

Abstract
Induction of ischemic preconditioning (IPC) represents a potential therapy against cerebral ischemia by activation of adaptive pathways and modulation of mitochondria to induce ischemic tolerance to various cells and tissues. Mitochondrial dysfunction has been ascribed to contribute to numerous neurodegenerative conditions and cerebral ischemia. Nuclear erythroid 2-related factor 2 (Nrf2) is a transcription factor that has traditionally been involved in upregulating cellular antioxidant systems to combat oxidative stress in the brain; however, the association of Nrf2 with mitochondria in the brain remains unclear. In the present study, we investigated the effects of Nrf2 on (i) IPC-induced protection of astrocytes; (ii) OXPHOS protein expression; and (iii) mitochondrial supercomplex formation.
Oxygen-glucose deprivation (OGD) was used as an in vitro model of cerebral ischemia and IPC in cultured rodent astrocytes derived from WT C57Bl/6J and Nrf2−/− mice. OXPHOS proteins were probed via western blotting, and supercomplexes were determined by blue native gel electrophoresis.
IPC-induced cytoprotection in wild-type, but not Nrf2−/− mouse astrocyte cultures following a lethal duration of OGD. In addition, our results suggest that Nrf2 localizes to the outer membrane in non-synaptic brain mitochondria, and that a lack of Nrf2 in vivo produces altered supercomplex formation in mitochondria.
Our findings support a role of Nrf2 in mediating IPC-induced protection in astrocytes, which can profoundly impact the ischemic tolerance of neurons. In addition, we provide novel evidence for the association of Nrf2 to brain mitochondria and supercomplex formation. These studies offer new targets and pathways of Nrf2, which may be heavily implicated following cerebral ischemia.
Keywords: Preconditioning, Nrf2, Mitochondria, Reactive oxygen species
Introduction
A potential therapy that could mitigate the morbidity and mortality of cerebral ischemic injury may be conceptualized as ischemic preconditioning (IPC). Previous studies from our group and others have shown that IPC treatment induced neuroprotection in rodent models of global cerebral ischemia [1–5]. However, preconditioning has historically focused mainly on neuronal physiology and amelioration of neuronal cell death following in vitro and in vivo models of ischemia. As a result, the role of astrocytes in mediating IPC-induced cytoprotection has not been thoroughly characterized, despite the well-known functions of astrocytes in mediating several neuroprotective mechanisms [6].
In addition, mitochondria have gained much recognition as a potential contributor to neurodegenerative diseases. In many of these diseases, disorders in mitochondrial quality control, bioenergetics, and reactive oxygen species production have been ascribed as the causative dysfunction in neurodegenerative conditions, including cerebral ischemia (reviewed in [7]). As a result, a better understanding of the role of mitochondria in neurodegenerative diseases may help to identify potential therapeutic targets to intervene and restore normal functioning in the brain.
The antioxidant transcription factor, nuclear erythroid 2-related factor 2 (Nrf2), has been extensively studied in the context of ischemia [8, 9]. Nrf2 has been suggested to be highly expressed in astrocytes as opposed to neurons [10]. As an antioxidant transcription factor, Nrf2 has been shown to translocate to the nucleus in various cell types after exposure to oxidative stress. Upon nuclear translocation, Nrf2 increases gene transcription of some common antioxidants, such as catalase, superoxide dismutase, and glutathione [11]. As mitochondria are organelles that contribute to a substantial portion of ROS production in the cell, an interaction of Nrf2 with mitochondria may serve to keep the nucleus apprised of mitochondrial functioning. However, the relationship of Nrf2 to mitochondria in the brain has not been extensively investigated.
As Nrf2 has a well-documented role in mediating nuclear gene transcription, its involvement with mitochondria may suggest that Nrf2 could facilitate mitochondrial-nuclear communication. Mitochondrial-nuclear communication and coordination, particularly in oxidative phosphorylation (OXPHOS) regulation, are critical to maintaining normal bioenergetics within a cell. In addition, key functions like mitochondrial biogenesis and mitophagy also rely on the coordination of nuclear transcription factors and mitochondrial-targeted downstream pathways [12]. Therefore, Nrf2 may represent a novel transcription factor that can coordinate nuclear-mitochondrial communication and OXPHOS regulation.
Thus, the focus of the present study was to determine if IPC treatment could induce cytoprotection in astrocytes through Nrf2. We found that Nrf2 was enriched in astrocytes compared to neurons, and that IPC was able to activate Nrf2-dependent gene transcription in rat astrocytes. Absence of functional Nrf2 protein reduced IPC-induced cytoprotection in astrocyte cultures following an in vitro model of cerebral ischemia. Finally, we provide novel evidence which suggests that Nrf2 associates with the outer mitochondrial membrane in non-synaptic mitochondria, and that Nrf2 plays a role in OXPHOS supercomplex association. These studies highlight the contribution of Nrf2 to IPC-induced protection of astrocytes and novel interactions of Nrf2 with mitochondria.
Methods
Materials
Minimum Essential Medium (MEM), Hank’s balanced salt solution (HBSS), and fetal bovine serum (FBS) were purchased from Gibco/Life Technologies (Grand Island, NY). All other reagents were purchased from Sigma Aldrich Co. (St. Louis, MO, USA) unless otherwise noted.
Animal Use
All animal protocols were approved by the Animal Care and Use Committee of the University of Miami. Experiments were conducted in accordance to ARRIVE guidelines. 16–17 day-pregnant Sprague-Dawley rats were purchased from Charles Rivers Laboratories and were housed in a temperature-controlled environment with 12-h light/12-h dark cycle and ad libitum food and water. Five week old male and female homozygous knockout mice (Nrf2−/−, Jackson Laboratories) were bred to establish homozygous Nrf2−/− colonies. Genotyping was performed by Transnetyx, Inc. (Cordova, TN, USA) to verify mouse colonies (see Supplementary Fig. 1).
Russ Lenth’s Power analysis [13] revealed that n = 6 for in vivo studies was sufficient, while n = 3–6 for in vitro studies was sufficient. This is similar to our group’s previous experience with similar types of studies. The operational user treated cells or harvested tissue. Prior to further processing, samples were blinded by another user. Experiments were performed as described, until samples were unblinded for final analysis.
Preparation of Embryonic and Post-Natal Astrocyte Cultures
Neuronal cultures were prepared as previously described [14] with slight modifications. Cortical tissue from embryonic pups was isolated and digested with 0.25% Trypsin and 1% DNase, titurated, and filtered through a 70-μM-mesh filter to remove any undigested tissue debris. Neurons were plated in neuronal plating media (MEM supplemented with 5% FBS, 1% GlutaMAX, and 20 mM glucose) at a density of 2 hemispheres for each 24 well plate, with each well containing a 12 mm poly-d lysine coated coverslip. Alternatively, for assessing levels of Nrf2 in naïve rat neurons via western blotting, 3–4 hemispheres were plated in 100-mm-culture dishes. Cultures were maintained 10–14 days, with ½ media exchanges occurring every 3–4 days. Proliferation of non-neuronal cells was inhibited by the addition of cytosine arabinoside (5 μM final) DIV 3.
Astrocyte cultures were prepared as previously described [14] with slight modifications. For rat astrocyte cultures, cortices from postnatal Sprague-Dawley rat pups (P2-P4) were harvested, followed by digestion with 0.25% trypsin and 1% DNase. Following trituration and filtration through a 70-μm filter, the resulting cell suspension was centrifuged at 200 × g for 5 min and was plated in MEM supplemented with 20 mmol/L glucose, 1% GlutaMAX, 1% Penicillin/ Streptomycin, and 10% FBS before plating. Complete media changes were performed every 2–3 days until cultures reached 70% confluency 7 days following the initial plating. Astrocyte cultures were then passaged and plated in appropriate culture vessels at a density of 50,000 cells/cm2, were allowed to reach full confluency, and were maintained for an additional 6–7 days before experimental use. A similar protocol was used for the culture of mouse astrocytes from WT and Nrf2−/− postnatal (P2-P4) pups. For immunofluorescence staining, rat astrocytes were plated onto poly-d lysine coated coverslips.
Oxygen Glucose Deprivation
To mimic IPC in vitro, astrocyte cultures were exposed to oxygen and glucose deprivation (OGD) as previously described [15] for 1 or 2 h. Through empirical testing, both 1 and 2 h were determined to be a sublethal duration of OGD that induced the highest degree of protection to astrocytes following a lethal OGD insult (6 h). The 6-h time point was chosen because greater than 50% cell death occurred along with minimal cell detachment from the tissue culture dishes, allowing for more accurate lactate dehydrogenase release assays. For mouse astrocyte cell cultures, OGD duration was decreased to 5 h for similar reasons, as near maximal cell death and cell detachment occurred in Nrf2−/− astrocyte cultures at OGD durations greater than 5 h. To induce OGD, cells were washed two times with glucose-free HBSS (in mmol/L) (CaCl2 1.26, KCl 5.37, KH2PO4 0.44, MgCl2 0.49, MgSO4 0.41, NaCl 136, NaHCO3 4.17, Na2HPO4 0.34, sucrose 20, pH 7.4) and exposed to an oxygen-free environment (90% nitrogen, 5% hydrogen, and 5% CO2, 37 °C) using a COY anaerobic chamber (COY Laboratory Products Inc., Lake Charter Township, MI, USA). OGD was terminated by placing the cells back into glucose-containing maintenance media and returning cultures to a 5% CO2, 37 °C incubator. Sham IPC was performed using similar number of washes and glucose-free HBSS, except glucose (20 mmol/L) was substituted for sucrose and cells were placed back into normoxic conditions.
Cell Death Assay
To assay cell death, astrocyte culture media was assayed for lactate dehydrogenase using a commercially available kit (Roche Inc.). Cell death assays were performed according to manufacturer’s instructions. Briefly, cell culture media was collected from astrocyte cultures prior to IPC or sham treatment, prior to lethal OGD treatment, 48 h-post lethal OGD treatment, and finally following complete lysis of cells with 2% triton in PBS solution from the same well. LDH release into cell culture media was measured colorimetrically at an absorbance of 490 nm. LDH release results were normalized to the total amount of LDH present in each corresponding well, thereby normalizing for slight differences in cell density across each individual well of the cell culture plate. Values are represented as a percentage of maximum LDH release.
Immunofluorescence
Immunofluorescence was conducted as previously described [16], with minor modifications. Briefly, coverslips containing either cultured rat neurons or astrocytes were washed with × 1 PBS three times and then were fixed for 15 min in 4% paraformaldehyde in PBS (pH 7.4). After aspiration and rinsing with × 1 PBS, samples were blocked in blocking buffer (5% donkey serum, 0.3% Triton X-100, × 1 PBS) for 1 h. Sections were incubated with primary antibodies (1:500 anti-GFAP, 1:500 anti-βIII-Tubulin, Cell Signaling Technology, Danvers, MA, USA) overnight at 4 °C and secondary antibodies (Alexa Fluor 488, 568 or 647, 1:500, Thermo Fisher Scientific) for 2 h at room temperature protected from light. Coverslips were placed on glass slides containing Prolong® Gold Antifade Reagent with Hoechst 33342 (for nucleus staining) and allowed to cure prior to imaging. Coverslips were imaged on an Olympus BX50 confocal microscope (Olympus, Center Valley, PA, USA) and rendered on Imaris software (Bitplane, Concord, MA, USA).
Isolation of Non-synaptic Mitochondria
Non-synaptic mitochondria were isolated from WT or Nrf2−/− mice as previously described [17]. Mouse cortex were homogenized in isolation medium (250 mM sucrose, 1 mg/ml bovine serum albumin (fraction V essentially fatty acid-free, BSA), 1.0 mM ethylenediaminetetra-acetic acid (EDTA), and 0.25 mM dithiothreitol, pH 7.4) Tissue was minced with a pair of scissors and rinsed thoroughly with the isolation medium. The minced tissue was homogenized in a hand-operated Teflon glass homogenizer by 7–8 strokes. The homogenate was diluted to yield 10% (w/v) homogenate and centrifuged at 720 × g for 5 min using a Sorvall (Newton, CT, USA) RC5 centrifuge. The supernatant was collected in another tube and centrifuged again at the same speed to reduce nuclear contamination of the eventual mitochondria sample. To isolate glial and neuronal cell body mitochondria, non-synaptic mitochondria was collected by layering the supernatant obtained from the final slow-speed centrifuge on a 24% (v/v) Percoll gradient (Percoll diluted in isolation media with BSA). The gradients were centrifuged at 32,500 × g for 5 min. The resulting pellet was washed once with isolation media and centrifuged at 15,000 × g for 10 min. The pellet was again washed with 0.25 M sucrose by centrifugation at 15,000 × g for 10min. The resulting pellet was resuspended in 0.25 M sucrose, and protein content was determined by bicinchoninic acid (BCA) assay. All mitochondrial isolation procedures were performed at 4 °C.
Western Blot
Cells and isolated mitochondria were lysed in RIPA Buffer (20 mmol/L Tris-HCl pH 7.5, 150 mmol/L NaCl, 1 mmol/L EDTA, 1% NP-40, 1% sodium deoxycholate, 2.5 mmol/L sodium pyrophosphate, 1 mmol/L Na 3VO4, and 1 mmol/L PMSF). Protein concentration was determined by BCA protein assay and 30 μg of protein was loaded onto a 12% SDS-polyacrylamide gel and electroblotted to nitrocellulose. Membranes were blocked in 5% dry milk/Tris-buffered saline and hybridized with primary antibodies overnight at 4 °C. Blots were probed with rabbit anti-Nrf2 (1:500, Santa Cruz Biotechnology, Dallas, TX, USA), rabbit anti-UCP2 (1:500, Calbiochem Inc.), rabbit anti-GAPDH (1:10,000, Cell Signaling Technology, Danvers, MA, USA), mouse anti-β-actin (1:10,000, Sigma), rabbit anti-MnSOD (1:2000, Cell Signaling Technology) or goat anti-Lamin-B (1:1000, Cell Signaling Technology). Membranes were washed with TBST followed by incubation with anti-mouse, anti-goat, or anti-rabbit HRP-conjugated secondary antibodies (1:5000, Pierce, Thermo Scientific; Rockford, IL, USA) for 1 h at room temperature. Proteins were detected using enhanced chemilumi-nescence (ECL) system (Pierce, Thermo Scientific). Western blot densitometry was analyzed using ImageJ (NIH) [18].
Submitochondrial Localization Experiments
For the proteinase K treatment, the mitochondria were prepared without protease inhibitor and were incubated with 0, 25, 50, 100, and 150 μg of proteinase K at 37 °C for 30 min. After this, mitochondrial samples were centrifuged at 13000 × g for 10 min. The supernatant was discarded, and proteinase K-treated mitochondria were subjected to SDS- PAGE and immunoblotting.
Blue Native Gel Electrophoresis
Separation of electron transport supercomplexes was performed using BN-PAGE as previously described [19]. Mitochondrial membranes were solubilized with digitonin in a buffer composed of 750 mM 6-aminohexanoic acid, 50 mM Bis-Tris, and 0.5 mM EDTA, pH 7.0 at 4 °C to a digitonin; protein ratio of 8:1 (w/w). This concentration of digitonin was empirically determined to solubilize mitochondrial membranes while still maintaining oligomeric association of respiratory supercomplexes. Following treatment with digitonin, samples were centrifuged for 10 min at 13000 × g at 4 °C. Serva Blue G was added to the resulting supernatants using a detergent-to-dye ratio of 8:1 (w/w). Proteins (100 μg aliquots per lane) were separated on a Native PAGE Novex 3–12% Bis-Tris gradient gel (Invitrogen, Carlsbad, CA, USA). Following electrophoresis, protein bands were visualized using Bio-Safe Coomassie-G250.
Separation of Individual Electron Transport Complexes by 2D SDS PAGE
Following blue native PAGE, supercomplexes were further resolved in the second dimension under reducing conditions to determine their composition, as previously described [19]. In brief, a lane from the blue native gel was excised and placed in a solution of SDS sample buffer. These lanes were microwaved briefly for 30 s until the SDS buffer began to boil. After a subsequent 10 min incubation in the sample buffer, the lane was placed atop a 12% SDS-PAGEgelandwasran under normal SDS page conditions. Following previously described immunoblotting methods, the composition of the supercomplexes were analyzed using an OXPHOS western blotting cocktail of antibodies which probed complexes I-V (Abcam, Inc., Cambridge, MA, USA). The bands were imaged with Kodak Film and were analyzed with ImageJ software. All analyses were done in blinded manner.
Statistical Analysis
All data are expressed as mean ± STDEV. Statistical analysis between two groups was performed using the unpaired Student’s t test. Statistical analysis between more than two groups was performed using a one-way ANOVA with Bonferroni’s multiple comparison post-hoc test unless other-wise specified. p ≤ 0.05 was considered statistically significant (GraphPad Prism v. 5.00 for Windows, GraphPad Software, San Diego, CA, USA).
Results
Nrf2 Is Predominately Located in Rat Cortical Astrocytes
Previous studies have shown that astrocytes contain a higher antioxidant potential than neurons [20]. Therefore, we first determined if basal Nrf2 levels were more abundant in astrocytes compared to that in neurons. Whole cell lysates prepared from rat astrocyte and rat neuronal cultures were immunoblotted for Nrf2. Whole-cell astrocyte lysates contained higher levels of Nrf2 protein as compared to neuronal cultures under naïve conditions (Fig. 1a). The results show that neurons had approximately 92% less basal Nrf2 levels compared to astrocytes (Fig. 1b, p < 0.005, n = 3). Representative images of astrocyte (Fig. 1c) and neuronal (Fig. 1d) cultures demonstrate the highly enriched population of each cell subtype via immunofluorescence staining. These results indicate that Nrf2 is predominantly found in cortical astrocytes.
Fig. 1.

The antioxidant transcription factor, Nrf2, is enriched in astrocytes compared to neurons. a Western blot analysis of whole cell lysates from astrocyte and neuronal cultures derived from postnatal and embryonic rat pups, respectively. Samples were immunoblotted with Nrf2 and Actin (whole-cell loading control). b Quantification of western blots from (a), **p ≤ 0.05. n = 3. Nrf2 protein levels were normalized to Actin. Data represented as a percentage of astrocyte Nrf2/Actin ratios. c Representative immunofluorescence images demonstrating high enrichment of rat neuronal cultures. d Rat astrocyte cultures. Green: BIII-Tubulin, neuronal marker; red: GFAP, astrocyte marker; blue: Hoechst 33,342, nuclear strain
IPC Protects Astrocytes against Lethal OGD
IPC has been described in many different cells and tissues [21], but has not been well-defined in rodent astrocytes. Therefore, we investigated if IPC could induce cytoprotection in postnatal astrocytes against lethal ischemia using an in vitro model of ischemia (oxygen glucose deprivation (OGD)). Following lethal OGD, astrocytes that were treated with IPC had 44 ± 5% cell death compared to 78 ± 7% cell death in sham-treated cultures (Fig. 2, n = 6, p < 0.05) as determined by LDH release assay. Our results suggest that IPC treatment was able to induce tolerance in rat astrocyte cultures against lethal ischemic injury.
Fig. 2.
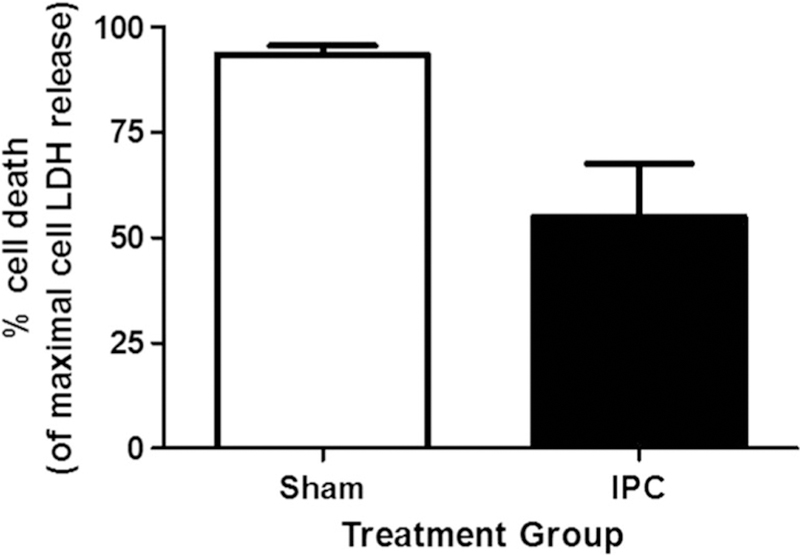
IPC treatment protects astrocytes against lethal oxygen glucose deprivation injury. LDH release assay results of rat astrocyte cultures following lethal OGD previously treated with sham, or IPC treatment. n = 6*p ≤ 0.05
IPC Treatment Activates Nrf2 in Rat Astrocyte Culture
After establishing a preconditioning paradigm in astrocyte cultures, we next investigated whether IPC treatment could activate Nrf2 in astrocytes. To determine if downstream targets of Nrf2 were increased following preconditioning, whole-cell astrocyte lysates were probed for NQO-1, a gene under strict Nrf2 transcriptional control. Forty-eight hours following IPC treatment, NQO-1 protein levels were significantly increased in astrocyte cultures compared to sham treatments (Fig. 3b). From these studies, the results indicate that IPC increased Nrf2-dependent gene transcription.
Fig. 3.

IPC treatment activates Nrf2 in astrocyte cultures. a Whole cell astrocyte lysates were probed for NQO-1 expression following IPC or sham treatment. b Quantification of western blot results. NQO-1 protein was normalized to actin (whole cell loading control). n = 4, *p ≤ 0.05
IPC-Induced Protection Is Abolished in Nrf2−/− Astrocyte Cultures Following Lethal OGD
To investigate the role of Nrf2 in IPC-induced cytoprotection, we subjected cortical mouse astrocytes cultured from WT and Nrf2−/− mice to IPC treatment 48 h prior to lethal OGD exposure. Based on results obtained from the LDH release assay, 1 and 2 h durations of OGD protected WT astrocytes against lethal (5 h OGD) ischemia as compared to sham preconditioning (1 h: 14 ± 11%; 2 h: 6 ±0.3%; sham: 32 ± 3.3%, p≤ 0.005, n = 4–6) but no cytoprotection was observed in Nrf2−/− cultures following IPC (Fig. 4). In addition, Nrf2−/− sham-treated cultures exhibited increased cell death compared to WT sham-treated astrocyte cultures following lethal OGD (52 ±1.2% vs. 32 ±3.3%, respectively, p ≤ 0.05). These results suggest that Nrf2−/− astrocytes are more susceptible to OGD injury, and that loss of Nrf2 protein significantly attenuated IPC-induced astrocyte protection.
Fig. 4.

IPC treatment of WT astrocyte cultures induces neuroprotection against lethal OGD, which is abrogated in Nrf2−/− astrocyte cultures. a Cell death assessment of WT and Nrf2−/− astrocyte cultures following lethal OGD. Cultures were treated with 1 and 2 h durations of OGD as preconditioning treatments, along with sham treatments. Cell death was determined using LDH release assay, and data is represented as a percent of the maximal LDH release n = 4–6, *p ≤ 0.05
Nrf2 Co-localizes to Non-synaptic Mitochondria in WT Mice
Previous studies from our group have suggested that Nrf2 plays a role in mediating mitochondrial bioenergetics and mitochondrial antioxidant protein levels in non-synaptic mitochondria [22]. A previous study suggested that Nrf2 can associate with mitochondria in a human cell line following overexpression of an outer mitochondrial membrane adapter protein, phosphoglycerate mutase family member 5 (PGAM 5) [23]. Therefore, we investigated whether Nrf2 is integrally associated with non-synaptic cortical mitochondria in vivo. Non-synaptic mitochondria were isolated from WT mice and purified on a Percoll gradient. We subjected 100 μg aliquots of WT non-synaptic mitochondria to increasing concentrations of proteinase K treatment, which degrades exposed outer mitochondrial membrane proteins [24]. In Fig. 5, we found that proteinase K was able to degrade Nrf2 in un-permeabilized mitochondria. As a positive control, Tom20, an integral outer mitochondrial protein, showed a similar pattern of degradation with increasing proteinase K treatment. Finally, a matrix protein, mtHSP70, did not show signs of degradation following proteinase K, indicating that proteinase K was limited to degradation of outer mitochondrial membrane proteins. As a further control, the control sample displayed in Fig. 5 shows the immunoblotting of proteins on unpermeabilized mitochondria. When mitochondria were lysed with 2% solution of triton and subjected to proteinase K, we observed almost complete degradation of protein by proteinase K as expected, since the enzyme now had access to proteins contained in all the compartments of mitochondria. These results suggest that Nrf2 is present on non-synaptic mitochondria, and that it is located on the outer mitochondrial membrane.
Fig. 5.

Nrf2 localizes to non-synaptic mitochondrial fraction of WT mice cortex. Non-synaptic mitochondria was isolated from WT C57 black mice and purified on a Percoll gradient. Mitochondria were then treated with or without proteinase K to determine submitochondrial localization of Nrf2. mtHsp70 (matrix protein) and Tom20 (outer mitochondrial membrane protein) were used as controls for proteinase K treatment. Ctrl: unpermeabilized non-synaptic mitochondria. Triton: treatment of mitochondria with 2% triton solution. n = 3
Nrf2 Knockout Mice Do Not Have Altered OXPHOS Protein Expression Compared to WT Mice
In mitochondria, OXPHOS protein expression is contributed by both mitochondrial and nuclear genomes [25]. From Fig. 5, given that Nrf2 appears to associate with WT mitochondria, we surmised that Nrf2 may coordinate nuclear and mitochondrial OXPHOS gene expression. To determine this, we measured the protein level ratio of a nuclear-encoded OXPHOS subunit, SDHA, and of a mitochondrial-encoded OXPHOS subunit, MCTO1 (COX-1) under basal conditions from isolated WT or Nrf2−/− non-synaptic mitochondria. The ratio of a nuclear-encoded OXPHOS protein subunit to a mitochondrial-encoded OXPHOS subunit has been suggested to be an indicator of mitochondria-nuclear OXPHOS balance and mitochondrial biogenesis [26]. From our results, there was a non-significant decrease in SDHA/MCTO1 ratio in Nrf2−/− non-synaptic mitochondria compared to WT non-synaptic mitochondria. These results suggest that Nrf2−/− mitochondria may have altered mitochondria-nuclear OXPHOS balance (Fig. 6a, b, p = 0.07). Once again, however, this decrease was statistically non-significant.
Fig. 6.

Nrf2 knockout mice do not have altered mitochondrial-nuclear ratio of OXPHOS protein expression compared to WT mice. One hunred micrograms of crude mitochondria from WT or Nrf2−/− mice were isolated and subjected to SDS-page electrophoresis. A subunit of COX-1 (MCTO1), a subunit of complex II (SDHA), and a cocktail of antibodies directed against the ETC complexes (I-V) were probed via western blotting. a Representative image is presented. b Quantification of SDHA/MCTO1 ratio, a measurement of mitochondrial biogenesis and mitochondrial-nuclear OXPHOS balance. N.S.: not significant. c Quantification of each of the ETC complexes (I-V), normalized to VDAC (mitochondrial loading control). n = 4–6
In addition, we investigated whether the protein levels of the electron transport chain complexes were altered by Nrf2. We therefore isolated non-synaptic mitochondria from naïve WT or Nrf2−/− mouse cortices and measured protein levels of individual electron transport chain complexes. From Fig. 6c, we did not observe any significant difference between the levels of complexes I-V between WT and Nrf2−/− mice. From these results, there appears to be no alterations in OXPHOS protein expression or mitochondrial-nuclear OXPHOS balance between WTand Nrf2−/− mice.
Nrf2 Knockout Mice Have Altered Expression of Electron Chain Supercomplexes Compared to WT Mice
A previous study suggested that electron transport chain complexes can aggregate to form “supercomplexes,” and that this structure confers a bioenergetic advantage and a reduction in ROS production in mitochondria [27]. Given that we did not observe any gross alterations in the expression of specific complexes in mitochondria from WT or Nrf2−/− mice, we investigated whether Nrf2 could play a role in regulating mitochondrial supercomplex formation. Non-reducing electrophoresis conditions and 2D separation of supercomplexes obtained from WT non-synaptic mitochondria resulted in the protein pattern observed in Fig. 7a. Based on western blotting of the OXPHOS complex subunits, we detected supercomplexes containing complexes I/III/IV/V, I/III/V, I/III/IV, I/V, III/IV, and IV/V. Complex II, as previously described [28], does not typically associate with other complexes to form a supercomplex.
Fig. 7.

Nrf2 knockout mice have altered expression of electron chain supercomplexes compared to WT mice. One hundred micrograms of crude mitochondria from WT (a) or Nrf2 KO (b) mice were isolated and subjected to BNGE and 2D SDS-page electrophoresis. For two-dimensional electrophoresis, bovine heart mitochondrial lysate was also included in the electrophoresis as a positive control to indicate the molecular weight of the respiratory complexes. Arrows in top left indicating direction of 1D native and 2D SDS electrophoresis. Representative image, n = 3
The profile of supercomplexes in Nrf2−/− exhibited marked differences (Fig. 7b). There was a decrease of supercomplex assemblies, as Nrf2−/− only contained supercomplexes composed of complexes I/III/V, I/V, and III/IV. Once again, complex II was isolated from interacting with other complexes. The results from Fig. 7 suggest that under naïve conditions, Nrf2 increased supercomplex formation.
Discussion
Our investigation sought to determine if Nrf2 plays a role in IPC-induced protection in astrocytes and if Nrf2 plays a role in mitochondrial OXPHOS complex expression. From the current studies, we developed a model of IPC that induces protection in rat astrocytes against lethal durations of oxygen-glucose deprivation (Fig. 2). In addition, this duration of IPC can activate Nrf2 as determined by measuring NQO-1, a downstream gene product under strict transcriptional control ofNrf2 (Fig. 3). Our focus on astrocytes was due to our results in Fig. 1 that highlighted the enriched presence of Nrf2 in astrocytes compared to neurons. Therefore, our results suggested that Nrf2, due to its previously reported regulatory roles, could be a key player in IPC-induced cytoprotection in astrocytes. This conjecture was supported since IPC-induced protection was significantly reduced in mouse astrocytes cultured from Nrf2−/− mice, suggesting that Nrf2 contributes to the protective effects of IPC treatment (Fig. 4). Next, we showed that Nrf2 can localize to the outer mitochondrial membrane of non-synaptic mitochondria from WT mice (Fig. 5). While we did not observe any differences in OXPHOS expression between WT and Nrf2−/− mice (Fig. 6), Nrf2−/− mice had fewer non-synaptic mitochondrial supercomplexes compared to WT mice (Fig. 7). These studies provide novel evidence that Nrf2 not only colocalizes with mitochondria in the brain, but may play an important role in mediating supercomplex formation.
Mitochondrial dysfunction, particularly high levels of mitochondrial ROS production, has been shown to be a precipitating factor in the pathogenesis of cerebral ischemia [29]. However, subinjurious levels of ROS serve as important signaling molecules that can be exploited to produce long-term adaptation in cells against a second lethal ischemic insult [30]. Studies have shown that treatment of cells with antioxidants prior to induction of IPC decreases the induction of ischemic tolerance and subsequent protection against lethal ischemic injury [31]. As a result, for our studies we focused on the Nrf2 pathway as this transcription factor is involved in cellular adaptation to oxidative stress through the upregulation of endogenous antioxidants.
Because mitochondrial function is critical to cellular adaptation and cellular dysfunction during cerebral ischemia, we sought to determine if there was a relationship between Nrf2 and brain mitochondria. Previous studies have implicated the role of Phosphoglycerate Mutase Family Member 5 (PGAM5) as functioning as a tether to which Nrf2 and its cytosolic repressor, Keap1, are retained on the outer mitochondrial membrane in HeLa cells [23]. Therefore, we first determined if Nrf2 could associate and co-localize with non-synaptic mitochondria from the mouse cortex. We utilized non-synaptic mitochondria because this population contained more glial and astrocytic mitochondria, as compared to the synaptic fraction which is essentially synaptosomes from neurons [32]. However, non-synaptic mitochondria also have cell body neuronal mitochondria; as we found Nrf2 levels to be several fold greater in astrocytes than in neurons (Fig. 1), neuronal mitochondrial contamination of the non-synaptic fraction was unlikely to obscure Nrf2’s mitochondrial function. A previous study by Lo et al. provided evidence that after overexpression of PGAM5, Nrf2 was able to associate with this outer mitochondrial membrane [23]. From our results from Fig. 5, our studies demonstrated that Nrf2 can co-localize to the outer mitochondrial membrane in non-synaptic mitochondria. To our knowledge, this is the first study to provide novel evidence of the localization of Nrf2 to mitochondria in the brain.
As Nrf2 is a nuclear transcription factor, our results with Nrf2’s localization to non-synaptic mitochondria may suggest a novel role of Nrf2 in coordinating mitochondria and nuclear OXPHOS expression. Therefore, we investigated the protein expression of a nuclear-encoded OXPHOS subunit (SDHA from complex II) and a mitochondrial-encoded OXPHOS subunit (MCTO-1 from complex I) in non-synaptic mitochondria. From Fig. 6, we found a non-significant trend suggestive of decreased SDHA/MCTO-1 ratio in Nrf2−/− mitochondria compared to WT mice. In addition, we found no significant differences between these two strains of mice in regards to protein expression of individual electron transport chain complexes.
In regards to the electron transport chain (ETC), previous studies have suggested that complexes can exist in oligomeric forms called supercomplexes [28]. These supercomplexes are assemblies of two or more ETC complexes, and have been suggested to provide more efficient bioenergetics and produce less ROS [28]. Therefore, we then investigated whether Nrf2 influenced the presence of supercomplexes in non-synaptic mitochondria. From Fig. 7, blue native gel electrophoresis and two-dimensional separation demonstrated that Nrf2−/− mice have decreased supercomplex formation compared to WT mice. While the mechanism of supercomplex formation is still largely unknown, Nrf2 may facilitate supercomplex formation in an effort to suppress ROS production from mitochondria. In addition to these findings, previous studies from our lab showed that Nrf2−/− mice have a lower respiratory control ratio compared to WT mice, suggestive of mitochondrial uncoupling [22]; therefore, the activation of Nrf2 following IPC could therefore facilitate more efficient mitochondrial bioenergetics and decreased ROS production, perhaps through mitochondrial supercomplex formation, ultimately serving to sustain cellular function in the face of lethal cerebral ischemia. This proposed mechanism of ROS signaling has never before been documented in previous literature, and could serve to unify ROS and mitochondrial-nuclear signaling for neuroprotection.
The association of Nrf2 to mitochondria may have several additional implications. It is possible that Nrf2 in close proximity to mitochondria can more readily sense excessive ROS formation, particularly since mitochondria are a significant contributor to intracellular ROS production under both basal and pathologic states. This association can in turn promote its translocation to the nucleus. Previous studies have shown that Nrf2, following nuclear translocation, can bind to p62 [33, 34]; p62 has been implicated in stimulation of mitochondrial autophagy (mitophagy) [35]. In addition, Nrf2 has been implicated in regulating mitochondrial biogenesis [36]; Nrf2 can also bind to nuclear respiratory factor 1 (NRF1), a prominent gene that can subsequently bind to a promoter found on the transcription factor of mitochondria (TFAM) [37]. TFAM translocates to mitochondria, where it initiates mitochondrial gene transcription. Therefore, Nrf2 may associate with mitochondria to serve as a messenger to relay the function of mitochondria, represented in its production of ROS, to the nucleus. Once in the nucleus, Nrf2 can promote increase in antioxidant enzymes to suppress ROS production, but may induce more long-term adaptation to oxidative stress by modulating mitochondrial gene transcription and biogenesis. While our studies only looked at one measurement of mitochondrial-nuclear OXPHOS balance (SDHA/MCTO1 ratio), future studies will further investigate this relationship. Investigating other markers of nuclear-mitochondrial balance, such as mitochondrial quality control (mitophagy and biogenesis) as well as apoptosis would further highlight the role of Nrf2 as a novel communicator between the mitochondrial and nuclear compartments.
In conclusion, our studies implicate the astrocyte-enriched transcription factor Nrf2 in mediating IPC-induced protection. Nrf2 may serve to not only protect astrocytes during cerebral ischemia, but by sustaining astrocyte function may also indirectly enhance neuronal ischemic tolerance. Finally, our studies provide novel evidence for the association of Nrf2 with brain mitochondria and implicate a role of Nrf2 in OXPHOS supercomplex formation. As a result, Nrf2 may represent a novel pathway in nuclear-mitochondrial communication and IPC-mediated neuroprotection.
Supplementary Material
Acknowledgments
Funding This study was funded by grants from the National Institutes of Health, National Institute of Neurological Disease and Stroke (NINDS) NS45676, NS054147, NS34773, F31 NS080344.
Footnotes
Compliance with Ethical Standards
Ethical Approval All applicable international, national, and/or institutional guidelines for the care and use of animals were followed. This article does not contain any studies with human participants performed by any of the authors.
Electronic supplementary material The online version of this article (https://doi.org/10.1007/s12975–017-0574-y) contains supplementary material, which is available to authorized users.
Conflict of Interest There authors declare that they have no conflicts of interest.
References
- 1.Murry CE, Jennings RB, Reimer KA Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74:1124–36. [DOI] [PubMed] [Google Scholar]
- 2.Schurr A, Reid KH, Tseng MT, West C, Rigor BM Adaptation of adult brain tissue to anoxia and hypoxia in vitro. Brain Res. 1986;374:244–8. [DOI] [PubMed] [Google Scholar]
- 3.Dave KR, Saul I, Prado R, Busto R Perez-Pinzon MA Remote organ ischemic preconditioning protect brain from ischemic damage following asphyxial cardiac arrest. Neurosci Lett. 2006;404: 170–5. [DOI] [PubMed] [Google Scholar]
- 4.Della-Morte D, Dave KR, DeFazio RA, Bao YC, Raval AP, Perez-Pinzon MA Resveratrol pretreatment protects rat brain from cerebral ischemic damage via a sirtuin 1-uncoupling protein 2 pathway Neuroscience. 2009;159:993–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim EJ, Raval AP, Hirsch N, Perez-Pinzon MA Ischemic preconditioning mediates cyclooxygenase-2 expression via nuclear factor-kappa b activation in mixed cortical neuronal cultures. Transl Stroke Res. 2010;1:40–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Perez-Alvarez A, Araque A. Astrocyte-neuron interaction at tripartite synapses. CurrDrug Targets. 2013;14:1220–4. [DOI] [PubMed] [Google Scholar]
- 7.Johri A, Beal MF Mitochondrial dysfunction in neurodegenerative diseases. J Pharmacol Exp Ther. 2012;342:619–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang B, Cao W, Biswal S, Dore S. Carbon monoxide-activated nrf2 pathway leads to protection against permanent focal cerebral ischemia. Stroke. 2011;42:2605–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhao X, Sun G, Zhang J, Strong R, Dash PK, Kan YW, et al. Transcription factor nrf2 protects the brain from damage produced by intracerebral hemorrhage. Stroke. 2007;38:3280–6. [DOI] [PubMed] [Google Scholar]
- 10.Bell KF, Fowler JH, Al-Mubarak B, Horsburgh K, Hardingham GE Activation of nrf2-regulated glutathione pathway genes by ischemic preconditioning. Oxidative Med Cell Longev. 2011;2011:689524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kitteringham NR, Abdullah A, Walsh J, Randle L, Jenkins RE, Sison R, et al. Proteomic analysis of nrf2 deficient transgenic mice reveals cellular defence and lipid metabolism as primary nrf2-dependent pathways in the liver. J Proteome. 2010;73:1612–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ryan MT, Hoogenraad NJ Mitochondrial-nuclear communications. Annu Rev Biochem. 2007;76:701–22. [DOI] [PubMed] [Google Scholar]
- 13.Lenth RV Statistical power calculations. J Anim Sci. 2007;85:E24–9. [DOI] [PubMed] [Google Scholar]
- 14.Kaech S, Banker G. Culturing hippocampal neurons. Nat Protoc. 2006;1:2406–15. [DOI] [PubMed] [Google Scholar]
- 15.Kim EJ, Raval AP, Perez-Pinzon MA Preconditioning mediated by sublethal oxygen-glucose deprivation-induced cyclooxygenase-2 expression via the signal transducers and activators of transcription 3 phosphorylation. J Cereb Blood Flow Metab. 2008;28:1329–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.SY X, YM W, Ji Z, Gao XY, Pan SYA modified technique for culturing primary fetal rat cortical neurons. J Biomed Biotechnol. 2012;2012:803930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dave KR, DeFazio RA, Raval AP, Torraco A, Saul I, Barrientos A, et al. Ischemic preconditioning targets the respiration of synaptic mitochondria via protein kinase c epsilon. J Neurosci. 2008;28: 4172–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Degasperi A, Birtwistle MR, Volinsky N, Rauch J, Kolch W, Kholodenko BN Evaluating strategies to normalise biological replicates of western blot data. PLoS One. 2014;9:e87293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sunderhaus S, Eubel H, Braun HP Two-dimensional blue native/blue native polyacrylamide gel electrophoresis for the characterization of mitochondrial protein complexes and supercomplexes. Methods Mol Biol. 2007;372:315–24. [DOI] [PubMed] [Google Scholar]
- 20.Shih AY, Johnson DA, Wong G, Kraft AD, Jiang L, Erb H, et al. Coordinate regulation of glutathione biosynthesis and release by nrf2-expressing glia potently protects neurons from oxidative stress. J Neurosci. 2003;23:3394–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stokfisz K, Ledakowicz-Polak A, Zagorski M, Zielinska M. Ischaemic preconditioning - current knowledge and potential future applications after 30 years of experience. Adv Med Sci. 2017;62: 307–16. [DOI] [PubMed] [Google Scholar]
- 22.Narayanan SV, Dave KR, Saul I, Perez-Pinzon MA Resveratrol preconditioning protects against cerebral ischemic injury via nuclear erythroid 2-related factor 2. Stroke. 2015; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lo SC, Hannink M. Pgam5 tethers a ternary complex containing keap1 and nrf2 to mitochondria. Exp Cell Res. 2008;314:1789–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tondera D, Czauderna F, Paulick K, Schwarzer R, Kaufmann J, Santel A. The mitochondrial protein mtp18 contributes to mitochondrial fission in mammalian cells. J Cell Sci. 2005;118:3049–59. [DOI] [PubMed] [Google Scholar]
- 25.Dennerlein S, Wang C, Rehling P. Plasticity of mitochondrial translation. Trends Cell Biol. 2017;27:712–21. [DOI] [PubMed] [Google Scholar]
- 26.Medeiros DM Assessing mitochondria biogenesis. Methods. 2008;46:288–94. [DOI] [PubMed] [Google Scholar]
- 27.Maranzana E, Barbero G, Falasca AI, Lenaz G, Genova ML Mitochondrial respiratory supercomplex association limits production of reactive oxygen species from complex i. Antioxid Redox Signal. 2013;19:1469–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Genova ML, Lenaz G. Functional role of mitochondrial respiratory supercomplexes. Biochim Biophys Acta. 2014;1837:427–43. [DOI] [PubMed] [Google Scholar]
- 29.Andrienko TN, Pasdois P, Pereira GC, Ovens MJ, Halestrap AP The role of succinate and ros in reperfusion injury—a critical appraisal. JMol Cell Cardiol. 2017;110:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vanden Hoek TL, Becker LB, Shao Z, Li C, Schumacker PT Reactive oxygen species released from mitochondria during brief hypoxia induce preconditioning in cardiomyocytes. J Biol Chem. 1998;273:18092–8. [DOI] [PubMed] [Google Scholar]
- 31.Sun JZ, Tang XL, Park SW, Qiu Y, Turrens JF, Bolli R. Evidence for an essential role of reactive oxygen species in the genesis of late preconditioning against myocardial stunning in conscious pigs. J Clin Invest. 1996;97:562–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stauch KL, Purnell PR, Fox HS Quantitative proteomics of synaptic and nonsynaptic mitochondria: Insights for synaptic mitochondrial vulnerability. J Proteome Res. 2014;13:2620–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jain A, Lamark T, Sjottem E, Larsen KB, Awuh JA, Overvatn A, et al. P62/sqstm1 is a target gene for transcription factor nrf2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J Biol Chem. 2010;285:22576–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Inami Y, Waguri S, Sakamoto A, Kouno T, Nakada K, Hino O, et al. Persistent activation of nrf2 through p62 in hepatocellular carcinoma cells. J Cell Biol. 2011;193:275–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ichimura Y, Waguri S, Sou YS, Kageyama S, Hasegawa J, Ishimura R, et al. Phosphorylation of p62 activates the keap1-nrf2 pathway during selective autophagy. Mol Cell. 2013;51:618–31. [DOI] [PubMed] [Google Scholar]
- 36.Piantadosi CA, Carraway MS, Babiker A, Suliman HB Heme oxygenase-1 regulates cardiac mitochondrial biogenesis via nrf2-mediated transcriptional control of nuclear respiratory factor-1. Circ Res. 2008;103:1232–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tufekci KU, Civi Bayin E, Genc S, Genc K. The nrf2/are pathway: a promising target to counteract mitochondrial dysfunction in Parkinson’s disease. Parkinson’s disease. 2011; 2011:314082. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.