

Abstract
The clinical applications of positron emission tomography (PET) imaging pharmaceuticals have increased tremendously over the past several years since the approval of 18fluorine-fluorodeoxyglucose (18F-FDG) by the Food and Drug Administration (FDA). Numerous 18F-labeled target-specific potential imaging pharmaceuticals, based on small and large molecules, have been evaluated in preclinical and clinical settings. 18F-labeling of organic moieties involves the introduction of the radioisotope by C-18F bond formation via a nucleophilic or an electrophilic substitution reaction. However, biomolecules, such as peptides, proteins, and oligonucleotides, cannot be radiolabeled via a C-18F bond formation as these reactions involve harsh conditions, including organic solvents, high temperature, and nonphysiological conditions. Several approaches, including 18F-labeled prosthetic groups, silicon, boron, and aluminum fluoride acceptor chemistry, and click chemistry have been developed, in the past, for 18F labeling of biomolecules. Linear and macrocyclic polyaminocarboxylates and their analogs and derivatives form thermodynamically stable and kinetically inert aluminum chelates. Hence, macrocyclic polyaminocarboxylates have been used for conjugation with biomolecules, such as folate, peptides, affibodies, and protein fragments, followed by 18F-AlF chelation, and evaluation of their targeting abilities in preclinical and clinical environments. The goal of this report is to provide an overview of the 18F radiochemistry and 18F-labeling methodologies for small molecules and target-specific biomolecules, a comprehensive review of coordination chemistry of Al3+, 18F-AlF labeling of peptide and protein conjugates, and evaluation of 18F-labeled biomolecule conjugates as potential imaging pharmaceuticals.
Graphical Abstract
■ INTRODUCTION
Traditional noninvasive imaging modalities such as Computed Tomography (CT) and Magnetic Resonance Imaging (MRI) are used for detecting anatomical and morphological changes associated with an underlying pathology. CT is the technique of choice for diagnosis and staging of malignant diseases and for monitoring response to treatment. However, it lacks necessary sensitivity and specificity for an early diagnosis of many cancers. More sensitive radioisotope-based molecular imaging techniques such as Positron Emission Tomography (PET) and Single-Photon Emission Computed Tomography (SPECT) are used to capture functional or phenotypic changes associated with pathology.1 PET is considered superior than SPECT due to availability of higher sensitivity instrumentations and better quantification of regional tissue concentrations of radioisotope-labeled molecular entities, i.e., imaging pharmaceuticals. Additionally, sensitivity and specificity for many applications are improved by the hybrid technologies, i.e., PET-CT and PET-MRI.
The PET technique has sufficient acquisition speed that allows determination of pharmacokinetics (PK) and distribution of imaging pharmaceuticals (i.e., biodistribution) and produces three-dimensional (3D) images of the functional processes in the body.2,3 When a positron-radioisotope based imaging pharmaceutical is injected into the body of a subject, it emits positrons. A positron collides with an electron in a tissue producing two gamma-ray photons with 511 keV energy at ∼180° apart by the annihilation process. The photons produced by the imaging pharmaceutical are detected by a PET imager. Three-dimensional images of the target tissue are reconstructed by a computer using an appropriate software. Various nonmetallic (11C, 13N, 15O, 18F, and 124I, etc.) and metallic (64Cu, 68Ga, and 89Zr, etc.) radionuclides are used for these applications in preclinical and clinical environments. A summary of the physical characteristics and the production methods for these PET radionuclides is given in Table 1.
Table 1.
Physical Properties and Production Methods for Some Cyclotron Produced Positron (β+) Emitting Radionuclides
| radionuclide | Production method | half-life | % decay mode | max, β+ energy, MeV | average energy, MeV |
|---|---|---|---|---|---|
| 11C | 14N(p/α)11C | 20.4 min | β+/99.8 | 0.98 | 0.39 |
| EC/0.2 | |||||
| 13N | 13C(p,n)13N | 10.0 min | β+/99.8 | 1.19 | 0.49 |
| 16O(p, α)13N | EC/0.2 | ||||
| 15O | 15N(p,n)15O | 2.03 min | β+/99.9 | 1.72 | 0.74 |
| EC/0.1 | |||||
| 18F | 18O(p,n)18F | 109.8 min | β+/96.9 | 0.635 | 0.25 |
| EC/3.1 | |||||
| 64Cu | 64Ni(p,n)64Cu | 12.7 h | β+/17.4 | 0.65 | 0.28 |
| EC/43.8 | |||||
| 64Ga | 68Ge/68Ga | 68 min | β+/88.9 | 1.9 | 0.84 |
| Generator | EC/11.1 | ||||
| 89Zr | 89Y(p,n)89Zr | 78.4 h | β+/22.7 | 0.9 | 0.4 |
| EC/77.3 | |||||
| 124I | 124Te (p,n)124I | 4.2 d | β+/23 | 2.15 | 0.97 |
| EC/77 |
The clinical applications of PET imaging pharmaceuticals have increased tremendously over the past several years since the availability of the Food and Drug Administration (FDA) approved 18fluorine-fluorodeoxyglucose (18F-FDG). Additionally, several 18F-labeled imaging pharmaceuticals (Table 2) for various applications, including neurology and oncology, are being used routinely in the clinic. A large number of other 18F-labeled small molecules have been evaluated in the past three decades as potential PET imaging pharmaceuticals in preclinical and clinical settings (under the approval of Radioisotope Drug Research Committee, RDRC, Institutional Review Board, IRB, and Investigational New Drug, IND, of the FDA etc.). Some of these potential imaging pharmaceuticals are listed in Table 3 and can be divided into several categories, (1) by clinical use category such as oncology, neurology, cardiology, (2) by the biological/biochemical process category such as protein synthesis, amino acid transport, nucleic acid or membrane component synthesis, and (3) by specific tracers, dealing with, for example, with receptors or gene expression and so forth.4–6
Table 2.
18F-Labeled Imaging Pharmaceuticals for PET Imaging Approved by the Food and Drug Administration (FDA)
| PET imaging pharmaceutical | year of approval | manufacturer | indication |
|---|---|---|---|
| [18F] Sodium Fluoride | 1972 | various | bone imaging |
| [18F]FDGa | 1994, 2004, 2005 | various | epileptic foci myocardial glucose metabolism tumor glucose metabolism |
| [18F]-Florbetapir | 2012 | Eli Lilly | β-amyloid, Alzheimer Disease |
| [18F]-Fluemetamol | 2013 | GE HealthCare | β-amyloid, Alzheimer Disease |
| [18F]-Florbet aben | 2014 | Piramal Imaging | β-amyloid, Alzheimer Disease |
| [18F]-Fluciclovine | 2016 | Blue Earth Diagnostics | prostate cancer |
[18F]FDG = [18F] Fluorodcoxyglucose.
Table 3.
18F-Labeled Molecular Entities in Pre-Clinical and Clinical Evaluation Environments
| imaging pharmaceutical | clinical application | biochemical process | mechanism of uptake or localization |
|---|---|---|---|
| [18F]FECH | oncology | membrane synthesis | choline kinase |
| [18F]FA | cardiology | fatty acid synthesis | Acetyl-CoA synthetase |
| [18F]FLT | oncology | DNA synthesis and cell proliferation | thymidine kinase (TK-l) in DNA synthesis |
| [18F]FMAU | |||
| [18F]FMISO | oncology | hypoxia | intracellular reduction and binding |
| [18F]FAZA | |||
| [18F]FETA | |||
| [18F]FES | oncology | receptor binding | estrogen receptors |
| [18F]MFES | |||
| [18F]FDHT | oncology | receptor binding | androgen receptors |
| [18F]FDOPA | neurology oncology | amino acid transport and protein synthesis | amino add transport and protein synthesis |
| [18F]FMT | |||
| [18F]FTYR | |||
| [18F]FET | |||
| [18F]Galacto-RGD | oncology | receptor binding for angiogenesis | αvβ3 integrin receptor |
| [18F] AH111585 | |||
| [18F]PSMA-1007 | oncology | receptor binding | prostate-specific membrane antigen |
| [18F]DCFPYL | |||
| [18pjFP | neuropsychiatry | dopaminergic system | dopamine D2/D3 receptor |
| [18F]FTP | |||
| [18F]FPCIT | neurology | dopaminergic neurons | dopamine transporter |
| [18F]FP-DTBZ | neurology | dopaminergic neurons | VMAT2 |
| [18F]MPPF | neurology | serotoninergic system | 5-HT1A receptors |
| [18F] Altanserin | neurology | serotoninergic system | 5-HT2A receptors |
| [18F] Setoperone | neurology | ||
| [18F] Flumazenil | neurology | GABAA receptor complex | benzodiazepine site |
| [18F]FEPPA | |||
| [18F]FMM | neurology | senile plaques | Aβ and NFTs |
| [18F]AZD-4694 | |||
| [18F]FDDNP | |||
| [18F]FHBG | gene therapy | gene expression | Herpes vims thymidine kinase |
The majority of clinical applications involve 18F-FDG as a PET imaging pharmaceutical; however, it has its own limitations and cannot be used for several neurological, oncological, and cardiological applications.7 For example, most prostate tumor lesions exhibit the low metabolic activity which results in limited uptake of 18F-FDG.8 Therefore, the need for receptor-targeted imaging pharmaceuticals has led to the discovery and development of numerous radiolabeled peptides and proteins that can target receptors which are known to overexpress on certain tumors.9–11 Some of the target-specific biomolecules, that are known to have high specificity and affinity for receptors associated with tumors and other pathological conditions, are folate, peptides (gastrin-releasing peptide, RGD, somatostatin etc.), antibodies, and antibody fragments.4,5 Developing an efficient method for radiolabeling of a biomolecule, with high specific activity, is the first step in the development of a potential imaging pharmaceutical. In this regard, thermodynamically stable and kinetically inert radiolabeled metal (including transition metals and lanthanides) chelates conjugated to target-specific biomolecules have been studied extensively for their potential applications as imaging pharmaceuticals.11–18
18F labeling of an organic moiety, such as a small molecule, involves a radioisotope introduction by a carbon−fluorine bond formation via a nucleophilic or an electrophilic substitution reaction.19–21 Extensive studies have been conducted, in the past, on numerous compounds to develop and optimize these substitution reactions leading to the routine production of some of these imaging pharmaceuticals (Tables 2 and 3).4–7,19–25 However, implementation of these processes still remains cumbersome, often involves multiple steps, dry organic solvents, nonphysiological and high-temperature conditions, and requires expensive, sophisticated, and automated synthesis modules. Moreover, 18F labeling of biomolecules, via carbon−fluorine bond formation, such as peptides, protein fragments, proteins, and oligonucleotides may not be able to handle such harsh conditions and requires alternate labeling methodologies.
Three methodologies have been developed for 18F-labeling of biomolecules in the past.26–37 These are (1) generation of 18F-labeled bifunctional agents or prosthetic groups followed by their reaction with biomolecules under mild conditions, (2) functionalization of a biomolecule via either a silicon- or a boron-acceptor group for 18F labeling by a displacement and an isotope exchange (IE) reaction or by a chelating group for 18F-AlF labeling, and (3) using click chemistry which involves Cu(I) mediated reaction of a functionalized peptide with a 18F-prosthetic group. A brief overview of these strategies for 18F-labeling of biomolecules is provided below.
The goal of the present report is to provide an overview of the 18F radiochemistry and 18F-labeling methodologies for small molecules, via carbon−fluorine bond formation, and target-specific biomolecules, a comprehensive review of coordination chemistry of Al3+, 18F-AlF labeling of peptide and protein conjugates, and evaluation of 18F-labeled biomolecule conjugates for various cancer targets in preclinical and clinical environments. This is the first report providing a thorough review of various areas that are essential for discovery and development of novel PET radioisotope-based imaging pharmaceuticals.
■ OVERVIEW OF 18F RADIOCHEMISTRY AND 18F-LABELING VIA CARBON−FLUORINE BOND FORMATION
Due to the desirable physical properties of fluorine (i.e., high electronegativity, small van der Waals radius, and ability to form strong C−F bond with carbon, 112 kcal/mol),38 favorable nuclear and radiochemical properties of 18F radioisotope39 (i.e., high, 96.9%, positron decay, ideal half-life, 109.8 min, low positron energy, 0.635 MeV, a short range in tissue, 2.4 mm, high specific activity production by cyclotrons), well developed chemistry for fluorination and radiofluorination (i.e., high labeling yield, 20–40%) of small organic molecules, and acceptable radiation dosimetry of 18F-labeled imaging pharmaceuticals, 18F-based imaging pharmaceuticals are now being used routinely in the clinic. The optimal physical half-life of 18F allows for more complex radiosynthesis, longer in vivo evaluations, and most importantly commercial distribution to clinical PET centers.
Only 2 h physical half-life of 18F radionuclide requires that production time of a PET imaging pharmaceutical must be as short as possible. Ideally, the synthesis and purification time for a tracer should be less than 2 to 3 times the half-life of 18F. It is preferred that the 18F introduction in the molecule should be in the last step of the radiosynthesis. In a synthesis procedure, a large excess of the precursor is usually necessary to enhance the rate and increase the extent of the reaction. Finally, the excess precursor and the side products are removed by using a prep High-Performance Liquid Chromatography (HPLC) method while the salts are removed by a Reversed-Phase Sep Pak (RP-Sep Pak) cartridge.
18F labeling via C−F bond formation is traditionally accomplished by electrophilic (18F−F2) and nucleophilic (18F−) substitution reactions.4–7,19–25 For the production of electrophilic 18F−F2, a passivated nickel target is loaded with neon gas with 0.1% natural fluorine gas and bombarded with 8 to 9 MeV deuterons for 1 to 2 h. This process produces <1 curie (Ci) of 18F−F2 radioactivity in the gaseous form with a low specific activity (10−20 mCi/μmol). The low specific activity achieved is because out of the two atoms in the 18F−F2, only one is radioactive, and because of the presence of fluorine gas as a carrier. Alternatively, 18F−F2 can also be produced by proton bombardment of 18O2 gas. The radiolabeled products produced from the electrophilic substitution reaction are also low specific activity, as the specific activity of the 18F−F2 is low. Therefore, the electrophilic process is less preferred and it is only used when nucleophilic substitution reactions are not appropriate, although 18F-FDOPA was originally synthesized using the electrophilic reaction. In general, 18F−F2 is converted into less reactive and more selective fluorination agents such as acetyl hypofluorite, xenon difluoride, and fluorosulfonamides and aryltrimethyltin precursor.
The most successful approach for preparing 18F-labeled compounds with high specific activity is by the nucleophilic substitution reaction of aliphatic and aromatic moieties. For the production of nucleophilic fluoride ions (18F−), a liquid (a silver or tungsten, or titanium target filled with 0.3 to 3 mL 18O-enriched water, H218O) target is bombarded with protons (10 to 19 MeV energy, 20 to 30 μA beam current). Several curies of the 18F-HF or 18F-fluoride ions in water (with high specific activity, ∼10 Ci/μmol) can be easily produced by this method.
In general, 18F-HF (18F Water) is converted to alkali metal halides, such as 18F-FK, either (1) by transferring the material from the target to a reaction vessel containing a base such as potassium carbonate or (2) by passing the 18F-HF (or 18F water) through an anion exchange resin (such as QMA-Sep Pak cartridges), followed by eluting with a base, K2CO3, to produce 18F-FK. A ligand with strong affinity for potassium (such as Kryptofix 2.2.2 in acetonitrile) is used for removing the K+ and providing free F− for the nucleophilic reaction. The acetonitrile/water mixture containing fluoride and potassium complex of Kryptofix 2.2.2 is evaporated by heating at 80 °C under vacuum.22 Dried residue in the reaction vessel is used further for nucleophilic reaction with the precursor. Using some other bases, e.g., tetrabutylammonium hydroxide (TBAH) for conversion to 18F-TBAF avoids the use of Kryptofix 2.2.2 and can be used directly into organic solvents for the nucleophilic reaction.
Fluoride ion is a poor nucleophile in an aqueous medium; therefore, dipolar aprotic solvents are traditionally used for fluorination reactions. The preferred solvent for nucleophilic substitution reactions of aliphatic compounds is acetonitrile, as it can easily be removed by evaporation. Removal of the solvent like acetonitrile is important as its presence could make HPLC purification very challenging. Moreover, the amount of acetonitrile also needs to be controlled in the final product. Alternatively, DMSO (dimethyl sulfoxide) and DMF (dimethylformamide) may be used for reactions that require higher temperatures. 18F-labeling chemistry using electrophilic and nucleophilic substitution reactions is well developed and optimized. Tables 2 and 3 list several small-molecule products that are either commercially available and are being used clinically or are being tested in preclinical and clinical environments. There are several excellent review articles related to their syntheses and clinical evaluations.4–7,19–25
■ OVERVIEW OF 18F-LABELING STRATEGIES FOR BIOMOLECULES
As discussed above, 18F-labeling of biomolecules, via C−F bond formation, is challenging as these labeling conditions are not compatible with their stability. Three methodologies for 18F-labeling of biomolecules involving (1) 18F-labeled bifunctional agents or prosthetic groups, (2) click chemistry, and (3) a silicon- or a boron-acceptor or a chelating group were developed.26–37
A series of 18F-prosthetic groups have been developed for labeling of biomolecules under mild reaction conditions. For example, 18F-fluorobenzaldehyde, 18F-FBA, has been shown to form a conjugate via oxime formation with the amine function in the peptide.40–43 Similarly, N-succinimidyl (e.g., N-succinimidyl-4-18F-fluorobenzoate, 18F-SFB)44,45 and maleimide (e.g., N-(2-(4-[18F]fluorobenzamido)ethyl) maleimide, [18F]FBEM, and 1-[3–2-[18F]fluoropyridine-3-yloxylpropylpyr-role-2,5dione, [18F]FPyME)46,47 containing 18F-prosthetic groups were used to conjugate with amine and thiol groups in biomolecules, respectively. However, these labeling techniques are also time-consuming, challenging, and not amenable to kit production. Moreover, some of these methodologies result in poor radiochemical yields for the 18F-labeling of proteins, lower site specificity of some prosthetic groups, and more lipophilic conjugates than the native biomolecule resulting increased biliary excretion.
Since benzenesulfonyl fluorides are more resistant to hydrolysis in aqueous media, several aryl 18F-sulfonyl fluorides were prepared and evaluated for their stability and for radiofluorination of biomolecules. 48–50 Inkster et al.48 prepared several aryl sulfonyl fluorides; however, 3-formyl-2,4,6-trimethylbenzenesulfonyl [18F]fluoride was coupled with a 9-amino-acid bombesin analog, BBN-NH2 in a good yield (64%). The conjugate was stable for >2 h in 10% DMSO in PBS under physiological temperature and pH but was only 55% intact after 15 min incubation in mouse serum. Matesic and co-workers50 prepared numerous sulfonyl fluorides and predicted that [18F]sulfonylfluorides functional groups with a combination of electron-donating groups and increased steric bulk near the sulfonyl group will be most stable in vivo. A new 18F-labeled 4-fluorophenylboronic acid prosthetic group was prepared and used for Pd-catalyzed labeling (RCY given in the parentheses) of a small molecule (83−87%), a peptide (33−48%), and a protein (∼2−5%).51
The click chemistry has become a powerful and versatile synthesis tool in the radiopharmaceutical chemistry.52 The reaction involves the 1,3-dipolar cycloaddition of an alkyne with an azide functional group via Cu(I) catalyzed reaction forming a triazole moiety. Marik and Sutcliffe53 radiolabeled, first time, azidopropionic acid derivatives of model peptides with various [18F]fluoroalkynes. In more recent work, acetylene-bearing 2-[18F]fluoropyridines, [18F]FPy5yne and PEG-[18F]FPyKYNE, were prepared via nucleophilic heteroaromatic [18F]fluorination of their corresponding precursors, and these groups were used to label azide-modified peptides and oligodeoxyribonucleotide.54–56 This technique requires careful dry down of the solvents.54–56 A 2-cyanobenzothiozole-based 18F, [18F]-FPyPEGCBT, and an ethynyl-4-[18F]fluorobenzene prosthetic groups were used for conjugation with the terminal cysteine group in a cRGDyK peptide (30 min reaction time, 7 ± 1% End of Bombardment yield) and in matrix-metalloproteinase inhibitor (70 min reaction time, 56 ± 12% yield), respectively.57,58
Several main group inorganic elements are known to form stronger fluorine bonds than a carbon−fluorine bond. For example, bond dissociation energies (given in the parentheses, kJ/mol) for some main group element-fluoride bonds in diatomic molecules are B−F (732), C−F (513.8 ± 10), Si−F (576.4 ± 17), and Al−F (675).38 Therefore, these inorganic elements have been used as carriers for 18F labeling of biomolecules in high specific activity but under mild conditions, i.e., in aqueous media and low temperature. The 18F labeled SiF4 and BF4 were prepared initially by isotope exchange reactions between F-metal fluoride (such as Li, K, Rb, and Cs) and SiF4 and by the reaction of 18F-metal fluoride and boron trifluoride, respectively.59,60 18F-flouorosilane was initially proposed as a labeling reagent by Rosenthal et al.;61 however, a preliminary in vivo evaluation revealed fast hydrolysis of the compound followed by bone uptake of free 18F, suggesting an unsuitable labeling reagent. Blower and co-workers and Schirrmacher and Jurkschat identified simultaneously that hydrolysis of 18F-silanes can be significantly reduced by the introduction of bulky substituents like t-butyl groups to the silicon moiety.62,63
Two novel methodologies, based on isotope exchange (IE) reaction, were invented in 2005 and 2006, i.e., RBF3− labeling by Perrin et al.64 and silicon fluoride acceptor (SiFA) by Schirrmacher et al.63 These two methodologies demonstrated that biomolecules can be 18F-labeled in aqueous solution and at room temperature and led to further research by Mu et al.65 based on the leaving group approach. Radiofluorination of RBF−3 and the 18F-SiFA using either leaving group displacement or IE methodologies has been used for labeling of several peptides (somatostatin, bombesin/gastrin-releasing, and RGD, etc.) and proteins. Radiolabeling conditions for RBF−3 and SiFA using IE methodologies are milder than those of displacement reactions, performed at room temperature under moderately acidic conditions.66–69 A successful application of Si−18F chemistry was demonstrated by a kit-like 18F-labeling of proteins70 and followed by the development of SiFAlin-based scaffolds71,72 and dioxaborolanes73 for radio-labeling. Several excellent review articles have been published in the past decade.26–37
The bond dissociation energy of Al−F is greater than any other main group metal fluoride bond making it as an attractive carrier for 18F. For example, some bond dissociation energies (given in the parentheses, kJ/mol) are: Al−F (675), Ga−F (584 ± 13), In−F (516 ± 13), and Tl−F (439 ± 21).38 The Al−F bond strength is reflected in the reported stability constants of various binary and ternary fluoro complexes of aluminum.74 Due to water sensitivity of the aluminum−carbon bond and the low hydrolysis constant of Al3+ (pKa = 5.52),75 18F-AlF itself cannot be used as a direct radiolabeling agent for biomolecules. Instead,18F-AlF is coordinated to a chelating agent-biomolecule conjugate.
Linear and macrocyclic polyaminocarboxylates, such as DTPA (diethylenetriaminepentaacetic acid), NOTA (1,4,7-triazacyclononane-1,4,7-triacetic acid), NODA-GA (1,4,7-triazacyclononane, 1-glutaric acid-4,7-acetic acid), and DOTA (1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid), structures 1–4 given in Figure 1, are known to form thermodynamically stable and kinetically inert metal chelates and to keep Al3+ in soluble form. Consequently, McBride and co-workers discovered and developed a versatile method which involved formation of a DTPA or NOTA conjugate of biomolecules followed by labeling with 18F-AlF.76–78
Figure 1.
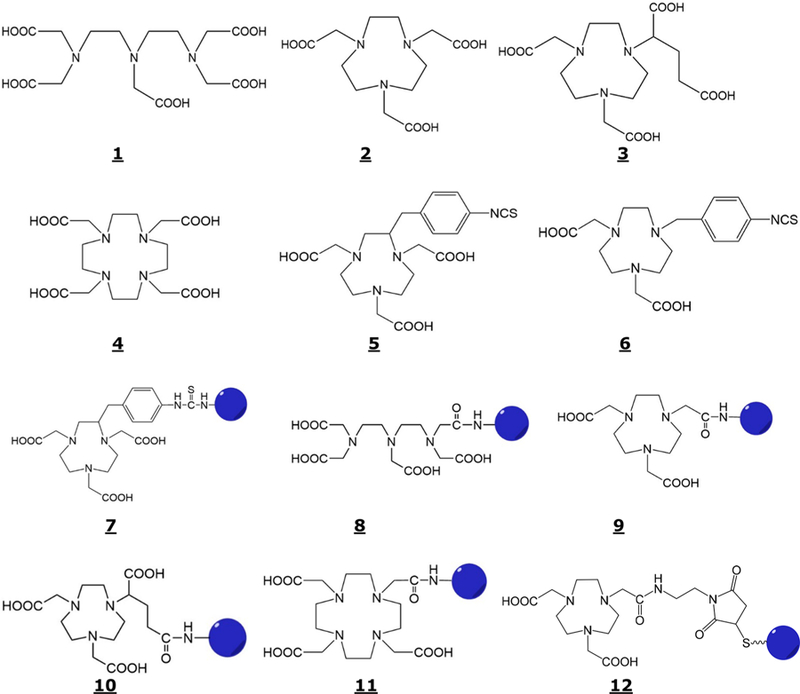
Structures of DTPA (1), NOTA (2), NODA-GA (3), DOTA (4), p-SCN-Bz-NOTA (5), SCN-Bz-NODA (6), p-SCN-Bz-NOTA-Biomolecule (7), DTTA-CH2CONH-Biomolecule (8), NODA-CH2CONH-Biomolecule (9), NODA-GA-CH2CONH-Biomolecule (10), DO3A-CH2CONH-Biomolecule (11), and NODA-MAL-CS-Biomolecule (12).
In general, a linear or a macrocyclic polyaminocarboxylate chelating agent is modified for conjugation by introducing a p-SCN benzyl group in the carbon backbone (e.g., S-2-(4-isothiocyanatobenzyl)-1,4,7-triazacyclononane-1,4,7-triacetic acid, p-SCN-Bz-NOTA, Structure 5, Figure 1) or at an amine function in the ring, (e.g., 4,7-bis(carboxymethyl)-1-(4-isothiocyanato-benzyl)-1,4,7-triazacyclononane, SCN-Bz-NODA, Structure 6, Figure 1) or by forming an N-hydroxysuccinimide (NHS) or a maleimide (MAL) ester of one of the several carboxylic acid functions followed by the reaction with an amine or a sulfhydryl group respectively, in the biomolecule. Some of these conjugation methods will result in a reduction of the number of carboxylic acid functions and formation of one amide function for coordination to the metal. For example p-SCN Bz-NOTA forms a conjugate by reacting with the primary amine via forming a thiourea bond (Structure 7, Figure 1). Similarly, the NHS esters of DTPA, NOTA, NODA-GA, and DOTA react with the amine functions in the biomolecules to form the conjugates via forming amide bonds (structures 8–11) and a MAL ester with a sulfhydryl group to form structure 12 given in Figure 1.
■ ALUMINUM COORDINATION CHEMISTRY
Aluminum, which belongs to Group 13 of the periodic table, is the third most abundant element in the earth’s crust (8.8%).79 It is usually bound to oxygen (alumina, Al2O3) or fluorine (cryolite, Na3AlF6) rather than existing in the free form. The most stable and common oxidation state of aluminum is +3; however, some compounds are known in which it has a low oxidation state of +1 and others with +2 oxidation state in the gas phase. The element forms acidic cationic complexes, such as Al(H2O)3+, with a Ka value of 1.12 × 10−5 for deprotonation of an axial water. Due to its low hydrolysis constant, Al3+ hydrolyzes into mono- and polyhydroxo species that precipitate (log Ksp = −33.5)80 around pH 5 and predominate over soluble complexes between pH 5 and 9. The precipitated Al(OH)3 dissolves again above pH 9 by forming soluble aluminates.
The effective ionic radius of Al3+ is 50 pm,80 it is highly electropositive, and does not polarize easily. Based on the Pearson’s HSAB (Hard and Soft Acids and Bases) classification,81 Al3+ behaves as a Lewis acid (electron pair acceptor; electrophile). Therefore, Lewis bases (electron pair donors; nucleophiles) are effective ligands to bind aluminum.82 Al3+ prefers to coordinate with hard Lewis bases which have neutral donor molecules or anions (such as H2O, ROH, RNH2, OH−, Cl−, F−, PO43−, SO42−, CH3COO−, RO−). Tetrahedral, trigonal bipyramidal, and octahedral molecular geometries are known for aluminum complexes of Cl−, F−, and H2O, and with four ([AlCl4]−), five ([AlF4(OH)]2−), and six ([Al(H2O)6]3+, and [AlF6]3−) coordination numbers, respectively (Figure 2).
Figure 2.
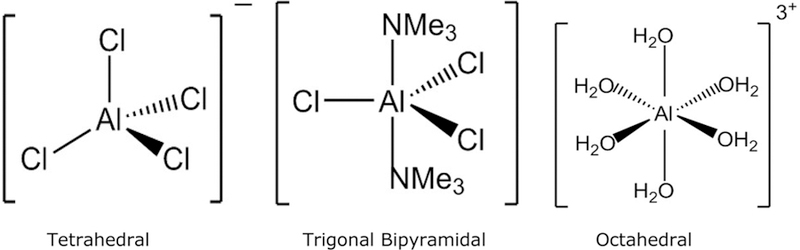
Tetrahedral, trigonal bipyramidal, and octahedral molecular geometries of Al3+ complexes.
A comparison of stability constants of fluoride complexes of various metal ions shows that the binding of F− to Al3+ is unusually strong.74 For most ligands including hydroxide the stability order for metal complexes is Fe3+ > Ga3+ > Al3+; however, it changes for the F− ion, i.e., Al3+ > Fe3+ > Ga3+. The stepwise stability constants (log Kn) for AlFn (where n = 1 to 5) complexes are 6.40, 5.21, 3.91, 2.63, and 1.35 (μ = 0.1 M). Aluminum ligand binding is a partially covalent interaction that otherwise involves ionic or electrostatic bonds. The most stable aluminum chelates are with multidentate ligands with negatively charged oxygen donor atoms (such as alkoxides, phenoxides, and carboxylates) which form chelate rings. A review by Martell and co-workers80 provides an excellent summary of stepwise protonation constants of various multi-dentate ligands and the stability constants of their Al3+ chelates. Affinities of these ligands for metal ions also increase with the basicity of the ligand donor groups. As observed for gadolinium and calcium chelates,83–86 the stability of aluminum chelates (log KML)80,87–89 also increases linearly (Figure 3) with the overall basicity of the ligand donor groups (i.e., a sum of pKa values for the neutral form of the ligand). This reflects that, like gadolinium and calcium chelates, the aluminum chelates are also primarily ionic in nature. Another factor that plays an important role in the formation of aluminum chelates is the chelate ring size, i.e., five-membered chelate rings prefer larger metal ions, while the six-membered chelate rings are preferred by smaller metal ions, providing the least strain.
Figure 3.
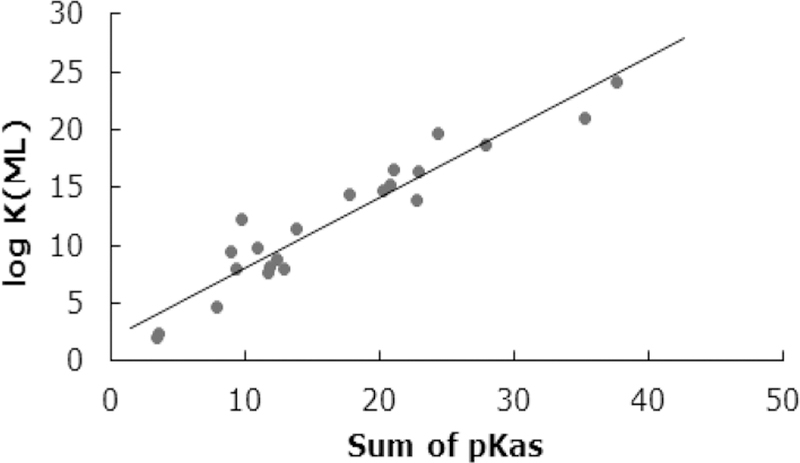
Correlation between log KML of Al3+ chelates and the sum of the pKa values of the neutral form of some linear and macrocyclic polyaminocarboxylates.
Depending on the reaction conditions, fluoride and hydroxide, with high affinity to Al3+, compete for a limited number of available binding sites in a multidentate ligand coordinated metal ion to form a ternary complex. The maximum coordination number of Al3+ is six. For example, the equilibrium or stability constant (log KF) for formation of Al(EDTA)F2− (where EDTA is ethylenediaminetetraacetic acid) ternary complex and the pKOH value (deprotonation of coordinated water) for Al(EDTA)− have been reported as 4.9590 and 5.83,80 respectively, suggesting that the hydroxo complex becomes the predominant species under neutral pH conditions. Similar trends were reported for other aluminum chelates of several other ligands (where L = NTA − nitrilotriacetic acid, HEDTA − hydroxyethyl ethylenediaminetriacetic acid, and CDTA − trans-1,2,-cyclohexyldiaminetetra-acetic acid). The log KF and pKOH values reported were 5.41, 5.53, 3.14, and 5.09, 4.89, 7.82 for NTA, HEDTA, and CDTA, respectively.80,90 Figure 4 shows a linear plot of log KF vs pKOH of Al3+ chelates of NTA, EDTA, HEDTA, and CDTA. An excellent linear correlation (log KF = −0.825 pKOH + 9.631) with r2 = 0.994 was observed. The inverse relationship between log KF and pKOH suggests that the fully formed chelates that are difficult to hydrolyze are likely to form a weak fluoro ternary complex of Al3+ from the reaction of aluminum chelate and fluoride.
Figure 4.
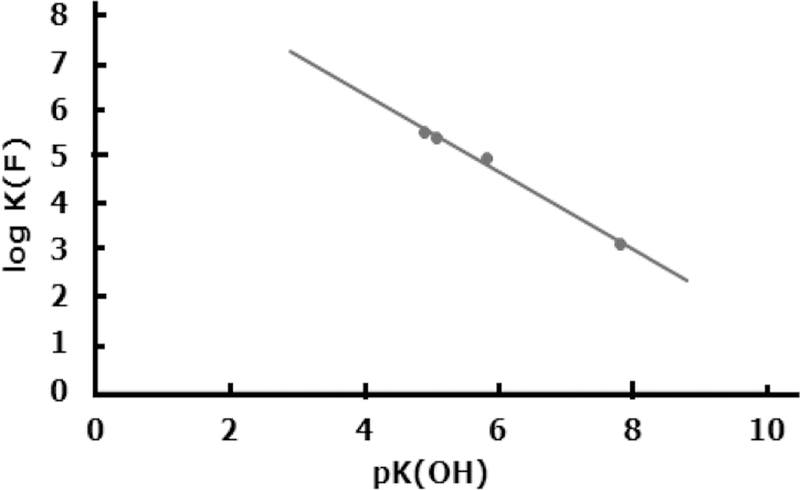
Plot of log KF (equilibrium constants for formation of fluoro ternary complexes of aluminum polyaminocarboxylates) vs pKOH (deprotonation constants of coordinated water of aluminum polyaminocarboxylates).
Since NTA is only a tetradentate ligand and aluminum prefers an octahedral geometry, a quaternary complex (OH)-Al(NTA)F2− is likely to form in neutral solution in the presence of fluoride. On the other hand, one of the coordinated carboxylates must be substituted by fluoride or hydroxide to form ternary complexes of EDTA, HEDTA, and CDTA ligands. Farkas et al.87 detected a metastable hydroxo complex of Al(NOTA), i.e., Al(NOTA)(OH) under basic conditions, by using pH-potentiometry and determined a pKOH value as 12.2. The metastable Al(NOTA)(OH) complex transforms slowly to Al(OH)4− and free NOTA. More than 6 orders of magnitude higher pKOH value for Al(NOTA) than Al(EDTA)− may be due to more inert Al-carboxylate bonds in the macrocyclic NOTA chelate than in the corresponding Al(EDTA)− chelate. The formation of the ternary complex of Al(NOTA) with F− was not detected during the reaction of Al(NOTA) with fluoride using a fluoride selective electrode and/or 19F-NMR. These results are not at all surprising as a log KF value of −0.435 (or KF = 0.37) can be calculated from the linear correlation between log KF and pKOH discussed above (Figure 4). However, the formation of the ternary complex was almost 100% complete when a mixture of Al3+, NOTA, and F− in 1:1 mixture of ethanol:water was heated at 100 °C for 15 min, presumably due to the preference of Al3+ for fluoride coordination over carboxylate coordination.
The rate of water exchange for Al(H2O)63+ is rather slow, i.e., 1.3 s−1 with a volume of activation as +5.7 cm3 M−1. The positive volume of activation suggests that the water-exchange reaction follows a dissociative interchange (Id) mechanism.91 The rate of the reaction increases significantly if one of the coordinated water molecules is deprotonated (kex = 3.1 × 104 s−1 for Al(H2O)5(OH)2+). A reduced charge on the deprotonated small Al3+ may be responsible for the increased water exchange rate. Limited kinetic data are available for Al3+ reactions (formation and dissociation) in aqueous medium.87,91–95 This is due to the fact that (1) stability of aluminum complexes is relatively low in strongly acidic medium (2) Al(H2O)63+ hydrolyzes at lower acidity or higher pH, and (3) there is a lack of specific UV/vis absorbance to monitor the progress of the reactions. However, it has been proposed that Al(H2O)63+ and Al(H2O)5(OH)2+ react via an Id mechanism with the latter being more reactive.93–95
Due to the sluggish nature of Al3+, the rates of complexation of aluminum with linear polyaminocarboxylates (such as EDTA and DTPA) have been rather slow with second-order rate constants (M−1 s−1) as 4.73 and 21.5 for H3EDTA− and H2EDTA2−, respectively, and 2.06 and 19.3 for H4DTPA− and H3DTPA2−, respectively.94 Both Al(H2O)63+ and Al-(H2O)5(OH)2+ were identified as reactive forms for various protonated forms of the ligands. The rates of formation and dissociation of Al(NOTA) are very slow in acidic medium. For example, only about 1.5% of the Al(NOTA) chelate converted to Al3+ in 16 days in 1 M HCl.87 Similarly, the hydrolysis of Al(NOTA) is slow under basic conditions demonstrating its inertness. Based on the reported first- and second-order rate constants by Farkas et al.,87 half-lives (t1/2) of base hydrolysis can be calculated as 71.8 and 21.8 h in 0.1 and 1.0 M sodium hydroxide, respectively. The formation kinetics of the Al-(EDTA)F2− ternary complex were studied by Nemes et al.96 using potentiometric and 19F NMR methods. Various simultaneous reactions between Al(EDTA)−, Al(EDTA)-(OH)2− and F− and HF were proposed. Two second-order rate constants, 20.7 ± 0.3 M−1 s−1 and 471 ± 93 M−1 s−1 for the reaction of F− and HF, respectively, with Al(EDTA)− were reported.96 Due to the kinetic inertia of Al(NOTA), there was no reaction observed between the chelate and the fluoride, however, the formation of Al(NOTA)F− was complete in 15 min by heating Al3+, NOTA, F− in 1:1 ethanol:water mixture at 100 °C (as given above). It appears that kinetics of formation of Al(NOTA)F− ternary complex is fairly complicated in the Al3+-NOTA-F−-H+ four-compartment system and requires more work to understand the chemistry.
■ 18F-ALF-LABELED BIOMOLECULES CONJUGATED TO CHELATING AGENTS
Radiolabeling of biomolecules with a metallic radionuclide (e.g., 64Cu, 68Ga, 89Zr, etc.) using a bifunctional chelating agent is a well-established methodology for development of potential imaging pharmaceuticals.11–18,97–100 Physicochemical properties and coordination chemistry of Al3+, i.e., forming thermodynamically stable and kinetically inert aluminum chelates with polyaminocarboxylates and unusually strong Al−F bond38,80–82,87–89 led to the discovery of a novel methodology for 18F-labeling of biomolecules that are conjugated to a chelating agent.76–78 Moreover, the AlFn complex is stable in vivo, since this is a part of the mechanism that the body uses to incorporate fluoride into tooth enamel.101 Hence, small doses of AlFn should be compatible for human use.102 Among suitable ligands, a hexadentate macrocyclic polyaminocarboxyate ligand, NOTA (Structure 2, Figure 1), and its analogs and derivatives have been found suitable for AlF2+ chelation.87,88 The following sections will provide a comprehensive review of the 18F-AlF labeling of several peptides, folate, and proteins that have high affinity for receptors which are overexpressed on tumors and their evaluation as potential imaging pharmaceuticals in preclinical and clinical environments.
Preclinical Evaluation of 18F-AlF-Labeled Peptide Conjugates.
Carcinoembryonic Antigen (CEA)-Specific Peptides.
Several hapten peptide conjugates were evaluated in the past for in vivo targeting of Carcinoembryonic Antigen (CEA) expressing tumors using a pretargeting technique.103–106 The pretargeting technique uses a bifunctional reagent (e.g., bispecific monoclonal antibody, bsMAb) with the affinity for a tumor and for a small hapten peptide. Typically, mice are implanted with CEA-expressing LS174T human colonic tumors, a bispecific monoclonal anti-CEA antihapten antibody is given to the mice, and 16 h later a 18F-labeled hapten peptide is administered.
Initial studies were conducted with the first-generation chelating agent−peptide conjugates that are capable of binding the 18F-AlF.76 At low fluoride concentrations, Al3+ formed a mono fluoro complex with a DTTA-peptide conjugate (8-Gln-Ala-Lys (HSG)-D-Tyr-Lys (HSG)-NH2, IMP 272) which included two hapten moieties (HSG is histamine-succinylglycine) on the lysine side chains. Upon heating, a mixture of 6 nmol each of Al3+, 18F−, and IMP 272 in a pH 4 buffer at 100 °C for 15 min showed only 7% incorporation of the radioactivity. However, when 26 nmols of the IMP 272 were added to the reaction mixture and heated for an additional 15 min the incorporation yield increased to 92%. Although the yield of 18F-AlF labeling was improved, the 18F-AlF-IMP 272 was unstable in water, i.e., 17% loss of 18F− within 40 min. Another DTTA conjugated analog, (8-Dpr(R)-3-amino-3-(2-bromophenyl)-propionyl)-D-Ala-D-Lys(HSG)-D-Ala-D-Lys-(HSG)-NH, IMP 375) was synthesized76 and evaluated for 18F-AlF labeling yield and stability. 18F-AlF-labeled IMP 375 was stable in water with 98% labeling yield, but human serum stability was not acceptable. Low in vitro stability of these 18F-AlF-labeled conjugates may be correlated with the nature of linear polyaminocarboxylate chelates.
The NOTA, a hexadentate macrocyclic ligand with three amines and three carboxylic acids (Structure 2, Figure 1), forms a thermodynamically stable and kinetically inert aluminum chelate87,88 with a known distorted octahedral geometry (with 2.067 and 1.846 Å bond distance for M-N and M-O, respectively) in the solid state.107,108 Thus, a commercially available p-SCN-Bz-NOTA (Structure 5, Figure 1) was conjugated to a pretargeting peptide (7-D-Ala-D-Lys(HSG)-D-Tyr-D-Lys(HSG)-NH2, IMP 449),76,77 and the conjugate was labeled with 18F-AlF by heating a mixture of Al3+, 18F−, and IMP 449 at 100 °C for 15 min followed by HPLC purification. The uncorrected labeling yield was 5% to 20% and the purified product was stable in serum at 37 °C for 4 h. 18F-AlF-IMP 449 along with 18F− alone and the 18F-AlF complex was evaluated in preclinical models using nude mice bearing the human colon cancer xenograft, LS174T.
As expected, 18F− alone and 18F-AlF accumulated in the bone. Tissue uptake of 18F-AlF-IMP 449 was significantly different than the tissue uptake of 18F− and 18F-AlF. Significantly lower uptake of 18F-AlF-IMP 449 in all tissues, except kidney, was observed suggesting that the intact material was eliminated via renal route. Similar to 18F-FDG, higher uptake (6.01 ± 1.72% Injected Dose or %ID/g) of 18F-AlF-IMP 449 resulted in the tumor upon pretargeting with TF2 anti-CEACAM5 BsmAb. TF2 is an engineered trivalent bispecific antibody with a humanized anti-HSG Fab fragment derived from the anti-HSG mAb. In vivo stability of 18F-AlF-IMP 449 could not be investigated due to its rapid clearance. However, the analysis of the urine sample from these animals showed that all of the activity was bound to the peptide which was supported by no bone uptake of the tracer. Imaging studies were conducted with 18F-AlF-IMP 449 with and without pretargeting and with 18F-FDG. Static images were taken 2 h post injection and the tumor was easily visualized in the pretargeted animals only. Targeting and biodistribution studies of a 68Ga-labeled hapten peptide conjugate (11-D-Tyr-D-Lys (HSG)-D-Glu-D-Lys (HSG)-NH2, IMP 288) showed similar results, i.e., 10.7 ± 3.6% ID/g tumor uptake in 1 h.109
Since Al3+ can only bind with six donor atoms, consequently, the chemistry of pentadentate chelating agents such as 1,4,7-triazacyclononane-1–4 diacetic acid (NODA, Structure 13, Figure 5) and its derivatives were explored for 18F-AlF coordination. Shetty et al.110 and D’Souza et al.111 determined the chemical structure, using X-ray crystallography, of an AlF-benzyl-1,4,7-triazacyclononane-1,4-diacetic acid (Bz-NODA, Structure 14, Figure 5) and AlF-1,4,7-triazacyclononane-1,4-diacetic acid with methyl phenylacetic acid (NODA-MPAA, Structure 15, Figure 5) chelates, respectively. Both studies showed very similar crystal structures, i.e., the Al3+ was found to be at the center of an octahedron, two nitrogens (one with acetate arm and another with benzyl or MPPA arm) and two oxygen from the acetates being in the equatorial positions, and one nitrogen from the ring and fluoride being in the axial positions. The 18F-AlF-14 was found to be stable in human serum at 37 °C and in sodium acetate buffer (pH 4) at room temperature for at least 2 h. In vivo stability of 18F-AlF-14 was studied by conducting biodistribution studies in balb/c mice. The material cleared from blood rapidly (i.e., >90% cleared in 60 min) and excreted via both the renal and hepatobiliary routes. Stability studies of AlF-15 and Al(OH)-15 at pH 7.4 (PBS buffer) suggested that the former was stable for over 24 h while the latter showed around 23% loss in 3 h.111
Figure 5.
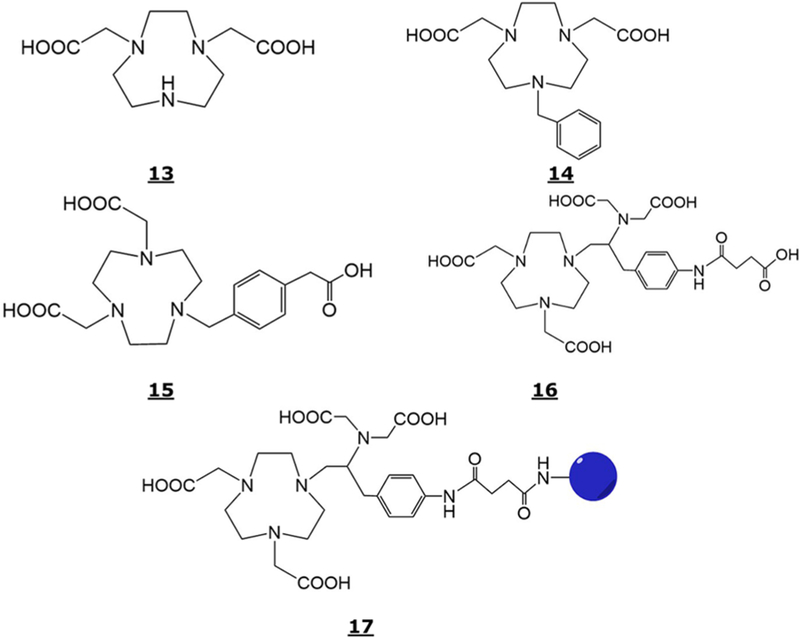
Structure of NODA (13), Bz-NODA (14), NODA-MPAA (15), C-NETA (16), and C-NETA-CONH-biomolecule (17).
A prototype kit formulation of the NODA-MPAA conjugated hapten peptide (NODA-MPAA-D-Lys(HSG)-D-Tyr-D-Lys-(HSG)-NH2, IMP 485) was prepared and optimized for pH, the peptide to Al3+ ratio, bulking agent, radioprotectant, and the buffer.112 The kit was reconstituted with an aqueous solution of Na18F and 1:1 mixture of ethanol and water. The mixture was heated at 100−110 °C for 15 min and purified by a solid-phase extraction (SPE) method. The 18F-AlF-labeled IMP 485 was isolated in high yield (45−97%) and high specific activity within 20 min. There was no evidence of defluorination when 18F-AlF-labeled IMP 485 was incubated in human serum at 37 °C for 4 h and in vivo, i.e., urine samples showed that the intact product was eliminated. Tumor targeting of the 18F-AlF-IMP 485 in nude mice bearing human colon cancer xenografts, pretargeted with an anti-CEACAMS bispecific antibody, showed 28.1 ± 4.5% ID/g tumor uptake at 1 h. Tumor to organ ratios were 9 ± 4, 123 ± 38, 110 ± 43, and 120 ± 105 for kidney, liver, blood, and bone, respectively. There was very low bone uptake (0.06 ± 0.02% ID/g) suggesting a good in vivo stability of the 18F-AlF-labeled IMP 485.
Three new peptide conjugates were developed by the reaction of NOTA (Structure 2, Figure 1), NODA-GA (Structure 3, Figure 1), and C-NETA (Structure 16, Figure 5) with hapten peptides to produce 9- and 10-D-Ala-D-Lys (HSG)-D-Tyr-D-Lys (HSG)-NH2, IMP 461, and IMP 460, and 17-D-Lys(HSG)-D-Tyr-D-Lys (HSG)-NH2, IMP467, respectively. These conjugates and IMP 449 were labeled with 18F-AlF and evaluated in the pretargeting model.113 The 18F-AlF labeling yields (% given in the parentheses) for the four chelate conjugates followed the order: IMP 467 (87%) > IMP 449 (44%) > IMP 461 (31%) > IMP 460 (5.8%). Significantly higher 18F-AlF labeling yield for IMP 467, containing C-NETA ligand, may be due to more rapid metal binding kinetics observed.114 In contrast to the IMP 460 and IMP 461, the IMP 467 formed two 18F-labeled complexes that interconverted at room temperature. The 18F-AlF-labeling of IMP 467 was optimized with a short processing time (30 min) and 52% yield with one SPE purification. In vitro stability studies of 18F-AlF-IMP 467 were conducted in PBS buffer and in fresh human serum. Approximately 2.3% and 0.5% free 18F− were observed in 5.5 and 5 h incubation in PBS and human serum, respectively. Biodistribution studies were performed in LS174T human colon cancer xenograft-bearing nude mice using a pretargeting method. The 18F-AlF-IMP 467 was stable in vivo and higher tumor uptake (11.8% at 1 h and 8.16% at 3 h) in TF2-pretargeted mice were observed than 0.23% at 1 h and 0.09% at 3 h in nonpretargeted animals. The 18F-AlF-IMP 467 eliminated in the urine and had identical Reversed-Phase HPLC elution profile as the administered material suggesting in vivo stability.
These studies have successfully demonstrated the feasibility of 18F-labeling of biomolecules, their potential as target-specific imaging pharmaceuticals, in the preclinical environment, using a pretargeting technique, and a prototype kit formulation for clinical use. However, there is no report related to human applications, possibly due to unacceptable in vivo stability of 18F-AlF-labeled biomolecules in preclinical models and regulatory challenges related to the technique.
Gastrin-Releasing Peptide Receptor-Specific Analogs.
The gastrin-releasing peptide receptor (GRPR), a subtype of the bombesin receptor family, is an attractive target for imaging tumors with neuroendocrine origin including prostate, breast, and small cell lung cancers. Especially for prostate cancer, high-affinity GRPR expression has been identified in tissue biopsy samples and immortalized cell lines.115 In a study by Markwalder and Reubi, GRPR expression in primary prostatic invasive carcinoma was present in 100% of the tissues tested. In 83% of these cases, the expression was determined to be either high or very high.116 Bombesin (BBN) is a 14-amino-acid amphibian peptide analog of the 27-amino-acid mammalian GRP. BBN and GRP share a homologous 7-amino-acid amidated C-terminus, Trp-Ala-Val-Gly-His-Leu-Met-NH2, which is necessary for binding to the GRPR.117 Synthetic BBNs are modified versions of the above peptide sharing the common 7-amino-acid C-terminus. The N-terminus is free for conjugation with appropriate radiolabeled metal chelate for various applications.
A NODA-conjugated BBN derivative, 9–8-Aoc-BBN (7–14)-NH2), was labeled with 18F-AlF,118 efficiently in one step, with 50% to 90% yield and was evaluated for its GRPR targeting properties in mice with subcutaneous PC-3 xenografts. The 68Ga-9–8-Aoc-BBN (7–14)-NH2 was used as a reference for comparison. The labeled peptide showed high in vitro serum stability, high binding affinity (IC50 value being 0.37 ± 0.15 nM), higher tumor uptake (2.15 ± 0.55% ID/g, 1 h post injection) than tumor uptake by 68Ga reference (1.24 ± 0.26% ID/g), and cleared rapidly from blood (i.e., <0.07% ID/g at 1 h after injection), mainly via kidneys. In addition to tumor uptake, 18F-AlF-labeled 9–8-Aoc-BBN (7–14) NH2 had significantly higher uptake in pancreas than 68Ga-labeled analog (27.09 ± 12.77% ID/g vs 5.93 ± 2.10% ID/g). Fused PET and CT images were consistent with the biodistribution data, i.e., PC-3 tumors could be visualized,118 with significant accumulation and retention in other organs also such as kidney, liver, intestines, and pancreas.
In an effort to develop a clinically translatable BBN-based imaging pharmaceutical, Liu and co-workers synthesized and evaluated 18F-AlF and 64Cu labeled NODA-GA-RM1 (10-RM1; where RM1 = G-4-aminobenzoyl-D-Phe-Gln-Trp-Ala-Val-Gly-His-StaLeu-NH2) and AMBA (where AMBA = G-4-aminobenzoyl-Gln-Trp-Ala-Val-Gly-His-Leu-Met-NH2) conjugates for their GRPR binding and for their potential application in PET imaging of prostate cancer in a PC-3 Xenograft model.119 Both 64Cu and 18F-AlF-labeled 10-RM1 conjugates showed comparable in vitro serum stability and in vivo tumor imaging properties. For example, tumor uptake values were as follows: 3.3 ± 0.38, 3.0 ± 0.76, and 3.5 ± 1.0% ID/g for 64Culabeled 10-RM1 and 4.6 ± 1.5, 4.0 ± 0.87, and 3.9 ± 0.48% ID/g for 18F-AlF-10-RM1 at 0.5, 1, and 2 h, respectively. The 18F-AlF-labeled 10-RM1 showed high GRPR binding (IC50 value of 0.25 ± 0.04 nM) and low serum stability (>90% of the tracer remained intact after 1 h incubation in mouse serum at 37 °C). On the contrary, 18F-AlF-NODA-GA-AMBA has weaker GRPR binding (IC50 value being 1.9 ± 0.5 nM), lower serum stability, and lower tumor uptake, 3.2 ± 0.6, 2.2 ± 0.33, and 1.8 ± 0.1% ID/g at 0.5, 1.5, and 4 h post injection, respectively.
A 18F-AlF-labeled antagonist analog of bombesin, NODA-P2-RM26 (9-PEG2-D-Phe-Gln-Trp-Ala-Val-Gly-His-Sta-Leu-NH2) showed a low nanomolar inhibition efficiency (IC50 = 4.4 ± 0.8 nM) and low internalization rate as less than 14% of the cell-bound radioactivity was internalized after 4 h.120 The biodistribution and PET imaging studies of 18F-AlF-9-P2-RM26 showed specificity in accumulating in the PC-3 tumor xenografts (5.5 ± 0.7% ID/g uptake, 3 h post injection) and the high tumor-to-blood ratio (87%). The tumors were clearly visible with high contrast after injection of the a new 18F-labeled GRPR antagonist, NOTA-MABBN,121 in PC-3 xenograft mice. For example, at 60 min post injection, the tumor uptake of 18F-AlF-NOTA-MATBBN and 18F-FDG was 4.59 ± 0.43 and 1.98 ± 0.3% ID/g, respectively. The radiotracer excreted mainly through the kidneys and was stable in PBS and in human serum for 2 h.121
In a more recent study, three GRPR-targeted peptides, 18F-AlF-JMV5132, 68Ga-JMV5132, and 68Ga-JMV4168 (where JMV 5132 = 15-βAla-βAla-[H-D-Phe-Gln-Trp-Ala-Val-Gly-His-Sta-Leu-NH2] and JMV 4168 = 11-βAla-βAla-[H-D-Phe-Gln-Trp-Ala-Val-Gly-His-Sta-Leu-NH2]) were evaluated in PC-3 xenografts.122 The IC50 values determined were 13.2, 3.0, and 3.2 for Ga-JMV5132. Ga-JMV4168, and AlF-JMV5132, respectively. In mice with subcutaneous PC-3 xenografts, all imaging pharmaceuticals cleared rapidly from blood, exclusively via the kidneys for 68Ga-JMV4168 and partially via liver for 68Ga-JMV5132 and 18F-AlF-JMV5132. All three imaging pharmaceuticals had 5−6% ID/g tumor uptake at 2 h post injection.
Two novel 18F-AlF-labeled lanthionine-stabilized BBN analogs, designated 18F-AlF-NOTA-4,7-lanthionine-BBN and 18F-AlF-NOTA-2,6-lanthionine-BBN, were prepared and evaluated.123 The IC50 values were determined as 251 ± 8 nM, 114 ± 3, 23 ± 4, and 15 ± 2 for 4,7-lanthionine-BBN, Al19F-NOTA-4,7-lanthionine-BBN, 2,6-lanthionine-BBN, and Al19F-NOTA-2,6-lanthionine-BBN, respectively. Consistent with the low IC50 values, the tumor uptake of 0.82 ± 0.23 and 1.40 ± 0.81% ID/g were observed in PC-3 xenografts in nude mice for Al19F-NOTA-4,7-lanthionine-BBN, and Al19F-NOTA-2,6-lanthionine-BBN, respectively. In vitro stability studies of both tracers showed 90% and 75% intact compounds after 4 h incubation in saline and human plasma, respectively.
In summary, various 18F-AlF labeled bombesin peptides and their analogs showed nanomolar binding affinity to GRPR, however, their low tumor uptake and limited in vitro stability did not qualify them for further research and evaluation.
αvβ3 Integrin Specific Peptides.
Since angiogenesis plays an important role in tumor growth and metastasis, tumor angiogenesis could potentially be utilized for diagnosis of malignancies and for cancer treatment.124 One of the several approaches of angiogenesis imaging is a visualization of αvβ3 integrin, an angiogenic biomarker overexpressed in the endothelium of most solid tumors. Integrins are a family of glycoproteins that function in cellular adhesion, migration, and signal transduction. It is known that the αvβ3 integrin target binds to a variety of extracellular proteins through Arginine-Glycine-Aspartic Acid (i.e., RGD) amino acid sequence. Based on these findings, numerous peptides, including several cyclic peptides (e.g., cyclic RGD) with high affinity compared to corresponding linear peptide, were designed and evaluated for specificity and affinity in preclinical environments and eventually translating into clinic.125–135
An isothiocyanate-benzyl-NODA (SCN-Bz-NODA, Structure 6) chelating agent was conjugated to a αvβ3 targeting peptide, a monomeric cyclic RGDyK peptide (where RGDyK is cyclo Arg-Gly-Asp-D-Tyr-Lys).136 The final product was HPLC puKrified, lyophilized, and characterized by 1H NMR and ESI and FAB mass spectra. The conjugate was 18F-AlF-labeled136 with a good radiochemical yield and purity (97.1 ± 1.2%) in a short reaction and purification time (25 min). 18F-AlF-labeled conjugate showed in vitro and in vivo stability. The labeled conjugate was tested in αvβ3‑positive U87MG (human glioma cells) xenograft-bearing mice by conducting biodistribution and small animal micro-PET imaging studies. Both studies showed 4.41 ± 0.98% ID/g tumor uptake. High kidneys and liver uptake indicated that the imaging pharmaceutical excreted via both the renal and hepatobiliary routes. The tumor-to-muscle and tumor-to-blood ratios were 8.17 ± 0.50 and 4.95 ± 0.36% ID/g, respectively. The in vivo tumor uptake of the labeled conjugate was evaluated in U87MG tumor-bearing nude mice using dynamic small animal micro-PET scans also at 1 and 2 h post injection. The standardized uptake values (SUVs) were determined as 7.42 ± 0.49 and 3.77 ± 0.57 at 1 and 2 h post injection, respectively, which decreased to 0.72 ± 0.14 and 0.42 ± 0.15, respectively, after blocking with 3 mg/kg cRGDyK confirming that the imaging pharmaceutical is αvβ3 integrin-specific.
A 20-amino-acid peptide, A20FMDV2 (Asn-Ala-Val-Pro-Asn-Leu-Arg-Gly-Asp-Leu-Gln-Val-Leu-Ala-Gln-Lys-Val-Ala-Arg-Thr), which selectively targets the α β integrin, an epithelial-specific cell surface receptor that has been detected in a range of particularly challenging cancers, was conjugated with NODA chelating agent with a linker containing PEG28 (9-PEG28-A20FMDV2). The conjugate was labeled with 18F-AlF and evaluated for its stability and efficacy in vitro and in vivo in PBS/mouse serum and xenograft mice, respectively.137 For example, binding of 18F-AlF-9-PEG28-A20FMDV2 was evaluated using DX3puroβ6 cell, that expresses αvβ6 integrin, with DX3pro as a control. Binding of the radiotracer to the DX3puroβ6 was significantly higher (42.4 ± 1.2%) compared to 5.1 ± 0.4% to DX3puro cell lines after 1 h incubation. The radiotracer showed no decomposition after 12 h incubation in PBS and 2 h incubation in mouse serum. However, HPLC analysis of extracts of a homogenized DX3puroβ6 tumor, collected at 1 h post injection, showed 11% intact radiotracer and one major metabolite which eluted earlier than the main peak. Similarly, HPLC analysis of urine samples collected during biodistribution study at 1 h showed only 10% intact tracer and two metabolites. Biodistribution and small-animal PET/CT studies in DX3puroβ6 xenograft mouse model showed the tracer’s ability to target αvβ6 and rapid blood clearance. The tracer cleared via kidneys and tumor uptake was low 1.74 ± 0.38% ID/g at 1h post injection. Although, the potential imaging pharmaceutical has good in vitro properties but tumor uptake and in vivo stability are low.
To improve αvβ3 binding affinity, a dimeric cyclic RGD peptide, E[c(RGDyK)]2 (abbreviated as RGD2) was conjugated first with the NOTA ligand and the resulting bioconjugate, NODA-RGD2 (9-RGD2), was labeled with 18F-AlF.138 Integrin binding affinity of 18F-AlF-9-RGD2 was determined by using U87MG cell-based receptor binding assay and 125I-echistatin as a radio ligand. Biodistribution and imaging studies, to demonstrate the tumor targeting efficacy and in vivo profiling, were conducted with 18F-AlF-9-RGD2 and compared with 18F-labeled dimeric cyclic RGD peptide (18F-FP-RGD2) in αvβ3 integrin-expressing U87MG glioblastoma xenograft model.138 In general, both tracers showed similar characteristics. For example, U87MG tumors were clearly visualized with a good tumor to background ratio by using both 18F-AlF-9-RGD2 and 18F-FP-RGD2 tracers. Tumor uptakes were 5.7 ± 2.1, 5.3 ± 1.7, 1.9 ± 0.7, and 4.0 ± 1.1, 2.8 ± 0.7, 1.1 ± 0.2% ID/g at 0.5, 1, and 2 h for 18F-AlF-9-RGD2 and 18F-FP-RGD2, respectively. Both tracers excreted mainly via kidneys. There were no significant differences in liver, kidney, and muscle uptake at 2 h post injection for both tracers. Specificity of both tracers was demonstrated by conducting cyclic RGDyK blocking experiments. The IC50 values of 18F-FP-RGD2 and 18F-AlF-9-RGD2 were 42 ± 4.1 and 46 ± 4.4 nM (n = 4), respectively, and 95% 18F-AlF-9-RGD2 was found intact after serum incubation at 37 °C for 2 h.
The chelating agent NODA-GA-NHS ester was conjugated to a dimeric RGD peptide, E[c(RGDfK)]2 (where cRGDfK is cyclo Arg-Gly-Asp-D-Phe-Lys) to produce a conjugate containing a metal chelating agent 10-E[c(RGDfK)]2 with six donor atoms (i.e., three amines and three carboxylic acids).139 The 18F-AlF-labeled NODA-GA conjugate was evaluated in vitro and in vivo and compared with the corresponding 68Ga- and 111In-labeled analogs. 18F-AlF-10-E[c(RGDfK)]2 cleared rapidly from blood, i.e., 0.03 ± 0.01% ID/g in blood at 2 h post injection. Uptake of the imaging pharmaceutical in αvβ3 integrin-expressing SK-RC-52 tumors was significantly lower than its corresponding 68Ga- and 111In-labeled analogs. For example, tumor uptake values were 3.44 ± 0.2, 6.20 ± 0.76, and 4.99 ± 0.64% ID/g at 2 h post injection for 18F-AlF, 68Ga, and 111In-labeled 10-E[c(RGDfK)]2, respectively.
Synthesis of an FDA approved imaging pharmaceutical for clinical trials, 18F-FPPRGD2, a 18F-labeled dimeric cyclic RGDyK peptide with mini-PEGylation, for PET imaging of angiogenesis is time-consuming and requires multiple synthetic steps. Therefore, PRGD2 was conjugated to p-SCN-Benzyl NOTA (5) chelating agent and the conjugate (7-PRGD2) was labeled with 68Ga and 18F-AlF. The 18F-FPPRGD2, 68Ga, and 18F-AlF-labeled 7-PRGD2 were evaluated for comparative pharmacokinetics and tumor imaging properties using a small animal PET.140,141 All three tracers showed rapid and high uptake in U87MG glioblastoma tumors with a high target-to-background ratio, similar uptake in the liver, kidneys, and muscle, and rapid kidney clearance.140,141 The IC50 (nM) values were 175.4, 119.2, 82.7, and 91.4 for FPRGD2, AlF-7-PRGD2, Ga-7-PRGD2, and PRGD2, respectively. Tumor uptake values of the three tracers were in the range of 2.5−3.9% ID/g. 18F-AlF-labeled PRGD2 (also designated as 18F-Alfatide or Alfatide I) was identified as a potential αvβ3 imaging pharmaceutical for translation into clinic.
Three new dimeric cyclic RGDfK peptides with or without PEGylation (E[c(RGDfK)]2, PEG4-E[c(RGDfK)]2, and E-[PEG4-c(RGDfK)]2 were synthesized by Chen and co-workers.142 To eliminate any possibility of thiourea bond oxidation, these peptides were conjugated to a NOTA chelating agent to produce 9-E[c(RGDfK)]2, 9-PEG4-E[c(RGDfK)]2, and 9-E[PEG4-c(RGDfK)]2. The conjugates were labeled with 18F-AlF and screened in vitro for serum stability and receptor binding affinity and in vivo for tumor uptake and whole body distribution through biodistribution and PET imaging using U87MG tumor-bearing mice.142 The serum stability of these 18F-AlF-labeled dimers were comparable to the dimers of cRGDyK; however, the IC50 values were lower by 3- to 10-fold. For example, the measured IC50 (nM) values were reported as 200.49, 513.63, 393.85, and 127.93 for E[c(RGDfK)]2, 9-E[c(RGDfK)]2, 9-PEG4-E[c(RGDfK)]2, and 9-E[PEG4-c-(RGDfK)]2, respectively. From the PET imaging studies, the tumor uptake (with tumor-to-muscle ratio in the parentheses) at 60 min post injection were 2.75 ± 0.20 (4.40 ± 0.28), 2.33 ± 0.41 (3.70 ± 0.71), and 2.92 ± 0.4 (4.11 ± 0.73)% ID/g for 18F-AlF-labeled 9-E[c(RGDfK)]2, 9-PEG4-E[c(RGDfK)]2, and 9-E[PEG4-c(RGDfK)]2, respectively. Consistent with the PET imaging results, the tumor uptake of 18F-AlF-9-E[PEG4-c(RGDfK)]2 in biodistribution studies, at 60 min post injection, was 2.39 ± 0.54% ID/g and tracer accumulation in kidney, liver, and bone was 5.42 ± 1.44, 3.13 ± 0.51, and 0.72 ± 0.14% ID/g, respectively.
18F-AlF-9-E[PEG4-c(RGDfK)]2 (also known as 2PRGD2 or Alfatide II) and 18F-FDG were used to monitor the response of doxorubicin therapy in U87MG and MDA-MB435 xenograft mice.143 Dual-tracer dynamic imaging technique was used which involved an initial injection of Alfatide II followed by 18F-FDG injection 40 min later. The signal from each tracer was successfully separated from compartmental modeling. Dual-tracer single scan imaging was found to reflect tumor response, and quantitative kinetic parameters calculated from dynamic data were more sensitive than static imaging.
Both NRP-1 (Neuropilin-1) and αvβ3 are overexpressed in gliomas; therefore, a dual αvβ3 and NRP-1 targeted heterodimeric peptide RGD-ATWLPPR (where ATWLPPR = Ala-Thr-Trp-Leu-Pro-Pro-Arg), in which cRGDyK peptide was connected with ATWLPPR through a glutamate linker, was conjugated with the NOTA chelating agent. The dual αvβ3 integrin and NRP-1 receptor-binding affinities of RGD-ATWLPPR were determined using U87MG cells and compared with the cell binding affinities of RGD, 9-RGD, and 9-RGD-ATWLPPR.144 Using 125Iechistatin for competition binding studies the IC50 values for RGD, 9-RGD, RGD-ATWLPPR, and 9-RGD-ATWLPPR were 46.75 ± 4.40, 48.53 ± 6.95, 39.97 ± 5.97, and 43.75 ± 4.82 nM, respectively. The receptor-binding affinity (IC50) of ATWLPPR, 9-ATWLPPR, RGD-ATWLPPR, and 9-RGD-ATWLPPR were measured by using 125ITyr-ATWLPPR as a competition binding ligand as 68.78 ± 6.24, 72.82 ± 4.14, 62.96 ± 5.21, and 60.08 ± 6.54, respectively. The cellular uptake of RGD, ATWLPPR, and RGD-ATWLPPR was determined in U87MG cell lines which highly expresses αvβ3 and moderately expresses NRP-1. Percent binding for 18F-AlF-9-RGD, 18F-AlF-9-ATWLPPR, and 18F-AlF-9-RGD-ATWLPPR were 7.47 ± 0.73, 4.72 ± 0.82, and 9.04 ± 0.67, respectively, after 60 min incubation and 8.75 ± 0.77, 5.29 ± 0.81, and 10.02 ± 0.90, respectively, after 120 min incubation. Static micro-PET/CT scans were performed on a U87MG xenograft mouse. The U87MG tumors were clearly visible with tumor-to-muscle contrast after 30 min post injection of all three tracers.
Biodistribution studies of 18F-AlF-9-RGD, 18F-AlF-9-ATWLPPR, and 18F-AlF-9-RGD-ATWLPPR were conducted in U87MG tumor-bearing mice. Predominant kidney uptake by the three tracers suggests renal clearance although 18F-AlF-9-RGD-ATWLPPR had some liver and bone uptake. All three tracers cleared from blood rapidly, i.e., only 0.5% ID/g remaining 60 min post injection. Tumor uptake of 18F-AlF-9-RGD-ATWLPPR was 5.31 ± 0.16, 5.02 ± 0.14, and 4.54 ± 0.39% ID/g at 30, 60, and 120 min, respectively, post injection. These uptake values were significantly higher than for 18F-AlF-9-RGD (3.21 ± 0.29, 2.69 ± 0.21, and 2.02 ± 0.20% ID/g at 30, 60, and 120 min) and for 18F-AlF-9-ATWLPPR (2.66 ± 0.18, 2.22 ± 0.27, and 1.85 ± 0.08% ID/g at 30, 60, 120 min), respectively. 18F-AlF-9-RGD-ATWLPPR had a higher tumor-to-organ ratios (tumor-to-muscle, tumor-to-blood, and tumor-to-kidney) than for 18F-AlF-9-RGD or 18F-AlF-9-ATWLPPR.
Somatostatin Receptor Subtype-Selective Analogs.
The majority of human neuroendocrine tumors (NETs) overexpress multiple somatostatin receptor subtypes, i.e., sst1, sst2, sst3, sst4, and sst5, although these receptors are overexpressed on other tumor types also, such as non-Hodgkin’s lymphoma, melanoma, breast, pancreatic, gastric, colon, prostate, lung, and so forth.145 Several Somatostatin receptor specific imaging pharmaceuticals have been evaluated in nuclear medicine for tumor diagnosis, staging, and therapy (peptide receptor radionuclide therapy, PRRT). For example, the somatostatin analog, octreotide (Structure 18, Figure 6), binds with high affinity to the sstr2 and sstr5, a low affinity to the sstr3, and no binding with sstr1 and sstr4 subtypes. Consequently, 111In-DTPA-octreotide (OctreoSacn) has been used routinely in the clinic for primary and metastatic neuroendocrine tumors (NETs) imaging.146 A small change in the peptide structure or sequence or chelating agent type, linkers, and metal replacement has shown dramatic effects on the binding affinity of the radiolabeled peptide to individual somatostatin receptor subtypes. Therefore, more recently 68Ga and 177Lu/90Y chelates DOTA TOC (DOTA-Tyr3-octreotide, Structure 19, Figure 6) characterized by sstr2 affinity, DOTA-NOC with sstr2, sstr3, and sstr5 affinity (DOTA-1-Na13-octreotide, Structure 20, Figure 6), and DOTA TATE with sstr2 affinity (DOTA-Tyr3-octreotate, Structure 21, Figure 6) have been used for imaging and therapy, respectively.147,148 Other radiolabeled imaging agents for sstr2 positive tumors involved 64Cu and 68Ga antagonists conjugated to different chelators (4,11-bis-(carboxymethyl)-1,4,8,11-tetraazabicyclo [6.6.2]hexadecane (CB-TE2A), NODA-GA, and DOTA.149
Figure 6.
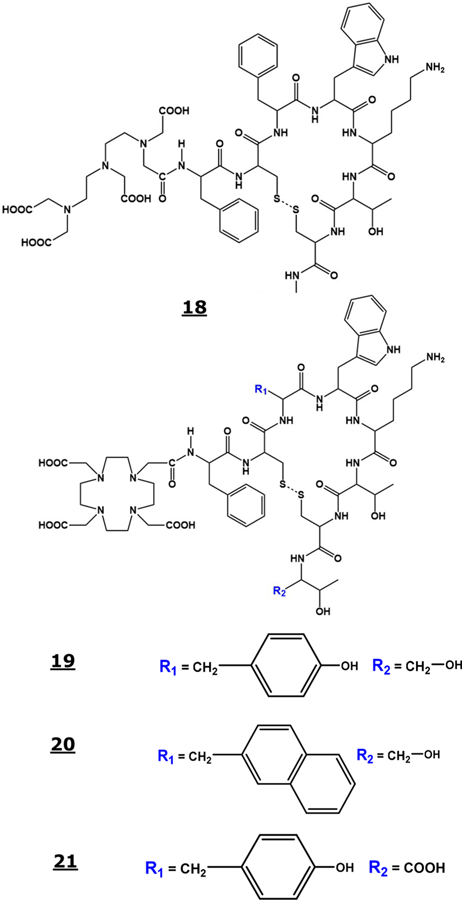
Structure of octreotide (18), DOTA-TOC (19), DOTA-NOC (20), and DOTA-TATE (21).
18F-AlF-labeling of the octreotide peptide analog (9-D-Phe-cyclo[Cys-Phe-D-Trp-Lys-Thr-Cys]-Throl, IMP 466) in aqueous medium produced stereoisomers.150 The effect of buffers and amount of IMP 466 on the labeling yield was studied and maximum yield observed was 50%. The apparent IC50 values for the somatostatin receptor binding on AR42J cells were determined in a competition binding assay using 18F-AlF-IMP 466 along with 68Ga-IMP 466, and 111In-DTTA-Octreotide. The IC50 values (in nM) determined were as 3.6 ± 0.6 (18F-AlF-IMP466), 13 ± 3 (68Ga-IMP466), and 6.3 ± 0.9 (111In-DTPA-Octreotide). The stability of 18F-AlF-IMP466 was tested in human serum at 37 °C and no release of 18F-AlF was observed in 4 h. The PET imaging and biodistribution of 18F-AlF-IMP466 in AR42J tumor-bearing Balb/c mice (n = 5) showed 28.3 ± 5.7% ID/g tumor uptake of 18F-IMP466 at 2 h post injection and reduced to 8.6 ± 0.7% ID/g in the presence of large excess of unlabeled IMP 466 suggesting that the uptake was receptor mediated. Uptake in normal tissues, except kidneys, including bone, was low. Further optimization of the labeling process increased 18F-AlF labeling yield up to 97% when a cosolvent, such as 80% ethanol or acetonitrile, was used in the reaction.151
Prostate-Specific Membrane Antigen-Specific Peptides.
Prostate cancer (PCa) is a most common cancer in men;152 therefore, early detection of primary disease and its metastases is critical for clinical staging, prognosis, and therapy management. Several radiotracers have been proposed for molecular imaging of prostate cancer, including choline (11C-Choline and 18F-Choline) as a marker of membrane cell proliferation, 11C-Acetate as a radiotracer for PCa imaging via incorporation into intracellular phosphatidylcholine membrane, and 18F-FACBC (18F-fluciclovine;1-amino-3-fluorocyclo-butane-1-carboxylic acid) that is used to monitor amino acid transport. 18F-FACBC has been found to be successful and superior to 11C-Choline in the assessment of primary and metastatic prostate cancer,153–155 although numerous studies reported limited sensitivity and specificity of these tracers for imaging PCa in patients with low PSA levels.156
The prostate-specific membrane antigen (PSMA) is a transmembrane protein that has significantly elevated expression in prostate cancer cells than in the benign prostatic tissues.
Several PSMA-targeted PET tracers have been developed and evaluated in the past. This includes, 68Ga-labeled PSMA 11,157,158 PSMA 617,159,160 PSMA I&T,161 THP-PSMA,162 and 18F-labeled DCFBC,163,164 DCFPyL,165 and PSMA-1007.166–168 An excellent review related to PSMA-based theranostics radiotracers was published recently.169 The most widely used radiotracer for PET imaging in Europe is PSMA 11 or 68Ga PSMA HBED-CC. HBED-CC (N,N′-bis [2-hydroxy-5-(carboxyethyl) benzyl] ethylenediamine-N,N′-diacetic acid, Figure 7, Structure 22) is an acyclic chelating agent to bind 68Ga and is conjugated to PSMA inhibitor, Glu-NH−CO-NH-Lys(Ahx). 68Ga-PSMA-11 PET/CT detects tumor lesions in a high percentage of patients with recurrent prostate cancers.170
Figure 7.
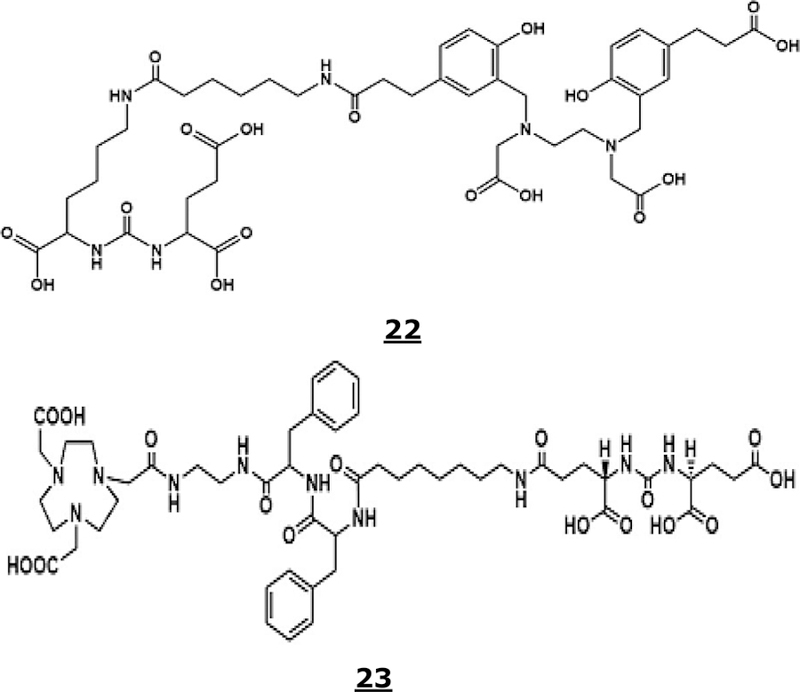
Structures of HBED-CC or PSMA 11 (22) and NOTA-DUPA-Pep (23).
Short half-life and nonideal energies of 68Ga, cost, and the limited number of doses available from 68Ge/68Ga generators motivated researchers to investigate the potential of 18F-labeled PSMA analogs for PET imaging of prostate cancer. PSMA 11 was labeled with18F-AlF by heating a mixture of 18F-spiked AlCl3 and PSMA 11 under various conditions.171–173 The crude product was purified by a Sep-Pak C18 light or an HLB cartridge. The yield of radiolabeling varied between 30% to 90% depending on the reaction conditions with >98% radiochemical purity. The total synthesis time was between 45 and 50 min.
For in vitro stability studies, the 18F-AlF-labeled PSMA 11 was incubated in mouse and human serum at 37 °C for 2 to 4 h, analyzed by radio-TLC and -HPLC.172,173 No significant decomposition was observed in 2 h. After 3 h, 18F-AlF-labeled PSMA 11 had >97% and 80% radiochemical purity in pH 6.8 buffered and unbuffered solutions, respectively. The in vitro stability of the radiotracer was also determined in mixtures of ethanol/saline, ethanol/acetate, and ethanol/PBS.173 Radio-chemical purities of the materials were 99% and 22% after 4 h in a mixture of 1% ethanol and 99% saline and 10% ethanol and PBS, respectively.174 A Kd (binding coefficient) value was determined in a PSMA-positive cell line, LnCap, as 10.3 ± 2.2 nM171 which is comparable to 12.0 ± 2.8 nM for 68Ga-labeled PSMA-11157 and 6.7 ± 1.7 nM for 18F-labeled PSMA 1007.166,171 18F-AlF-labeled PSMA 11 exhibited uptake in LnCap cell lines, i.e., 3.4% to 3.5%.172,174 18F-AlF-labeled PSMA 11 biodistribution studies were conducted using LnCap and PC-3 tumor-bearing wild-type C57BL6 mice and by using micoPET/CT imaging. Tumor uptake of the tracer in LnCap cell tumor-bearing mice was high.172
A new PSMA-ligand, NOTA-DUPA-Pep (where DUPA is 2-[3-(1,3-dicarboxypropyl)-ureido]pentanedioic acid, Figure 7, Structure 23) was synthesized and labeled with 18F-AlF.174 Reaction kinetics (dependence on the concentrations of the precursor and AlCl3 and temperature) was examined. Highest radiochemical yield (83 ± 1.1%) was obtained at 105 °C after 15 min of reaction time. At the end of the synthesis and purification (55 min) the 18F-AlF labeling yield was 79 ± 0.7% (uncorrected, n = 3) with >98% radiochemical purity.
Since 18F-AlF labeling of NOTA or NODA peptide conjugates requires heating at 100 to 120 °C, a series of novel acyclic chelating agents, which are capable of binding with Al3+ at low temperature (i.e., 40 °C), were synthesized and evaluated.175 One of the several chelating agents, an HBED analog, showed some potential. The rat serum stability of its 18F-AlF labeled chelate was found to be comparable to that of the previously reported 18F-AlF-labeled NODA analog, i.e., up to 60 min. Additionally, no defluorination was observed during biodistribution studies in normal mice since no significant bone uptake was observed. As a proof of concept, 18F-AlF-chelate was conjugated with the urea-based PSMA inhibitor, Glu-NH-CO-NH-Lys and a biodistribution study in healthy mice was performed. In summary, the acyclic chelators may have some potential, however, there is still room for improvement.
Since 18F-labeled PSMA-1007 has comparable IC50 and tumor uptake values than 18F-AlF-labeled PSMA 11, has a GMP-compliant production process, similar to 18F-FDG, and it is already going through human clinical trials in Europe and under discussions for clinical trials in the US,166–168 it is doubtful if 18F-AlF labeled PSMA 11 will be commercially available in the future.
MMP2 and MMP9 Specific Peptides.
Matrix metalloproteinase, MMP2 and MMP9, overexpression has been associated with tumor progression, invasion, and metastasis. Targeted imaging of these MMPs would be a useful strategy to noninvasively detect and characterize solid tumors. A NODA conjugate of a cyclic decapeptide (c(Lys-Ala-His-Trp-Gly-Phe-Thr-Leu-Asp)NH2 or C6, (9-C6) was labeled with 18F-AlF (i.e, 18F-AlF-9-C6) and tested in vitro and in vivo.176 The probe, 18F-AlF-9-C6, was stable (>95% remained) after 4 h incubation in physiological saline at room temperature or in human serum at 37 °C. The MMP2 binding affinity, IC50, of 18F-AlF-9-C6, using 9-C6 as a competing ligand, was determined as 0.18 nM. In vivo PET imaging and biodistribution data suggested low uptake of 18F-AlF-9-C6 in the SKOV-3 tumor-bearing mice, i.e., 1.20 ± 0.24%, 0.75 ± 0.25%, and 0.27 ± 0.14% ID/g after 30, 60, 120 min post injection, respectively, and cleared by renal route. Low tumor uptake and stability of the probe makes it unsuitable for further evaluation.
Follicle-Simulating Hormone Receptor (FSHR) Specific Peptides.
Overexpression of FSHR (Follicle-Simulating Hormone Receptor) has been detected in vascular endothelium of numerous human cancer tumors, such as prostate, breast, kidney, and lung cancers. FSH is a glycoprotein hormone with two subunits (α and β chains). Several receptor binding domains of FSHβ chain have been identified, including FSH1 (with 33−53-amino-acid sequence, Tyr-Thr-Arg-Asp-Leu-Val-Tyr-Lys-Asp-Pro-Ala-Arg-Pro-Lys-Ile-Gln-Lys-Thr-Cys-Thr-Phe). A 18F-AlF-labeled maleimide-NOTA conjugate of FSH1 (12-FSH1) was evaluated in preliminary studies for PET imaging of FSHR-positive tumors.177 Low PC3 cells uptake (20%) and low cell binding (i.e., 252 ± 1.12 nM) of 18F-AlF-12-FSH1 tracer were observed. Biodistribution and PET imaging studies using PC3 tumor-bearing mice demonstrated 4.21 ± 0.69% ID/g accumulation in the tumor at 10 min post injection. Clearance of the tracer from the normal organs was faster than the tumor resulting in increased contrast over time. High levels of radioactivity in the kidney at 10 min post injection suggested renal clearance.
Glucagon-Like Peptide Receptor (GLP1) Binding Peptide.
The GLP-1 receptor (Glucagon-like peptide receptor) is overexpressed in insulinoma, a neuroendocrine tumor of the pancreas. Exendin-4, an agonist of glucagon-like peptide (GLP1) receptor, is an incretin mimetic peptide which is composed of 39 amino acids. Two 18F-labeled analogs of Exendin-4 were prepared by conjugating [18F]FBEM (N-[2-(4-[18F]fluorobenzamide)ethyl]maleimide) prosthetic group with GLP1. The tracers showed good tumor uptake but the synthesis of the tracers was challenging and time-consuming. To overcome this challenge,178 a 18F-AlF-NOTA conjugate analog of exendin-4 (i.e., 18F-AlF-12-cys40-exendin-4 was prepared. The binding affinities (IC50 values) of exendin-4, FBEM-cys40-exendin-4, and 12-cys40-exendin-4 were determined as 0.98, 1.10, and 2.84 nM, respectively, via a competition cell binding assay using 125I-GLP (7−36) and INS-1 rat cells. Tumor uptake of the 18F-AlF-labeled conjugate in INS-1 insulinoma xenografts reached its maximum (16.9 ± 1.8% ID/g, n = 4) after 5 min post injection and remained constant during the study. Kidney uptake of the radioactivity was high. Tumor and plasma extract samples, 60 min post injection, were analyzed by a radio HPLC analytical method and showed 74% and 64% intact parent compound, the remaining material being a polar radioactive metabolite. On the contrary, analysis of the kidney and urine extract samples showed only one polar radioactive metabolite. These data suggest low stability, and hence these compounds are unsuitable as potential imaging pharmaceuticals. Similar in vitro stability and binding affinity and tumor uptake were observed in a study reported recently.179
Annexin 1 (Anxa 1) Specific Peptide.
Annexin 1 (Anxa 1) is a novel biomarker expressed on the surface of endothelial cells that are part of tumor vasculature. Anxa 1 expression in the tumor vasculature is universal in several tumor types in mice and humans.180 Therefore, it is an attractive target for imaging. A peptide, Ile-Phe-Leu-Leu-Trp-Gln-Arg, designated as IF7, was found to bind Anxa 1 with high affinity and specificity. For in vitro stability study of the 18F-AlF-labeled 7-IF7 conjugate, the tracer was incubated in PBS and mouse serum at 37 °C for 2 h. After 2 h incubation, the radiochemical purities were determined as >94% and 90.7% in PBS and mouse serum, respectively.181 Biodistribution and micro-PET imaging studies involving nude mice bearing A431 xenografts showed low tumor uptake making it unsuitable for further evaluation.
Urokinase-Type Plasminogen Activator Receptor (uPAR) Binding Peptide.
Urokinase-type plasminogen activator receptors (uPAR) are overexpressed in various human cancers including prostate, colorectal, and stomach cancers. The expression of uPAR is either very low or undetectable in normal tissues. Therefore, a linear peptide with high affinity for uPAR, AE105 (Asp-Cha-Phe-(d)Ser-(d)Arg-Tyr-Leu-Trp-Ser-CONH2), was considered to be a promising ligand for detection and imaging of cancerous tissues that overexpress uPAR. The uPAR binding peptide, AE 105, was conjugated via amine function in aspartate moiety with one of the carboxylic acids in NOTA (9-AE105), labeled with 18F-AlF, and was evaluated as a potential PET imaging pharmaceutical for uPAR positive prostate tumors.182 18F-AlF labeling of the conjugate was optimized and it was observed that the addition of 33% ethanol gave best yield and purity (92.7% with >92% radiochemical purity). The inhibitory effects (IC50) on the uPAR:uPA interaction of AE105, the conjugate of AE105, and 18F-AlF-9-AE105 were determined as 14.1, 24.5, and 21.0 nM, respectively. The in vivo PET imaging studies were conducted in mice bearing PC-3 tumors and scans were performed at 0.5, 1.0, and 2 h post injection. Reconstructed images showed tumor lesions with tumor-specific uptake, 5.9 ± 0.35, 4.22 ± 0.13, and 2.54 ± 0.24% ID/g at 0.5, 1.0, and 2 h post injection, respectively. Biodistribution data at the end of imaging studies confirmed the in vivo PET imaging results.
Preclinical Studies with Folate-Receptor-Specific Analog Conjugates.
Expression of the folate receptor, a glycosylphosphatidylinositol-anchored cell surface receptor, is limited in healthy tissues and organs although is overexpressed on the vast majority of cancer tissues, including epithelial, ovarian, cervical, breast, lung, kidney, colorectal, and brain tumors. On the contrary sarcomas, lymphomas, and cancers of the pancreas, testicles, bladder, prostate, and liver often do not show elevated levels of folate receptors. Folic acid, a small molecule with 441 Da molecular weight, has a high binding affinity to the folate receptor and can be conjugated with drugs or diagnostic imaging agents. Various modalities including, optical, magnetic resonance imaging (MRI), computed tomography, ultrasound imaging, single-photon emission computed tomography (SPECT), and positron emission tomography (PET) can be utilized.183–188 Several folate conjugates have been developed and evaluated for SPECT and PET imaging, including 68Ga- and 18F-labeled folate receptor-targeted conjugates; however, synthesis and preclinical evaluation of 18F-AlF-NOTA-labeled folate conjugate for PET imaging of folate-receptor-positive tumors was not reported until recently.189 Binding of the 18F-AlF-NOTA-Folate was measured in homogenates of KB and Cal 51 tumor xenografts in the presence and absence of folic acid. A Kd value of 18.7 nM was determined, which is weaker than binding of free folic acid (4.6 nM). In vivo imaging and ex vivo biodistribution studies were performed using folate receptor positive (KB cell) and folate receptor negative (A549 cell) tumor xenograft-bearing nu/nu mice. The study demonstrated high folate receptor-mediated uptake in the folate receptor positive tumor (i.e., 10.9 ± 2.7% ID/g) and the kidney (78.6 ± 5.1% ID/g) and low liver uptake (5.3 ± 0.3% ID/g).
Preclinical Evaluation of 18F-AlF-Labeled Conjugates of Proteins and Protein Fragments.
EGFR, HER2, and HER3 Overexpression.
The Epidermal Growth Factor Receptor (EGFR, ErbB1, HER1 in humans), a transmembrane protein and a member of ErbB family of receptors, is highly expressed in a variety of human cancers including nonsmall-cell lung cancer (NSCLS). The overexpression of EGFR has been observed in both premalignant lesions and malignant tumors of the lung and occurs in 40−80% patients with NSCLS. Human epidermal growth factor receptor 2 (HER2) and 3 (HER3), transmembrane proteins that belong to the human epidermal growth factor tyrosine kinase receptor family (EGFR or ErbB), are found in patients with NSCLS and other tumors. For example, HER2 is overexpressed in 18−25% of all breast cancer carcinoma and in subsets of ovarian, lung, prostate, and gastric cancers.190,191 Breast cancers overexpressing HER2 have been associated with aggressive tumor growth, high relapse, poor prognosis, and more resistance to endocrine therapy and chemotherapy.
Monoclonal antibody trastuzumab and tyrosine kinase inhibitor lapatinib have been developed as therapeutic agents for targeting HER2, specifically trastuzumab for breast and gastric cancer patients with HER2-overexpressing tumors.192 PET and SPECT techniques using radiolabeled antibodies, including trastuzumab, pertuzumab, and trastuzumab fragment, were able to detect HER2 expression; however, their large size resulted in slow tumor uptake and clearance from circulation.193,194 A new class of targeting proteins, based on 58 amino acids with 7 kDa molecular weight and with high affinity for various tumor-associated antigens, Affibodies, have been evaluated recently. Affibody molecules are small proteins engineered to bind a large number of target proteins with high affinity, imitating monoclonal antibodies. Their small size allows rapid extravasation in tumors and blood clearance that provides higher contrast within several hours post injection. For example, 111In- and 68Ga-labeled 11-ZHER2:342‑pep2 in a clinical pilot study have shown that it is possible to visualize HER-2 expressing tumors in patients with metastatic breast cancer.195 Additionally, 111In-labeled 11-ZHER2:2395 (a variant of ZHER2:342) showed discrimination between high (SKOV-3) and low (LS174T) HER2 expression xenografts.196
PET radionuclides, such as 18F-labeled affibody, may improve imaging of HER2 expression because of higher sensitivity and improved quantification. Therefore, the 18F-AlF labeled NODA-MAL conjugated affibody molecule, 18F-AlF-12-ZHER2:2395, was evaluated as a suitable agent for HER2 expression in a mouse model for ovarian cancer.197 The tumor-targeting capabilities of various radionuclide-, 18F, 68Ga, and 111In, labeled ZHER2:2395 affibody were compared in mice with HER2-expressing SKOV-3 xenografts. As expected 18F-AlF labeling of the NODA-MAL conjugate gave a rather low yield, 21.0 ± 5.7%, as compared to 84.0 ± 0.9% and 94.0% for 68Ga and 111In labeling, respectively. Stability studies showed that 18F-AlF-12-ZHER2:2395 did not release 18F-AlF after 4 h incubation in human or mouse serum at 37 °C. The IC50 values for 12-ZHER2:2395 were determined in a competitive cell binding assay using SKOV-3 cells as 5.0, 6.3, and 5.3 nM for Al19F, 69Ga, and 115In labeled affibody, respectively. Biodistribution and imaging studies (1 and 4 h post injection) were conducted by injecting 18F-AlF labeled 12-ZHER2:2395 in mice bearing subcutaneous SKOV-3 xenografts. PET/CT and SPECT/CT images clearly showed the HER2-expressing SKOV-3 xenografts with good contrast to normal tissue. High kidney uptake and tumor-to-liver ratios of the 18F-AlF labeled affibody were observed. Tumor uptake were 4.4 ± 0.8, 5.6 ± 1.6, and 7.1 ± 1.4% ID/g with tumor-to-blood ratios of 7.4 ± 1.8, 8.0 ± 1.3, and 4.8 ± 1.3 for 18F-AlF, 68Ga, 111In-labeled 12-ZHER2:2395 conjugate, respectively.
Three different 18F labeling strategies, i.e., silicon-fluoride acceptor approach (18F-SiFA), 18F-AlF-NOTA, and 4-18F-fluorobenzaldehyde (18F-FBA), for radiolabeling of the HER2-specific affibody molecule ZHER2:2891, were investigated. The non-decay-corrected radiochemical yield using 18F-AlF method was low, i.e., 11 ± 4% (n = 6). The radiolabeled affibody molecules were evaluated in a preclinical model involving CD-1 nude mice bearing high and low HER2-expressing NCI-N87 and A431 tumors, respectively.198 In non-tumor-bearing mice, a significant (73.8 ± 3.0%ID) kidney uptake of 18F-AlF-12-ZHER2:2891 post 90 min injection was observed. Significantly lower kidney uptake of 18F-FBA-ZHER2:2891 and 18F-SiFA-ZHER2:2891 (4.8 ± 0.6 and 10.1 ± 0.7% ID, respectively, post 90 min injection) was observed. All radiolabeled affibody molecules showed increased uptake by the high-HER2-expressing NCI-187 tumors compared with the low-HER2-expressing A431 tumors. For example, % ID/g NCI-187 tumor uptake at 90 min post injection were 7.15 ± 0.69%, 4.79 ± 1.26%, and 3.49 ± 0.74% for 18F-FBA-, 18F-AlF-, and 18F-SiF-labeled affibody molecules, respectively. 18F-SiF-labeled affibody showed high bone retention over time suggesting defluorination. 18F-AlF-labeled affibody molecule showed high tumor-to-muscle (28.89) and tumor-to-liver (2.83) ratios in NCI-187 biodistributions. The dual-flank A431/NCI-187 tumor mouse model was used to perform PET/CT study using 18F-AlF-labeled affibody and images demonstrated its elimination through kidneys and bladder. Additionally, higher retention of the 18F-AlF-labeled tracer was seen in the high-HER2-expressing NCI-187 compared to low-HER2-expressing A431tumors.
Another EGFR targeting affibody (ZEGFR:1907), conjugated with DOTA and radiolabeled with 64Cu, showed high specificity, sensitivity, and tumor contrast in EGFR positive tumors as early as 1 h post injection.199 Two new radiolabeling approaches, conjugating ZEGFR:1907 with NOTA and labeling with 18F-AlF and conjugating the prosthetic group 18F-labeled-2-cyanobenzothiozol (18F-CBT) with Cys-ZEGFR:1907, were reported recently.200 Binding affinity and specificity of both tracers were evaluated using A431 cells and biodistribution and PET imaging studies were conducted on mice bearing A431 xenografts. Both tracers showed nanomolar affinity to EGFRs in A431 cells, i.e., Kd values of 18F-AlF-12-ZEGFR:1907 and 18F-CBT-ZEGFR:1907 were 12.72 ± 1.25 and 25.82 ± 3.62 nM, respectively. 18F-AlF-12-ZEGFR:1907 was relatively more stable than 18F-CBT-ZEGFR:1907 in in vitro stability studies. The former remained intact after 1 to 2 h of incubation in mouse serum; on the contrary, the latter degraded 25% in the same period. Relatively high tumor uptakes, at 3 h post injection, of both tracers was observed in the biodistribution studies, i.e., 4.77 ± 0.36 and 4.08 ± 0.54% ID/g for 18F-AlF-12-ZEGFR:1907 and 18F-CBT-ZEGFR:1907, respectively. Higher kidney and liver uptake for 18F-AlF-12-ZEGFR:1907 (112.26 ± 12.57 and 13.31 ± 0.80% ID/g) than 18F-CBT-ZEGFR:1907 (8.12 ± 1.0% and 3.08 ± 0.15% ID/g) were seen at 3 h post injection. In contrast, bone uptake of 18F-AlF-12-ZEGFR:1907 was lower than 18F-CBT-ZEGFR:1907 (1.75 ± 0.35 vs 12.99 ± 2.37% ID/g). 18F-AlF-12-ZEGFR:1907 provided higher tumor-to-blood, tumor-to-lung, tumor-to-muscle, and tumor-to-bone ratios than 18F-CBT-ZEGFR:1907 except for tumor-to-liver and tumor-to-kidney ratios. Small-animal PET imaging studies demonstrated that both tracers clearly visualized EGFR-expressing A431 xenografts. Additionally, 18F-AlF-12-ZEGFR:1907 showed better tumor-to-background contrast and high uptake of the tracer in liver and kidneys than 18F-CBT-ZEGFR:1907.
HER3 imaging is challenging due to modest receptor numbers (<5000 receptors/cell) in overexpressing cancer cells. An affibody molecule (ZHER3:8698) with HER3 targeting specificity was conjugated to NODA-MAL (12-ZHER3:8698) and labeled with 18F-AlF using two different strategies. The conventional labeling of 12-ZHER3:8698 at pH 4, 100 °C for 15 min using ethanol as organic cosolvent (50% v/v) gave 38.8 ± 5.8% radiochemical yield. However, this procedure resulted in the radiolabeled product with variable purity attributed to thermolysis.201 An alternate technique for 18F-AlF-labeling of ZHER3:8698 by reacting a novel tetrazine functionalized 1,4,7-triazacyclononane-1,4-diacetate and the trans-cyclooctene (TCO) functionalized affibody, at room temperature, was developed. The 18F-AlF-labeled 12-ZHER3:8698 and NODA-ZHER3:8698 conjugates showed specific uptake at 1 h post injection in high HER3-expressing MCF-7 tumors in mice, i.e., 4.36 ± 0.92% 4.96 ± 0.6% ID/g, respectively. Both conjugates showed high renal excretion which was supported by PET imaging studies. In vitro cell binding studies in HER3-expressing MCF-7 cells suggested Kd values as 0.44 ± 0.04 and 1.01 ± 0.28 nM for 18F-AlF-labeled 12-ZHER3:8698 and NODA-ZHER3:8698, respectively. The stability of both conjugates was determined by incubating the radiolabeled conjugates in mouse serum at 37 °C. By HPLC analysis it was found that 97.9 ± 0.5% of 12-ZHER3:8698 and 91.5 ± 1.2% of NODA-ZHER3:8698 remained intact after 1 h. The blood clearance of the affibody is fast; however, the stability of the conjugates may have an impact on the suitability for further development.
A new restrained complexing agent, (±)-H3RESCA, an acyclic N2O3 donor atom containing pentadentate ligand, that allows efficient 18F-AlF labeling using mild conditions, was developed recently175 and conjugated to HSA, to a nanobody (NbV4m119) as Kupffer cell marker, and an affibody (PEP04314 also known as ZHER2:2891) for targeting HER2.202 The conjugates were labeled with 18F-AlF at 37 °C, in less than 35 min, successfully with good radiochemical yields 52−63%, 35−53%, and 20 ± 7% for HSA, nanobody, and affibody conjugate, respectively. For comparison, 12-ZHER2:2891 was also 18F-AlF labeled at 100 °C giving a much lower yield of the reaction as 8 ± 6% (n = 4) which was comparable to previously reported value, 11 ± 4% (n = 6).198 Both tracers were evaluated in healthy rhesus monkey for pharmacokinetics and distribution profile by using whole-body PET/CT. Biexponential blood clearance for both tracers was observed and alpha and beta clearance half-lives were: 0.08 ± 0.05 h, 1.09 ± 0.23 and 0.04 ± 0.01, 2.70 ± 0.43 h for 18F-AlF-12-ZHER2:2891 and 18F-AlF-(±)-H3RESCA-ZHER2:2891, respectively. The sum of % ID/g in kidney and urinary bladder, after 120−180 min post injection, were comparable for both tracers.
Small proteins such as Fab′ fragments of humanized MN-14 anti CEACAM5 IgG antibody have been labeled with 18F-AlF and evaluated in a preclinical model as potential imaging pharmaceuticals.203 N-(2-Aminoethyl)maleimide (EM) was conjugated to NODA-MPAA (15) to form a NODA-MPAEM. The NODA-MPAEM chelating agent was labeled with 18F-AlF, conjugated to hMN-14Fab′, purified by using a Sephadex G50−80 spin column and tested for immunoreactivity. CaPan-1 cells are known to express elevated levels of EGFR and do not express the SMAD4 protein. 18F-AlF labeled protein conjugate was administered to CaPan-1 human pancreatic adenocarcinoma (HTB-79) xenograft nude mice. At 3 h post injection, the 18F-AlF-labeled hMn-Fab′ showed elevated uptake in the kidneys suggesting renal clearance of Fab′. Blood concentration of the tracer was low with corresponding elevated uptake in liver and spleen. The faster blood clearance showed lower tumor uptake but higher tumor-to-blood ratio (5.9 ± 1.3). 18F-AlF-NODA-MPAEM-hMN-14Fab was stable after incubation in human serum for 3 h, which was supported by bone uptake data in biodistribution studies.
Clinical Experience with 18F-AlF-Labeled Peptide Conjugates.
A simple lyophilized kit for rapid 18F-AlF-labeling of the PRGD2 peptide (20 min radiosynthesis and purification time) to produce 18F-Alfatide, an imaging agent for integrin αvβ3, was developed.204 Under optimized conditions, 18F-Alfatide (also known as 18F-Alfatide I now) was prepared in high yield 42.1 ± 2.0% (decay corrected) with 95% radiochemical purity. A clinical study using 18F-Alfatide, along with 18F-FDG, was conducted involving nine patients with primary diagnosis of lung cancer and one patient with tuberculosis.204 PET imaging identified all primary tumors with the mean uptake of 2.90 ± 0.10. The tumor-to-muscle and tumor-to-blood ratios were 5.87 ± 2.02 and 2.71 ± 0.92, respectively. Major uptake of 18F-Alfatide was observed in kidneys and bladder indicating renal clearance. Liver, spleen, and intestine also showed moderate uptake. Similar observations were made in other studies recently.205,206
In another study, 18F-Alfatide I and 18F-FDG were used to compare detection of lymph node metastasis in Differentiated Thyroid Cancer (DTC) involving 20 patients with presumptive lymph node metastasis.207 Sixteen patients undergoing fine needle aspiration biopsy (FNAB) were evaluated by cytology results. A total of 39 presumptive lymph node metastasis were visualized in PET/CT images. Thirty five lesions were confirmed as malignant by FNAB technique and other clinical findings. Although most DTC lymph node metastasis showed abnormal uptake of 18F-Alfatide I; however, it was a less effective diagnostic agent than 18F-FDG. There was no correlation between 18F-Alfatide and 18F-FDG uptake to suggest that the two tracers are complementary to each other in detecting DTC lesions.
In a recent study, 18F-Alfatide II (18F-AlF-NOTA-E[PEG4-c(RGDfK)]2) was evaluated for safety, estimated absorbed dose, and its value in patients with brain metastases.208 The study involved five healthy volunteers (3 male and 2 female) and nine patients (5 male and 4 female) with 20 metastases brain tumors as confirmed by MRI or CT. Safety data included vital signs, physical examination, ECG, laboratory parameters, and adverse reaction. No adverse events or effects were observed following 18F-Alfatide II injection and no obvious changes in vital signs or clinical laboratory tests were found before and after the injection of 18F-Alfatide II. 18F-Alfatide II was quickly eliminated via urinary system although moderate uptake was observed in liver and spleen while other organs had low levels of radioactivity. In the imaging study involving nine patients, all brain lesions were visualized by 18F-Alfatide II, while only 10 by 18F-FDG, and 13 by CT. Of the brain lesions detected by 18F-FDG or CT, all were visible by using 18F-Alfatide II. Despite the overall higher uptake of 18F-FDG, 18F-Alfatide II showed better tumor-to-background ratio, i.e., 18.9 ± 14.1 for 18F-Alfatide II vs 1.5 ± 0.5 for 18F-FDG demonstrating the value of 18F-Alfatide in detecting metastases as a biomarker of angiogenesis.
A pilot study was conducted to verify the efficacy of 18F-Alfatide II, for detecting bone metastasis in humans, in comparison with 18F-FDG.209 The study involved 36 patients and final diagnosis of bone lesions was established based on the data analysis and clinical follow up. It was found that 18F-Alfatide II can detect bone metastasis lesions with good contrast and higher sensitivity than 18F-FDG, i.e., positive rate of 92% vs 77%. Especially, 18F-Alfatide II was superior to 18F-FDG in detecting osteoblastic (77% vs 53%) and bone marrow metastatic lesions (98% vs 77%). Overall, skeletal and bone marrow metastases can be detected with 100% sensitivity in osteolytic and bone marrow lesions using 18F-Alfatide PET/CT. The sensitivity of 18F-Alfatide PET/CT in osteoblastic metastases is relatively low, however, still significantly higher than 18F-FDG PET/CT. In summary, 18F-Alfatide II may be useful in the future in metastatic lesion detection, patient management, and drug therapy response monitoring.
■ SUMMARY
In this report, we have highlighted an overview of the 18F radiochemistry and 18F-labeling methodologies for small molecules, via carbon−fluorine bond formation, and target-specific biomolecules, a comprehensive review of coordination chemistry of Al3+, 18F-AlF labeling of peptide and protein conjugates, and evaluation of 18F-labeled biomolecule conjugates for various cancer targets in preclinical and clinical environments. Since the first report in 2009 related to the 18F-AlF labeling technique for biomolecules, numerous studies have been completed related to labeling and evaluation of target-specific peptides and proteins. The labeling method is a versatile procedure that can be used for biomolecules labeling while retaining their binding affinities. The procedure is fast and simple, and 18F-AlF labeling can be accomplished in one or two steps in aqueous solution, although it may need organic solvents for improved reaction yield, and may require high temperature, up to 100 °C, and pH 4. Numerous target-specific biomolecules have been radiolabeled and have shown good potential as PET imaging pharmaceuticals, but also showed limited in vivo stability. The in vivo stability is specifically important for 18F-AlF labeled proteins which have longer circulation time than the small peptides. Three kits containing lyophilized powder of the peptide conjugates for 18F-labeling have been prepared successfully. Two kits containing PRGD2 and 2PRGD2 conjugates for preparation of 18F-Alfatide I and 18F-Alfatide II, respectively, were introduced into the clinic and feasibility was demonstrated in specific imaging of αvβ3 expression in lung cancer patients, detection of metastasis in lymph nodes of differentiated thyroid cancer, and brain cancer. More studies are needed to move Alfatide I or II into phase II and III clinical trials. Other two novel approaches, using 18F-silicon and 18F-boron chemistry to label peptides and proteins, may provide additional novel PET imaging pharmaceuticals in the future.
ACKNOWLEDGMENTS
This work was supported by the Ohio Third Frontier TECH 13-060, TECH 09-028, R01EB022134, and the Wright Center of Innovation Development Fund. The authors are grateful to Professor Michael V. Knopp (Director and Principal Investigator of the Wright Center of Innovation in Biomedical Imaging) for his encouragement and support during this work. Additionally, we thank Drs. Michael Tweedle and Adam Pippin for reviewing the manuscript and making some valuable suggestions.
ABBREVIATIONS
- [18F]FECH
[18F]Fluoroethylcholine
- [18F]FA
[18F]-Fluoroacetate
- [18F]FLT
[18F]Fluorodeoxythymidine
- [18F]FMAU
[18F]Fluoromethylarabinofuranosyluracil
- [18F]-FMISO
[18F]Fluoromisonidazole
- [18F]FAZA
[18F]-F l uoroazomycinarab inoside
- [ 18 F]FE TA
[ 18 F]-Fluoroetanidazole
- [18F]FES
[18F]Fluoroestradiol
- [18F]MFES
[18F]Methoxyfluoroestradiol
- [18F]FDHT
[18F]-Fluorodihydrotestosterone
- [ 18 F]FDOPA
[ 18 F]-Fluorodihydroxyphenylalanine
- [18F]FMT
[18F]Fluoro-α-methyltyrosine
- [18F]FET
[18F]Fluoroethyltyrosine
- [18F]FTYR
[18F]Fluorotyrosine
- [18F]Galacto-RGD
[18F]-Galacto-cyclo(Ar-Gly-Asp-D-Tyr-Lys)
- [18F]AH111585
[18F]-Fluciclatide
- [18F]DCFPYL
Dicarboxypropylcarboamoylfluor-opyridinyllysine
- [18F]FP
[18F]Fallypride
- [18F]FP-CIT
[18F]-Fluoropropylcarbomethoxyiodophenylnortropane
- [18F]FTP
Fluortriopride
- [18F]DTBZ
[18F]-Fluoropropyldihydrotetrabenazine
- [18F]MPPF
[18F] Methoxyphenylpyridinyl fluorobenzamidoethylpiperazine
- [18F]-FEPPA
Fluoroethoxybenzylphenylpyridinylacetamide
- [18F]FMM
[18F]Flutemetamol
- [18F]AZD4694
[18F]-Flutafuranol
- [18F]FDDNP
[18F]-Fluoroethylmethylaminonaphthylethylidenemalonitrile
- [18F]FHBG
[18F]Fluorohydroxy methylbutylguanine
Footnotes
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).Hargreaves RJ, and Rabiner EA (2014) Translational PET Imaging Research. Neurobiol. Dis 61, 32–38. [DOI] [PubMed] [Google Scholar]
- (2).Ollinger JM, and Fessler JA (1997) Positron-Emission Tomography. IEEE Signal Proc. Mag 14, 43–55. [Google Scholar]
- (3).Muehllehner G, and Karp JS (2006) Positron Emission Tomography. Phys. Med. Biol 51, R117–R137. [DOI] [PubMed] [Google Scholar]
- (4).Alauddin MM (2012) Positron Emission Tomography (PET) Imaging with 18F-Based Radiotracers. Am. J. Nucl. Med. Mol. Imaging 2, 55–76. [PMC free article] [PubMed] [Google Scholar]
- (5).Vallabhajosula S (2007) 18F-Labeled Positron Emission Tomographic Radiopharmaceuticals in Oncology: An Overview of Radiochemistry and Mechanisms Of Tumor Localization. Semin. Nucl. Med 37, 400–419. [DOI] [PubMed] [Google Scholar]
- (6).Couturier O, Luxen A, Chatal JF, Vuillez JP, Rigo P, and Hustinx R (2004) Fluorinated Tracers for Imaging Cancer with Positron Emission Tomography. Eur. J. Nucl. Med. Mol. Imaging 31, 1182–1206. [DOI] [PubMed] [Google Scholar]
- (7).Nanni C, Fantini L, Nicolini S, and Fanti S (2010) Non FDG PET. Clin. Radiol 65, 536–548. [DOI] [PubMed] [Google Scholar]
- (8).Fanti S, Nanni C, Ambrosini V, Gross MD, Rubello D, and Farsad M (2007) PET In Genitourinary Tract Cancers. Q. J. Nucl. Med. Mol. Im 51, 260–271. [PubMed] [Google Scholar]
- (9).Lee S, Xie J, and Chen X (2010) Peptide-Based Probes for Targeted Molecular Imaging. Biochemistry 49, 1364–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Schottelius M, and Wester H-J (2009) Molecular Imaging Targeting Peptide Receptors. Methods 48, 161–177. [DOI] [PubMed] [Google Scholar]
- (11).Fani M, and Maecke HR (2012) Radiopharmaceutical Development of Radiolabeled Peptides. Eur. J. Nucl. Med. Mol. Imaging 39, S11–S30. [DOI] [PubMed] [Google Scholar]
- (12).Correia JDG, Paulo A, Raposinho PD, and Santos I (2011) Radiometallated Peptides for Molecular Imaging and Targeted Therapy. Dalton Trans 40, 6144–6167. [DOI] [PubMed] [Google Scholar]
- (13).Tornesello AL, Buonaguro L, Tornesello ML, and Buonaguro FM (2017) New Insights in the Design of Bioactive Peptides and Chelating Agents for Imaging and Therapy in Oncology. Molecules 22, 1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Tweedle MF (2009) Peptide-Targeted Diagnostics and Radiotherapeutics. Acc. Chem. Res 42, 958–968. [DOI] [PubMed] [Google Scholar]
- (15).Fani M, Maecke HR, and Okarvi SM (2012) Radiolabeled Peptides: Valuable Tools for The Detection and Treatment of Cancer. Theranostics 2, 481–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Smith CJ, Volkert WA, and Hoffman TJ (2005) Radiolabeled Peptide Conjugates for Targeting of Bombesin Receptor Superfamily Subtypes. Nucl. Med. Biol 32, 733–740. [DOI] [PubMed] [Google Scholar]
- (17).Moreno P, Ramos-Alvarez I, Moody TW, and Jensen RT (2016) Bombesin Related Peptides/Receptors and Their Promising Therapeutic Roles in Cancer Imaging, Targeting And Treatment. Expert Opin. Ther. Targets 20, 1055–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Deri MA, Zeglis BM, Francesconi LC, and Lewis JS (2013) PET Imaging with 89Zr: From Radiochemistry to the Clinic. Nucl. Med. Biol 40, 3–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Ametamey SM, Honer M, and Schubiger PA (2008) Molecular Imaging with PET. Chem. Rev 108, 1501–1516. [DOI] [PubMed] [Google Scholar]
- (20).Gu Y, Huang D, Liu Z, Huang J, and Zeng W (2011) Labeling Strategies with F-18 for Positron Emission Tomography Imaging. Med. Chem 7, 334–344. [DOI] [PubMed] [Google Scholar]
- (21).Jacobson O, Kiesewetter DO, and Chen X (2015) Fluorine-18 Radiochemistry, Labeling Strategies and Synthetic Routes. Bioconjugate Chem 26, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Shao X, Hoareau R, Hockley BG, Tluczek LJM, Henderson BD, Padgett HC, and Scott PJH (2011) Highlighting the Versatility of the Tracer Lab Synthesis Modules. Part 1: Fully Automated Production of [18F] Labelled Radiopharmaceuticals Using a Tracer Lab FXFN. J. Labelled Compd. Radiopharm 54, 292–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Varagnolo L, Stokkel MPM, Mazzi U, and Pauwels EKJ (2000) 18F-Labeled Radiopharmaceuticals for PET in Oncology, Excluding FDG. Nucl. Med. Biol 27, 103–112. [DOI] [PubMed] [Google Scholar]
- (24).Elsinga PH (2002) Radiopharmaceutical Chemistry for Positron Emission Tomography. Methods 27, 208–217. [DOI] [PubMed] [Google Scholar]
- (25).Rice SL, Roney CA, Daumar P, and Lewis JS (2011) The Next Generation of Positron Emission Tomography Radiopharmaceuticals in Oncology. Semin. Nucl. Med 41, 265–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Smith GE, Sladen HL, Biagini CG, and Blower PJ (2011) Inorganic Approaches for Radiolabeling Biomolecules with Fluorine-18 for Imaging with Positron Emission Tomography. Dalton Trans 40, 6196–6205. [DOI] [PubMed] [Google Scholar]
- (27).Smith TAD (2012) [18F]Fluoride Labeling of Macro-molecules in Aqueous Conditions: Silicon and Boroaryl-Based [18] Fluorine Acceptors, [18F]FDG Conjugation and Al18F Chelation. J. Labelled Compd. Radiopharm 55, 281–288. [Google Scholar]
- (28).Bernard-Gauthier V, Wangler C, Schirrmacher E, Kostikov A, Jurkschat K, Wangler B, and Schirrmacher R (2014) 18F-Labeled Silicon-Based Fluoride Acceptors: Potential Opportunities for Novel Positron Emitting Radiopharmaceuticals. BioMed Res. Int 2014, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Burke BP, Clemente GS, and Archibald SJ (2015) Boron-18F Containing Positron Emission Tomography Radiotracers: Advances and Opportunities. Contrast Media Mol. Imaging 10, 96–110. [DOI] [PubMed] [Google Scholar]
- (30).Bernard-Gauthier V, Bailey JJ, Liu Z, Wangler B, Wangler C, Jurkschat K, Perrin DM, and Scirrmacher R (2016) From Unorthodox To Established: The Current Status of 18F-Trifluor-oborate- and 18F-SiFA-Based Radiopharmaceuticals in PET Nuclear Imaging. Bioconjugate Chem 27, 267–279. [DOI] [PubMed] [Google Scholar]
- (31).Chansaenpak K, Vabre B, and Gabbai FP (2016) [18F]-Group 13 Fluoride Derivatives as Radiotracers for Positron Emission Tomography. Chem. Soc. Rev 45, 954–971. [DOI] [PubMed] [Google Scholar]
- (32).Wangler C, Kostikov A, Zhu J, Chin J, Wangler B, and Schirrmacher R (2012) Silicon-[18F] Fluorine Radiochemistry: Basics, Applications, and Challenges. Appl. Sci 2, 277–302. [Google Scholar]
- (33).Zeng JL, Wang J, and Ma JA (2015) New Strategies for Rapid 18F-Radiolabeling of Biomolecules for Radionuclide-Based In Vivo Imaging. Bioconjugate Chem 26, 1000–1003. [DOI] [PubMed] [Google Scholar]
- (34).Mu L, August Schubiger P, and Ametamey SM (2010) [18F]Fluorosilicon- and [18F] Fluoroboron-Based Biomolecules for PET Imaging. Curr. Radiopharm 3, 224–242. [Google Scholar]
- (35).Kuhnast B, and Dolle F (2010) The Challenge of Labeling Macromolecules with Fluorine-18: Three Decades of Research. Curr. Radiopharm 3, 174–201. [Google Scholar]
- (36).Liu S, Shen B, Chin FT, and Cheng Z (2011) Recent Progress in Radiofluorination of Peptides For PET Imaging. Curr. Org. Synth 8, 584–592. [Google Scholar]
- (37).Richter S, and Wuest F (2014) 18F-Labeled Peptides: The Future is Bright. Molecules 19, 20536–20556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Luo YR (2007) Comprehensive Handbook of Chemical Bond Energies, CRC Press, Boca Raton, FL. [Google Scholar]
- (39).IAEA (2009) Cyclotron Produced Radionuclides: Physical Characteristics and Production Methods, Technical Report Series No 468, International Atomic Energy Agency (IAEA), Vienna. [Google Scholar]
- (40).Poethko T, Schottelius M, Thumshirn G, Hersel U, Herz M, Henriksen G, Kessler H, Schwaiger M, and Wester HJ (2004) Two-Step Methodology for High-Yield Routine Radiohalogenation of Peptides: 18F-Labeled RGD and Octreotide Analogs. Radiochim. Acta 45, 892–902. [PubMed] [Google Scholar]
- (41).Battle MR, Goggi JL, Allen L, Barnett J, and Morrison MS (2011) Monitoring Tumor Response to Antiangiogenic Sunitinib Therapy with 18F-Fluciclatide, An 18F-Labeled αvβ3-Integrin and αvβ5-Integrin Imaging Agent. J. Nucl. Med 52, 424–430. [DOI] [PubMed] [Google Scholar]
- (42).Li XG, Haaparanta M, and Solin O (2012) Oxime Formation for Fluorine-18 Labeling of Peptides and Proteins for Positron Emission Tomography (PET) Imaging: A Review. J. Fluorine Chem 143, 49–56. [Google Scholar]
- (43).Edgar FG, Hansen HD, Leth-Peterson S, Ettrup A, Kristensen JL, Knudsen GM, and Herth MM (2017) Synthesis, Radiofluorination, and Preliminary Evaluation of the Potential 5-HT2A Receptor Agonist [18F]Cimbi-92 And [18F]Cimbi-150. J. Labelled Compd. Radiopharm 60, 586–591. [DOI] [PubMed] [Google Scholar]
- (44).Lang L, and Eckelman WC (1994) One-Step Synthesis of 18F Labeled [18F]-N-Succinimidyl 4-(Fluoromethyl)Benzoate for Protein Labeling. Appl. Radiat. Isot 45, 1155–1163. [DOI] [PubMed] [Google Scholar]
- (45).Bejot R, Elizarov AM, Ball E, Zhang J, Miraghaie R, Kolb HC, and Gouverneur V (2011) Batch-Mode Microfluidic Radiosynthesis of N-Succinimidyl-4-[18F]Fluorobenzoate for Protein Labeling. J. Labelled Compd. Radiopharm 54, 117–122. [Google Scholar]
- (46).Kramer-Marek G, Kiesewetter DO, Martiniova L, Jagoda E, Lee SB, and Capala J (2008) [18F]FBEM-ZHER2:342−Affibody Molecule—A New Molecular Tracer for In Vivo Monitoring of HER2 Expression by Positron Emission Tomography. Eur. J. Nucl. Med. Mol. Imaging 35, 1008–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Denholt CL, Kuhnast B, Dolle F, Hinnen F, Hansen PR, Gillings N, and Kjaer A (2010) Fluorine-18 Labelling of a Series of Potential EGFRvIII Targeting Peptides With A Parallel Labelling Approach Using [18F]FPyME. J. Labelled Compd. Radiopharm 53, 774–778. [Google Scholar]
- (48).Inkster JAH, Liu K, Ait-Mohand S, Schaffer P, Guerin B, Ruth TJ, and Storr T (2012) Sulfonyl Fluoride-Based Prosthetic Compounds as Potential 18F Labeling Agents. Chem. - Eur. J 18, 11079–11087. [DOI] [PubMed] [Google Scholar]
- (49).Fiel SA, Yang H, Schaffer P, Weng S, Inkster JAH, Wong MCK, and Li PCH (2015) Magnetic Droplet Microfluidics as A Platform for The Concentration Of [18F]Fluoride and Radiosynthesis Of Sulfonyl [18F]Fluoride. ACS Appl. Mater. Interfaces 7, 12923–12929. [DOI] [PubMed] [Google Scholar]
- (50).Matesic L, Wyatt NA, Fraser BH, Roberts MP, and Pham TQ (2013) Ascertaining the Suitability of Aryl Sufonyl Fluorides for [18F]Radiochemistry Applications: A Systematic Investigation Using Microfluidics. J. Org. Chem 78, 11262–11270. [DOI] [PubMed] [Google Scholar]
- (51).Gao Z, Gouverneur V, and Davis BG (2013) Enhanced Aqueous Suzuki-Miyaura Allows Site-Specific Polypeptide 18F-Labeling. J. Am. Chem. Soc 135, 13612–13615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Wangler C, Schirrmacher R, Bartenstein P, and Wangler B (2010) Click-Chemistry Reaction in Radiopharmaceutical Chemistry: Fast & Easy Introduction of Radiolabels into Biomolecules for In Vivo Imaging. Curr. Med. Chem 17, 1092–1116. [DOI] [PubMed] [Google Scholar]
- (53).Marik J, and Sutcliffe JL (2006) Click for PET: Rapid Preparation of [18]Fluoropeptides Using CuI Catalyzed 1,3-Dipolar Cycloaddition. Tetrahedron Lett 47, 6681–6684. [Google Scholar]
- (54).Inkster JAH, Guerin B, Ruth TJ, and Adam MJ (2008) Radiosynthesis and Bioconjugation of [18F]FPy5yne, A Prosthetic Group For The 18F Labeling Of Bioactive Peptides. J. Labelled Compd. Radiopharm 51, 444–452. [Google Scholar]
- (55).Inkster JAH, Adam MJ, Storr T, and Ruth TJ (2009) Labeling of an Antisense Oligonucleotide with [18F]FPy5yne. Nucleosides, Nucleotides Nucleic Acids 28, 1131–1143. [DOI] [PubMed] [Google Scholar]
- (56).Inkster J, Lin KS, Ait-Mohand S, Gosselin S, Benard F, Guerin B, Pourghiasian M, Ruth T, Scaffer P, and Storr T (2013) 2-Fluoropyridine Prosthetic Compounds For The 18F Labeling of Bombesin Analogs. Bioorg. Med. Chem. Lett 23, 3920–3926. [DOI] [PubMed] [Google Scholar]
- (57).Inkster JAH, Colin DJ, and Seimbille Y (2015) A Novel 2-Cyanobenzothiazole-Based 18F Prosthetic Group for Conjugation to 1,2-Aminothiol-Bearing Targeting Vectors. Org. Biomol. Chem 13, 3667–3676. [DOI] [PubMed] [Google Scholar]
- (58).Roberts MP, Pham TQ, Doan J, Jiang CD, Hambley TW, Greguric I, and Fraser BH (2015) Radiosynthesis and ‘Click’ Conjugation of Ethynyl-4-[18F]Fluorobenzene An Improved [18F]-Synthon For Indirect Radiolabeling. J. Labelled Compd. Radiopharm 58, 473–478. [DOI] [PubMed] [Google Scholar]
- (59).Gens TA, Wethongton JA, and Brosi AR (1958) The Exchange of F18 Between Metallic Fluorides And Silicon Tetra-fluoride. J. Phys. Chem 62, 1593–1593. [Google Scholar]
- (60).Entzian W, Aronow S, Soloway AH, and Sweet WH (1964) A Preliminary Evaluation of F18-Labeled Tetrafluoroborate as A Scanning Agent for Intracranial Tumors. J. Nucl. Med 5, 542–550. [PubMed] [Google Scholar]
- (61).Rosenthal MS, Bosch AL, Nickles RJ, and Gatley SJ (1985) Synthesis and Some Characteristics of No-Carrier Added [18F]Fluorotrimethylsilane. Int. J. Appl. Radiat. Isot 36, 318–319. [DOI] [PubMed] [Google Scholar]
- (62).Choudhry U, Martin KE, Biagini S, and Blower PJ (2006) Alkoxysilane Groups For Instant Labeling of Biomolecules With 18F. Nucl. Med. Commun 27, 293. [Google Scholar]
- (63).Schirrmacher R, Bradtmoller G, Schirrmacher E, Thews O, Tillmanns J, Siessmeier T, Buchholz HG, Bartenstein P, Wangler B, and Niemeyer CM (2006) 18F-Labeling of Peptides by Means of an Organosilicon-Based Fluoride Acceptor. Angew. Chem., Int. Ed 45, 6047–6050. [DOI] [PubMed] [Google Scholar]
- (64).Ting R, Adam MJ, Ruth TJ, and Perrin DM (2005) Arylfluoroborates and Alkylfluorosilicates as Potential PET Imaging Agents: High-Yielding Aqueous Biomolecular 18F-Labeling. J. Am. Chem. Soc 127, 13094–13095. [DOI] [PubMed] [Google Scholar]
- (65).Mu L, Hohne A, Schubiger PA, Ametamey SM, Graham K, Cyr JE, Dinkelborg L, Stellfeld T, Srinivasan A, Voigtmann U, et al. (2008) Silicon-Based Building Blocks for One-Step 18F-Radiolabeling of Peptides For PET Imaging. Angew. Chem., Int. Ed 47, 4922–4925. [DOI] [PubMed] [Google Scholar]
- (66).Li Z, Chansaenpak K, Liu S, Wade CR, Conti PS, and Gabbai FP (2012) Harvesting 18F-Fluoride Ions In Water Via Direct 18F-19F Isotope Exchange: Radiofluorination of Zwitterionic Aryltri-fluoroborates and In Vivo Stability Studies. MedChemComm 3, 1305–1308. [Google Scholar]
- (67).Liu Z, Lin KS, Benard F, Pourghiasian M, Kiesewetter DO, Perrin DM, and Chen X (2015) One-Step 18F Labeling Of Biomolecules Using Organotrifluoro-borates. Nat. Protoc 10, 1423–1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Wangler C, Niedermoser S, Chin J, Orchowski K, Schirrmacher E, Jurkschat K, Iovkova-Berends L, Kostikov AP, Schirrmacher R, and Wangler B (2012) One-Step 18F-Labeling of Peptides for Positron Emission Tomography Imaging Using SIFA Methodology. Nat. Protoc 7, 1946–1955. [DOI] [PubMed] [Google Scholar]
- (69).Wangler B, Kostikov AP, Niedermoser S, Chin J, Orchowski K, Schirrmacher E, Iovkova-Berends L, Jurkschat K, Wangler C, and Schirrmacher R (2012) Protein Labeling With the Labeling Precursor [18F]SiFA-SH for Positron Emission Tomography. Nat. Protoc 7, 1964–1969. [DOI] [PubMed] [Google Scholar]
- (70).Wangler B, Quandt G, Iovkova L, Schirrmacher E, Wangler C, Boening G, Hacker M, Schmoeckel M, Jurkschat K, Bartenstein P, et al. (2009) Kit-Like 18F-Labeling of Proteins: Synthesis of 4-(Di-Tert-Butyl[18F]Fluorosilyl)Benzenethiol (Si[18F]-FA-SH) Labeled Rat Serum Albumin For Blood Pool Imaging With PET. Bioconjugate Chem 20, 317–321. [DOI] [PubMed] [Google Scholar]
- (71).Litau S, Niedermoser S, Vogler N, Roscher M, Schirrmacher R, Fricker G, Wangler B, and Wangler C (2015) Next Generation of SiFAlin-Based TATE Derivatives for PET Imaging of SSTR-Positive Tumors: Influence Of Molecular Design on In Vitro SSTR Binding and In Vivo Pharmacokietics. Bioconjugate Chem 26, 2350–2359. [DOI] [PubMed] [Google Scholar]
- (72).Niedermoser S, Chin J, Wangler C, Kostikov A, Bernard-Gauthier V, Vogler N, Soucy J-P, McEwan AJ, Schirrmacher R, and Wangler B (2015) In Vitro Evaluation of 18F-SiFAlin-Modified TATE: A Potential Challenge for 68Ga-DOTATATE, the Clinical Gold Standard for Somatostatin Receptor Imaging with PET. J. Nucl. Med 56, 1100–1105. [DOI] [PubMed] [Google Scholar]
- (73).Rodriguez EA, Wang Y, Crisp JL, Vera DR, Tsien RY, and Ting RA (2016) New Dioxaborolane Chemistry Enables [18F]-Positron-Emitting, Fluorescent [18F]-Multimodality Biomolecules Generation from Solid State. Bioconjugate Chem 27, 1390–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Martin RB (1996) Ternary Complexes of Al3+ and F− with a Third Ligand. Coord. Chem. Rev 149, 23–32. [Google Scholar]
- (75).Oehman L (1988) Equilibrium and Structural Studies of Silicon(IV) and Aluminum(III) in Aqueous Solution. 17. Stable and Metastable Complexes in the, System Hydrogen (+)-Aluminum (3+)-Citric Acid. Inorg. Chem 27, 2565–2570. [Google Scholar]
- (76).McBride WJ, Sharkey RM, Karacay H, D’Souza CA, Rossi EA, Laverman P, Chang C−H, Boerman OC, and Goldenberg DM (2009) A Novel Method of 18F Radiolabeling for PET. J. Nucl. Med 50, 991–998. [DOI] [PubMed] [Google Scholar]
- (77).McBride WJ, Sharkey RM, and Goldenberg DM (2013) Radiofluorination Using Aluminum-Fluoride (Al18F). EJNMMI Res 3, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Laverman P, McBride WJ, Sharkey RM, Goldenberg DM, and Boerman OC (2014) Al18F Labeling Of Peptides and Proteins. J. Labelled Compd. Radiopharm 57, 219–223. [DOI] [PubMed] [Google Scholar]
- (79).Cotton FA, and Wilkinson G (1980) Advanced Inorganic Chemistry, 4th ed., John Wiley and Sons, Inc., New York. [Google Scholar]
- (80).Martell AE, Hancock RD, Smith RM, and Motekaitis RJ (1996) Coordination of Al(III) in the Environment and in Biological Systems. Coord. Chem. Rev 149, 311–328. [Google Scholar]
- (81).Pearson RG (1963) Hard and Soft Acids and Bases. J. Am. Chem. Soc 85, 3533–39. [Google Scholar]
- (82).Hancock RD, and Martell AE (1989) Ligand Design for Selective Complexation of Metal Ions In Aqueous Solution. Chem. Rev 89, 1875–1914. [Google Scholar]
- (83).Kumar K, Chang CA, and Tweedle MF (1993) Equilibrium and Kinetic Studies of Some Lanthanide Complexes of Macrocyclic Polyamino Carboxylates. Inorg. Chem 32, 587–593. [Google Scholar]
- (84).Kumar K, and Tweedle MF (1993) Macrocyclic Polyaminocarboxylate Complexes of Lanthanides As Magnetic Resonance Imaging Contrast Agents. Pure Appl. Chem 65, 515–520. [Google Scholar]
- (85).Kumar K, Tweedle MF, Malley M, and Gougoutas JZ (1995) Synthesis, stability, and crystal structure studies of some Ca2+, Cu2+, and Zn2+ complexes of macrocyclic polyaminocarboxylates. Inorg. Chem 34, 6472–6480. [Google Scholar]
- (86).Kumar K (1997) Macrocyclic Polyamino Carboxylate Complexes of Gd(III) as Magnetic Resonance Imaging Contrast Agents. J. Alloys Compd 249, 163–172. [Google Scholar]
- (87).Farkas E, Fodor T, Kalman FK, Tirsco G, and Toth I (2015) Equilibrium and Dissociation Kinetics of [Al (NOTA)] Complex (NOTA = 1,4,7-Triazacyclononane-1,4,7-triacetate). React. Kinet., Mech. Catal 116, 19–33. [Google Scholar]
- (88).Andre JP, Macke H, Kaspar A, Kunnecke B, Zehnder M, and Macko L (2002) In Vivo and In Vitro 27Al NMR Studies Of Aluminum(III) Chelates of Triazacyclononane Polycarboxylate Ligands. J. Inorg. Biochem 88, 1–6. [DOI] [PubMed] [Google Scholar]
- (89).Andre JP, Macke H, Zehnder M, Macko L, and Akyel KG (1998) 1,4,7-Triazacyclononane-1-Succinic Acid-4, 7-Diacetic Acid (NODASA): A New Bifunctional Chelator for Radio Gallium-Labeling of Biomolecules. Chem. Commun, 1301–1302.
- (90).Yuchi A, Hotta H, Wada H, and Nakagawa G (1987) Mixed Ligand Complexes of Trivalent Metal Ions with An Amine-N-Polycarboxylate And Fluoride. Bull. Bull. Chem. Soc. Jpn 60, 1379–1382. [Google Scholar]
- (91).Hugi-Cleary D, Helm L, and Merbach AE (1985) Variable-Temperature and Aariable-Pressure 17O-NMR Study of Water Exchange of Hexaaqua Aluminum (III). Helv. Chim. Acta 68, 545–554. [Google Scholar]
- (92).Nordin JP, Sullivan DJ, Phillips ML, and Casey WH (1998) An 17O-NMR Study of the Exchange of Water on AlOH(H2O)52+(Aq). Inorg. Chem 37, 4760–4763. [DOI] [PubMed] [Google Scholar]
- (93).Crumbliss AL, and Garrison JM (1988) A Comparison of Some Aspects of the Aqueous Coordination Chemistry of Aluminum (III) and Iron (III). Comments Inorg. Chem 8, 1–26. [Google Scholar]
- (94).Tomany CT, and Hynes MJ (1999) Kinetics and Mechanisms of the Reaction of Aluminum (III) with Polyamino-carboxylic Acids. BioInorg. React. Mech 1, 137–144. [Google Scholar]
- (95).O’ Coinceanainn M, and Hynes MJ (2001) The Kinetics and Mechanisms of the Reactions of Aluminum (III) with Gallic Acid, Gallic Acid Methyl Ester and Adrenaline. J. Inorg. Biochem 84, 1–12. [DOI] [PubMed] [Google Scholar]
- (96).Nemes J, Toth I, and Zekany L (1998) Formation Kinetics of an Aluminum(III)-Ethylenedinitrotetraacetate-Fluoride Mixed Ligand Complex. J. Chem. Soc., Dalton Trans, 2707–2713.
- (97).Smith CJ, Volkert WA, and Hoffman TJ (2003) Gastrin Releasing Peptide (GRP) Receptor Targeted Radiopharmaceuticals: A Concise Update. Nucl. Med. Biol 30, 861–868. [DOI] [PubMed] [Google Scholar]
- (98).Rogers BE, Brechbiel MW, Kirkman RL, Clarkson M, and Buchsbaum DJ (1999) In Vitro Binding and Internalization of An Indium-111 Labeled Bombesin Derivative to Cells Expressing The Gastrin Releasing Peptide Receptor. Technetium, Rhenium and other metals in chemistry and nuclear medicine Italy: SGE editoriali, 519–525. [Google Scholar]
- (99).Smith CJ, Gali H, Sieckman GL, Hayes DL, Owen NK, Mazuru DG, Volkert WA, and Hoffman TJ (2003) Radiochemical Investigations of 177Lu-DOTA-8-Aoc-BBN[7−14]-NH2: An In Vitro/In Vivo Assessment of the Targeting Ability of This New Radiopharmaceutical for PC-3 Human Prostate Cancer Cells. Nucl. Med. Biol 30, 101–109. [DOI] [PubMed] [Google Scholar]
- (100).Heppeler A, Froidevaux S, Eberle AN, and Maecke HR (2000) Receptor Targeting for Tumor Localization and Therapy with Radio Peptides. Curr. Med. Chem 7, 971–994. [DOI] [PubMed] [Google Scholar]
- (101).Li L (2003) The Biochemistry and Physiology of Metallic Fluoride: Action, Mechanism and Implications. Crit. Rev. Oral Biol. Med 14, 100–114. [DOI] [PubMed] [Google Scholar]
- (102).Anthony B, and Chabre M (1992) Characterization Of the Aluminum and Beryllium Fluoride Species Which Activate Transducin. J. Biol. Chem 267, 6710–6718. [PubMed] [Google Scholar]
- (103).Boerman OC, van Schaijk FG, Oyen WJ, and Corstens FH (2003) Pretargeted Radioimmunotherapy of Cancer: Progress Step by Step. J. Nucl. Med 44, 400–411. [PubMed] [Google Scholar]
- (104).Sharkey RM, Karacay H, Litwin S, Rossi EA, McBride WJ, Chang C−H, and Goldenberg DM (2008) Improved Therapeutic Results by Pretargeted Radioimmunotherapy of Non-Hodgkin’s Lymphoma with A New Recombinant, Trivalent, Anti-CD20, Bispecific Antibody. Cancer Res 68, 5282–5290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (105).Goldenberg DM, Sharkey RM, Paganelli G, Barbet J, and Chatal J-M (2006) Antibody Pretargeting Advances Cancer Radioimmunodetection and Radioimmunotherapy. J. Clin. Oncol 24, 823–834. [DOI] [PubMed] [Google Scholar]
- (106).Sharkey RM, Cardillo TM, Rossi EA, Chang C−H, Karacay H, McBride WJ, Hansen HJ, Horak ID, and Goldenberg DM (2005) Signal Amplification in Molecular Imaging by Pretargeting A Multivalent, Bispecific Antibody. Nat. Med 11, 1250–1255. [DOI] [PubMed] [Google Scholar]
- (107).Jyo A, Kohno T, Terazono Y, and Kawano S (1990) Crystal Structure of the Al(III) Complex of 1,4,7-Triazacyclononane-N,N′,N″-triacetate. Anal. Sci 6, 629–630. [Google Scholar]
- (108).Bossek U, Hanke D, Wieghardt K, and Nuber B (1993) Pendent Arm Macrocyclic Complexes: Crystal Structures of Al-(TCTA) and In(TS-TACN). Polyhedron 12, 1–5. [Google Scholar]
- (109).Schoffelen R, Sharkey RM, Goldenberg DM, Franssen G, McBride WJ, Rossi EA, Chang C−H, Laverman P, Disselhorst JA, Eek A, et al. (2010) Pre Targeted Immuno-Positron Tomography Imaging of Carcinoembryonic Antigen-Expressing Tumors With Bispecific Antibody and A 68Ga- And 18F-Labeled Hapten Peptide in Mice with Human Tumor Xenografts. Mol. Cancer Ther 9, 1019–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (110).Shetty D, Choi SY, Jeong JM, Lee JY, Hoigebazar L, Lee Y−S, Lee DS, Chung J−K, Lee MC, and Chung YK (2011) Stable Aluminum Fluoride Chelates with Triazacyclononane Derivatives Proved by X-Ray Crystallography and 18F-Labeling Study. Chem. Commun 47, 9732–9734. [DOI] [PubMed] [Google Scholar]
- (111).D’Souza CA, McBride WJ, Sharkey RM, Todaro LJ, and Goldenberg DM (2011) High-Yielding Aqueous 18F-Labeling of Peptides Via Al18F Chelation. Bioconjugate Chem 22, 1793–1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (112).McBride WJ, D’Souza CA, Karacay H, Sharkey RM, and Goldenberg DM (2012) New Lyophilized Kit for Rapid Radiofluorination of Peptides. Bioconjugate Chem 23, 538–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (113).McBride WJ, D’Souza CA, Sharkey RM, Karacay H, Rossi EA, Chang C−H, and Goldenberg DM (2010) Improved 18F Labeling of Peptides With A Fluoride-Aluminum-Chelate Complex. Bioconjugate Chem 21, 1331–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (114).Chong H−S, Garmestani K, Ma D, Milenic DE, Overstreet T, and Brechbiel MW (2002) Synthesis and Biological Evaluation of Novel Macrocyclic Ligands with Pendent Donor Groups As Potential Yttrium Chelators for Radioimmunotherapy with Improved Complex Formation Kinetics. J. Med. Chem 45, 3458–3464. [DOI] [PubMed] [Google Scholar]
- (115).Pinski J, Halmos G, Yano T, Szepeshazi K, Qin Y, Ertl T, and Schally AV (1994) Inhibition of Growth of MKN45 Human Gastric-Carcinoma Xenografts in Nude Mice By Treatment With Bombesin/Gastrin-Releasing-Peptide Antagonist (RC-3095) and Somatostatin Analogue RC-160. Int. J. Cancer 57, 574–580. [DOI] [PubMed] [Google Scholar]
- (116).Markwalder R, and Reubi JC (1999) Gastrin-Releasing Peptide Receptors in the Human Prostate: Relation to Neoplastic Transformation. Cancer Res 59, 1152–1159. [PubMed] [Google Scholar]
- (117).Moody TW, Crawley JN, and Jensen RT (1982) Pharmacology and Neurochemistry of Bombesin-Like Peptides. Peptides 3, 559–563. [DOI] [PubMed] [Google Scholar]
- (118).Dijkgraaf I, Franssen GM, McBride WJ, D’Souza CA, Laverman P, Smith CJ, Goldenberg DM, Oyen WJG, and Boerman OC (2012) PET of Tumors Expressing Gastrin-Releasing Peptide Receptor with An 18F-Labeled Bombesin Analog. J. Nucl. Med 53, 947–952. [DOI] [PubMed] [Google Scholar]
- (119).Liu Y, Hu X, Liu H, Bu L, Ma X, Cheng K, Li J, Tian M, Zhang H, and Cheng Z (2013) A Comparative Study of Radiolabeled Bombesin Analogs for the PET Imaging of Prostate Cancer. J. Nucl. Med 54, 2132–2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (120).Varasteh Z, Aberg O, Velikyan I, Lindeberg G, Sorensen J, Larhed M, Antoni G, Sandstrom M, Tolmachev V, and Orlova A (2013) In Vitro and In Vivo Evaluation of A 18F-Labeled High Affinity NOTA Conjugated Bombesin Antagonist As A PET Ligand for GRPR-Targeted Tumor Imaging. PLoS One 8, e81932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (121).Pan D, Yan Y, Yang R, Xu YP, Chen F, Wang L, Luo S, and Yang M (2014) PET Imaging of Prostate Tumors with 18F-Al-NOTA-MATBBN. Contrast Media Mol. Imaging 9, 342–348. [DOI] [PubMed] [Google Scholar]
- (122).Chatalic KLS, Franssen GM, van Weerden WM, McBride WJ, Laverman P, de Blois E, Hajjaj B, Brunel L, Goldenberg DM, Fehrentz JA, et al. (2014) Preclinical Comparison of Al18F-and 68Ga-Labeled Gastrin-Releasing Peptide Receptor Antagonists for PET Imaging of Prostate Cancer. J. Nucl. Med 55, 2050–2056. [DOI] [PubMed] [Google Scholar]
- (123).Carlucci G, Kuipers A, Ananias HJ, de Paula FD, Dierckx RA, Helfrich W, Rink R, Moll GN, de Jong IJ, and Elsinga PH (2015) GRPR-Selective PET Imaging of Prostate Cancer Using [18F]-Lanthionine-Bombesin Analogs. Peptides 67, 45–54. [DOI] [PubMed] [Google Scholar]
- (124).Folkman J (1995) Angiogenesis in Cancer, Vascular, Rheumatoid and Other Disease. Nat. Med 1, 27–30. [DOI] [PubMed] [Google Scholar]
- (125).Haubner R, Wester HJ, Reuning U, Senekowitsch-Schmidtke R, Diefenbach B, Kessler H, Stocklin G, and Schwaiger M (1999) Radiolabeled αvβ3 Integrin Anatgonists: a New Class of Tracers for Tumor Targeting. J. Nucl. Med 40, 1061–1071. [PubMed] [Google Scholar]
- (126).Haubner R, Wester HJ, Burkhart F, Senekowitsch-Schmidtke R, Weber W, Goodman SI, Kessler H, and Schwaiger M (2001) Glycosylated RGD-Containing Peptides: Tracer for Tumor Targeting and Angiogenesis Imaging with Improved Biokinetics. J. Nucl. Med 42, 326–336. [PubMed] [Google Scholar]
- (127).Haubner R, Kuhnast B, Mang C, Weber WA, Kessler H, Wester HJ, and Schwaiger M (2004) [18F]Galacto-RGD: Synthesis, Radiolabeling, Metabolic Stability, and Radiation Dose Estimates. Bioconjugate Chem 15, 61–69. [DOI] [PubMed] [Google Scholar]
- (128).Haubner R (2006) αvβ3-Integrin Imaging: A New Approach to Characterize Angiogenesis? Eur. J. Nucl. Med. Mol. Imaging 33, 54–63. [DOI] [PubMed] [Google Scholar]
- (129).Janseen ML, Oyen WJ, Dijkgaraaf I, Massuger LF, Frielink C, Edwards DS, Rajopahye M, Boonstra H, Corsens FH, and Boerman OC (2002) Tumor Targeting with Radiolabeled αvβ3 Integrim Binding Peptides in Nude Mouse Model. Cancer Res 62, 6146–6151. [PubMed] [Google Scholar]
- (130).Stollman TH, Ruers TJM, Oyen WJG, and Boerman OC (2009) New Targeted Probes for Radioimaging of Angiogenesis. Methods 48, 188–192. [DOI] [PubMed] [Google Scholar]
- (131).Niu G, and Chen X (2009) PET imaging of angiogenesis. PET Clinics 4, 17–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (132).Liu S (2015) Radiolabeled Cyclic RGD Peptides Bioconjugates as Radiotracers Targeting Multiple Integrins. Bioconjugate Chem 26, 1413–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (133).Cai W, and Chen X (2007) Nanoplatforms for Targeted Molecular Imaging in Living Subjects. Small 3, 1840–54. [DOI] [PubMed] [Google Scholar]
- (134).Chen K, and Conti PS (2010) Target-Specific Delivery of Peptide-Based Probes for PET Imaging. Adv. Drug Delivery Rev 62, 1005–1022. [DOI] [PubMed] [Google Scholar]
- (135).Haubner R, Maschauer S, and Prante O (2014) PET Radiopharmaceuticals for Imaging Integrin Expression: Tracers in Clinical Studies and Recent Developments. BioMed Res. Int 2014, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (136).Shetty D, Jeong JM, Kim YJ, Li JY, Hoigebazar L, Lee Y, Lee DS, and Chung JK (2012) Development of A Bifunctional Chelating Agent Containing Isothiocyanate Residue for One Step F-18 Labeling of Peptides and Application for RGD Labeling. Bioorg. Med. Chem 20, 5941–5947. [DOI] [PubMed] [Google Scholar]
- (137).Hausner SH, Bauer N, and Sutcliffe JL (2014) In Vitro and In Vivo Evaluation of The Effects of Aluminum [18F] Fluoride Radiolabeling on An Integrin αvβ6-Specific Peptide. Nucl. Med. Biol 41, 43–50. [DOI] [PubMed] [Google Scholar]
- (138).Liu S, Liu H, Jiang H, Xu Y, Zhang H, and Cheng Z (2011) One-Step Radiosynthesis of 18F-AlF-NOTA-RGD2 for Tumor Angiogenesis PET Imaging. Eur. J. Nucl. Med. Mol. Imaging 38, 1732–1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (139).Dijkgraaf I, Terry SYA, McBride WJ, Goldenberg DM, Laverman P, Franssen GM, Oyen WJG, and Boerman OC (2013) Imaging Integrin αvβ3 Expression in Tumors with an 18F-Labeled Dimeric RGD Peptide. Contrast Media Mol. Imaging 8, 238–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (140).Lang L, Li W, Guo N, Ma Y, Zhu L, Kiesewetter DO, Shen B, Niu G, and Chen X (2011) Comparison Study of [18F]AlF-NOTA-PRGD2, [18F]FPPRGD2 and [68Ga]Ga-NOTA-PRGD2 for PET Imaging of U87MG Tumors in Mice. Bioconjugate Chem 22, 2415–2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (141).Guo N, Lang L, Li W, Kiesewetter DO, Gao H, Niu G, Xie Q, and Chen X (2012) Quantitative Analysis and Comparison Study of [18F]AlF-NOTA-PRGD2, [18F]FPPRGD2 and [68Ga]Ga-NOTA-PRGD2 Using A Reference Tissue Model. PLoS One 7, e37506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (142).Guo J, Lang L, Hu S, Guo N, Zhu L, Sun Z, Ma Y, Kiesewetter DO, Niu G, Xie Q, and Chen X (2014) Comparison of Three Dimeric 18F-AlF-NOTA-RGD Tracers. Mol. Imag. Biol 16, 274–283. [DOI] [PubMed] [Google Scholar]
- (143).Guo J, Guo N, Lang L, Kiesewetter D, Xie Q, Li Q, Eden HS, Niu G, and Chen X (2014) 18F-Alfatide II and 18F-FDG Dual-Tracer Dynamic PET for Parametric, Early Prediction of Tumor Response to Therapy. J. Nucl. Med 55, 154–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (144).Wu H, Chen H, Pan D, Ma Y, Liang S, Wan Y, and Fang Y (2014) Imaging Integrin αvβ3 and NRP-1 Positive Gliomas with A Novel Fluorine-18 Labeled RGD-ATWLPPR Heterodimeric Peptide Probe. Mol. Imag. Biol 16, 781–792. [DOI] [PubMed] [Google Scholar]
- (145).Ruzza P, and Calderan A (2011) Radiolabeled Peptide-Receptor Ligands in Tumor Imaging. Expert Opin. Med. Diagn 5, 411–424. [DOI] [PubMed] [Google Scholar]
- (146).Krenning EP, Kwekkeboom DJ, Bakker WH, Breeman AP, Kooij PPM, Oei HY, van Hagen M, Postema PTE, de Jong M, Reubi JC, et al. (1993) Somatostatin Receptor Scintigraphy with [111n-DTPA-D-Phel]- and [123I-Tyr3]-Octreotide: The Rotterdam Experience with More Than 1000 Patients. Eur. J. Nucl. Med 20, 716–731. [DOI] [PubMed] [Google Scholar]
- (147).Fani M, Nicolas GP, and Wild D (2017) Somatostatin Receptor Antagonists for Imaging and Therapy. J. Nucl. Med 58 (Supplement2), 61S–66S. [DOI] [PubMed] [Google Scholar]
- (148).Kwekkeboom DJ, Kam BL, van Essen M, Teunissen JJ, van Eijck CH, Valkema R, de Jong M, de Herder WW, and Krenning FP (2010) Somatostatin Receptor-Based Imaging and Therapy of Gastroenteropancreatic Neuroendocrine Tumors. Endocr.-Relat. Cancer 17, R53–R73. [DOI] [PubMed] [Google Scholar]
- (149).Fani M, Del Pozzo LD, Abiraj K, Mansi R, Tamma ML, Cescato R, Waser B, Weber WA, Reubi JC, and Maecke HR (2011) PET of Somatostatin Receptor-Positive Tumors Using 64Cu- and 68Ga-Somatostatin Antagonists: The Chelate Makes A Difference. J. Nucl. Med 52, 1110–1118. [DOI] [PubMed] [Google Scholar]
- (150).Laverman P, McBride WJ, Sharkey RM, Eek A, Joosten L, Oyen WJG, Goldenberg DM, and Boertman OC (2010) A Novel Facile Method of Labeling Octreotide With 18F-Fluorine. J. Nucl. Med 51, 454–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (151).Laverman P, D’Souza CA, Eek A, McBride WJ, Sharkey RM, Oyen WJG, Goldenberg DM, and Boerman OC (2012) Optimized Labeling of NOTA-Conjugated Octreotide with F-18. Tumor Biol 33, 427–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (152).Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, and Jemal A (2015) Global Cancer Statistics, 2012. Ca-Cancer J. Clin 65, 87–108. [DOI] [PubMed] [Google Scholar]
- (153).Schuster DM, Taleghani PA, Nieh PT, Master VA, Amzat R, Savir-Brauch B, Halkar RK, Fox T, Osunkoya AO, Moreno CS, et al. (2013) Characterization of Primary Prostate Carcinoma by Anti-A-Amino-2-[(18)F]Fluorocyclobutane-1-Carbox-ylic Acid (Anti-3-[(18)F]FACBC) uptake. Am. J. Nucl. Med. Mol. Imag 3, 85–96. [PMC free article] [PubMed] [Google Scholar]
- (154).Schuster DM, Savir-Baruch B, Nieh PT, Master VA, Halkar RK, Rossi PJ, Lewis MM, Nye JA, Yu W, Bowman FD, et al. (2011) Detection of Recurrent Prostate Carcinoma with Anti-1-Amino-3-18F-Fluorocyclobutane-1-Carboxylic Acid PET/CT And 111In-Capromab Pendetide SPECT/CT. Radiology 259, 852–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (155).Nanni C, Schiavina R, Brunocilla E, Boschi S, Borghesi M, Zanoni L, Pettinato C, Martorana G, and Fanti S (2015) 18F-Fluciclovine PET/CT for the Detection of Prostate Cancer Relapse: a Comparison to 11C-Choline PET/CT. Clin. Nucl. Med 40, e386–91. [DOI] [PubMed] [Google Scholar]
- (156).Yu CY, Desai B, Ji L, Groshen S, and Jadvar H (2014) Comparative Performance of PET Tracers in Biochemical Recurrence of Prostate Cancer: A Critical Analysis of Literature. Am. J. Nucl. Med. Mol. Imag 4, 580–601. [PMC free article] [PubMed] [Google Scholar]
- (157).Eder M, Schafer M, Bauder-Wust U, Hull W-E, Wangler C, Mier W, Haberkorn U, and Eisenhut M (2012) 68Ga-complex Lipophilicity and Targeting Property of A Urea Based PSMA Inhibitor for PET Imaging. Bioconjugate Chem 23, 688–697. [DOI] [PubMed] [Google Scholar]
- (158).Eder M, Neels O, Muller M, Bauder-Wust U, Remde Y, Schafer M, Hennrich U, Eisenhut M, Afshar-Oromieh A, Haberkorn U, et al. (2014) Novel Preclinical and Radiopharmaceutical Aspects of [68Ga]Ga-PSMA-HBED-CC: A New PET Tracer for Imaging Prostate Cancer. Pharmaceuticals 7, 779–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (159).Benesova M, Schafer M, Bauder-Wust U, Schafer M, Klika KD, Mier W, Haberkorn U, Kopka K, and Eder M (2016) Linker Modification Strategies to Control the Prostate-Specific Membrane Antigen (PSMA)-Targeting and Pharmacokinetic Properties of DOTA-Conjugated PSMA Inhibitors. J. Med. Chem 59, 1761–1775. [DOI] [PubMed] [Google Scholar]
- (160).Benesova M, Schafer M, Bauder-Wust U, Afshar-Oromieh A, Kratochwil C, Mier W, Haberkorn U, Kopka K, and Eder M (2015) Preclinical Evaluation of A Tailor-Made DOTA-Conjugated PSMA Inhibitor with Optimized Linker Moiety for Imaging and Endoradiotherapy of Prostate Cancer. J. Nucl. Med 56, 914–920. [DOI] [PubMed] [Google Scholar]
- (161).Weineisen M, Schottelius M, Simecek J, Baum RP, Yildiz A, Beykan S, Kulkarni HR, Lassmann M, Klette I, Eiber M, et al. (2015) 68Ga- and 177Lu-Labeled PSMA I &T: Optimization of A PSMA-Targeted Theranostics Concept and First Proof-of-Concept Human Studies. J. Nucl. Med 56, 1169–1176. [DOI] [PubMed] [Google Scholar]
- (162).Young JD, Abbate V, Imberti C, Meszaros L, Ma MT, Terry SYA, Hider RC, Mullen GE, and Blower PJ (2017) 68GaTHP-PSMA: A PET Imaging Agent for Prostate Cancer Offering Rapid, Room-Temperature, One-Step Kit-Based Radiolabeling. J. Nucl. Med 58, 1270–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (163).Mease RC, Dusich CL, Foss CA, Ravert HT, Dannals RF, Seidel J, Prideaux A, Fox JJ, Sqouros G, Kozikowski AP, et al. (2008) N-[N-[(S)-1,3-Dicarboxypropyl]Carbamoyl]-4-[18F]-Fluorobemzyl-L-Cysteine), [18F]DCFBC: A New Imaging Probe for Prostate Cancer. Clin. Cancer Res 14, 3036–3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (164).Rowe SP, Gage KL, Faraj SF, Macura KJ, Cornish TC, Gonzalez-Roibon N, Guner G, Munari E, Partin AW, Pavlovich CP, et al. (2015) 18F-DCFBC PET/CT for PSMA-Based Detection and Characterization of Primary Prostate Cancer. J. Nucl. Med 56, 1003–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (165).Chen Y, Pullambhatla M, Foss CA, Byun Y, Nimmagadda S, Senthamizhchelvan S, Sgouros G, Mease RC, and Pomper MG (2011) 2-(3-{1-Carboxy-5-[(6-[18F]Fluoro-Pyridine-3-Carbonyl)-Amino]-Pentyl}-Ureido)-Pentanedioic Acid, [18F]DCFPyL, A PSMA-Based PET Imaging Agent for Prostate Cancer. Clin. Cancer Res 17, 7645–7653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (166).Cardinale J, Schafer M, Benesova M, Bauder-Wust U, Leotta K, Eder M, Neels OC, Haberkorn U, Giesel FL, and Kopka K (2017) Preclinical Evaluation of 18F-PSMA-1007: A New PSMA Ligand for Prostate Cancer Imaging. J. Nucl. Med 58, 425–431. [DOI] [PubMed] [Google Scholar]
- (167).Giesel FL, Hadaschik B, Cardinale L, Radtke J, Vinsensia M, Lehnert W, Kesch C, Tolstov Y, Singer S, Grabe N, et al. (2017) F-18 Labeled PSMA-1007: Biodistribution, Radiation Dosimetry and Histopathological Validation Of Tumor Lesions in Prostate Cancer Patients. Eur. J. Nucl. Med. Mol. Imaging 44, 678–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (168).Cardinale J, Martin R, Remde Y, Schaefer M, Hienzsch A, Hubner S, Zerges AM, Marx H, Hesse R, Weber K, et al. (2017) Procedure for the GMP-Compliant Production and Quality Control Of [18F]PSMA-1007: A Next Generation Radiofluorinated Tracer for The Detection of Prostate Cancer. Pharmaceuticals 10, 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (169).Kopka K, Benesova M, Barinka C, Haberkorn U, and Babich J (2017) Glu-Ureido-Based Inhibitors of Prostate-Specific Membrane Antigen: Lessons Learned During The Development of A Novel Class of Low-Molecular-Weight Theranostics Radiotracers. J. Nucl. Med 58, 17S–26S. [DOI] [PubMed] [Google Scholar]
- (170).Afshar-Oromieh A, Holland-Letz T, Giesel FL, Kratochwil C, Mier W, Haufe S, Debus N, Eder M, Eisenhut M, Schafer M, et al. (2017) Diagnostic Performance of 68Ga-PSMA-11 (HBED-CC) PET/CT in Patients With Recurrent Prostate Cancer: Evaluation in 1007 Patients. Eur. J. Nucl. Med. Mol. Imaging 44, 1258–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (171).Malik N, Baur B, Winter G, Reske SN, Beer AJ, and Solbach C (2015) Radiofluorination of PSMA-HBED Vial Al18F2+Chelation and Biological Evaluations In Vitro. Mol. Imag. Biol 17, 777–785. [DOI] [PubMed] [Google Scholar]
- (172).Boschi S, Lee JT, Beykan S, Slavik R, Wei L, Spick C, Eberlein U, Buck AK, Lodi F, Cicoria G, et al. (2016) Synthesis and Preclinical Evaluation of An Al18F Radiofluorinated Glu-Urea-Lys(AHX)-HBED-CC PSMA Ligand. Eur. J. Nucl. Med. Mol. Imaging 43, 2122–2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (173).Al-Momani E, Israel I, and Samnick S (2017) Validation of A (Al18F]PSMA-11 Preparation for Clinical Applications. Appl. Radiat. Isot 130, 102–106. [DOI] [PubMed] [Google Scholar]
- (174).Malik N, Zlatopolskiy B, Machulla HJ, Reske SN, and Solbach C (2012) One Pot Radiofluorination of A New Potential PSMA Ligand [Al18F]NOTA-DUPA-Pep. J. Labelled Compd. Radio-pharm 55, 320–325. [Google Scholar]
- (175).Cleeren F, Lecina J, Billaud EMF, Ahamed M, Verbruggen A, and Bormans GM (2016) New Chelators for Low Temperature Al18F-Labeling of Biomolecules. Bioconjugate Chem 27, 790–798. [DOI] [PubMed] [Google Scholar]
- (176).Liu Q, Pan D, Cheng C, Zhang D, Zhang A, Wang L, Jiang H, Wang T, Liu H, Xu Y, et al. (2015) Development of A Novel PET Tracer [18F]AlF-NOTA-C6 Targeting MMP2 for Tumor Imaging. PLoS One 10, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (177).Xu Y, Pan D, Zhu C, Xu Q, Wang L, Chen F, Yang R, Luo S, Yang M, and Yan Y (2014) Pilot Study of Novel 18F-Labeled FSHR Probe for Tumor Imaging. Mol. Imag. Biol 16, 578–585. [DOI] [PubMed] [Google Scholar]
- (178).Kiesewetter D, Guo N, Guo J, Gao H, Zhu L, Ma Y, Niu G, and Chen X (2012) Evaluation of An [18F]AlF-NOTA Analog of Exendin-4 For Imaging of GLP-1 Receptor in Insulinoma. Theranostics 2, 999–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (179).Xu Q, Zhu C, Xu Y, Pan D, Liu P, Yang R, Wang L, Chen F, Sun X, Luo S, et al. (2015) Preliminary Evaluation of [18F]AlF-NOTA-MAL-Cys39-Exendin-4 in Insulinoma with PET. J. Drug Target 23, 813–820. [DOI] [PubMed] [Google Scholar]
- (180).Chen X, Fan Z, Chen Y, Fang X, and Sha X (2013) Retro-Inverso Carbohydrate Mimetic Peptides with Annexin1-Binding Selectivity, are Stable In Vivo, and Target Tumor Vasculature. PLoS One 8, e80390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (181).Gu X, Jiang M, Pan D, Cai G, Zhang R, Zhou Y, Ding Y, Zhu B, and Lin X (2016) Preliminary Evaluation of Novel 18F-AlF-NOTA-IF7 as Tumor Imaging Agent. J. Radioanal. Nucl. Chem 308, 851–856. [Google Scholar]
- (182).Persson M, Liu H, Madsen J, Cheng Z, and Kjaer A (2013) First 18F-Labeled Ligand for PET Imaging of uPAR: In Vivo studies in Human Prostate Cancer Xenografts. Nucl. Med. Biol 40, 618–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (183).Muller C, and Schibli R (2011) Folic Acid Conjugates for Nuclear Imaging Of Folate Receptor-Positive Cancer. J. Nucl. Med 52, 1–4. [DOI] [PubMed] [Google Scholar]
- (184).Ke CY, Mathias CJ, and Green MA (2004) Folate-Receptor-Targeted Radionuclide Imaging Agents. Adv. Drug Delivery Rev 56, 1143–1160. [DOI] [PubMed] [Google Scholar]
- (185).Sega E, and Low P (2008) Tumor Detection Using Folate-Receptor-Targeted Imaging Agents. Cancer Metastasis Rev 27, 655–664. [DOI] [PubMed] [Google Scholar]
- (186).Low PS, and Kularatne SA (2009) Folate-Targeted Therapeutics and Imaging Agents for Cancer. Curr. Opin. Chem. Biol 13, 256–262. [DOI] [PubMed] [Google Scholar]
- (187).Muller C (2012) Folate Based Radiopharmaceuticals for Imaging and Therapy of Cancer and Inflammation. Curr. Pharm. Des 18, 1058–83. [DOI] [PubMed] [Google Scholar]
- (188).Zwicke GL, Ali Mansoori G, and Jeffery CJ (2012) Utilizing the Folate Receptor for Active Targeting of Cancer Nanotherapeutics. Nano Rev 3, 18496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (189).Chen Q, Meng X, McQuade P, Rubins D, Lin S−A, Zeng Z, Haley H, Miller P, Trotter DG, and Low PS (2016) Synthesis and Preclinical Evaluation of Folate-NOTA-Al18F for PET Imaging of Folate-Receptor-Positive Tumors. Mol. Pharmaceutics 13, 1520–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (190).Baselga J, and Swain SM (2009) Novel Anticancer Targets: Revisiting ERBB2 and Discovering ERBB3. Nat. Rev. Cancer 9, 463–475. [DOI] [PubMed] [Google Scholar]
- (191).Meric-Bernstein F, and Hung MC (2006) Advances in Targeting Human Epidermal Growth Factor Receptor-2 Signaling for Cancer Therapy. Clin. Cancer Res 12, 6326–6330. [DOI] [PubMed] [Google Scholar]
- (192).Harris L, Fritsche H, Mennel R, Norton L, Ravdin P, Taube S, Somerfield MR, Hayes DF, and Bast RC Jr (2007) American Society of Clinical Oncology 2007 Update of Recommendations for the Use of Tumor Markers in Breast Cancer. J. Clin. Oncol 25, 5287–5312. [DOI] [PubMed] [Google Scholar]
- (193).Lub-de Hooge MN, Kosterink JGW, Perik PJ, Nijnuis H, Tran L, Bart J, Suurmeijer AJH, de Jong S, and de Vries EGE (2004) Preclinical Characterization of 111InDTPA-Trastuzumab. Br. J. Pharmacol 143, 99–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (194).Paudyal P, Paudyal B, Hanaoka H, Oriuchi N, Iida Y, Yoshioka H, Tominaga H, Watanabe S, Watanabe S, Ishioka NS, et al. (2010) Imaging and Biodistribution of Her2/neu Expression in Non-Small Cell Lung Cancer Xenografts with Cu-Labeled Trastuzumab PET. Cancer Sci 101, 1045–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (195).Baum RP, Prasad V, Muller D, Schuchardt C, Orlova A, Wennborg A, Tolmachev V, and Feldwisch J (2010) Molecular Imaging of HER2-Expressimng Maliganant Tumors in Breast Cancer Patients Using Synthetic 111In- or 68Ga-Labeled Affibody Molecules. J. Nucl. Med 51, 892–895. [DOI] [PubMed] [Google Scholar]
- (196).Tolmachev V, Wallberg H, Sandstrom M, Hansson M, Wennborg A, and Orlova A (2011) Optimal Specific Radioactivity of Anti-HER2 Affibody Molecules Enables Discrimination Between Xenografts with High and Low HER2 Expression Levels. Eur. J. Nucl. Med. Mol. Imaging 38, 531–539. [DOI] [PubMed] [Google Scholar]
- (197).Heskamp S, Laverman P, Rosik D, Boschetti F, van der Graaf WTA, Oyen WJG, van Laarhoven HWM, Tolmachev V, and Boerman OC (2012) Imaging of Human Epithelial Growth Factor Receptor Type 2 Expression With 18F-Labeled Affibody Molecule ZHER2:2395 in A Mouse Model for Ovarian Cancer. J. Nucl. Med 53, 146–153. [DOI] [PubMed] [Google Scholar]
- (198).Glaser M, Iveson P, Hoppmann S, Indrevoll B, Wilson A, Arukwe J, Danikas A, Bhalla R, and Hiscock D (2013) Three Methods for 18F Labeling of The HER2-Binding Affibody Molecule Z(HER2:2891) Including Preclinical Assessment. J. Nucl. Med 54, 1981–1988. [DOI] [PubMed] [Google Scholar]
- (199).Miao Z, Ren G, Liu H, Jiang L, and Cheng Z (2010) Small-Animal PET Imaging of Human Epidermal Growth Factor Receptor Positive Tumor With a 64Cu Labeled Affibody Protein. Bioconjugate Chem 21, 947–954. [DOI] [PubMed] [Google Scholar]
- (200).Su X, Cheng K, Jeon J, Shen B, Venturin GT, Hu X, Rao J, Chin FT, Wu H, and Cheng Z (2014) Comparison of Two Site-Specifically 18F-Labeled Affibodies for PET Imaging of EGFR Positive Tumors. Mol. Pharmaceutics 11, 3947–3956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (201).Da Pieve C, Allott L, Martins CD, Vardon A, Ciobota DM, Kramer-Marek G, and Smith G (2016) Efficient [18F]AlF Radiolabeling of ZHER3:8698 Affibody Molecule for Imaging of HER3 Positive Tumors. Bioconjugate Chem 27, 1839–1849. [DOI] [PubMed] [Google Scholar]
- (202).Cleeren F, Lecina J, Ahamed M, Raes G, Devoogdt N, Caveliers V, McQuade P, Rubins DJ, Li W, Verbruggen A, et al. (2017) Al18F-Labeling of Heat-Sensitive Biomolecules for Positron Emission Tomography Imaging. Theranostics 7, 2924–2939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (203).McBride WJ, D’Souza CA, Sharkey RM, and Goldenberg DM (2012) The Radiolabeling of Proteins by The [18F]AlF Method. Appl. Radiat. Isot 70, 200–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (204).Wan W, Guo N, Pan D, Yu C, Weng Y, Luo S, Ding H, Xu Y, Wang L, Lang L, Xie Q, Yang M, and Chen X (2013) First Experience of 18F-Alfatide in Lung Cancer Patients Using a New Lyophilized Kit for Rapid Radiofluorination. J. Nucl. Med 54, 691–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (205).Gao S, Wu H, Li W, Zhao S, Teng X, Lu H, Hu X, Wang S, Yu J, and Yuan S (2015) A avβ3 with RGD PET/CT in Suspected Lung Cancer Patients. Eur. J. Nucl. Med. Mol. Imaging 42, 2029–2037. [DOI] [PubMed] [Google Scholar]
- (206).Luan X, Huang Y, Gao S, Sun X, Wang S, Ma L, Teng X, Lu H, Yu J, and Yuan S (2016) 18F-Alfatide PET/CT May Predict Short-Term Outcome of Concurrent Chemo Radiotherapy in Patients with Advanced Non-Small Cell Lung Cancer. Eur. J. Nucl. Med. Mol. Imaging 43, 2336–2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (207).Cheng W, Wu Z, Liang S, Fu H, Wu S, Tang Y, Ye Z, and Wang H (2014) Comparison of 18F-AlF-NOTA-PRGD2 and 18F-FDG Uptake in Lymph Node Metastasis of Differentiated Thyroid Cancer. PLoS One 9, e100521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (208).Yu C, Pan D, Mi B, Xu Y, Lang L, Niu G, Yang M, Wan W, and Chen X (2015) 18F-Alfatide II PET/CT in Healthy Human Volunteers and Patients with Brain Metastases. Eur. J. Nucl. Med. Mol. Imaging 42, 2021–2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (209).Mi B, Yu C, Pan D, Yang M, Wan W, and Chen X (2015) Pilot Prospective Evaluation of 18F-Alfatide II for Detection of Skeletal Metastases. Theranostics 5, 1115–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]