

In this study, Hou et al. analyzed large-scale cancer genome data sets to identify USP21 as one of the most commonly amplified genes (22%) in PDAC. Using in vivo approaches, they go on to show USP21 deubiquitinates TCF7, a member of the Wnt pathway, promoting pancreatic cancer cell proliferation and stemness.
Keywords: pancreatic cancer, TCF7, USP21, Wnt pathway, cancer stemness, deubiquitinase
Abstract
The ubiquitin-specific protease (USP) family is the largest group of cysteine proteases. Cancer genomic analysis identified frequent amplification of USP21 (22%) in human pancreatic ductal adenocarcinoma (PDAC). USP21 overexpression correlates with human PDAC progression, and enforced expression of USP21 accelerates murine PDAC tumor growth and drives PanIN to PDAC progression in immortalized human pancreatic ductal cells. Conversely, depletion of USP21 impairs PDAC tumor growth. Mechanistically, USP21 deubiquitinates and stabilizes the TCF/LEF transcription factor TCF7, which promotes cancer cell stemness. Our work identifies and validates USP21 as a PDAC oncogene, providing a potential druggable target for this intractable disease.
Current therapeutic options are ineffective in PDAC (Adamska et al. 2017). Genomic profiling of PDAC has provided a comprehensive atlas of recurrent genetic aberrations that may promote PDAC tumorigenesis (Biankin et al. 2012; Waddell et al. 2015; Witkiewicz et al. 2015; Bailey et al. 2016; The Cancer Genome Atlas Research Network 2017). These genetic events include known oncogenes and tumor suppressor genes and numerous novel genetic alterations. Moreover, classification of PDAC based on molecular signatures suggests the existence of distinct potential oncogenic drivers for different PDAC subtypes. These observations prompted us to explore newly characterized genetic alterations in PDAC with the goal of identifying and understanding new oncogenes that may expand therapeutic strategies.
Protein ubiquitination is one of the most common posttranslational modifications that can affect protein function in myriad ways including regulation of protein stability, subcellular localization, and activity (Swatek and Komander 2016). Ubiquitin modification is controlled by the opposing actions of E3 ligases and deubiquitinases to add or remove ubiquitin, respectively. Ubiquitination regulators are deregulated in PDAC, including tumor suppressors FBXW7 and USP9X (Pérez-Mancera et al. 2012; Jin et al. 2017), and potential oncogenes TRIM31 and RNF13 (Zhang et al. 2009; Yu et al. 2018). Our analysis of PDAC genomic data revealed frequent amplification (22%) of the protein deubiquitinase USP21, prompting exploration of its potential role as an oncogene for PDAC.
In this study, we show that USP21 overexpression and its nuclear localization correlate positively with PDAC disease progression in patient samples. USP21 promotes mouse PDAC growth and tumor-initiating capacity. Moreover, USP21 accelerates colony formation and cell proliferation of immortalized human pancreatic ductal epithelial cells (hTERT-HPNE E6/E7 cells) in vitro and promotes PanIN-to-tumor progression of the hTERT-HPNE E6/E7 xenografts in vivo. Mechanistically, USP21 interacts with, deubiquitinates and stabilizes the Wnt pathway transcription factor TCF7 to activate gene expression in the Wnt network. Together, these studies establish USP21 as an oncogene and potential therapeutic target in PDAC.
Results and Discussion
USP21 is highly amplified in PDAC
The USP family, consisting of 58 members in human, is the largest group of the cysteine proteases that belong to deubiquitinases (Reyes-Turcu et al. 2009). Genomic analysis of all the USPs in human PDAC samples micro-dissected for cancer cell enrichment (Cerami et al. 2012; Gao et al. 2013; Witkiewicz et al. 2015) revealed USP21 amplification in 22% of cases (Fig. 1A) with the minimal common region of ∼0.23 Mb, within which there were no other known oncogenes (Supplemental Fig. S1A). Analysis of USP21 and PDAC signature mutations did not reveal co-occurrence or exclusive patterns of significance (Supplemental Fig. S1B). USP21 amplification was also observed in several other cancer types (Fig. 1B), which is consistent with previous reports that USP21 regulates cell proliferation and metastasis in bladder cancer (Chen et al. 2017), hepatocellular carcinoma (Liu et al. 2017; Li et al. 2018), triple-negative breast cancer and renal cell carcinoma (Peng et al. 2016). However, there were no molecular mechanistic insights as to whether and how USP21 may contribute to oncogenesis.
Figure 1.
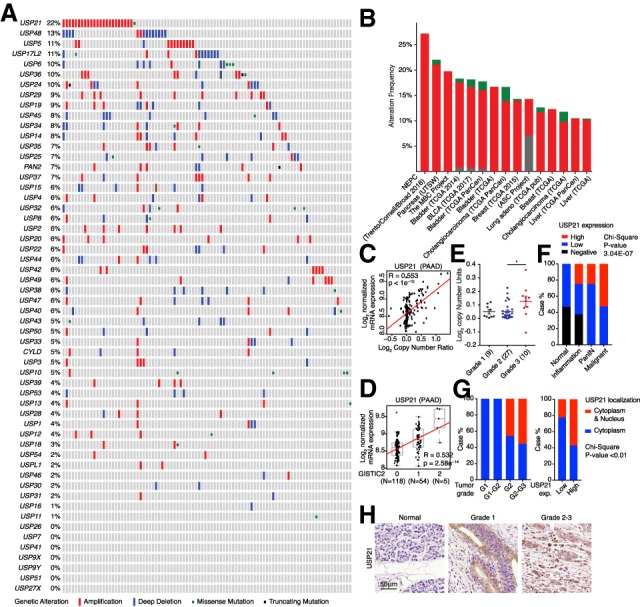
USP21 is up-regulated in PDAC. (A,B) Genetic alternations of USPs in UTSW PDAC data set (A) and pan-cancer analysis (B). (C,D) USP21 mRNA level positively correlates with copy number (C) and GISTIC scores (D) in TCGA PAAD data set. (E) USP21 copy number positively correlates with PDAC tumor grade analyzed in Oncomine. (F) USP21 expression positively correlates with PDAC progression by TMA analysis. (G) USP21 nuclear localization positively correlates with tumor grade and USP21 expression by TMA analysis. (H) Representative IHC images of USP21 in TMA tissues.
USP21 protein is highly conserved between human and mouse (97% similarity), containing an N-terminal nuclear export signal and a C-terminal USP domain (García-Santisteban et al. 2012), which deubiquitinates numerous cytoplasmic and nuclear substrates (Nakagawa et al. 2008; Zhang et al. 2013; Fan et al. 2014). USP21 amplification correlated positively with elevated mRNA levels (Fig. 1C,D) and higher histologic tumor grade (Fig. 1E) in the TCGA pancreatic adenocarcinoma (PAAD) data set. Correspondingly, tissue microarray (TMA) analysis of human PDAC samples showed a positive correlation between high USP21 expression and tumor progression (Fig. 1F). Especially, nuclear localized USP21 correlated positively with higher tumor grades and with higher expression levels of USP21 (Fig. 1G,H), suggesting that the nuclear functions of USP21 may regulate tumor progression.
Catalytically active USP21 promotes PDAC tumor growth
To test the oncogenic function of USP21, we observed that, while enforced USP21 did not alter pancreas cancer cell proliferation in vitro (Supplemental Fig. S2A), USP21 overexpression (but not enzyme dead USP21, ED-USP21) promoted tumor growth of PDAC cells derived from the murine iKPC model (Fig. 2A; Ying et al. 2012). Moreover, USP21 overexpression promoted cell proliferation (Fig. 2B; Supplemental Fig. S2B) and anchorage-independent colony formation (Fig. 2C,D) of hTERT-HPNE E6/E7 and hTERT-HPNE E6/E7-KRASG12D cells. We examined the tumorigenic role of USP21 in vivo by orthotopically transplanting hTERT-HPNE E6/E7 cells with USP21 or GFP into the pancreases of NSG mice (designated as GFP-HPNE and USP21-HPNE, n = 4 each), which were collected after 8 wk. Whereas only one of four GFP-HPNE pancreases possessed a small mass, three of four USP21-HPNE pancreases possessed sizeable masses. The GFP-HPNE mass consisted of small PanIN lesions with limited proliferation activity, while the USP21-HPNE masses consisted of advanced adenocarcinomas with high-proliferative indices (Fig. 2E), indicating that USP21 overexpression drives tumor progression of human pancreas epithelial cells from PanIN to PDAC. Conversely, genetic deletion of Usp21 by CRISPR/Cas9 in iKPC cells impaired tumorigenicity that could be rescued by re-expression of USP21 (Fig. 2F; Supplemental Fig. S2C–E). Together, these results suggest that USP21 is a PDAC oncogene that depends on its catalytic function.
Figure 2.
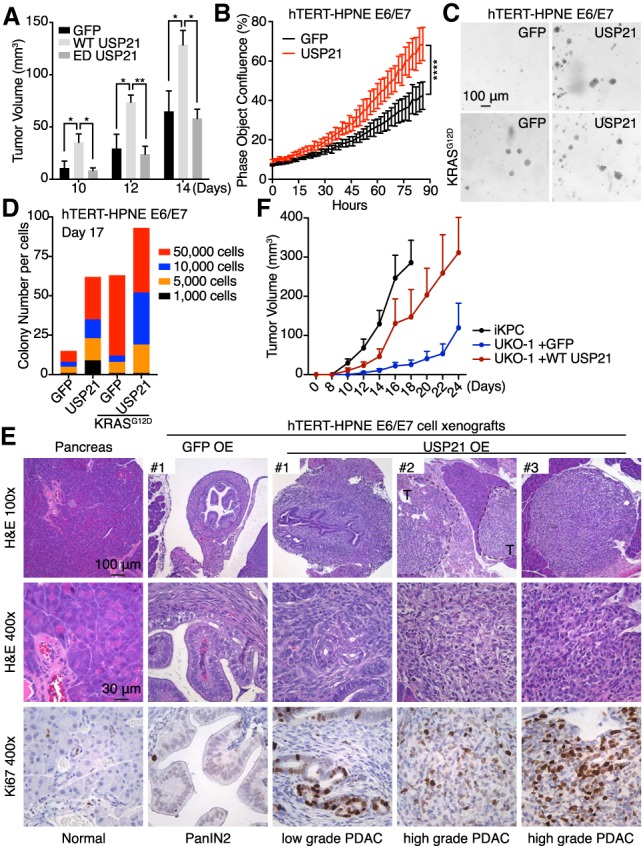
USP21 promotes pancreatic tumor growth. (A) Tumor growth comparison of GFP, WT-, and ED-USP21 OE iKPC cells (n = 5). Two-hundred-thousand cells were injected subcutaneously in nude mice. Tumor sizes were measured on days 10, 12, and 14. (B) Cell proliferation comparison of GFP-HPNE and USP21-HPNE cells (n = 6). (C,D) Representative images (C) and colony quantification (D) of anchorage-independent colony formation of hTERT-HPNE E6/E7 and hTERT-HPNE E6/E7-KRASG12D cells overexpressing WT-USP21 and GFP. (E) Characterization of xenograft masses from orthotopically injected GFP-HPNE and USP21-HPNE cells in NSG mice (n = 4). Proliferative cells were positively stained by Ki67. (T) Tumor. (F) Knockout of Usp21 impaired iKPC tumor growth (n = 5). For A, B, and F, data are represented as mean ± SEM.
Nuclear USP21 activates Wnt/β-catenin pathway and promotes cancer cell stemness
To assess the biological relevance of USP21 subcellular localization, USP21 was engineered with nuclear localization (NLS) or nuclear export (NES) signals (Fig. 3A–C). While USP21 or USP21 variants did not impact colony formation, cell proliferation, migration, or invasion in vitro (Supplemental Fig. S3A–D), nuclear NLS-USP21, but not cytoplasmic NES-USP21, promoted the iKPC tumor growth (Fig. 3D).
Figure 3.
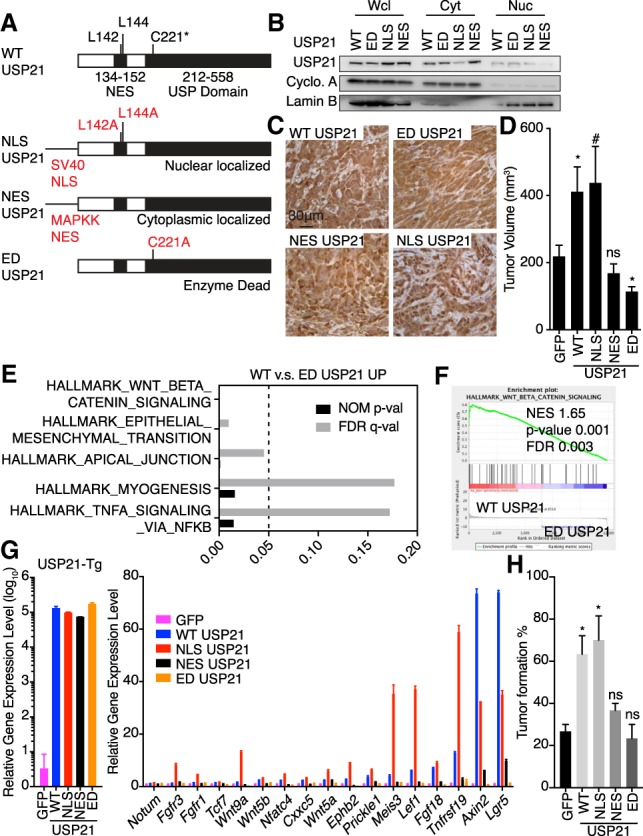
Nuclear function of USP21 activates Wnt pathway and increases cancer cell stemness. (A) Plasmid construction for specific subcellular localization of USP21. (B) Validation of subcellular localization of USP21 variants. (C) Representative IHC images of USP21 localization in iKPC cells overexpressing USP21 variants. (D) Comparison of tumor growth between iKPC cells overexpressing GFP and USP21 variants (n = 5). (E) GSEA of pathways enriched in WT-USP21 versus ED-USP21 OE iKPC cells. (F) Enrichment of Wnt pathway in WT-USP21 versus ED-USP21 OE iKPC cells. (G) RT-PCR validation of Wnt pathway genes comparing iKPC cells overexpressing GFP and USP21 variants (n = 3). Data are represented as mean ± SD. (H) Limited dilution assay comparing iKPC cells overexpressing GFP and USP21 variants. n = 3 independent experiments. For D and H, data are represented as mean ± SEM. Comparison is between GFP control with other groups as indicated.
To dissect the oncogenic mechanisms governed by USP21, we compared the transcriptome of iKPC cells overexpressing wild-type (WT) USP21 and ED-USP21. Gene set enrichment analysis (GSEA) showed that the Wnt/β-catenin pathway was the top up-regulated pathway by USP21 (Fig. 3E,F), which was validated in both iKPC cells and tumors by semiquantitative real-time (RT)-PCR (Fig. 3G; Supplemental Fig. S3E). Correspondingly, we found that NLS-USP21, but not NES-USP21 or ED-USP21, up-regulated Wnt pathway genes (Fig. 3G).
In cancer, the Wnt/β-catenin pathway has been shown to regulate tumor cell stemness in multiple cancer types (Reya and Clevers 2005; Vermeulen et al. 2010), prompting us to conduct limited dilution tumor-initiating assays. We found that WT-USP21 and NLS-USP21 supported efficient tumor formation at 50%–80% with five iKPC cells compared with 10%–20% efficiency for five iKPC cells overexpressing ED-USP21 or GFP controls (Fig. 3H; Supplemental Fig. S3F). Conversely, Usp21 depletion decreased the tumor formation efficiency from about 40% to 5%–10% with 20 iKPC cells (Supplemental Fig. S3G). Thus, nuclear USP21 up-regulates Wnt pathway signaling and promotes tumor-initiating potential in PDAC cells.
USP21 promotes cancer cell stemness via TCF7
Consistent with the known function of USP21 (Nakagawa et al. 2008), overexpression of NLS-USP21 dramatically decreased H2AK119ub1 in iKPC cells (Supplemental Fig. S4E). Knockdown of Rnf2, the E3 ligase of Polycomb-repressive complex 1 that monoubiquitinates H2AK119 (Di Croce and Helin 2013), decreased monoubiquitination of H2AK119 (Supplemental Fig. S4A,B) but did not alter the Wnt pathway gene expression (Supplemental Fig. S4C), suggesting that the up-regulation of Wnt pathway genes by USP21 may involve mechanisms other than H2AK119ub1 deubiquitination.
Since only nuclear USP21 activated the Wnt pathway genes, we next explored the Wnt pathway components that either reside in the nucleus or shuttle between the cytoplasm and nucleus in iKPC cells, including β-catenin, Dishevelled proteins and TCF/LEF transcription factors (Supplemental Fig. S4D). We observed that overexpression of NLS-USP21 dramatically increased protein level of the long isoform of TCF7 in iKPC cells but not those of β-catenin, Dishevelled proteins, and TCF7L2 (Fig. 4A; Supplemental Fig. S4E,F).
Figure 4.
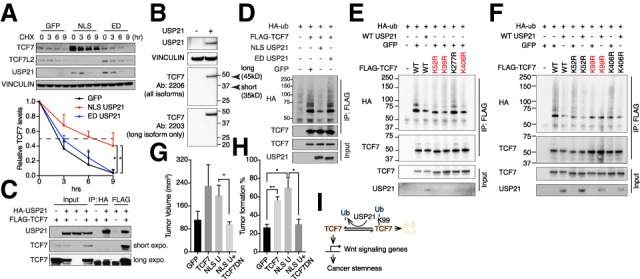
TCF7 mediates tumor-promoting function of USP21. (A) CHX protein chasing assay of TCF7 and TCF7L2 in GFP, NLS-USP21, and ED-USP21 OE iKPC cells. Data are represented as mean ± SD (n = 3). (B) Western blot analysis of TCF7 isoforms in iKPC cells. (C) Co-IP analysis between WT-USP21 and TCF7 in 293T cells. Arrowhead indicates the correct band size. (D) Ubiquitination assay of TCF7 in GFP, NLS- USP21, and ED-USP21 OE 293T cells. (E) Ubiquitination assay of TCF7 in 293T cells overexpressing WT TCF7 and TCF7 mutants. (F) Ubiquitination assay in 293T cells overexpressing WT TCF7 and TCF7 mutants with and without USP21 cotransfection. (G) Tumor growth comparison of iKPC cells overexpressing indicated ORFs (n = 4). (H) Limited dilution assay comparing iKPC cells overexpressing indicated ORFs. n = 3 independent experiments. (I) Schematic graph of the USP21 regulation of TCF7 to promote cancer stemness in PDAC. Statistical analysis was performed between indicated groups. For G and H, data are represented as mean ± SEM.
TCF7 belongs to the TCF/LEF family, which has at least two major isoforms with different β-catenin-binding capacities (Shiokawa et al. 2017). The long isoform of TCF7 has a β-catenin-binding domain at the N terminus, while the short isoform of TCF7 lacks it and instead represses Wnt signaling. Only the long isoform of TCF7 is expressed in iKPC cells (Fig. 4B). Protein stability analysis showed that expression of NLS- or WT-USP21 increased the half-life of the long isoform of TCF7 to ∼6 h from a baseline of 3 h in cells with ED-USP21 or GFP controls (Fig. 4A; Supplemental Fig. S4G). Consistent with the known ability of TCF7 to activate its own promoter (Wu et al. 2012), Tcf7 was also transcriptionally up-regulated in WT-USP21 and nuclear NLS-USP21 overexpressed (OE) iKPC cells (Supplemental Fig. S4H). Conversely, Usp21 depletion decreased TCF7 protein expression and stability (Supplemental Fig. S5A,B) as well as Tcf7 mRNA levels (Supplemental Fig. S5C). MG132 treatment blunted USP21 regulation of TCF7 protein stability (Supplemental Figs. S4I, S5D), indicating that USP21 prevents TCF7 from proteasome-mediated protein degradation. Furthermore, the pan-DUB inhibitor PR-619 decreased TCF7 protein levels in USP21-OE iKPC cells in a dose-dependent manner (Supplemental Fig. S4J).
USP21 and TCF7 were shown to interact physically by coimmunoprecipitation (co-IP) assay (Fig. 4C). Overexpression of WT-USP21 or NLS-USP21 decreased the ubiquitination levels of TCF7 in iKPC cells compared with ED-USP21 or GFP controls (Fig. 4D–F). To determine the specific sites of TCF7 protein that are deubiquitinated by USP21, we ranked the predicted ubiquitinated lysine residues of TCF7 conserved in human and mouse by three bioinformatic tools (UbiSite, BDM-PUB, and UbPred), the top four of which (K52, K99, K277, and K406) with high confidence scores were mutated from lysine to arginine (K to R) individually to block the site-specific ubiquitination. Mutations of K52R, K99R, and K406R deceased the TCF7 ubiquitination levels compared with WT control (Fig. 4E), while USP21 was unable to further deubiquitinate K99R TCF7 (Fig. 4F), suggesting that the Lys99 of TCF7 is the major residue deubiquitinated by USP21. Accordingly, overexpression of USP21 increased the protein stability of the exogenous WT TCF7 (Supplemental Fig. S5E) but did not affect the protein stability of K99R mutated TCF7 (Supplemental Fig. S5F).
To assess the functional role of Tcf7 in tumorigenesis, overexpression of the long isoform of TCF7 promoted tumor growth of iKPC cells to a level similar to that of NLS-USP21 (Fig. 4G), while the dominant-negative TCF7 lacking the β-catenin-binding domain (TCF7DN) blocked the tumor-promoting actions of NLS-USP21 (Fig. 4G; Supplemental Fig. S4K). Moreover, enforced TCF7 promoted iKPC cell “stemness” similar to that described above by limited dilution assay and, conversely, TCF7DN blocked NLS-USP21-mediated stemness (Fig. 4H). Thus, TCF7 is a nuclear substrate of USP21, which participates in the oncogenic actions of USP21 in PDAC (Fig. 4I).
USP21 regulation of TCF7 is conserved in human cells
Consistent with our findings in mouse cells, we observed dramatically up-regulated TCF7 protein levels in USP21-HPNE xenograft tumors compared with GFP-HPNE controls (Fig. 5A). USP21 also increased TCF7 protein half-life from ∼1.5 to 3 h in human MIA PaCa-2 PDAC cells compared with GFP controls (Fig. 5B), while USP21 depletion decreased TCF7 protein expression and stability (Supplemental Fig. S6A,B). Functionally, overexpression of USP21 promoted MIA PaCa-2 xenograft tumor growth, and TCF7DN neutralized the oncogenic impact of USP21 (Fig. 5C). Thus, USP21 stabilizes TCF7 to promote tumor growth in human cells.
Figure 5.
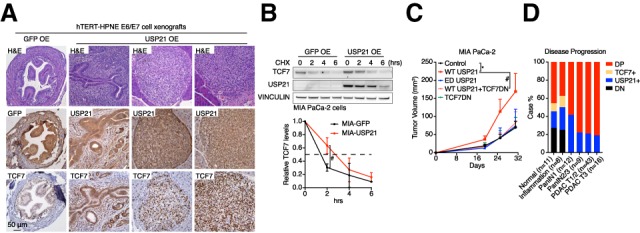
USP21 stabilizes TCF7 and promotes tumor growth via TCF7 in human PDAC cells. (A) Comparison of TCF7 protein expression in GFP-HPNE and USP21-HPNE xenografts by IHC. (B) CHX assay of TCF7 in GFP and USP21 OE Mia PaCa-2 cells (n = 2). Data are represented as mean ± SD. (C) Tumor growth comparison of MIA PaCa-2 cells overexpressing indicated ORFs (n ≥ 4 for each group). Data are represented as mean ± SEM. (D) Coexpression of TCF7 and USP21 positively correlates with PDAC progression by TMA analysis. (T) Tumor grade; (DP) TCF7 and USP21 double-positive samples; (USP21+ or TCF7+) USP21 or TCF7 single-positive samples; (DN) TCF7 and USP21 double-negative samples. Statistical analysis was performed between indicated groups.
Consistent with functional studies, we observed that increased coexpression of TCF7 and USP21 correlated positively with disease progression in human PDAC TMA analysis (Fig. 5D; Supplemental Fig. S6C) and that USP21 overexpression positively correlated with the hallmark of Wnt pathway signature in Moffitt PDAC data set (Supplemental Fig. S6D; Moffitt et al. 2015) as well as with poor patient survival in both Moffitt and Stratford PDAC data sets (Supplemental Fig. S6E,F; Stratford et al. 2010; Moffitt et al. 2015). Additionally, the negative impact of USP21 on patient survival was more significant in the PDAC basal-like subtype—in which the Wnt pathway activation is one of the major molecular characteristics (Nicolle et al. 2017)—than in the classical subtype (Supplemental Fig. S6G,H), reinforcing the role of USP21 in regulating the Wnt pathway.
In summary, our study identified USP21 as a frequently amplified gene in PDAC with the capacity to promote tumor progression and growth, and enhance cancer stem cell self-renewal. USP21 executes its oncogenic functions via its deubiquitination activity, which serves to stabilize TCF7 that conveys the biological actions of an activated Wnt pathway, including enhancement of cancer cell stemness (Fig. 4I). Thus, we conclude that USP21 is an oncogene for PDAC.
USP21 has been shown to exhibit oncogenic functions in other cancer types, in which whether USP21 promotes cancer cell stemness via TCF7 will require further investigation. Additionally, examination of USP21 overexpression in spontaneous PDAC mouse models may further enhance our understanding of its oncogenic role at tumor initiation stage and its cooperative effect with oncogenic KRAS.
The precise role of TCF7 in tumorigenesis may relate to the differential expression of its long or short isoforms. In normal colon cells, the short isoform of TCF7 is expressed and antagonizes the tumorigenic function of TCF4 (Najdi et al. 2009), while the long isoform of TCF7 is required for colon tumorigenesis and promotes R-spondin-independent organoid growth of normal colon stem cells (Shiokawa et al. 2017). In our study, only the long isoform of TCF7 is expressed and stabilized by USP21 in PDAC cells, which provides further support of a tumorigenic role for the long isoform of TCF7 in cancer.
β-Catenin protein levels increased in more than 65% PDAC tumors (Zeng et al. 2006), and high nuclear localized β-catenin correlates positively with poor clinical outcome in PDAC patients. Consistently, activation of Wnt/β-catenin signaling promotes PDAC tumorigenesis, while inhibition of the pathway suppresses PDAC development (Sano et al. 2016). Our work identifies USP21 as a therapeutically trackable target for disruption of TCF7-mediated Wnt signaling activation and impairment of PDAC.
Materials and methods
Transgenic mice
All mouse-related experiments were approved by MD Anderson Cancer Center's Institutional Animal Care and Use Committee (IACUC). The iKPC mouse model was described previously (Ying et al. 2012).
Establishment of primary iKPC PDAC cell lines and 2D and 3D cell culture
The iKPC tumors were dissociated using tumor dissociation kit (Miltenyi Biotec). Cells were cultured in RPMI1640 + 10% HI FBS (Gibco) with 1 µg/mL doxycycline (Clontech). For 3D culture, 400–500 iKPC cells were seeded in 50 µL of GR-Matrigel (Corning) droplets in 24-well low attachment cell culture plates (Thermo) and cultured in the same medium above.
Human cell culture and colony formation assay
The hTERT-HPNE E6/E7 and hTERT-HPNE E6/E7-KRASG12D cells were cultured as American Type Culture Collection indicated. MIA PaCa-2 cells were cultured in DMEM with 10% FBS. For colony formation assay, 0.6% soft agar was used as the bottom layer, and cells were suspended in 0.3% soft agar as the top layer. Culture medium was added on top of agar layers and changed every 3 d.
Plasmid construction, genetic manipulation, and lentivirus production
DNA sequence information for protein tags, shRNAs, sgRNA, and primers are listed in Supplemental Table 1. USP21 (BC090946.1) and Tcf7 (XM_006532800) were cloned in pHAGE lentivirus vector. HA and Flag tags were added at the N-terminus of USP21 and Tcf7 ORFs, respectively. HA-tagged ubiquitin was from Addgene. Site mutations of Tcf7 ORF were generated by the QuikChange site-directed mutagenesis kit. Viruses were packaged using second-generation lentiviral systems.
In vivo cell transplant and limited dilution assay
Nude mice and NSG SCID mice were purchased from Taconic and the Department of Experimental Radiation Oncology of MDACC. For transplantation, cells were resuspended in mixture of Opti-MEM (Gibco) and GR-Matrigel (1:1). We transplanted 200,000 iKPC cells subcutaneously in nude mice fed by doxycycline water immediately. We transplanted 1,000,000 USP21-HPNE or GFP-HPNE cells into the tails of pancreases and MIA PaCa-2 cells subcutaneously into NSG SCID mice. For limited dilution assay, serially diluted iKPC cells were transplanted subcutaneously in NSG SCID mice.
Cell proliferation, migration, invasion, and colony formation assays
Cell proliferation was measured every 4 h by IncuCyte (Essen BioScience). Cell migration was determined by cell scratch assays. Cell invasion assay was performed on Matrigel in Boyden chambers. Invaded cells were stained by crystal violet and quantified by ImageJ. For colony formation, 500 cells per well were seeded in 6-well plates and stained with crystal violet after 1 wk.
RT-PCR, mRNA sequencing, and GSEA
RT-PCR was run using 7500 Fast real-time PCR system, and data were analyzed by GraphPad Prism 7.0c. The parameter for mRNA sequencing was NGS-75-nt paired end. Data were processed as described previously (Conesa et al. 2016) and analyzed by GSEA software (Mootha et al. 2003; Subramanian et al. 2005). The data accession number is GSE134686.
Antibody information and protein assays
Antibody information is in Supplemental Table 2. Western blot, IHC, co-IP, and ubiquitination assay were performed as described previously (Choo and Zhang 2009; Ying et al. 2012). TMA slides were from Biomax. Cells were fractionated using Subcellular Protein Fractionation Kit (Thermo). For protein chasing assay, cells were treated with 100 µg/mL cycloheximide (CHX) (Sigma) for serial time lengths before collection. Cells were treated with 10 µM MG132 (Sigma) for 6 h. Protein levels were quantified by ImageJ.
Bioinformatic analysis
TCGA PAAD data set was obtained from the GDAC data portal (2016-01-26 archive). Moffitt and Stratford PDAC data sets were obtained from GEO databases. Pearson's correlation and patient survival were analyzed in R.
Statistical analysis
Statistical analysis was performed using the unpaired Student's t-test to generate two-tailed P-values. Asterisk labeling for P-values is as follows: P > 0.1 (ns), 0.05 < P ≤ 0.1 (#), 0.01 < P ≤ 0.05 (*), 0.001 < P ≤ 0.01(**), 0.0001 < P ≤ 0.001 (***), and P ≤ 0.0001 (****).
Supplementary Material
Acknowledgments
We thank Zhaohui Xu and Ivonne Flores for mouse breeding, Howard Chang for technical assistance, and Haoqiang Ying, Guocan Wang, and Jian Hu for project discussions. P.H. is supported by the Seed Grant Program of the Hirshberg Foundation for Pancreatic Cancer Research. MD Anderson's Sequencing & Microarray Facility is supported by a Cancer Center Support Grant CA016672. This study is supported by National Institutes of Health R01 CA231349 grant (Y.A.W.) and National Cancer Institute P01 CA117969 grant (R.A.D.). C.G. was supported by the Cancer Prevention and Research Institute of Texas Research Training Program (RP170067).
Author contributions: R.A.D., Y.A.W., and P.H. conceived the study, performed the methodology, and acquired funding. P.H., X.M., Q.Z., W.L., H.W., J.Z., X.Z., J.A., and C.G. performed the investigation and validated the results. P.H. and Y.A.W. wrote the original draft of the manuscript. R.A.D., Y.A.W., D.J.S., and P.H. reviewed and edited the manuscript. C.W., J.L., and J.Z were responsible for the software used. S.J. obtained the resources for the study. R.A.D. and Y.A.W. supervised the study.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.326314.119.
References
- Adamska A, Domenichini A, Falasca M. 2017. Pancreatic ductal adenocarcinoma: current and evolving therapies. Int J Mol Sci 18: 1338 10.3390/ijms18071338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey P, Chang DK, Nones K, Johns AL, Patch AM, Gingras MC, Miller DK, Christ AN, Bruxner TJ, Quinn MC, et al. 2016. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 531: 47–52. 10.1038/nature16965 [DOI] [PubMed] [Google Scholar]
- Biankin AV, Waddell N, Kassahn KS, Gingras MC, Muthuswamy LB, Johns AL, Miller DK, Wilson PJ, Patch AM, Wu J, et al. 2012. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 491: 399–405. 10.1038/nature11547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research Network. 2017. Integrated genomic characterization of pancreatic ductal adenocarcinoma. Cancer Cell 32: 185–203.e13. 10.1016/j.ccell.2017.07.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. 2012. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2: 401–404. 10.1158/2159-8290.CD-12-0095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Zhou B, Chen D. 2017. USP21 promotes cell proliferation and metastasis through suppressing EZH2 ubiquitination in bladder carcinoma. Onco Targets Ther 10: 681–689. 10.2147/OTT.S124795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choo YS, Zhang Z. 2009. Detection of protein ubiquitination. J Vis Exp 10.3791/1293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conesa A, Madrigal P, Tarazona S, Gomez-Cabrero D, Cervera A, McPherson A, Szcześniak MW, Gaffney DJ, Elo LL, Zhang X, et al. 2016. A survey of best practices for RNA-seq data analysis. Genome Biol 17: 13 10.1186/s13059-016-0881-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Croce L, Helin K. 2013. Transcriptional regulation by Polycomb group proteins. Nat Struct Mol Biol 20: 1147–1155. 10.1038/nsmb.2669 [DOI] [PubMed] [Google Scholar]
- Fan Y, Mao R, Yu Y, Liu S, Shi Z, Cheng J, Zhang H, An L, Zhao Y, Xu X, et al. 2014. USP21 negatively regulates antiviral response by acting as a RIG-I deubiquitinase. J Exp Med 211: 313–328. 10.1084/jem.20122844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. 2013. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 6: pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Santisteban I, Bañuelos S, Rodríguez JA. 2012. A global survey of CRM1-dependent nuclear export sequences in the human deubiquitinase family. Biochem J 441: 209–217. 10.1042/BJ20111300 [DOI] [PubMed] [Google Scholar]
- Jin X, Yang C, Fan P, Xiao J, Zhang W, Zhan S, Liu T, Wang D, Wu H. 2017. CDK5/FBW7-dependent ubiquitination and degradation of EZH2 inhibits pancreatic cancer cell migration and invasion. J Biol Chem 292: 6269–6280. 10.1074/jbc.M116.764407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Cui K, Prochownik EV, Li Y. 2018. The deubiquitinase USP21 stabilizes MEK2 to promote tumor growth. Cell Death Dis 9: 482 10.1038/s41419-018-0523-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Kruswick A, Dang H, Tran AD, Kwon SM, Wang XW, Oberdoerffer P. 2017. Ubiquitin-specific protease 21 stabilizes BRCA2 to control DNA repair and tumor growth. Nat Commun 8: 137 10.1038/s41467-017-00206-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffitt RA, Marayati R, Flate EL, Volmar KE, Loeza SG, Hoadley KA, Rashid NU, Williams LA, Eaton SC, Chung AH, et al. 2015. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat Genet 47: 1168–1178. 10.1038/ng.3398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstråle M, Laurila E, et al. 2003. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 34: 267–273. 10.1038/ng1180 [DOI] [PubMed] [Google Scholar]
- Najdi R, Syed A, Arce L, Theisen H, Ting JH, Atcha F, Nguyen AV, Martinez M, Holcombe RF, Edwards RA, et al. 2009. A Wnt kinase network alters nuclear localization of TCF-1 in colon cancer. Oncogene 28: 4133–4146. 10.1038/onc.2009.271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa T, Kajitani T, Togo S, Masuko N, Ohdan H, Hishikawa Y, Koji T, Matsuyama T, Ikura T, Muramatsu M, et al. 2008. Deubiquitylation of histone H2A activates transcriptional initiation via trans-histone cross-talk with H3K4 di- and trimethylation. Genes Dev 22: 37–49. 10.1101/gad.1609708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolle R, Blum Y, Marisa L, Loncle C, Gayet O, Moutardier V, Turrini O, Giovannini M, Bian B, Bigonnet M, et al. 2017. Pancreatic adenocarcinoma therapeutic targets revealed by tumor-stroma cross-talk analyses in patient-derived xenografts. Cell Rep 21: 2458–2470. 10.1016/j.celrep.2017.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng L, Hu Y, Chen D, Linghu R, Wang Y, Kou X, Yang J, Jiao S. 2016. Ubiquitin specific protease 21 upregulation in breast cancer promotes cell tumorigenic capability and is associated with the NOD-like receptor signaling pathway. Oncol Lett 12: 4531–4537. 10.3892/ol.2016.5263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-Mancera PA, Rust AG, van der Weyden L, Kristiansen G, Li A, Sarver AL, Silverstein KA, Grützmann R, Aust D, Rümmele P, et al. 2012. The deubiquitinase USP9X suppresses pancreatic ductal adenocarcinoma. Nature 486: 266–270. 10.1038/nature11114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reya T, Clevers H. 2005. Wnt signalling in stem cells and cancer. Nature 434: 843–850. 10.1038/nature03319 [DOI] [PubMed] [Google Scholar]
- Reyes-Turcu FE, Ventii KH, Wilkinson KD. 2009. Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes. Annu Rev Biochem 78: 363–397. 10.1146/annurev.biochem.78.082307.091526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sano M, Driscoll DR, DeJesus-Monge WE, Quattrochi B, Appleman VA, Ou J, Zhu LJ, Yoshida N, Yamazaki S, Takayama T, et al. 2016. Activation of WNT/β-catenin signaling enhances pancreatic cancer development and the malignant potential via up-regulation of Cyr61. Neoplasia 18: 785–794. 10.1016/j.neo.2016.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiokawa D, Sato A, Ohata H, Mutoh M, Sekine S, Kato M, Shibata T, Nakagama H, Okamoto K. 2017. The induction of selected Wnt target genes by Tcf1 mediates generation of tumorigenic colon stem cells. Cell Rep 19: 981–994. 10.1016/j.celrep.2017.04.017 [DOI] [PubMed] [Google Scholar]
- Stratford JK, Bentrem DJ, Anderson JM, Fan C, Volmar KA, Marron JS, Routh ED, Caskey LS, Samuel JC, Der CJ, et al. 2010. A six-gene signature predicts survival of patients with localized pancreatic ductal adenocarcinoma. PLoS Med 7: e1000307 10.1371/journal.pmed.1000307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. 2005. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci 102: 15545–15550. 10.1073/pnas.0506580102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swatek KN, Komander D. 2016. Ubiquitin modifications. Cell Res 26: 399–422. 10.1038/cr.2016.39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermeulen L, De Sousa EMF, van der Heijden M, Cameron K, de Jong JH, Borovski T, Tuynman JB, Todaro M, Merz C, Rodermond H, et al. 2010. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat Cell Biol 12: 468–476. 10.1038/ncb2048 [DOI] [PubMed] [Google Scholar]
- Waddell N, Pajic M, Patch AM, Chang DK, Kassahn KS, Bailey P, Johns AL, Miller D, Nones K, Quek K, et al. 2015. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 518: 495–501. 10.1038/nature14169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witkiewicz AK, McMillan EA, Balaji U, Baek G, Lin WC, Mansour J, Mollaee M, Wagner KU, Koduru P, Yopp A, et al. 2015. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat Commun 6: 6744 10.1038/ncomms7744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JQ, Seay M, Schulz VP, Hariharan M, Tuck D, Lian J, Du J, Shi M, Ye Z, Gerstein M, et al. 2012. Tcf7 is an important regulator of the switch of self-renewal and differentiation in a multipotential hematopoietic cell line. PLoS Genet 8: e1002565 10.1371/journal.pgen.1002565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying H, Kimmelman AC, Lyssiotis CA, Hua S, Chu GC, Fletcher-Sananikone E, Locasale JW, Son J, Zhang H, Coloff JL, et al. 2012. Oncogenic KRAS maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 149: 656–670. 10.1016/j.cell.2012.01.058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu C, Chen S, Guo Y, Sun C. 2018. Oncogenic TRIM31 confers gemcitabine resistance in pancreatic cancer via activating the NF-κB signaling pathway. Theranostics 8: 3224–3236. 10.7150/thno.23259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng G, Germinaro M, Micsenyi A, Monga NK, Bell A, Sood A, Malhotra V, Sood N, Midda V, Monga DK, et al. 2006. Aberrant Wnt/β-catenin signaling in pancreatic adenocarcinoma. Neoplasia 8: 279–289. 10.1593/neo.05607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Meng Y, Zhang L, Chen J, Zhu D. 2009. RNF13: a novel RING-type ubiquitin ligase over-expressed in pancreatic cancer. Cell Res 19: 348–357. 10.1038/cr.2008.285 [DOI] [PubMed] [Google Scholar]
- Zhang J, Chen C, Hou X, Gao Y, Lin F, Yang J, Gao Z, Pan L, Tao L, Wen C, et al. 2013. Identification of the E3 deubiquitinase ubiquitin-specific peptidase 21 (USP21) as a positive regulator of the transcription factor GATA3. J Biol Chem 288: 9373–9382. 10.1074/jbc.M112.374744 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.