regina Schinner (LMU, Munich) for reviewing the statistical analysis, Cheryl Gray (LMU, Munich) for the assistance in preparing the figures and for proof reading the manuscript, and Andreas Mittermaier (LMU, Munich) for preparing heatmaps. The authors wish to thank all patients who took part to the SORAMIC trial.
Abbreviations
- HCC
hepatocellular cancer
- cfDNA
circulating free DNA
- ctDNA
circulating tumor DNA
- NGS
next generation sequencing
- AFP
alpha-fetoprotein
- SNV
single nucleotide variants
- InDel
insertions and deletions
Authors’ contributions
MA-F, PM and JR: study design; MA-F and KR: data analysis and manuscript writing; TH: imaging analysis; JM, MP, BB, CV, JB, MS, CCC: patients recruitment, clinical data collection, data analysis. All authors read and approved the final manuscript.
Funding
This study was supported by the Otto-von-Guericke University Magdeburg, Magdeburg, Germany.
Availability of data and materials
The data supporting the conclusions of this article are included within the article.
Ethics approval and consent to participate
The study was approved by all the involved ethical boards and conducted in accordance with the Declaration of Helsinki. Before entering the trial, all patients gave their written informed consent.
Consent for publication
Not applicable.
Competing interests
JR was Principal Investigator of the SORAMIC trial, supported by Bayer GA and Sirtex Europe. PM was co-Principal Investigator of the SORAMIC trial, supported by Bayer GA and Sirtex Europe. All the other authors declare that they have no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Marianna Alunni-Fabbroni, Email: Marianna.Alunni@med.uni-muenchen.de.
Kerstin Rönsch, Email: KerstinRoensch@eurofins.com.
Thomas Huber, Email: thomas.huber@medma.uni-heidelberg.de.
Clemens C. Cyran, Email: Clemens.Cyran@med.uni-muenchen.de
Max Seidensticker, Email: Max.Seidensticker@med.uni-muenchen.de.
Julia Mayerle, Email: Julia.Mayerle@med.uni-muenchen.de.
Maciej Pech, Email: maciej.pech@med.ovgu.de.
Bristi Basu, Email: Bristi.Basu@cruk.cam.ac.uk.
Chris Verslype, Email: chris.verslype@uzleuven.be.
Julia Benckert, Email: julia.benckert@charite.de.
Peter Malfertheiner, Email: Peter.Malfertheiner@med.uni-muenchen.de.
Jens Ricke, Email: Jens.Ricke@med.uni-muenchen.de.
Supplementary information
Supplementary information accompanies this paper at 10.1186/s12967-019-2079-9.
References
- 1.Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136(5):E359–E386. doi: 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- 2.Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- 3.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61(2):69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 4.El-Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology. 2007;132(7):2557–2576. doi: 10.1053/j.gastro.2007.04.061. [DOI] [PubMed] [Google Scholar]
- 5.Bruix J, Sherman M, American Association for the Study of Liver D Management of hepatocellular carcinoma: an update. Hepatology. 2011;53(3):1020–1022. doi: 10.1002/hep.24199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cheng AL, Kang YK, Chen Z, Tsao CJ, Qin S, Kim JS, et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: a phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009;10(1):25–34. doi: 10.1016/S1470-2045(08)70285-7. [DOI] [PubMed] [Google Scholar]
- 7.Raja R, Kuziora M, Brohawn PZ, Higgs BW, Gupta A, Dennis PA, et al. Early reduction in ctDNA predicts survival in patients with lung and bladder cancer treated with durvalumab. Clin Cancer Res. 2018;24(24):6212–6222. doi: 10.1158/1078-0432.CCR-18-0386. [DOI] [PubMed] [Google Scholar]
- 8.Sorenson GD, Pribish DM, Valone FH, Memoli VA, Bzik DJ, Yao SL. Soluble normal and mutated DNA sequences from single-copy genes in human blood. Cancer Epidemiol Biomarkers Prev. 1994;3(1):67–71. [PubMed] [Google Scholar]
- 9.Vasioukhin V, Anker P, Maurice P, Lyautey J, Lederrey C, Stroun M. Point mutations of the N-ras gene in the blood plasma DNA of patients with myelodysplastic syndrome or acute myelogenous leukaemia. Br J Haematol. 1994;86(4):774–779. doi: 10.1111/j.1365-2141.1994.tb04828.x. [DOI] [PubMed] [Google Scholar]
- 10.Leon SA, Shapiro B, Sklaroff DM, Yaros MJ. Free DNA in the serum of cancer patients and the effect of therapy. Cancer Res. 1977;37(3):646–650. [PubMed] [Google Scholar]
- 11.Shu Y, Wu X, Tong X, Wang X, Chang Z, Mao Y, et al. Circulating tumor DNA mutation profiling by targeted next generation sequencing provides guidance for personalized treatments in multiple cancer types. Sci Rep. 2017;7(1):583. doi: 10.1038/s41598-017-00520-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Iizuka N, Sakaida I, Moribe T, Fujita N, Miura T, Stark M, et al. Elevated levels of circulating cell-free DNA in the blood of patients with hepatitis C virus-associated hepatocellular carcinoma. Anticancer Res. 2006;26(6C):4713–4719. [PubMed] [Google Scholar]
- 13.Tokuhisa Y, Iizuka N, Sakaida I, Moribe T, Fujita N, Miura T, et al. Circulating cell-free DNA as a predictive marker for distant metastasis of hepatitis C virus-related hepatocellular carcinoma. Br J Cancer. 2007;97(10):1399–1403. doi: 10.1038/sj.bjc.6604034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Diehl F, Schmidt K, Choti MA, Romans K, Goodman S, Li M, et al. Circulating mutant DNA to assess tumor dynamics. Nat Med. 2008;14(9):985–990. doi: 10.1038/nm.1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lo YM, Zhang J, Leung TN, Lau TK, Chang AM, Hjelm NM. Rapid clearance of fetal DNA from maternal plasma. Am J Hum Genet. 1999;64(1):218–224. doi: 10.1086/302205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Okajima W, Komatsu S, Ichikawa D, Miyamae M, Ohashi T, Imamura T, et al. Liquid biopsy in patients with hepatocellular carcinoma: circulating tumor cells and cell-free nucleic acids. World J Gastroenterol. 2017;23(31):5650–5668. doi: 10.3748/wjg.v23.i31.5650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yin CQ, Yuan CH, Qu Z, Guan Q, Chen H, Wang FB. Liquid biopsy of hepatocellular carcinoma: circulating tumor-derived biomarkers. Dis Markers. 2016;2016:1427849. doi: 10.1155/2016/1427849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ricke J, Bulla K, Kolligs F, Peck-Radosavljevic M, Reimer P, Sangro B, et al. Safety and toxicity of radioembolization plus Sorafenib in advanced hepatocellular carcinoma: analysis of the European multicentre trial SORAMIC. Liver Int. 2015;35(2):620–626. doi: 10.1111/liv.12622. [DOI] [PubMed] [Google Scholar]
- 19.Ricke J, Sangro B, Amthauer H, Bargellini I, Bartenstein P, De Toni E, et al. The impact of combining Selective Internal Radiation Therapy (SIRT) with Sorafenib on overall survival in patients with advanced hepatocellular carcinoma: The SORAMIC trial palliative cohort. J Hepatol. 2018;68:S102. [Google Scholar]
- 20.Colombo M. Screening for cancer in viral hepatitis. Clin Liver Dis. 2001;5(1):109–122. doi: 10.1016/s1089-3261(05)70156-2. [DOI] [PubMed] [Google Scholar]
- 21.Bialecki ES, Di Bisceglie AM. Diagnosis of hepatocellular carcinoma. HPB (Oxford). 2005;7(1):26–34. doi: 10.1080/13651820410024049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.World Medical A World Medical Association Declaration of Helsinki: ethical principles for medical research involving human subjects. JAMA. 2013;310(20):2191–2194. doi: 10.1001/jama.2013.281053. [DOI] [PubMed] [Google Scholar]
- 23.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim P, Yoon S, Kim N, Lee S, Ko M, Lee H, et al. ChimerDB 2.0–a knowledgebase for fusion genes updated. Nucleic Acids Res. 2010;38(Database issue):D81–D85. doi: 10.1093/nar/gkp982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Allin DM, Shaikh R, Carter P, Thway K, Sharabiani MTA, Gonzales-de-Castro D, et al. Circulating tumour DNA is a potential biomarker for disease progression and response to targeted therapy in advanced thyroid cancer. Eur J Cancer. 2018;103:165–175. doi: 10.1016/j.ejca.2018.08.013. [DOI] [PubMed] [Google Scholar]
- 26.Chen H, Sun LY, Zheng HQ, Zhang QF, Jin XM. Total serum DNA and DNA integrity: diagnostic value in patients with hepatitis B virus-related hepatocellular carcinoma. Pathology. 2012;44(4):318–324. doi: 10.1097/PAT.0b013e328353a24c. [DOI] [PubMed] [Google Scholar]
- 27.Shibata T, Aburatani H. Exploration of liver cancer genomes. Nat Rev Gastroenterol Hepatol. 2014;11(6):340–349. doi: 10.1038/nrgastro.2014.6. [DOI] [PubMed] [Google Scholar]
- 28.Dawson SJ, Tsui DW, Murtaza M, Biggs H, Rueda OM, Chin SF, et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N Engl J Med. 2013;368(13):1199–1209. doi: 10.1056/NEJMoa1213261. [DOI] [PubMed] [Google Scholar]
- 29.Tang JC, Feng YL, Guo T, Xie AY, Cai XJ. Circulating tumor DNA in hepatocellular carcinoma: trends and challenges. Cell Biosci. 2016;6:32. doi: 10.1186/s13578-016-0100-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Garcia-Murillas I, Schiavon G, Weigelt B, Ng C, Hrebien S, Cutts RJ, et al. Mutation tracking in circulating tumor DNA predicts relapse in early breast cancer. Sci Transl Med. 2015;7(302):302ra133. doi: 10.1126/scitranslmed.aab0021. [DOI] [PubMed] [Google Scholar]
- 31.Chaudhuri AA, Chabon JJ, Lovejoy AF, Newman AM, Stehr H, Azad TD, et al. Early detection of molecular residual disease in localized lung cancer by circulating tumor DNA profiling. Cancer Discov. 2017;7(12):1394–1403. doi: 10.1158/2159-8290.CD-17-0716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chaiteerakij R, Addissie BD, Roberts LR. Update on biomarkers of hepatocellular carcinoma. Clin Gastroenterol Hepatol. 2015;13(2):237–245. doi: 10.1016/j.cgh.2013.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lok AS, Sterling RK, Everhart JE, Wright EC, Hoefs JC, Di Bisceglie AM, et al. Des-gamma-carboxy prothrombin and alpha-fetoprotein as biomarkers for the early detection of hepatocellular carcinoma. Gastroenterology. 2010;138(2):493–502. doi: 10.1053/j.gastro.2009.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Giannini EG, Marenco S, Borgonovo G, Savarino V, Farinati F, Del Poggio P, et al. Alpha-fetoprotein has no prognostic role in small hepatocellular carcinoma identified during surveillance in compensated cirrhosis. Hepatology. 2012;56(4):1371–1379. doi: 10.1002/hep.25814. [DOI] [PubMed] [Google Scholar]
- 35.Shim JH, Yoon DL, Han S, Lee YJ, Lee SG, Kim KM, et al. Is serum alpha-fetoprotein useful for predicting recurrence and mortality specific to hepatocellular carcinoma after hepatectomy? A test based on propensity scores and competing risks analysis. Ann Surg Oncol. 2012;19(12):3687–3696. doi: 10.1245/s10434-012-2416-1. [DOI] [PubMed] [Google Scholar]
- 36.Tornesello ML, Buonaguro L, Tatangelo F, Botti G, Izzo F, Buonaguro FM. Mutations in TP53, CTNNB1 and PIK3CA genes in hepatocellular carcinoma associated with hepatitis B and hepatitis C virus infections. Genomics. 2013;102(2):74–83. doi: 10.1016/j.ygeno.2013.04.001. [DOI] [PubMed] [Google Scholar]
- 37.Chaudhary K, Poirion OB, Lu L, Huang S, Ching T, Garmire LX. Multimodal meta-analysis of 1494 hepatocellular carcinoma samples reveals significant impact of consensus driver genes on phenotypes. Clin Cancer Res. 2019;25(2):463–472. doi: 10.1158/1078-0432.CCR-18-0088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Johnson GG, Lin K, Cox TF, Oates M, Sibson DR, Eccles R, et al. CYP2B6*6 is an independent determinant of inferior response to fludarabine plus cyclophosphamide in chronic lymphocytic leukemia. Blood. 2013;122(26):4253–4258. doi: 10.1182/blood-2013-07-516666. [DOI] [PubMed] [Google Scholar]
- 39.Song Q, Zhou X, Yu J, Dong N, Wang X, Yang H, et al. The prognostic values of CYP2B6 genetic polymorphisms and metastatic sites for advanced breast cancer patients treated with docetaxel and thiotepa. Sci Rep. 2015;5:16775. doi: 10.1038/srep16775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hofmann MH, Blievernicht JK, Klein K, Saussele T, Schaeffeler E, Schwab M, et al. Aberrant splicing caused by single nucleotide polymorphism c.516G>T [Q172H], a marker of CYP2B6*6, is responsible for decreased expression and activity of CYP2B6 in liver. J Pharmacol Exp Ther. 2008;325(1):284–292. doi: 10.1124/jpet.107.133306. [DOI] [PubMed] [Google Scholar]
- 41.Nakajima M, Komagata S, Fujiki Y, Kanada Y, Ebi H, Itoh K, et al. Genetic polymorphisms of CYP2B6 affect the pharmacokinetics/pharmacodynamics of cyclophosphamide in Japanese cancer patients. Pharmacogenet Genomics. 2007;17(6):431–445. doi: 10.1097/FPC.0b013e328045c4fb. [DOI] [PubMed] [Google Scholar]
- 42.Eichelbaum M, Ingelman-Sundberg M, Evans WE. Pharmacogenomics and individualized drug therapy. Annu Rev Med. 2006;57:119–137. doi: 10.1146/annurev.med.56.082103.104724. [DOI] [PubMed] [Google Scholar]
- 43.Ingelman-Sundberg M. Pharmacogenetics of cytochrome P450 and its applications in drug therapy: the past, present and future. Trends Pharmacol Sci. 2004;25(4):193–200. doi: 10.1016/j.tips.2004.02.007. [DOI] [PubMed] [Google Scholar]
- 44.Campbell KJ, Tait SWG. Targeting BCL-2 regulated apoptosis in cancer. Open Biol. 2018;8:180002. doi: 10.1098/rsob.180002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rampino N, Yamamoto H, Ionov Y, Li Y, Sawai H, Reed JC, et al. Somatic frameshift mutations in the BAX gene in colon cancers of the microsatellite mutator phenotype. Science. 1997;275(5302):967–969. doi: 10.1126/science.275.5302.967. [DOI] [PubMed] [Google Scholar]
- 46.Wilhelm SM, Adnane L, Newell P, Villanueva A, Llovet JM, Lynch M. Preclinical overview of sorafenib, a multikinase inhibitor that targets both Raf and VEGF and PDGF receptor tyrosine kinase signaling. Mol Cancer Ther. 2008;7(10):3129–3140. doi: 10.1158/1535-7163.MCT-08-0013. [DOI] [PubMed] [Google Scholar]
- 47.Hao H, Zhang D, Shi J, Wang Y, Chen L, Guo Y, et al. Sorafenib induces autophagic cell death and apoptosis in hepatic stellate cell through the JNK and Akt signaling pathways. Anticancer Drugs. 2016;27(3):192–203. doi: 10.1097/CAD.0000000000000316. [DOI] [PubMed] [Google Scholar]
- 48.Cereghini S, Yaniv M, Cortese R. Hepatocyte dedifferentiation and extinction is accompanied by a block in the synthesis of mRNA coding for the transcription factor HNF1/LFB1. EMBO J. 1990;9(7):2257–2263. doi: 10.1002/j.1460-2075.1990.tb07396.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zeng X, Lin Y, Yin C, Zhang X, Ning BF, Zhang Q, et al. Recombinant adenovirus carrying the hepatocyte nuclear factor-1alpha gene inhibits hepatocellular carcinoma xenograft growth in mice. Hepatology. 2011;54(6):2036–2047. doi: 10.1002/hep.24647. [DOI] [PubMed] [Google Scholar]
- 50.Luo Z, Li Y, Wang H, Fleming J, Li M, Kang Y, et al. Hepatocyte nuclear factor 1A (HNF1A) as a possible tumor suppressor in pancreatic cancer. PLoS ONE. 2015;10(3):e0121082. doi: 10.1371/journal.pone.0121082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Takashima Y, Horisawa K, Udono M, Ohkawa Y, Suzuki A. Prolonged inhibition of hepatocellular carcinoma cell proliferation by combinatorial expression of defined transcription factors. Cancer Sci. 2018;109(11):3543–3553. doi: 10.1111/cas.13798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hechtman JF, Abou-Alfa GK, Stadler ZK, Mandelker DL, Roehrl MHA, Zehir A, et al. Somatic HNF1A mutations in the malignant transformation of hepatocellular adenomas: a retrospective analysis of data from MSK-IMPACT and TCGA. Hum Pathol. 2019;83:1–6. doi: 10.1016/j.humpath.2018.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang Y, Waters J, Leung ML, Unruh A, Roh W, Shi X, et al. Clonal evolution in breast cancer revealed by single nucleus genome sequencing. Nature. 2014;512(7513):155–160. doi: 10.1038/nature13600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Navin N, Kendall J, Troge J, Andrews P, Rodgers L, McIndoo J, et al. Tumour evolution inferred by single-cell sequencing. Nature. 2011;472(7341):90–94. doi: 10.1038/nature09807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tan SH, Sapari NS, Miao H, Hartman M, Loh M, Chng WJ, et al. High-throughput mutation profiling changes before and 3 weeks after chemotherapy in newly diagnosed breast cancer patients. PLoS ONE. 2015;10(12):e0142466. doi: 10.1371/journal.pone.0142466. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Figure S1. Size distribution of extracted cfDNA after capillary electrophoresis (CE). DNA extracted from all the plasma samples were fractionated by CE and analyzed. cfDNA displays a size of approximately 150 base pairs (bp), corresponding to one nucleosome, and multiple of this size, while gDNA has a higher molecular weight of several kilobases (kb). Samples displaying a unique peak around 150 bp were further analyzed, while samples showing a peak several kb were no further processed. Panels A and B report examples of cfDNA and gDNA as determined by CE, respectively (RFU, relative fluorescence unit; LM, lower marker; UM, upper marker).
Additional file 2: Table S1. Depth of coverage. An average coverage of at least 1000 times or more was reached for all the samples (average 1743.63, range 1182.17–2870.64), except one (sample F4) which showed a lower depth (840.41).
Additional file 3: Table S2. List of variants as detected in genomic DNA extracted from buffy coats for each patient. Chromosome location, gene name and mutation frequency are indicated. The analysis was done for all the patients except for patient E, whose corresponding cellular sample was not available. For this patient we set arbitrarily a mutation frequency threshold corresponding to the average of the other samples. Genes presenting a mutation frequency of 50 (± 5) and 100 (± 5) % were considered carrying a germline mutation and therefore were excluded from the following analysis.
Data Availability Statement
The data supporting the conclusions of this article are included within the article.