

In this study, Hustedt et al. use CRISPR-based genetic screens to build a clear picture of the postsynaptic steps of homologous recombination in mammalian cells. They report the identification of C17orf53/HROB, a factor required for cell survival after exposure to a variety of replication stress-inducing genotoxins and for the resolution but not formation of Rad51 foci.
Keywords: CRISPR screens, cisplatin, DNA damage, DNA repair, DNA synthesis, germ cells, helicase, homologous recombination, infertility
Abstract
DNA repair by homologous recombination (HR) is essential for genomic integrity, tumor suppression, and the formation of gametes. HR uses DNA synthesis to repair lesions such as DNA double-strand breaks and stalled DNA replication forks, but despite having a good understanding of the steps leading to homology search and strand invasion, we know much less of the mechanisms that establish recombination-associated DNA polymerization. Here, we report that C17orf53/HROB is an OB-fold-containing factor involved in HR that acts by recruiting the MCM8–MCM9 helicase to sites of DNA damage to promote DNA synthesis. Mice with targeted mutations in Hrob are infertile due to depletion of germ cells and display phenotypes consistent with a prophase I meiotic arrest. The HROB–MCM8–MCM9 pathway acts redundantly with the HELQ helicase, and cells lacking both HROB and HELQ have severely impaired HR, suggesting that they underpin two major routes for the completion of HR downstream from RAD51. The function of HROB in HR is reminiscent of that of gp59, which acts as the replicative helicase loader during bacteriophage T4 recombination-dependent DNA replication. We therefore propose that the loading of MCM8–MCM9 by HROB may similarly be a key step in the establishment of mammalian recombination-associated DNA synthesis.
In mammals, homologous recombination (HR) is essential for tumor suppression and cellular viability due to its critical role in enforcing genomic integrity (San Filippo et al. 2008). HR plays a critical role during the repair of DNA double-stand breaks (DSBs) formed in postreplicative chromatin following the collapse of replication forks or when the replisome encounters fork-blocking lesions such as interstrand cross-links (ICLs) (San Filippo et al. 2008; Kottemann and Smogorzewska 2013). HR is also integral to meiosis, since it promotes homologous chromosome pairing and genetic information exchange following SPO11-mediated DSBs (San Filippo et al. 2008).
In its simplest embodiment, HR is a copying mechanism that uses DNA synthesis to regenerate a sequence surrounding a break. Canonical HR reactions involve recombinases related to bacterial RecA: RAD51 and DMC1 (San Filippo et al. 2008). RAD51 is the only recombinase operating during mitotic recombination, whereas both RAD51 and DMC1 are active during meiosis (San Filippo et al. 2008). Recombinases replace the RPA trimer on single-stranded DNA (ssDNA) formed after DNA end resection to promote homology search and strand invasion of the homologous dsDNA. The result is a displacement loop (or D loop) where the 3′ end of the invading strand is used to initiate DNA synthesis (San Filippo et al. 2008). Strand exchange and DNA synthesis produce joint molecule intermediates that are finally processed by helicases, topoisomerases, and nucleases to restore unlinked DNA duplexes via processes such as Holliday junction (HJ) dissolution or resolution (West et al. 2015).
Exactly how DNA synthesis is initiated and terminated during recombination is not entirely clear and relies largely on in vitro studies or studies in genetically tractable systems such as budding yeast (Sebesta et al. 2011; Sneeden et al. 2013; McVey et al. 2016). The current view is that the 3′ end of the invading ssDNA is extended by DNA polymerase δ with the help of PCNA. However, this view is likely to be incomplete for mammalian HR, since additional factors such as ATRX and the MCM8–MCM9 complex have been implicated (Natsume et al. 2017; Juhasz et al. 2018). ATRX is a multifunctional motor protein related to RAD54 that promotes long-range DNA synthesis during HR, possibly by coupling histone deposition/exchange with DNA synthesis (Juhasz et al. 2018). The MCM8–MCM9 complex, on the other hand, forms a helicase that is related to the MCM2–MCM7 replicative helicase (Liu et al. 2009) and plays a poorly understood role in HR (Lutzmann et al. 2012; Nishimura et al. 2012; Park et al. 2013; Lee et al. 2015). A potential clue as to the function of the MCM8–MCM9 complex emerged recently when cells with conditionally inactivated MCM2 were found to synthesize DNA in an MCM8- and RAD51-dependent manner (Natsume et al. 2017). These results suggested that the MCM8–MCM9 complex promotes DNA synthesis during HR, perhaps to promote restart of broken replication forks.
Here, we present the identification of C17orf53/HROB as a factor that acts downstream from RAD51 during HR. HROB is a DNA-binding OB-fold containing protein whose function is to recruit the MCM8/MCM9 helicase to DNA lesions. Loss-of-function mutation in HROB phenocopies nearly all of the phenotypes associated with loss of the MCM8–MCM9 helicase, including ICL sensitivity, defective gametogenesis, and impaired DNA synthesis following MCM2 inactivation. HROB and MCM8 mutations are epistatic to each other in human cells, confirming that they act together in a single pathway. Finally, we observed that the combined inactivation of the HROB–MCM8–MCM9 pathway and the HELQ helicase results in severe HR deficiency, suggesting that most mitotic HR reactions in human cells involve one of these two redundant helicases.
Results
To identify new recombination factors, we mined a set of CRISPR/Cas9 screens that probed the genetic architecture of the response to ATR or PARP inhibition in human cells (Zimmermann et al. 2018; Hustedt et al. 2019; Wang et al. 2019). We reasoned that gene mutations leading to sensitivity to both ATR and PARP inhibitors would be enriched in HR factors. To start with, we used a recently characterized 117-gene set involved in promoting normal cellular resistance to ATR inhibition based on seven independent CRISPR/Cas9 screens from two groups (Hustedt et al. 2019; Wang et al. 2019) and asked which of those genes were also comprised in a list of 182 genes involved in promoting cellular resistance to PARP inhibition in SUM149PT cells (Zimmermann et al. 2018). This analysis yielded 25 genes (Fig. 1A,B) enriched in loci encoding factors for ICL repair (GO:0036297) and HR (GO:0000724; Fig. 1C). The products of these 25 genes were also highly connected within a protein–protein interaction network, suggesting that they are functionally related (Fig. 1D).
Figure 1.

Genes promoting resistance to ATR and PARP inhibition are enriched for HR and ICL repair factors. (A) Venn diagram of the core set of gene mutations sensitizing cells to ATR inhibition (ATRi) and gene mutations sensitizing SUM149PT cells to olaparib. (B) Scatter plot of drugZ analysis result of the SUM149PT olaparib CRISPR screen. Genes that were both hits (normZ ≤ −2.5) in the SUM149PT olaparib screen and part of the core set sensitizing cells to ATRi are labeled and color-coded according to the number of ATRi screens in which they were found. (C) Gene ontology (GO) terms significantly enriched (>10-fold enrichment; P < 0.05, binomial test with Bonferroni correction) among hits common between both data sets. Circle size indicates the number of genes from the common gene hit list included in each GO term, color indicates negative log P-value, and X-axis position indicates the fold enrichment compared with the whole-genome reference set. (D) easyN network analysis of interactions between hits common to the SUM149PT olaparib screen, and ATRi core set. (DDR) DNA damage response; (DSB) DNA double-strand break; (HR) homologous recombination; (ICL) interstrand cross-link. See also Supplemental Figure S1.
The uncharacterized C17orf53 gene within this set caught our attention. Loss of C17orf53 increased ATR inhibitor sensitivity in six out of seven screens (Hustedt et al. 2019; Wang et al. 2019) and led to strong sensitivity to the PARP inhibitor olaparib in SUM149PT cells in competitive growth assays (Supplemental Fig. S1A; Zimmermann et al. 2018). The C17orf53 protein was identified previously as a protein interacting with the single-strand DNA (ssDNA)-binding protein RPA (Tkac et al. 2016), and we confirmed this interaction, which was independent of DNA, by coimmunoprecipitation studies and in vitro pull-down experiments using proteins purified from insect cells (Supplemental Fig. S1B–D). These observations strengthened the possibility that C17orf53 may be directly involved in a process relating to genome stability.
C17orf53 homologs are present in nearly all vertebrate genomes and are present in the genome of the slime mold Dictyostelium discoideum (DDB_G0282151), plants, and mosses (such as LOC112279930 from Physcomitrella patens) but are not detected in commonly used genetically tractable model organisms such a Caenorhabditis elegans, Drosophila melanogaster, or prokaryotes (Supplemental Fig. S2). In human cells, the mRNA isoform that is most widely expressed (ENST00000319977.4) codes for a protein of 647 amino acid residues. The protein contains a domain of unknown function, DUF4539, which bears homology with the oligosaccharide-binding (OB) fold (Fig. 2A), a domain present in many DNA repair proteins such as BRCA2, RPA, and SHLD2 and often involved in interacting with ssDNA (Bochkarev and Bochkareva 2004). The OB-fold-encompassing region is also the most conserved among C17orf53 homologs (Fig. 2A). Based on the presence of an OB-fold and other observations described below, we renamed the C17orf53 protein HROB, for “homologous recombination OB-fold.”
Figure 2.

HROB is a genome stability factor. (A) Analysis of conservation (Consurf) and predicted domains (Pfam and HHPred) of HROB. HROB contains predicted OB-fold domains in its C terminus. Color code indicates conservation score of amino acid residues. (B) Immunoblot of whole-cell extracts (WCEs) from the indicated wild-type (WT) and HROB-KO clones probed for HROB. The asterisk indicates the nonspecific immunoreactive band. GAPDH was used as a loading control. (C) Quantification of γH2AX mean nuclear intensity in individual EdU-negative and EdU-positive (nucleotide-incorporating) cells. The red line indicates the median of the population (n = 3). (A.U.) Arbitrary units. (D) Quantification of colony formation of RPE1-hTERT TP53-KO (WT) and HROB-KO cells after treatment with the indicated DNA damage-inducing agents. (ATRi) ATR inhibitor; (CPT) camptothecin; (HU) hydroxyurea; (IR) ionizing radiation; (MMC) mitomycin C; (TMZ) temozolomide; (UV) ultraviolet light. (*) P ≤ 0.05; (***) P ≤ 0.001; (****) P ≤ 0.0001. P-values in C are from unpaired two-tailed t-tests. P-values in D are from paired two-tailed t-tests. Error bars indicate standard deviation (n ≥ 3 biologically independent experiments). See also Supplemental Figures S1–S3.
The presence of the OB-fold domain also prompted us to test whether HROB can interact with DNA in vitro. We observed that recombinant murine HROB expressed and purified from insect cells was able to interact with DNA, with a preference for ssDNA, in electrophoretic mobility shift assays (Supplemental Fig. S1E,F). These results suggested that HROB might be directly involved in DNA transactions, such as DNA repair.
HROB promotes DNA repair downstream from RAD51
To study the role of HROB, we generated knockout (KO) cell lines of the HROB gene in RPE1-hTERT TP53-KO and RPE1-hTERT Cas9 TP53-KO cells and generated a polyclonal antibody against the protein that detects a band that is no longer present in clones with frameshifting indel mutations (Fig. 2B; Supplemental Fig. S3A). We selected two clones, RPE1-hTERT TP53-KO HROB-KO #1 and HROB-KO #2, for our initial analyses. HROB-deficient cells were viable but showed evidence of spontaneous DNA lesions using γ-H2AX immunofluorescence (Fig. 2C; Supplemental Fig. S3B). We next carried out a survey of the genotoxin sensitivity of HROB-deficient cells in clonogenic survival assays. We found that HROB loss caused the strongest sensitivity to the DNA cross-linking agents mitomycin C (MMC) and cisplatin (Fig. 2D) and, to a lesser extent, sensitivity to other agents, including ATR inhibitors (VE-821 and AZD6738), temozolomide (TMZ), and camptothecin (CPT). HROB-deficient cells were not sensitive to ionizing radiation (IR). The TMZ sensitivity of HROB-depleted cells was noted in a recently reported set of CRISPR screens undertaken in glioblastoma cells (MacLeod et al. 2019). The genotoxin sensitivity profile of HROB-null cells suggests a role in the repair of replication fork-blocking lesions.
We next analyzed the cellular phenotypes associated with exposure of HROB-KO cells to cisplatin. Wild-type and HROB-KO cells were treated with cisplatin for 24 h, and, following release from treatment, samples were collected at regular intervals for analysis by flow cytometry, immunoblotting, or microscopy (Fig. 3A). HROB-KO cells accumulated in G2/M phase following release from cisplatin (Fig. 3B; Supplemental Fig. S4), which was accompanied by CHK1 S345 and RPA S33 phosphorylation (Fig. 3C), markers of ATR-dependent signaling and DNA damage checkpoint activation. These results hinted at the presence of ongoing unresolved DNA damage in HROB-deficient cells. In support of this possibility, we observed a striking persistence of γ-H2AX and RAD51 foci as long as 34 h after release from cisplatin treatment (Fig. 3D,E). HROB-KO cells accumulated micronuclei and multinucleated cells following release from cisplatin (Fig. 3F,G) and exhibited aberrant mitoses characterized by anaphase bridges or lagging chromosomes (Fig. 3H,I). These results suggest that HROB-deficient cells fail to segregate damaged chromosomes, which are the likely source of micronucleation.
Figure 3.

HROB is involved in interstrand cross-link repair. (A) Schematic of the experimental setup for B–I. (B) Representative flow cytometry analysis of RPE1-hTERT TP53-KO (WT) and the indicated HROB-KO cells for DNA content and EdU incorporation before and after cisplatin treatment. The box indicates the gate for G2-phase cells. Numbers indicate the percentage of G2-phase cells. Data are mean ± standard deviation (n = 3 biologically independent experiments). (C) RPE1-hTERT TP53-KO (WT) and the indicated HROB-KO cells show elevated ATR signaling after cisplatin treatment. Immunoblot of whole-cell extracts for ATR targets. (pCHK1) phosphoCHK1 (S345); (pRPA32) phosphoRPA (S33). Total CHK1, RPA32, and HROB served as controls. (D) Representative images of RPE1-hTERT TP53-KO (WT) and HROB-KO #1 cells untreated or treated with 4 µM cisplatin for 24 h and stained with the indicated antibodies after the indicated time of recovery. (E) Quantification of cells with more than five γH2AX (left) or RAD51/γH2AX (right) colocalizing foci after the indicated time of recovery from cisplatin treatment. Lines connect means. (F,G) Nuclear morphology in RPE1-hTERT TP53-KO (WT) and the indicated HROB-KO cells stained with DAPI. (F) Representative images of a normal cell nucleus, a cell with a micronucleus (arrowhead), and a multinucleated cell, as indicated. (G) Quantification of micronuclei and multinucleated cells after the indicated time of recovery from cisplatin treatment in RPE1-hTERT TP53-KO (WT) and HROB-KO cells. (UT) Untreated. Lines connect means. (H) Representative images of a normal mitotic cell and cells with mitotic aberrations. (I) Quantification of mitotic aberrations after the indicated time of recovery from treatment with cisplatin in RPE1-hTERT TP53-KO (WT) and HROB-KO cells. Lines connect means. (J) Representative images of RPE1-hTERT TP53-KO HROB-KO #1 + GFP-HROB cells transfected with the indicated siRNAs, treated with 8 µM cisplatin for 4 h followed by 20 h of recovery in drug-free medium, and stained with the indicated antibodies. (K) Quantification of RPE1-hTERT TP53-KO HROB-KO #1 + GFP-HROB cells with more than five GFP-HROB/γH2AX colocalizing foci after transfection with the indicated siRNAs with or without cisplatin treatment (8 µM for 4 h followed by 20 h of recovery in drug-free medium). (siCTRL) Nontargeting siRNA. The red line indicates the mean. (**) P ≤ 0.01; (***) P ≤ 0.001. P-values are from paired two-tailed t-tests. Error bars in E, G, and I indicate standard deviation (n ≥ 3 biologically independent experiments). See also Supplemental Figures S4 and S5.
The replication fork block caused by cisplatin treatment can be sensed by the Fanconi anemia pathway, which orchestrates nucleolytic incision of the stalled fork, creating a DSB that requires HR for repair (Long et al. 2011). We therefore assessed whether HROB acts upstream of or downstream from the initiation of the FA pathway by monitoring the ubiquitylation of FANCD2 (Garcia-Higuera et al. 2001). We found that this step was unaffected in HROB-KO cells, suggesting that HROB is not involved in the early stages of ICL repair (Supplemental Fig. S5A).
We next assessed whether HROB localizes to sites of DNA damage. We expressed GFP-HROB in HROB-KO cells and found that exogenously expressed HROB localizes to cisplatin-induced foci in a manner dependent on the presence of FANCD2 or CtIP (Fig. 3J,K; Supplemental Fig. S5B,C). Together, these experiments indicate that HROB directly promotes the repair of ICL-induced lesions downstream from FANCD2 ubiquitylation. Furthermore, the persistence of RAD51 foci in HROB-KO cells (Fig. 3D,E) suggests that HROB may be acting during HR after the formation of the RAD51 nucleofilament.
HROB-deficient mice are infertile
In order to study the phenotype of HROB loss in vivo, we designed a strategy to target the mouse HROB homolog BC030867 (Hrob) by CRISPR/Cas9 in zygotes. Thirteen mice were born with alterations at the Hrob locus detected by nested PCR. These founders showed no overt phenotypic changes up to 6 mo of age. However, six out of seven females and three out of six males were infertile, as they failed to produce offspring during a 4-mo breeding trial with wild-type partners. Every infertile mouse had at least one Hrob allele containing a frameshifting (fs) mutation, while the four fertile mutant mice all showed germline transmission of in-frame (if) deletion alleles, leaving the conserved HROB protein regions intact (Fig. 4A; Supplemental Table S1). These observations suggest that disruption of the Hrob reading frame causes infertility.
Figure 4.

Mutation of Hrob in mice causes infertility and loss of germ cells. (A, top) Analysis of conservation (ConSurf) and predicted domains (Pfam and HHPred) of BC030867, the mouse homolog of HROB (mHROB). (Bottom) Predicted protein products of germline-transmitted mutant mHROB in-frame deletion (Hrobif) alleles in four male (M) and two female (F) founder mice. Color code indicates the conservation score of amino acid residues. Arrowheads indicate sgRNA target sites. Numbers above each protein schematic indicate deletions (−) or insertions (+). Red represents the insertion site. (B) Quantification of collected normal oocytes, oocytes with fragmented nuclei, and lysed oocytes after hormone-induced hyperovulation. Data are from four age-matched control C57BL6 and six mutant mHROB females with frameshifting alleles (Hrobfs). (C) Representative image of ovary sections stained with hematoxylin and eosin. The top image from the control female shows follicles in various maturation stages. The bottom image shows an ovary with benign tubular adenomas (dark purple) from an Hrobfs female. (D) Representative images of testis sections stained with hematoxylin and eosin from age-matched C57BL6 control, Hrobif, and Hrobfs males. Hrobfs males show smaller seminiferous tubules that are mostly devoid of germ cells. Original images were acquired with a 20× objective. (E) Quantification of percentage of normal seminiferous tubules, tubules containing meiotic cells in prophase I, tubules containing spermatogonical stem cells (SSC), and tubules containing only sertoli cells (SC) from individual animals as indicated on the X-axis. (F) Quantification of testis size. The red line indicates the mean. (G,H) Representative images of testis sections from age-matched C57BL6 control and Hrobfs males stained with the indicated antibodies. Yellow arrowheads indicate pachytene cells with γH2AX staining of the sex body. (I) Quantification of the percentage of tubules with cells showing γH2AX staining of the sex body from individual animals as indicated on the X-axis. See also Supplemental Figure S6.
As the infertility of the founder mice precluded us from having a renewable source of Hrobfs animals, we further analyzed a cohort of the founder Hrobfs females to determine the cause of infertility. We assessed ovarian reserve at 25 wk of age, after priming females with exogenous gonadotropins. While we obtained 21 ± 2.4 ovulated metaphase II-arrested oocytes from age-matched wild-type females, none of the female Hrobfs mice produced any oocytes (Fig. 4B). Moreover, by visual inspection, all Hrobfs females showed atrophic uteri and abnormally small-sized ovaries, sometimes with a fluid-filled cyst in one of the ovaries (seen in two of six Hrobfs females) (Supplemental Fig. S6). Ovaries from age-matched control females contained numerous follicles of different maturation stages as well as corpus lutei. In contrast, ovaries from Hrobfs mice were completely devoid of any follicles (Fig. 4C). Instead, we observed benign tubular adenomas with extensive invagination of surface ovarian epithelium into the stromal compartment, a phenomenon commonly observed in ovaries devoid of germ cells (Murphy 1972; Lutzmann et al. 2012). This phenotype is consistent with defects in oogonial survival, early follicular formation, or their maintenance.
The analysis of the male founder cohort showed that age-matched control and Hrobif males had normal appearance of seminiferous tubules with germ cells at various stages of development. In contrast, Hrobfs animals showed mostly empty tubules lined only with supporting sertoli cells, suggesting a defect in spermatogonial proliferation or maintenance (Fig. 4D,E). Only 11%–28% of the tubules contained any germ cells, with the most advanced stages corresponding to cells with condensed chromatin consistent with the leptotene/zygotene stage of meiotic prophase I. This drastically diminished number of germ cells was accompanied by smaller tubule diameter, an increase in interstitial tissue between tubules, and an overall reduction in testis size of Hrobfs compared with Hrobif or wild-type animals (Fig. 4D,F).
Based on these observations, we hypothesized that the remaining germ cells in Hrobfs males arrested during prophase I. To narrow down the stage of arrest, we stained testis sections from Hrobfs and age-matched wild-type animals for several prophase I markers. We found that the remaining germ cells in Hrobfs animals showed elevated levels of pan-nuclear γH2AX, SCP3, RAD51, and DMC1 compared with the surrounding cells (Fig. 4G,H). The γH2AX-, SCP3-, and RAD51-staining pattern and nuclear morphology were most consistent with leptotene/zygotene cells in wild-type tubules. Strikingly, none of the few remaining germ cells in Hrobfs testes showed γH2AX sex body staining typical for pachytene cells (Fig. 4G,I; Mahadevaiah et al. 2001). These results suggest that the remaining germ cells found in Hrobfs animals arrest during the leptotene/zygotene stage of meiosis prophase I and almost never progress to the pachytene stage.
HROB is epistatic to MCM8–MCM9 and acts in parallel to HELQ
In order to provide insights into the function of HROB in HR, we searched for genes that modify the cisplatin sensitivity phenotype of HROB-deficient cells. We undertook two parallel CRISPR screens in which wild-type (RPE1-hTERT Cas9 TP53-KO) and HROB-KO cells were infected with the TKOv3 sgRNA library (Hart et al. 2017) and then split into two populations that were either left untreated or treated with an equitoxic dose of cisplatin that killed ∼20% of cells, essentially as done previously (Zimmermann et al. 2018). We calculated a gene-level depletion score using drugZ (Colic et al. 2019) and then compared the scores as shown in Figure 5A. The results, in tabular format, are in Supplemental Table S2. The genes contributing to cellular cisplatin resistance irrespective of genotype (n = 37 at normZ value less than −7.5) were highly enriched in ICL repair proteins, as expected (Supplemental Fig. S7). However, we noted a few outlier genes that responded differently depending on the presence or absence of HROB. First, the HELQ gene had a depletion score (normZ) that was much greater in the HROB-KO versus parental cell line, suggesting that HROB and HELQ act in parallel pathways in the response to cisplatin (Fig. 5A). Conversely, a group of three genes had cisplatin sensitivity in WT cells that was dependent on the presence of a functional HROB gene (Fig. 5A). These genes were HROB, MCM8, and MCM9. The presence of HROB in this group can be explained by the fact that sgRNAs against HROB do not cause cisplatin sensitivity in cells already lacking HROB. Similarly, these results suggested that mutations in MCM8 and MCM9 no longer caused cisplatin sensitivity in HROB-KO cells, hinting that HROB, MCM8, and MCM9 act together in a pathway.
Figure 5.

HROB-KO is synergistic with HELQ-KO and epistatic with MCM8-KO. (A) CRISPR/Cas9 genome-wide screens for mutations that sensitize cells to cisplatin were performed in RPE1-hTERT Cas9 TP53-KO (WT) and RPE1-hTERT Cas9 TP53-KO HROB-KO #1 (HROB-KO) cells. Results from drugZ analysis of a screen in WT cells were plotted against results of a screen in HROB-KO cells. Lower normZ scores indicated stronger sensitivity to cisplatin in the screen. CRISPR targeting of HELQ (blue) induced much stronger sensitivity in HROB-KO cells than in WT cells. Targeting of MCM8, MCM9, and HROB (yellow) induced stronger sensitivity in WT than in HROB-KO cells. (B) Cisplatin sensitivity of RPE1-hTERT Cas9 TP53-KO cells with the indicated genotypes. (Left) Quantification of colony formation. Lines show a nonlinear least-squares fit to a normalized dose response model with a variable slope. (Right) EC50 values calculated from fitted curves. Lines indicate the mean (n = 4 biologically independent experiments). (C) Chromosomal aberrations were quantified in metaphase spreads from RPE1-hTERT Cas9 TP53-KO cells with the indicated genotypes untreated or following treatment with 8 µM cisplatin for 1 h. (Left) Representative images of a normal chromosome, a chromosome with breaks, and a radial chromosome. (Right) Quantification of chromosomal aberrations (radial or with breaks) per metaphase from three biologically independent experiments. At least 10 metaphases were scored per experiment. (DKO) HROB-KO HELQ-KO double knockout. (D) Normalized gene conversion in RPE1-hTERT Cas9 TP53-KO cells with the indicated genotypes. Data are from four biologically independent experiments. (E) Quantification of cells with more than five γH2AX (left) or RAD51/γH2AX (right) colocalizing foci after the indicated time of cisplatin treatment or recovery. Lines connect means. Data are from three biologically independent experiments. (F) Cisplatin sensitivity was determined as in B. (G) Normalized gene conversion was determined as in D. Data are from four to five biologically independent experiments. (*) P ≤ 0.05; (**) P ≤ 0.01; (***) P ≤ 0.001. P-values in B, D, F, and G are from paired two-tailed t-tests. P-values in C are from unpaired two-tailed t-tests. Error bars indicate standard deviation. See also Supplemental Figures S7–S10.
HELQ is a helicase of the HEL308 family that is present in archaea and eukaryotes (Adelman and Boulton 2010). Loss of HELQ in mouse or human cells causes germ cell attrition and ICL sensitivity (Adelman et al. 2013; Luebben et al. 2013; Takata et al. 2013). Eukaryotic HELQ is involved in the postsynaptic steps of recombination, as the loss of HELQ results in persistent RAD51 foci in C. elegans (Ward et al. 2010) and mammalian cells (Adelman et al. 2013) due to defective remodeling of RAD51-bound DNA. To explore the genetic interactions suggested by the screens, we generated HROB/HELQ double-knockout (DKO) cells (Supplemental Fig. S8A,B) and assessed their genotoxin sensitivity by determining the effective concentration causing a 50% decrease in viability (EC50). We observed that the HROB/HELQ DKO cells were much more sensitive to cisplatin than the single-mutant cell lines (Fig. 5B), which was accompanied by an increase in chromosome aberrations (Fig. 5C; Supplemental Fig. S8C,D). The synergistic decrease in EC50 values in the HROB/HELQ DKO cell line was also observed with MMC and with the PARP inhibitor olaparib (Supplemental Fig. S8E,F). Strikingly, the EC50 values for the DKO cells in response to cisplatin and MMC (702 nM ± 145 nM and 21 nM ± 6 nM, respectively) were equivalent to those of an isogenic BRCA1-KO cell line (Zimmermann et al. 2018) tested under the same conditions (512 nM ± 334 nM and 22 nM ± 9 nM). While the sensitivity to PARP inhibition of the DKO cells was not as severe as that of the mutated BRCA1 cells (Supplemental Fig. 8G–I), these results nevertheless suggested that combined deficiency in HELQ and HROB leads to a severe defect in HR. To test this idea directly, we measured gene conversion, an HR reaction (Verma and Greenberg 2016), using the traffic light reporter assay (Kuhar et al. 2014) and similarly observed a large reduction in gene conversion in the DKO cell line (Fig. 5D). The HROB/HELQ DKO cells also showed a reduction in sister chromatid exchanges after cisplatin treatment, further supporting the idea that recombination is impaired in the double-mutant cells (Supplemental Fig. S9). This severe HR deficiency was accompanied by a defect in the resolution of both DNA lesions and RAD51 foci following cisplatin treatment, suggesting that both HELQ and HROB make major contributions to the postsynaptic steps of HR in parallel pathways (Fig. 5E).
We also generated independent HROB/MCM8 DKO clones and subjected them to most of the same analyses as above. We observed that in all assays, including genotoxin sensitivity and gene conversion measurements, the loss of MCM8 did not modify the phenotypes of HROB-null cells (Fig. 5F,G; Supplemental Fig. S10). We conclude that HROB and MCM8 act together during HR, in parallel to HELQ.
HROB acts upstream of MCM8 to promote repair synthesis
To genetically order HROB vis-à-vis the MCM8–MCM9 helicase, we examined whether MCM8 and MCM9 are required for the accumulation of HROB at cisplatin-induced DNA damage sites or vice versa. First, we observed that in contrast to the depletion of CtIP or RAD51, both of which impair the accumulation of HROB at DNA damage sites, the depletion of MCM8 or MCM9 did not affect HROB localization following cisplatin treatment (Fig. 6A; Supplemental Fig. S11A–D). However, in the reverse set of experiments, we found that HROB depletion by siRNA abrogated the recruitment of GFP-MCM8 to MMC-induced foci and sites of laser microirradiation without impacting MCM8–MCM9 expression levels (Fig. 6B,C; Supplemental Fig. S11E–J). These results indicate that HROB acts upstream of MCM8 and, by inference, the MCM8–MCM9 complex. We also observed that endogenous HROB and MCM8 physically interact in coimmunoprecipitation studies and in vitro pull-down experiments (Fig. 6D; Supplemental Fig. S12A,B), which strongly suggests that HROB acts as the recruiter of MCM8–MCM9 to DNA damage sites.
Figure 6.

HROB recruits MCM8 to sites of DNA interstrand cross-links and promotes DNA synthesis in the absence of MCM2. (A,E,F) RPE1-hTERT TP53-KO HROB-KO #1 + GFP-HROB cells were transfected with the indicated siRNAs, treated or not with 8 µM cisplatin for 4 h, and recovered in drug-free medium for 20 h. (A) Quantification of RPE1-hTERT TP53-KO HROB-KO #1 + GFP-HROB cells with more than five GFP-HROB/γH2AX colocalizing foci following transfection with the indicated siRNAs and with or without cisplatin treatment. The red line indicates the mean (n = 4 biologically independent experiments). (B,C) HeLa Flp-In GFP-MCM8 cells were transfected with the indicated siRNAs and subjected to treatment with 60 nM MMC for 24 h. (B) Representative images of cells transfected with the indicated siRNAs, treated with MMC, and stained with the indicated antibodies. (C) Quantification of cells with more than five GFP-MCM8/γH2AX colocalizing foci (left) or more than five γH2AX foci (right). (D) HCT116 cells with endogenously tagged MCM8-Stag-3Flag (MCM8-SF) were untreated or treated with 1 µg/mL MMC for 15 h, and their lysates were subjected to anti-Flag immunoprecipitation and immunoblotting with Flag, HROB, and MCM9 antibodies. (E) Representative images of cells transfected with the indicated siRNAs, treated with cisplatin, and stained with the indicated antibodies. (F) Quantification of RPE1-hTERT TP53-KO HROB-KO #1 + GFP-HROB cells with more than five GFP-HROB/γH2AX colocalizing foci following siRNA transfection and cisplatin treatment as indicated. The red line indicates the mean (n = 5 biologically independent experiments). (G,H) HCT116 MCM2-mAID (WT) or HCT116 MCM2-mAID MCM9-KO cells with or without additional HROB-KO as indicated were initially arrested in G1 using lovastatin (0 h). When most of the cells were in mid-S phase (13 h after release), MCM2 was degraded by auxin addition, and EdU incorporation was analyzed 21 h after release. (G) Representative flow cytometry analysis of DNA synthesis. EdU signal was plotted against DNA content (PI). Dashed vertical lines indicate 2n and 4n DNA content. The black solid square indicates the gate for EdU-positive cells. (H) Quantification of EdU-positive cells gated as shown in E. The red line indicates the mean (n = 3 biologically independent experiments). (*) P ≤ 0.05; (**) P ≤ 0.01. P-values are from paired two-tailed t-tests. See also Supplemental Figures S11–S13.
Given the genetic relationship between HELQ and HROB, we next assessed whether HELQ depletion impacted HROB localization to damage sites. In those experiments, we also investigated the role of RFWD3, since it has been proposed that RFWD3 acts during the late phase of HR to promote MCM8 loading on chromatin by ubiquitylating RPA and/or RAD51 (Feeney et al. 2017; Inano et al. 2017). While RFWD3 did not impact HROB recruitment to damage sites, we observed that loss of HELQ greatly increased the proportion of cells with HROB foci (Fig. 6E,F; Supplemental Fig. S12C–E). It therefore seems that in the absence of HELQ, DNA lesions are channeled toward the HROB–MCM8–MCM9 pathway.
Finally, given the proposed role of MCM8–MCM9 in promoting recombination-associated DNA synthesis, we sought to assess whether HROB also promoted DNA synthesis in a system where MCM2 is conditionally inactivated by an auxin–degron approach (MCM2-mAID) (Natsume et al. 2017). This loss of MCM2 resulted in replication fork-associated DNA double-strand breaks, and, in this system, MCM8–MCM9 were found to promote an alternative mode of DNA synthesis that was RAD51-dependent (Natsume et al. 2017). We found that depletion of HROB abrogated residual DNA synthesis following the acute depletion of MCM2, phenocopying the loss of MCM9 (Fig. 6G,H; Supplemental Fig. S13). Furthermore, loss of HROB does not further impair DNA synthesis following MCM2 inactivation in MCM9-KO cells (Fig. 6G,H), suggesting that they act together to promote RAD51-dependent DNA synthesis.
Discussion
The postsynaptic steps following RAD51-mediated strand invasion that lead to the establishment of recombination-associated DNA synthesis remain the least defined steps of HR. This lack of understanding is due to multiple factors, such as the various recombination pathways, the paucity of cellular markers downstream from RAD51, and the sheer number of proteins genetically implicated in HR but whose molecular functions remain poorly defined. The issue of genetic redundancy among HR-coding genes also likely explains the difficulty of building a clear picture of the postsynaptic steps of HR in mammalian cells. In that context, the observation that the dual inactivation of the HROB–MCM8–MCM9 and HELQ pathways results in a severe HR defect suggests that recombination intermediates are channeled in postsynaptic pathways underpinned by these helicases (Fig. 7). The biochemical basis of the remarkable redundancy between HELQ and HROB–MCM8–MCM9 remains to be defined, but one possibility could be that both the MCM8–MCM9 and HELQ helicases promote bubble migration independently of each other. In support of this possibility, both helicases are predicted to translocate on ssDNA in the same 3′–5′ direction (Marini and Wood 2002). We lastly note that HROB/HELQ DKO cells are viable and retain residual HR activity, indicating that additional pathways supporting recombination-dependent DNA synthesis must exist. Our successful use of somatic CRISPR screens to probe the cisplatin response in HROB-KO cells suggest that this approach may be fruitful in helping define the postsynaptic steps of HR.
Figure 7.
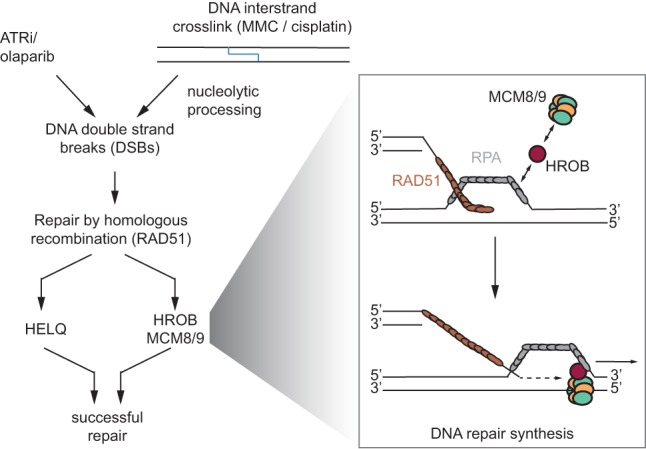
Model of HROB function in homologous recombination. HELQ and HROB–MCM8–MCM9 constitute two parallel pathways downstream from RAD51 filament formation. HROB interacts with the RPA complex and the MCM8–9 complex. HROB promotes MCM8–MCM9 localization to sites of homologous recombination. This may happen through the interaction of HROB with RPA on the displaced single strand of a displacement loop (D loop) after strand invasion. HROB may position MCM8–MCM9 correctly to facilitate bubble migration and thus DNA repair synthesis.
The most parsimonious model to explain our results is that HROB acts as a loader of MCM8–MCM9 (Fig. 7). This model is based on the observation that HROB directly interacts with MCM8–MCM9 and is necessary for its recruitment to DNA damage sites. It is also based on the observation that HROB has the ability to interact with ssDNA and RPA. However, while the MCM8 and MCM9 proteins are evolutionarily related to the replicative MCM2–MCM7 helicase (Liu et al. 2009), HROB bears no similarity to proteins that are part of the apparatus involved in loading MCM2–MCM7, such as ORC, CDT1, or CDC6 (Bleichert et al. 2017). Searching further afield, bacteriophage T4 uses a helicase loader to initiate recombination-dependent DNA replication (RDR), the gp59 protein (Kreuzer 2000). Remarkably, gp59 interacts with both the replicative helicase (gp41) and gp32, the ssDNA-binding protein analogous to RPA (Ishmael et al. 2001); this tripartite interaction is key for the initiation of RDR (Ma et al. 2004). The role of gp59 is to correctly position the helicase within the D loop to initiate bubble migration. The resonances between these systems are tantalizing, and it will be fascinating to determine whether HROB acts in this manner; i.e., by recruiting the MCM8–MCM9 complex to D loops to initiate bubble migration.
Further support for the role of HROB in HR is consistent with its phenotype in the mouse model, causing infertility due to severe depletion of germ cells in both sexes. The reported phenotypes of MCM8 knockout mice (Lutzmann et al. 2012) are strikingly similar to our observations in HROB-deficient mice and further suggest a common role for MCM8 and HROB during meiosis as well. Studies of meiotic recombination in yeast showed that strand invasion and extension of the invaded strand occur during zygotene and pachytene stages, respectively (Hunter and Kleckner 2001). A defect in DNA repair synthesis and therefore strand extension during meiotic recombination might prevent germ cells from progressing into the pachytene stage, a finding consistent with our observations.
Furthermore, it is noteworthy that the disruption of either Helq, Mcm8, Mcm9, or Hrob in mice leads to severe meiotic defects, whereas the impact of each single mutation on mitotic recombination is mild by comparison (Lutzmann et al. 2012; Adelman et al. 2013; Natsume et al. 2017). In all species examined, MCM8 does play an important role in meiosis, although the details of the phenotypic outcome of its loss vary for plants, Drosophila, and vertebrates. Indeed, in flies, the MCM8 homolog REC promotes crossover formation, whereas in Arabidopsis thaliana, the MCM8 and MCM9 homologs are instead involved in the repair of SPO11-induced breaks by a sister chromatid recombination pathway (Blanton et al. 2005; Crismani et al. 2013). Despite these differences, a potential role for MCM8–MCM9 in mediating recombination-associated DNA synthesis could underlie this range of phenotypes. In support of this possibility, analysis of meiotic recombination in rec mutant flies is consistent with a function of REC in DNA synthesis downstream from RAD51 but upstream of crossover formation stimulated by MEI-9 (XPF) (Blanton et al. 2005). A detailed analysis of HROB localization during gametogenesis will likely provide additional clues as to which recombination structures are the substrate of this pathway during meiosis. Nevertheless, given that loss-of-function mutations in MCM8 and MCM9 cause premature gonadal dysfunction in humans (Yatsenko and Rajkovic 2015), our work indicates that HROB is a candidate gene for this condition as well.
Materials and methods
Cell culture
RPE1-hTERT, HCT116, and 293T cells were purchased from American Type Culture Collection. HeLa T-Rex Flp-In cells orginated from the laboratory of Stephen Taylor and were a kind gift from Laurence Pelletier. RPE1-hTERT-derived cell lines, 293T-derived cell lines, and HeLa T-Rex Flp-In-derived cell lines were grown in Dulbecco's modified Eagle medium (DMEM) (Gibco/Thermo Fisher, 11965092) supplemented with 10% fetal bovine serum (FBS) (Wisent), 1× GlutaMAX, 1× nonessential amino acids (both Gibco/Thermo Fisher), and 100 U/mL penicillin and 100 µg/mL streptomycin (Pen/Strep) (Wisent). SUM149PT Cas9 cell origin and culture conditions were as described previously (Zimmermann et al. 2018). HCT116-derived cell lines were grown in McCoy's 5A (Gibco, 1660-082) + 10% FBS (Gibco) + 1% penicilllin–streptomycin mix (Nakalai Tesque, 09367-34) + 1× L-glutamine (final 2 mM; Wako Chemical, 073-05391). RPE1-hTERT Cas9 TP53-KO BRCA1-KO were cultured at 37°C and 3% O2. All other cell lines were grown at 37°C and atmospheric O2. All cell lines were routinely authenticated by STR and tested negative for mycoplasma.
Plasmids
DNA corresponding to sgRNAs was cloned into LentiGuide-Puro (Addgene, 52963), LentiCRISPRv2 (Addgene, 52961), or a modified form of LentiGuide-Puro in which Cas9 was replaced by NLS-tagged GFP or mCherry using AgeI and BamHI (designated as LentiGuide-NLS-GFP or -mCherry), as described (Noordermeer et al. 2018). LentiGuide-NLS-GFP containing MCM8 targeting sgRNA was a kind gift from the laboratory of Stephane Angers. The pcDNA5-FRT/TO-GFP plasmid was a kind gift from Anne-Claude Gingras. The coding sequence for HROB was obtained from the ORFeome collection (http://horfdb.dfci.harvard.edu.myaccess.library.utoronto.ca), archived in the Lunenfeld-Tanenbaum Research Institute's OpenFreezer (Olhovsky et al. 2011), and cloned into pcDNA5-FRT/TO-GFP using AscI and EcoRI. The resulting GFP-HROB coding sequence was subcloned into pCW57.1 (Addgene, 41393) using NheI and SalI. pcDNA3-Flag-MCM8 was a kind gift from Stephane Angers. The MCM8-coding sequence was subcloned into pcDNA5-FRT/TO-GFP using AscI and BamHI. The resulting GFP-MCM8-coding sequence was subcloned into pCW57.1 using NheI and AgeI. The traffic light reporter plasmids pCVL.TrafficLightReporter.Ef1a.Puro and pCVL.SFFV.d14GFP.Ef1a.HA.NLS.Sce(opt).T2A.TagBFP were obtained from Addgene (31482 and 32627, respectively).
Cell lines
Generation of RPE1-hTERT TP53-KO and RPE1-hTERT Cas9 TP53-KO is described elsewhere (Zimmermann et al. 2018). Cas9-expressing cell lines were maintained in the presence of 2 µg/mL blasticidin. RPE1-hTERT TP53-KO HROB-KO, RPE1-hTERT Cas9 TP53-KO HROB-KO, MCM8-KO, and HELQ-KO gene knockouts were generated by electroporation of LentiGuide or LentiCRISPRv2 vectors using a Lonza Amaxa II nucleofector. Sequences of the sgRNAs used in this study were as follows: HROB (CACAGCACCAGGTCTCAAGG), HELQ (ATGATTGGTGAAGGAAGCCG), and MCM8 (CATTGCTCTAAGAGGGACAG). Twenty-four hours after transfection, cells were selected with 15 µg/mL puromycin for 24–48 h followed by single-clone isolation. RPE1-hTERT Cas9 TP53-KO HELQ-KO HROB-KO double knockouts were generated starting from the HELQ-KO single knockout. RPE1-hTERT Cas9 TP53-KO HROB-KO MCM8-KO double knockouts were generated starting from the HROB-KO #1 single knockout. Clones were screened by TIDE analysis (Brinkman et al. 2014), and loss of protein expression was confirmed by immunoblotting and qPCR (for MCM8 knockouts). The cell line stably expressing GFP-HROB was generated by transducing RPE1-hTERT TP53-KO HROB-KO #1 with pCW57.1-GFP-HROB lentivirus and subsequent selection using 15 µg/mL puromycin for 48 h. Cells were maintained in the presence of 5 µg/mL puromycin and 1 µg/mL doxycyclin. The cell line stably expressing GFP-MCM8 was generated by transducing RPE1-hTERT Cas9 TP53-KO and RPE1-hTERT Cas9 TP53-KO HROB-KO #1 with pCW57.1-GFP-MCM8 lentivirus and subsequent selection using 15 µg/mL puromycin for 48 h. Cells were maintained in the presence of 5 µg/mL puromycin and 1 µg/mL doxycyclin. To generate HeLa cells stably expressing inducible GFP-MCM8, HeLa T-Rex Flp-In cells were transfected with pcDNA5-FRT/TO-GFP-MCM8 and a plasmid encoding Flp recombinase (pOG44; Invitrogen). Stable transfectants were selected with and maintained in the presence of 2 µg/mL blasticidin and 200 µg/mL hygromycin B. HCT116 MCM8-Stag-3Flag cells were generated using CRISPR-based C-terminal tagging of both endogenous MCM8 alleles (Yesbolatova et al. 2019). Generation of HCT116 MCM2-mAID(Neo) CMV-AtAFB2(Puro) MCM8-mCherry MCM9+/+ (HCT 116 MCM2-mAID) and HCT116 MCM2-mAID(Neo) CMV-AtAFB2(Puro) MCM8-mCherry MCM9−/− (HCT116 MCM2-mAID MCM9-KO) is described elsewhere (Natsume et al. 2017). In these cells, the HROB-KO mutation was generated by transfection of CRISPR guide RNA (targeting sequence: TGATGGACGGCTGTCAGCAC) and donor plasmids as described in Supplemental Figure S13 to target exon 3 of the HROB gene. The cells were selected in the presence of 5 µg/mL blasticidin, and clones were screened by PCR for integration of the donor sequence. Relevant primer sequences used for PCR reactions as described in Supplemental Figure S13 were #1972 (5′-GAGTACAGTGGCACCATCTCAG-3′), #1973 (5′-GAGTGGGAACTGTTAAGGCAGT-3′), and #1075 (5′-CTAAAGCGCATGCTCCAGAC-3′). PCR products were analyzed by MultiNA (Shimadzu). Two different DNA markers were used: 2-log DNA ladder (0.1–10.0 kb) (New England Biolabs) was used for the #1972/#1973 primer set, and pGEM DNA markers (Promega) were used for #1972/#1075. Finally, HROB-KO clones were confirmed by SDS-PAGE analysis and immunoblotting with antibodies to HROB.
Gene ontology term enrichment and network interaction analysis
Hits that were common between core ATRi-sensitizing mutations (Hustedt et al. 2019) and the SUM149PT olaparib screen (normZ ≤ −2.5) (Zimmermann et al. 2018) were analyzed with the “statistical overrepresentation test” (gene ontology database released December 1, 2018; annotation data set “GO biological process complete”) with Bonferonni correction for multiple testing. Mapping of the hits on the HumanMine protein interaction network was performed using the esyN interface (http://www.esyn.org). The network was then exported and visualized in Cytoscape v.3.4.0 (http://www.cytoscape.org).
Expression analysis
Assessment of HROB transcript isoform expression was performed via the Genotype–Tissue Expression (GTEx) Portal on January 31, 2019.
Domain and conservation analysis
Conservation was analyzed using the Consurf server (http://consurf.tau.ac.il/2016) using human or mouse HROB amino acid sequence as input without providing structure information, with automated sequence alignment generation and default settings. Conservation scores for each amino acid position were visualized according to color scale. Protein domains were predicted using InterPro release 70.0 (https://www.ebi.ac.uk/interpro) and HHPred with PDB_mmCIF70 (September 2018) as a reference database (https://toolkit.tuebingen.mpg.de/#/tools/hhpred).
Drugs
The following drugs were used in this study: AZD6738 (Selleckchem), VE-821 (Selleckchem), olaparib (Selleckchem), camptothecin (CPT) (Sigma), cisplatin (Sigma), mitomycin C (Sigma), hydroxyurea (Sigma), and temozolomide (Sigma).
Clonogenic survival assays
Cells were seeded in 10-cm dishes (500–3000 cells per plate, depending on genotype) into drug-free medium or medium containing the appropriate drug. Cells were treated with drugs either for 24 h followed by an additional 10–14 d of growth in drug-free medium (cisplatin, mitomycin C, hydroxyurea, or temozolomide) or continuously for 11–15 d (olaparib, camptothecin, AZD6738, and VE-821). For ionizing radiation and UV irradiation (254 nm), cells were seeded and irradiated 24 h later. For UV treatment, medium was replaced with PBS, and medium was refreshed after irradiation. Cultures were incubated at atmospheric oxygen except for experiments involving BRCA1-KO and respective control cell lines that were incubated at 3% O2. At the end of the experiment, medium was removed, and cells were rinsed with PBS and stained with 0.4% (w/v) crystal violet in 20% (v/v) methanol for 30 min. The stain was aspirated, and plates were rinsed twice in deionized water and air-dried. Colonies were counted manually. To calculate IC50 values, the data were fit to a three-parameter dose response model [log(inhibitor) vs. normalized response, variable slope] using the nonlinear regression function in Graphpad Prism.
RNAi
For siRNA-mediated knockdown, cells were transfected with 10 nmol of the respective siGENOME pools using Lipofectamine RNAiMax (Thermo Fisher). The medium was refreshed 6–8 h after transfection. For immunoblotting and qPCR, cells were harvested 96 h after siRNA transfection. For immunofluorescence, treatment with cisplatin or mitomycin C was started 48 h after siRNA transfection, and cells were subsequently processed for immunofluorescence staining 96 h after transfection. The following siGENOME siRNA pools were purchased from Dharmacon: nontargeting pool #2 (siCTRL; D-001206-14-05), siC17orf53/HROB (M-018061-00-0005), siCTIP (M-018061-00-0005), siFANCD2 (M-016376-02-0005), siMCM8 (M-017291-01-0005), siMCM9 (M-017615-01-0005), siRAD51 (M-003530-04-0005), siRAD54 (M-004592-01-0005), siHELQ (M-015379-01-0005), and siRFWD3 (M-017095-01-0005). Silencer Select Negative Control No.1 (Thermo) was used as siCTRL in Supplemental Figure S13.
Antibodies
The following primary antibodies were used in this study at the indicated dilutions for immunoblotting (IB), immunofluorescence (IF), or fluorescence-activated cell sorting (FACS): rabbit anti-HROB (1:5000–1:10,000 for IB; this study, see below), rabbit anti-GAPDH (1:20,000 for IB; Sigma, G9545), mouse anti-γH2AX (1:2000 or 1:2500 for IF on human cells and mouse tissues, respectively, and 1:2000 for FACS; Millipore, 05-636), rabbit antiphospho-histone H3 (S10) (1:1600 for FACS; Cell Signaling, 3377), rabbit anti-pCHK1 (S345) (1:1000 for IB; Cell Signaling, 2348), mouse anti-CHK1 (1:500 for IB; Santa Cruz Biotechnology, sc8408), rabbit anti-pRPA32 (S33) (1:20,000 for IB; Bethyl, A300-246A-3), mouse anti-RPA32 (1:500 for IB and IF; Abcam, ab2175), rabbit anti-RPA70 (1:500 for IB; Abcam, ab97338), rabbit anti-RAD51 (1:10,000 for IB and 1:16,000 or 1:100 for IF on human cells or mouse tissues, respectively; Bioacademia, 70-001), goat anti-GFP (1:10,000 for IB and 1:10,000 for IF; a kind gift from Laurence Pelletier), mouse anti-Flag-HRP (1:1000 for IB; Sigma, F3165), rabbit anti-FANCD2 (1:200 for IB; Santa Cruz Biotechnology, sc-20022), rabbit anti-CTIP (1:1000 for IB; Abcam, ab70163), rabbit anti-KAP1 (1:5000 for IB; Bethyl, A300-274A), rabbit anti-HELQ (1:1000 for IB; a kind gift from R. Wood), mouse anti-α-tubulin (1:2000 for IB; Calbiochem, CP06), rabbit anti-MCM8 (1:1000–1:3000 for IB), rabbit anti-MCM9 (1:2000 for IB), mouse anti-RAD54 (1:100 for IB; Santa Cruz Biotechnology, 374598), rabbit anti-RFWD3 (1:1000 for IB; Abcam, 138030), mouse anti-DMC1 (1:200 for IHC; Abcam, 11054), and rabbit anti-SCP3 (1:100 for IHC; Proteintech, 23024-1AP). For immunoblotting, primary antibody incubation was performed in 5% milk for all antibodies except anti-pCHK1(S345) and anti-pRPA32 (S33), where 5% bovine serum albumin (BSA) was used.
Secondary antibodies for immunoblotting used in this study were as follows: peroxidase-conjugated AffiniPure goat antirabbit IgG (Jackson ImmunoResearch, 111-035-144), peroxidase-conjugated AffiniPure bovine antigoat IgG (Jackson ImmunoResearch, 805-035-180), and peroxidase-conjugated sheep antimouse IgG (GE Healthcare, NA931V). All peroxidase-conjugated secondary antibodies were used at a dilution of 1:5000. Secondary antibodies for immunofluorescence and FACS analysis in this study were as follows: Alexa fluor 488-conjugated goat antimouse IgG (Life Technologies, A11029), Alexa fluor 647-conjugated goat antirabbit IgG (Life Technologies, A21244), Alexa fluor 488-conjugated goat antirabbit IgG (Life Technologies, A11034), Alexa fluor 555-conjugated goat antimouse IgG (Life Technologies, A21424), Alexa fluor 488-conjugated donkey antigoat IgG (Life Technologies, A11055), Alexa fluor 555-conjugated donkey anti-mouse IgG (A31570), and Alexa fluor 647-conjugated donkey antirabbit IgG (Life Technologies, A31573). All Alexa fluor-conjugated secondary antibodies were used at a dilution of 1:1000.
The rabbit anti-HROB antisera were produced by immunization of rabbits (Covance) with a bacterially expressed recombinant protein consisting of glutathione S-transferase fused to amino acids 293–527 of HROB.
High-content imaging
Cells were seeded in 96-well plates and cultured for at least 24 h. EdU (5-ethynyl-2-deoxyuridine; 20 µmol) (Life Technologies) was added 30 min prior to washing with PBS and fixation with 4% paraformaldehyde (PFA) in PBS for 10 min. Cells were rinsed with PBS and permeabilized using 0.3% Triton X-100/PBS for 30 min. Cells were washed with PBS and incubated in blocking buffer (0.2% fish skin gelatin, 0.5% BSA/PBS) for 30 min. Fresh blocking buffer containing mouse anti-γH2AX (1:5000) was added for 2 h. Cells were rinsed three times with PBS and blocking buffer with Alexa fluor 488-coupled goat antimouse antibody (1:1000), and 0.8 µg/mL DAPI (4,6-diamidino-2-phenylindole) (Sigma) was added for 1 h. After rinsing with PBS, immunocomplexes were fixed using 4% PFA/PBS for 5 min. Cells were rinsed with PBS and incubated with EdU staining buffer (150 mM Tris/HCl at pH 8.8, 1 mM CuSO4, 100 mM ascorbic acid, 10 µM Alexa fluor azide [Life Technologies]) for 30 min. After rinsing with PBS, images were acquired on an InCell Analyzer 6000 automated microscope (GE Life Sciences) with a 60× objective. Image analysis was performed using Columbus (PerkinElmer).
Anti-MCM8 immunoprecipitation
To detect endogenous MCM8 in RPE1-hTERT-derived cell lines, we performed MCM8 immunoprecipitation/immunoblotting using the TrueBlot system (Rockland). Cells (4 × 106) were lysed in 400 µL of MCM8 IP buffer (50 mM HEPES at pH 7.4, 100 mM KCl, 0.5% NP-40, 2 mM EDTA, 10 mM NaF, Complete protease inhibitor cocktail [Roche]) for 30 min on ice and cleared by centrifugation. Five microliters of rabbit anti-MCM8 antibody was added, and samples were incubated for 2 h at 4°C with rotation. Fifty microliters of pre-equilibrated TrueBlot antirabbit Ig IP beads (Rockland) was added to each sample prior to 1 h of rotation at 4°C. Beads were washed three times with MCM8 IP buffer and boiled in SDS sample buffer, and the supernatant was used for immunoblotting with rabbit anti-MCM8 and horseradish peroxidase-conjugated rabbit IgG TrueBlot (Rockland) as primary and secondary antibodies, respectively.
MCM8–MCM9-HROB coimmunoprecipitation
Thirty microliters of protein G Dynabeads (Veritas) was coupled with 3 µg of anti-Flag M2 antibody (Sigma, F1804) for 30 min at room temperature. Beads were then washed once with Flag IP buffer (20 mM HEPES at pH 7.4, 150 mM NaCl, 1 mM EDTA, 0.5% NP-40, 5% glycerol) and blocked with 1% BSA in PBS for 30 min at 4°C prior to another wash with Flag IP buffer. HCT116 and HCT116 MCM8-Stag-3xFlag cells (15-cm dish for each condition) were harvested by scraping into cold PBS. Cell pellets were washed once with cold PBS and resuspended in 1 mL of Flag IP buffer supplemented with Complete protease inhibitor cocktail (Roche), PhosSTOP phosphatase inhibitor cocktail (Sigma-Aldrich), 3 mM MgCl2, and 100 U of benzonase (Merck Millipore). Samples were incubated for 30 min on ice prior to centrifugation. Supernatant was added to uncoupled protein G Dynabeads for preclearing for 30 min at 4°C. An input sample was taken from the supernatant, and the residual supernatant was added to Flag antibody-coupled beads and incubated for 2 h with rotation at 4°C. The beads were then washed three times with 0.3 M NaCl in Flag IP buffer using a magnet. Bound protein complexes were eluted by incubation with 0.25 mg/mL Flag peptide (Sigma-Aldrich) in Flag IP buffer for 5 min on ice. Supernatant was collected using a magnet, and the elution was repeated another two times. Pooled eluates were subjected to protein precipitation using ice-cold acetone overnight at −20°C. Samples were centrifuged, and pellets were rinsed once with ice-cold methanol. Dried pellets were resuspended in NuPAGE LDS sample buffer (Thermo Fisher), heated for 10 min to 70°C, and analyzed by SDS-PAGE and immunoblotting with MCM8, MCM9, and HROB antibodies.
Cell cycle analysis (FACS)
Cells were either left untreated or treated with 4 µM cisplatin for 24 h. Cisplatin-treated cells were washed three times with PBS and allowed to recover in fresh medium for the indicated amounts of time. For each time point, 20 µM EdU was added to the medium 30 min prior to fixation. To harvest cells, the culture supernatant was collected, and cells were rinsed with PBS, trypsinized, and pooled with their respective culture supernatant and PBS wash to avoid losing weakly attached mitotic cells. Cells were spun down, and pellets were washed twice with cold PBS prior to fixation using 4% PFA/PBS for 10 min. Fixed cells were rinsed three times with PBS. For immunostaining, cells were permeabilized by dropwise addition of ice-cold MeOH to cell pellets while vortexing, followed by a 30-min incubation on ice. Cells were then rinsed twice in FACS buffer (PBS + 2 mM EDTA + 3% FBS) and incubated with primary antiphospho-histone H3 and anti-γH2AX diluted in FACS buffer for 1 h. Cells were then washed with FACS buffer and incubated with secondary antibodies (Alexa fluor 647-conjugated goat antirabbit and Alexa fluor 488-conjugated goat antimouse, both 1:1000) along with 0.5 µg/mL DAPI in FACS buffer for 30 min. Cells were then washed once with FACS buffer and once with PBS prior to a 30-min incubation with EdU staining buffer (150 mM Tris/HCl at pH 8.8, 1 mM CuSO4, 100 mM ascorbic acid, 10 µM Alexa fluor 555-conjugated azide [Life Technologies]). Finally, cells were washed with PBS and then analyzed on a Fortessa X-20s (BD Biosciences) flow cytometer. Flow cytometry data were analyzed using FlowJo software. Live cells and subsequently single cells were selected and gated into G1, S, G2/M, phospho-histone H3-positive (M), and γH2AX-positive cells as shown in Supplemental Figure S2, A–E.
Micronuclei, multinucleated cells, and mitotic aberration analysis
Cells were seeded on coverslips and either left untreated or treated with 4 µM cisplatin for 24 h. Cisplatin-treated cells were washed three times with PBS and then allowed to recover in fresh medium for the indicated times. To fix cells, coverslips were transferred to ice-cold methanol for 10 min, washed with PBS, and mounted using Prolong Gold with DAPI (Life Technologies). Images were acquired on a Zeiss LSM 780 laser scanning microscope equipped with a 63× objective. Micronuclei and normal nuclei were quantified by manual counting to determine the ratio of micronuclei to normal nuclei. Multinucleated cells were quantified by manual counting. For mitotic aberration analysis, Z-stack images (0.5-µm steps, 10 µm total) were acquired using a spinning-disk microscope (Olympus) equipped with a 100× objective. Aberrations of anaphase cells were quantified by manual counting on maximum projections that were generated using Fiji/ImageJ software (Schindelin et al. 2012).
Immunofluorescence
For γH2AX and RAD51 time-course experiments, cells were seeded on coverslips and, 24 h later, subjected to treatment with 4 µM cisplatin for 24 h or left untreated. Cisplatin was then removed, and cells incubated in medium without cisplatin. After the indicated amounts of time, cells were incubated with nuclear extraction buffer (NuEx; 20 mM HEPES at pH 7.4, 20 mM NaCl, 5 mM MgCl2, 0.5% NP-40, 1 mM DTT, Complete protease inhibitors [Roche]) for 10 min on ice, rinsed with ice-cold PBS, and subsequently fixed with 4% PFA for 10 min at room temperature. Cells were blocked in blocking buffer (0.2% fish skin gelatin, 0.5% BSA in PBS) for 30 min and incubated with primary antibodies diluted in blocking buffer for 2 h at room temperature. Cells were then washed three times with PBS for 5 min and stained with fluorescent secondary antibodies (Alexa fluor 488-conjugated goat antirabbit IgG and Alexa fluor 555-conjugated goat antimouse IgG, 1:1000 in blocking buffer) and 0.5–0.8 µg/mL DAPI for 1 h at room temperature. Cells were washed as above, mounted in ProLong Gold mounting medium (Life Technologies), and imaged used a Zeiss LSM780 laser scanning microscope. Cells with more than five colocalizing γ-H2AX and RAD51 foci were quantified by manual counting. All samples for GFP-MCM8 localization studies (see below) were counted blind. At least 100 cells per condition were analyzed in each experiment. For GFP-HROB localization, cells were seeded on coverslips and transfected with 10 nM siRNA using RNAiMax. Forty-eight hours after siRNA transfection, cells were treated with 8 µM cisplatin for 4 h with a 20-h recovery, which we determined was optimal for these experiments. Cells were stained using primary antibodies against GFP, γH2AX, and RAD51 diluted in blocking buffer as indicated above (see “Antibodies”). For secondary antibodies, Alexa fluor 488-conjugated donkey antigoat IgG, Alexa fluor 555-conjugated donkey antimouse IgG, and Alexa fluor 647-conjugated donkey antirabbit IgG (1:1000 in blocking buffer) were used. To detect GFP-MCM8 localization, HeLa Flp-In GFP and HeLa Flp-In GFP-MCM8 cells were seeded on coverslips in the presence of 5 µg/mL doxycyclin. After 24 h, cells were transfected with 10 nM siRNA using RNAiMax. Forty-eight hours after siRNA transfection, 60 nM mitomycin C was added, and cells were incubated for another 24 h prior to nuclear extraction, fixation, and immunostaining as described above for GFP-HROB localization.
Mouse Hrob (BC030867) deletion mutants
The BC030867 mutants were generated by direct delivery of Cas9 reagents to C57BL/NCrl (Charles River Laboratories, strain code 027) mouse zygotes essentially as described previously (Gertsenstein and Nutter 2018) by the Centre for Phenogenomics (Toronto, Ontario, Canada). Briefly, five single-guide RNAs (sgRNAs) with the desired spacer sequence (Supplemental Table S3) were synthesized by primer extension and in vitro transcription using EnGen sgRNA synthesis kit (New England Biolabs, E3322). An electroporation mix of 6 µM Cas9 protein (Integrated DNA Technologies, 1074182) and 22 ng/µL each sgRNA (Supplemental Table S3) was electroporated into C57BL/6NCrl zygotes with 12 30-V 1-msec pulses with 100-msec intervals. Electroporated zygotes were incubated in KSOMAA medium (Zenith Biotech, ZEKS-50) at 37°C with 6% CO2 until same-day transfer into CD-1 (Charles River Labs, strain code 022) surrogate host mothers. PCR primers flanking the sgRNA target sites (Supplemental Table S3) were used to amplify the region of interest from founder progeny or founders directly.
Fertility testing
At the age of 8.3 wk, males (seven mutant and one wild type as determined by initial PCR analysis) were set up for breeding with two control C57BL/6NCrl females each. Similarly, females (seven mutant and one wild type, by PCR) were set up for breeding with one control male each. Breeding was allowed for a period of 17 wk. One mutant female was found dead during the breeding study and therefore had to be excluded from this and further analyses. Similarly, one mutant male was excluded from the analysis retrospectively; in histological sections, it showed mosaicism, with about one-fifth of the testis appearing normal, while the remaining part was devoid of germ cells. All mutant animals that had not produced a litter after 17 wk were defined as infertile, and mutant alleles were analyzed by PCR and sequencing from genomic DNA (gDNA) derived from tissue samples. For mutant animals that produced litters, gDNA was isolated from tissue samples of N1 pups and analyzed by PCR and sequencing to determine the transmitted allele. Sequencing results are in Supplemental Table S1.
Histology and analysis of mouse tissues
Testes and ovaries were fixed in 10% buffered formalin, embedded in paraffin, and sectioned to examine using standard hematoxylin/eosin counterstain. Images were processed using Adobe Photoshop software. Specifically, background was whitened, brightness and contrast were adjusted, and the sharpening edges filter was applied. Slides were deparaffinized in xylene and rehydrated in a descending alcohol gradient. Antigen retrieval was performed using 10 mM citrate buffer (pH 6.0) for 10 min. Sections were allowed to cool down to room temperature and washed in PBS. To quench endogenous fluorescence, slides were incubated in a solution of 0.1% Sudan Black in 70% ethanol for 15 min, washed in PBS, and blocked with 10% horse serum in PBS for 1 h at room temperature. Primary antibodies were diluted as described above and incubated overnight at 4°C. The following day, slides were washed in PBS, then incubated for 1 h at room temperature with a host-specific secondary antibody conjugated with Alexa fluor dyes (Invitrogen Life Technologies and Bethyl), and counterstained with blue fluorescent 4′,6-diamidino-2-phenylindole (DAPI). Images (Z-stacks with 0.6-µm optical sections, 16 sections total) were acquired using a Zeiss LSM780 laser scanning microscope. Maximum intensity projections were generated using ImageJ software.
CRISPR screens
RPE1-hTERT Cas9 TP53-KO and RPE1-hTERT Cas9 TP53-KO HROB-KO #1 cells were transduced with the lentiviral TKOv3 library (Hart et al. 2017) at a low MOI (∼0.35), and puromycin-containing medium was added the next day to select for transductants. Selection was continued until 72 h after transduction, which was considered the initial time point, t0. To identify genes whose deletion caused cisplatin sensitization, a negative-selection screen was performed by subculturing cells at days 3 and 6 (t3 and t6), at which point the cultures were split into two populations. One was left untreated, while the second was treated with 1.5 µM cisplatin. Cells were subcultured, and medium with and without cisplatin was refreshed every 3 d. Cell pellets were frozen at day 18 for gDNA isolation. Screens were performed in technical duplicates, and library coverage of ≥375 cells per sgRNA was maintained at every step. gDNA from cell pellets was isolated using the QIAamp Blood Maxi kit (Qiagen), and genome-integrated sgRNA sequences were amplified by PCR using Q5 Mastermix (New England Biolabs Next UltraII, M5044S). i5 and i7 multiplexing barcodes were added in a second round of PCR, and final gel-purified products were sequenced on Illumina NextSeq500 systems to determine sgRNA representation in each sample. DrugZ (Colic et al. 2019) was used to identify gene knockouts that were depleted from cisplatin-treated t18 populations but not depleted from untreated cells.
Traffic light reporter assays
Cells were infected with a pCVL.TrafficLightReporter.Ef1a.Puro lentivirus at a low MOI (0.3–0.5) and selected with 15 µg/µL puromycin. Cells (7 × 105) were nucleofected with 5 µg of the pCVL.SFFV.d14GFP.Ef1a.HA.NLS.Sce(opt).T2A.TagBFP donor plasmid DNA in 100 µL of electroporation buffer (25 mM Na2HPO4 at pH 7.75, 2.5 mM KCl, 11 mM MgCl2), using program T23 on a Nucleofector 2b (Lonza). After 72 h, GFP and mCherry fluorescence was assessed in BFP-positive cells using a Fortessa X-20 (BD Biosciences) flow cytometer.
Chromosomal aberrations and sister chromatid exchanges
Cells (0.5 × 106) were seeded in 10-cm dishes, and BrdU (final concentration 10 µM) was added the next day. BrdU-containing medium was refreshed 24 h later, and cells were grown for another 24 h (48 h of BrdU incubation in total). For cisplatin treatment, 24 h after the first BrdU pulse, cells were exposed to 8 µM cisplatin in BrdU-containing medium for 1 h and then washed with PBS, and fresh medium with 10 µM BrdU was added. KaryoMAX colcemid (100 ng/mL; Gibco/Thermo Fisher) was added for 2 h after 48 h of BrdU exposure, and cells were harvested as follows: Growth medium was stored in a conical tube. Cells were washed with 1 mL of trypsin and incubated again for 5 min with 1 mL of trypsin. Growth medium and the 2 mL of trypsinized cells were centrifuged at 233g for 5 min at 4°C. Cells were then washed with PBS and resuspended in 75 mM KCl for 15 min at 37°C. Cells were centrifuged again, the supernatant was removed, and cells were fixed by drop-wise addition of 1 mL of fixative (ice-cold methanol:acetic acid, 3:1) while gently vortexing. An additional 9 mL of fixative was then added, and cells were fixed for at least 16 h at 4°C. Once fixed, metaphases were dropped on glass slides and air-dried overnight while protected from light. To visualize chromosomal aberrations, slides were dehydrated in a 70%, 95%, and 100% ethanol series (5 min each); air-dried; and mounted in DAPI-containing ProLong Gold mounting medium (Molecular Probes/Thermo Fisher). To visualize sister chromatid exchanges (SCE), slides were rehydrated in PBS for 5 min and stained with 2 µg/mL Hoechst 33342 (Thermo Fisher) in 2× SSC (final 300 mM NaCl, 30 mM sodium citrate at pH 7.0) for 15 min. Stained slides were placed in a plastic tray, covered with a thin layer of 2× SSC, and irradiated with 254-nm UV light (∼5400 J/m2). Slides were subsequently dehydrated in a 70%, 95%, and 100% ethanol series (5 min each); air-dried;and mounted in DAPI-containing ProLong Gold mounting medium (Molecular Probes/Thermo Fisher). Images were captured on a Zeiss LSM780 laser scanning microscope.
Laser microirradiation
RPE1-hTERT Cas9 TP53-KO and RPE1-hTERT Cas9 TP53-KO HROB-KO #1 cells were transduced with pCW-57.1-GFP-MCM8 and selected with 20 µg/mL puromycin for at least 2 d. Cells seeded on glass coverslips were presensitized with 1 µg/mL Hoechst 33342 for 15 min at 37°C. DNA damage was introduced with a 355-nm laser (40 mW; Coherent) focused through a plan-apochromat 40× oil objective to yield a spot size of 0.5–1 mm using a Zeiss LSM780 confocal microscope and the following laser settings: 100% power, one iteration, frame size 128 × 128, line step 7, pixel dwell 25.21 µsec. Four hours after irradiation, cells were pre-extracted using NuEx pre-extraction buffer (see “Immunofluorescence,” above) for 10 min on ice followed by fixation in 2% PFA/PBS. Antibody staining and blocking were performed in PBS + 0.1% Tween-20 and 5% BSA using GFP and γH2AX antibodies and imaged on an LSM780 confocal microscope.
Two-color competitive growth assays
Two-color competitive growth assays were performed as described previously (Hustedt et al. 2019). Cells were transduced with virus particles expressing either NLS-mCherry LacZ-sgRNA (control) or one of two NLS-GFP-sgRNAs targeting HROB (sgRNA1: CACAGCACCAGGTCTCAAGG, or sgRNA2: GAATTTAGCCAGAGCACCAT). Twenty-four hours after transduction virally transduced cells were selected using 15–20 µg/mL puromycin for 48 h. At this time, mCherry- and GFP-expressing cells were mixed 1:1 (2500 cells + 2500 cells) and plated in a 12-well format. Cells were imaged for GFP and mCherry signals 24 h after initial plating (t = 0) and then left untreated or exposed to 200 nM olaparib. During the course of the experiment, cells were subcultured when near confluency was reached and imaged on days 3, 6, 9, 12, and 15. An InCell Analyzer system (GE Healthcare Life Sciences) equipped with a 4× objective was used for imaging. Segmentation and counting of the number of GFP-positive and mCherry-positive cells were performed using an Acapella script (PerkinElmer).
Insect cell culture and protein expression
The Spodoptera frugiperda (Sf9) and Trichoplusia ni High Five (Thermo Fisher) insect cells were used to generate recombinant protein-coding baculovirus and to express recombinant proteins, respectively. Sf9 cells were cultured in Ex-Cell 420 serum-free medium (Sigma), and High Five cells were cultured in Sf900 II serum-free medium (Thermo Fisher) in shake flasks. Baculoviruses were generated by cotransfecting (Cellfectin II; Thermo Fisher) Sf9 cells with linearized Bac10:KO1629-derived viral DNA (Zhao et al. 2003) and a pAC8-derived transfer vector (Abdulrahman et al. 2009) (a kind gift from Nicolas Thomä) coding for N-terminally Flag-tagged codon-optimized mouse HROB (mHROB), human MCM8, or human MCM9. The resulting baculovirus underwent two rounds of amplification and was used to infect 2 × 109 High Five cells. The cells were harvested 48 h after infection. For MCM8/9 complex expression, High Five cells were coinfected with both the MCM8 and MCM9 baculoviruses.
Protein expression and purification
Recombinant human RPA was overexpressed and purified from Escherichia coli using the p11d-tRPA vector as described previously using Affi-Gel Blue resin (Bio-Rad), a prepacked hydroxyapatite column (Bio-Rad), and a Mono Q 5/50 GL anion exchange column (GE) (Henricksen et al. 1994). Recombinant Flag-mHROB was purified from High Five cells infected with baculovirus as described above. Cells were lysed by sonication in lysis buffer (50 mM Tris-Cl at pH 8.0, 150 mM NaCl, 1 mM TCEP, 5% glycerol, SigmaFAST protease inhibitor cocktail without EDTA) containing 250 U of benzonase. Lysates were precleared at 31,000g for 45 min at 4°C. Cleared lysates were incubated with 1-mL bed volume of anti-Flag M2 agarose (Sigma) for 1.5 h at 4°C. The Flag resin was collected and washed with 30 bed volumes of lysis buffer and 20 bed volumes of wash buffer (50 mM Tris-Cl at pH 8.0, 150 mM NaCl, 1 mM TCEP, 5% glycerol). Bound protein was eluted by incubating resin with wash buffer containing 100 µg/mL 3xFlag peptide for 15 min at 4°C (Glpbio). Eluate fractions containing mHROB were pooled and concentrated by ultrafiltration using 10 kDa MWCO Amicon columns (Millipore) and injected into a Superdex 200 Increase size exclusion chromatography column (GE) in wash buffer. Fractions containing mHROB were pooled and concentrated by ultrafiltration, frozen in liquid nitrogen, and stored at −80°C. Recombinant human MCM8/9 complex was purified from High Five cells coinfected with baculoviruses encoding N-terminally Flag-tagged MCM8 and MCM9. The Flag affinity purification step was essentially identical to that for Flag-mHROB except that the lysis buffer contained 200 mM NaCl. 3xFlag peptide eluate fractions containing the MCM8/9 complex were incubated overnight at 4°C with His6-tagged TEV protease to cleave off the Flag tag. TEV protease was removed by incubation with Ni-NTA agarose (Qiagen) after adding imidazole to a final concentration of 50 mM. The MCM8/9-containing flowthrough was diluted with wash buffer lacking NaCl to a final concentration of 75 mM NaCl. The flowthrough was applied to a HiTrap heparin column (GE) and eluted using a linear gradient from 75 to 1000 mM NaCl. Fractions containing MCM8/9 were concentrated by ultrafiltration and injected into a Superose 6 Increase size exclusion chromatography column (GE) containing wash buffer. Fractions containing MCM8/9 complex were concentrated and frozen in liquid nitrogen for storage at −80°C.
Immunoprecipitation of HROB with human RPA or MCM8/9 complex
Four-hundred-twenty nanograms of recombinant Flag-mHROB was incubated with either 850 ng of recombinant human RPA or ∼100 ng of recombinant human MCM8/9 complex in 200 µL of HROB IP buffer (50 mM Tris-Cl at pH 8.0, 200 mM NaCl, 5% glycerol, 0.1% Tween 20). The mixture was incubated for 30 min on ice. Ten microliters of anti-Flag M2 agarose resin was then added, and the mixture was further incubated for 30 min with gentle agitation at 4°C. The Flag resin was collected by centrifugation and washed three times with 500 µL of HROB IP buffer for 5 min each. Bound proteins were eluted by incubation with 20 µL of HROB IP buffer containing 100 µg/mL 3xFlag peptide for 30 min at 4°C with gentle agitation.
Immunoprecipitations in the presence of ssDNA and dsDNA were done identically with the addition of 100 nM single-stranded or double-stranded DNA (59 nt/bp) (see “HROB DNA-binding assays” for the full sequence) or 10 U of benzonase (Merck Millipore). The HROB IP buffer was supplemented with 5 mM MgCl2 to facilitate benzonase activity.
HROB DNA-binding assays
5′-32P-labeled ssDNA probes were prepared by T4 polynucleotide kinase (New England Biolabs) phosphorylation of an HPLC-purified 59-nti ssDNA (BioBasic; TACGTTAGTATGCGTTCTTCCTTCCAGAGGTTTTTTTTTTTTTTTTTTTTTTTTTTTTT) using [γ-32P]ATP (PerkinElmer). Labeled dsDNA probe was prepared by incubating the ssDNA probe with the complementary ssDNA (BioBasic; AAAAAAAAAAAAAAAAAAAAAAAAAAAAACCTCTGGAAGGAAGAACGCATACTAACGTA) for 10 min at 80°C and gradually cooling overnight to room temperature.
Recombinant Flag-mHROB was incubated with 1 nM 5′-32P-labeled ssDNA or dsDNA probes in EMSA buffer (150 mM NaCl, 50 mM Tris at pH 8.0, 1 mM TCEP, 5% glycerol, 0.1 mg/mL BSA) at room temperature for 30 min. The reactions were then resolved by native polyacrylamide gel electrophoresis using a 6% acrylamide gel (19:1 acrylamide:bis-acrylamide) in 0.5× TBE (44.5 mM Tris-borate at pH 8.3, 1 mM EDTA) running ice-cold at 135 V for ∼40 min. The gels were subsequently wrapped in clear plastic, directly exposed to a storage phosphor screen (GE) overnight, and visualized using a Typhoon FLA 9500 biomolecular imager (GE). Band intensities were quantified by ImageJ, and binding affinity curves were fitted using a one-site-specific binding model in GraphPad Prism.
EdU incorporation assay
HCT 116 MCM2-mAID, HCT 116 MCM2-mAID HROB-KO, HCT 116 MCM2-mAID MCM9-KO, and HCT 116 MCM2-mAID MCM9-KO HROB-KO were seeded into wells of a six-well plate (4.5 × 106 cells per well) and cultured for 1 d. Lovastatin was added to 20 µM for 24 h. Cells were washed once with lovastatin-free medium and then grown in medium containing 2 mM mevalonic acid (Sigma Aldrich) for 13 h. Indole-3-acetic acid (IAA) (Nacalai Tesque) was added to 0.5 mM, and cells were incubated another 7 h to allow for MCM2 degradation. EdU (10 mM) was added for 1 h prior to cell harvesting by trypsinization. Cells were washed once with 1% BSA/PBS and fixed with 4% PFA/PBS for 15 min at room temperature. Cells were then centrifuged, resuspended in 0.5% Triton X-100 and 1% BSA/PBS, and incubated for 10 min at room temperature. After centrifugation, cells were then resuspended in Click-iT reaction cocktail (Click-iT Plus Alexa fluor 647, Thermo Fisher) and incubated at room temperature for 30 min. After one wash with 1% BSA/PBS, cellular DNA was stained using 40 µg/mL propidium iodide. EdU and DNA signals were measured in 30,000 cells using an Accuri C6 flow cytometer (BD Biosciences). Data were analyzed using FCS Express 4 software (De Novo Software).
Competing interest statement
D.D. is a shareholder and advisor of Repare Therapeutics.
Supplementary Material
Acknowledgments
We thank Rachel Szilard for critical reading of the manuscript. We also thank Jason Moffat for TKO libraries and cell lines, as well as Nico Thomä for baculovirus expression plasmids. N.H. was supported by a Human Frontier Science Program Long-Term Fellowship, A.A.-Q. is a recipient of a long-term EMBO fellowship, D.S. is supported by a postdoctoral fellowship from the Canadian Institutes of Health Research (CIHR), and S.A. is a Banting postdoctoral fellow. D.D. is a Canada Research Chair (Tier I), and the work was supported by grants from the CIHR (FDN143343 to D.D., and FRN123518 and FRN156081 to A.J.) and Canadian Cancer Society grant 705644 (to D.D.) with additional support to D.D. from the Krembil Foundation.
Author contributions: N.H. designed, performed, and analyzed most experiments; made all figures; and helped write the manuscript. Y.S. performed MCM8 immunoprecipitation and EdU incorporation assays. M.Z. initiated the project and helped with immunofluorescence experiments. A.A.-Q. performed chromosomal aberration and sister chromatid exchange analysis. D.S. purified proteins and performed in vitro binding assays. S.A. performed laser microirradation experiments. A.M. analyzed CRISPR screen data. J.Y.Y., M.O., and Y.Z. helped with experiments supervised by N.H. M.T.K. supervised Y.S. A.J. helped design, performed, and analyzed mouse phenotypic studies and helped write the manuscript. D.D. designed and supervised experiments and wrote the manuscript.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.329508.119.
Freely available online through the Genes & Development Open Access option.
References
- Abdulrahman W, Uhring M, Kolb-Cheynel I, Garnier JM, Moras D, Rochel N, Busso D, Poterszman A. 2009. A set of baculovirus transfer vectors for screening of affinity tags and parallel expression strategies. Anal Biochem 385: 383–385. 10.1016/j.ab.2008.10.044 [DOI] [PubMed] [Google Scholar]
- Adelman CA, Boulton SJ. 2010. Metabolism of postsynaptic recombination intermediates. FEBS Lett 584: 3709–3716. 10.1016/j.febslet.2010.05.023 [DOI] [PubMed] [Google Scholar]
- Adelman CA, Lolo RL, Birkbak NJ, Murina O, Matsuzaki K, Horejsi Z, Parmar K, Borel V, Skehel JM, Stamp G, et al. 2013. HELQ promotes RAD51 paralogue-dependent repair to avert germ cell loss and tumorigenesis. Nature 502: 381–384. 10.1038/nature12565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanton HL, Radford SJ, McMahan S, Kearney HM, Ibrahim JG, Sekelsky J. 2005. REC, Drosophila MCM8, drives formation of meiotic crossovers. PLoS Genet 1: e40 10.1371/journal.pgen.0010040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleichert F, Botchan MR, Berger JM. 2017. Mechanisms for initiating cellular DNA replication. Science 355: eaah6317 10.1126/science.aah6317 [DOI] [PubMed] [Google Scholar]
- Bochkarev A, Bochkareva E. 2004. From RPA to BRCA2: lessons from single-stranded DNA binding by the OB-fold. Curr Opin Struct Biol 14: 36–42. 10.1016/j.sbi.2004.01.001 [DOI] [PubMed] [Google Scholar]
- Brinkman EK, Chen T, Amendola M, van Steensel B. 2014. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res 42: e168 10.1093/nar/gku936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colic M, Wang G, Zimmermann M, Mascall K, McLaughlin M, Bertolet L, Lenoir WF, Moffat J, Angers S, Durocher D, et al. 2019. Identifying chemogenetic interactions from CRISPR screens with drugZ. Genome Med 11: 52 10.1186/s13073-019-0665-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crismani W, Portemer V, Froger N, Chelysheva L, Horlow C, Vrielynck N, Mercier R. 2013. MCM8 is required for a pathway of meiotic double-strand break repair independent of DMC1 in Arabidopsis thaliana. PLoS Genet 9: e1003165 10.1371/journal.pgen.1003165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feeney L, Munoz IM, Lachaud C, Toth R, Appleton PL, Schindler D, Rouse J. 2017. RPA-mediated recruitment of the E3 ligase RFWD3 is vital for interstrand crosslink repair and human health. Mol Cell 66: 610–621.e4. 10.1016/j.molcel.2017.04.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Higuera I, Taniguchi T, Ganesan S, Meyn MS, Timmers C, Hejna J, Grompe M, D'Andrea AD. 2001. Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Mol Cell 7: 249–262. 10.1016/S1097-2765(01)00173-3 [DOI] [PubMed] [Google Scholar]
- Gertsenstein M, Nutter LMJ. 2018. Engineering point mutant and epitope-tagged alleles in mice using Cas9 RNA-guided nuclease. Curr Protoc Mouse Biol 8: 28–53. 10.1002/cpmo.40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart T, Tong AHY, Chan K, Van Leeuwen J, Seetharaman A, Aregger M, Chandrashekhar M, Hustedt N, Seth S, Noonan A, et al. 2017. Evaluation and design of genome-wide CRISPR/SpCas9 knockout screens. G3 7: 2719–2727. 10.1534/g3.117.041277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henricksen LA, Umbricht CB, Wold MS. 1994. Recombinant replication protein A: expression, complex formation, and functional characterization. J Biol Chem 269: 11121–11132. [PubMed] [Google Scholar]
- Hunter N, Kleckner N. 2001. The single-end invasion: an asymmetric intermediate at the double-strand break to double-holliday junction transition of meiotic recombination. Cell 106: 59–70. 10.1016/S0092-8674(01)00430-5 [DOI] [PubMed] [Google Scholar]
- Hustedt N, Álvarez-Quilón A, McEwan A, Yuan JY, Cho T, Koob L, Hart T, Durocher D. 2019. A consensus set of genetic vulnerabilities to ATR inhibition. Open Biol 10.1098/rsob.190156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inano S, Sato K, Katsuki Y, Kobayashi W, Tanaka H, Nakajima K, Nakada S, Miyoshi H, Knies K, Takaori-Kondo A, et al. 2017. RFWD3-mediated ubiquitination promotes timely removal of both RPA and RAD51 from DNA damage sites to facilitate homologous recombination. Mol Cell 66: 622–634.e8. 10.1016/j.molcel.2017.04.022 [DOI] [PubMed] [Google Scholar]
- Ishmael FT, Alley SC, Benkovic SJ. 2001. Identification and mapping of protein–protein interactions between gp32 and gp59 by cross-linking. J Biol Chem 276: 25236–25242. 10.1074/jbc.M100783200 [DOI] [PubMed] [Google Scholar]
- Juhasz S, Elbakry A, Mathes A, Lobrich M. 2018. ATRX promotes DNA repair synthesis and sister chromatid exchange during homologous recombination. Mol Cell 71: 11–24.e7. 10.1016/j.molcel.2018.05.014 [DOI] [PubMed] [Google Scholar]
- Kottemann MC, Smogorzewska A. 2013. Fanconi anaemia and the repair of Watson and Crick DNA crosslinks. Nature 493: 356–363. 10.1038/nature11863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreuzer KN. 2000. Recombination-dependent DNA replication in phage T4. Trends Biochem Sci 25: 165–173. 10.1016/S0968-0004(00)01559-0 [DOI] [PubMed] [Google Scholar]
- Kuhar R, Gwiazda KS, Humbert O, Mandt T, Pangallo J, Brault M, Khan I, Maizels N, Rawlings DJ, Scharenberg AM, et al. 2014. Novel fluorescent genome editing reporters for monitoring DNA repair pathway utilization at endonuclease-induced breaks. Nucleic Acids Res 42: e4 10.1093/nar/gkt872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KY, Im JS, Shibata E, Park J, Handa N, Kowalczykowski SC, Dutta A. 2015. MCM8-9 complex promotes resection of double-strand break ends by MRE11–RAD50–NBS1 complex. Nat Commun 6: 7744 10.1038/ncomms8744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Richards TA, Aves SJ. 2009. Ancient diversification of eukaryotic MCM DNA replication proteins. BMC Evol Biol 9: 60 10.1186/1471-2148-9-60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long DT, Raschle M, Joukov V, Walter JC. 2011. Mechanism of RAD51-dependent DNA interstrand cross-link repair. Science 333: 84–87. 10.1126/science.1204258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luebben SW, Kawabata T, Akre MK, Lee WL, Johnson CS, O'Sullivan MG, Shima N. 2013. Helq acts in parallel to Fancc to suppress replication-associated genome instability. Nucleic Acids Res 41: 10283–10297. 10.1093/nar/gkt676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutzmann M, Grey C, Traver S, Ganier O, Maya-Mendoza A, Ranisavljevic N, Bernex F, Nishiyama A, Montel N, Gavois E, et al. 2012. MCM8- and MCM9-deficient mice reveal gametogenesis defects and genome instability due to impaired homologous recombination. Mol Cell 47: 523–534. 10.1016/j.molcel.2012.05.048 [DOI] [PubMed] [Google Scholar]
- Ma Y, Wang T, Villemain JL, Giedroc DP, Morrical SW. 2004. Dual functions of single-stranded DNA-binding protein in helicase loading at the bacteriophage T4 DNA replication fork. J Biol Chem 279: 19035–19045. 10.1074/jbc.M311738200 [DOI] [PubMed] [Google Scholar]
- MacLeod G, Bozek DA, Rajakulendran N, Monteiro V, Ahmadi M, Steinhart Z, Kushida MM, Yu H, Coutinho FJ, Cavalli FMG, et al. 2019. Genome-wide CRISPR–Cas9 screens expose genetic vulnerabilities and mechanisms of temozolomide sensitivity in glioblastoma stem cells. Cell Rep 27: 971-986.e9. 10.1016/j.celrep.2019.03.047 [DOI] [PubMed] [Google Scholar]
- Mahadevaiah SK, Turner JM, Baudat F, Rogakou EP, de Boer P, Blanco-Rodriguez J, Jasin M, Keeney S, Bonner WM, Burgoyne PS. 2001. Recombinational DNA double-strand breaks in mice precede synapsis. Nat Genet 27: 271–276. 10.1038/85830 [DOI] [PubMed] [Google Scholar]
- Marini F, Wood RD. 2002. A human DNA helicase homologous to the DNA cross-link sensitivity protein Mus308. J Biol Chem 277: 8716–8723. 10.1074/jbc.M110271200 [DOI] [PubMed] [Google Scholar]
- McVey M, Khodaverdian VY, Meyer D, Cerqueira PG, Heyer WD. 2016. Eukaryotic DNA polymerases in homologous recombination. Annu Rev Genet 50: 393–421. 10.1146/annurev-genet-120215-035243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy ED. 1972. Hyperplastic and early neoplastic changes in the ovaries of mice after genic deletion of germ cells. J Natl Cancer Inst 48: 1283–1295. [PubMed] [Google Scholar]
- Natsume T, Nishimura K, Minocherhomji S, Bhowmick R, Hickson ID, Kanemaki MT. 2017. Acute inactivation of the replicative helicase in human cells triggers MCM8–9-dependent DNA synthesis. Genes Dev 31: 816–829. 10.1101/gad.297663.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura K, Ishiai M, Horikawa K, Fukagawa T, Takata M, Takisawa H, Kanemaki MT. 2012. Mcm8 and Mcm9 form a complex that functions in homologous recombination repair induced by DNA interstrand crosslinks. Mol Cell 47: 511–522. 10.1016/j.molcel.2012.05.047 [DOI] [PubMed] [Google Scholar]
- Noordermeer SM, Adam S, Setiaputra D, Barazas M, Pettitt SJ, Ling AK, Olivieri M, Alvarez-Quilon A, Moatti N, Zimmermann M, et al. 2018. The Shieldin complex mediates 53BP1-dependent DNA repair. Nature 560: 117–121. 10.1038/s41586-018-0340-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olhovsky M, Williton K, Dai AY, Pasculescu A, Lee JP, Goudreault M, Wells CD, Park JG, Gingras AC, Linding R, et al. 2011. OpenFreezer: a reagent information management software system. Nat Methods 8: 612–613. 10.1038/nmeth.1658 [DOI] [PubMed] [Google Scholar]
- Park J, Long DT, Lee KY, Abbas T, Shibata E, Negishi M, Luo Y, Schimenti JC, Gambus A, Walter JC, et al. 2013. The MCM8–MCM9 complex promotes RAD51 recruitment at DNA damage sites to facilitate homologous recombination. Mol Cell Biol 33: 1632–1644. 10.1128/MCB.01503-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- San Filippo J, Sung P, Klein H. 2008. Mechanism of eukaryotic homologous recombination. Annu Rev Biochem 77: 229–257. 10.1146/annurev.biochem.77.061306.125255 [DOI] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. 2012. Fiji: an open-source platform for biological-image analysis. Nat Methods 9: 676–682. 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebesta M, Burkovics P, Haracska L, Krejci L. 2011. Reconstitution of DNA repair synthesis in vitro and the role of polymerase and helicase activities. DNA Repair 10: 567–576. 10.1016/j.dnarep.2011.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sneeden JL, Grossi SM, Tappin I, Hurwitz J, Heyer WD. 2013. Reconstitution of recombination-associated DNA synthesis with human proteins. Nucleic Acids Res 41: 4913–4925. 10.1093/nar/gkt192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takata K, Reh S, Tomida J, Person MD, Wood RD. 2013. Human DNA helicase HELQ participates in DNA interstrand crosslink tolerance with ATR and RAD51 paralogs. Nat Commun 4: 2338 10.1038/ncomms3338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tkac J, Xu G, Adhikary H, Young JT, Gallo D, Escribano-Diaz C, Krietsch J, Orthwein A, Munro M, Sol W, et al. 2016. HELB is a feedback inhibitor of DNA end resection. Mol Cell 61: 405–418. 10.1016/j.molcel.2015.12.013 [DOI] [PubMed] [Google Scholar]
- Verma P, Greenberg RA. 2016. Noncanonical views of homology-directed DNA repair. Genes Dev 30: 1138–1154. 10.1101/gad.280545.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Wang G, Feng X, Shepherd P, Zhang J, Tang M, Chen Z, Srivastava M, McLaughlin ME, Navone NM, et al. 2019. Genome-wide CRISPR screens reveal synthetic lethality of RNASEH2 deficiency and ATR inhibition. Oncogene 38: 2451–2463. 10.1038/s41388-018-0606-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward JD, Muzzini DM, Petalcorin MI, Martinez-Perez E, Martin JS, Plevani P, Cassata G, Marini F, Boulton SJ. 2010. Overlapping mechanisms promote postsynaptic RAD-51 filament disassembly during meiotic double-strand break repair. Mol Cell 37: 259–272. 10.1016/j.molcel.2009.12.026 [DOI] [PubMed] [Google Scholar]
- West SC, Blanco MG, Chan YW, Matos J, Sarbajna S, Wyatt HD. 2015. Resolution of recombination intermediates: mechanisms and regulation. Cold Spring Harb Symp Quant Biol 80: 103–109. 10.1101/sqb.2015.80.027649 [DOI] [PubMed] [Google Scholar]
- Yatsenko SA, Rajkovic A. 2015. Reproductive aging and MCM8/9. Oncotarget 6: 15750–15751. 10.18632/oncotarget.4589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yesbolatova A, Natsume T, Hayashi KI, Kanemaki MT. 2019. Generation of conditional auxin-inducible degron (AID) cells and tight control of degron-fused proteins using the degradation inhibitor auxinole. Methods 164–165: 73–80 10.1016/j.ymeth.2019.04.010 [DOI] [PubMed] [Google Scholar]
- Zhao Y, Chapman DA, Jones IM. 2003. Improving baculovirus recombination. Nucleic Acids Res 31: E6 10.1093/nar/gng006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann M, Murina O, Reijns MAM, Agathanggelou A, Challis R, Tarnauskaite Z, Muir M, Fluteau A, Aregger M, McEwan A, et al. 2018. CRISPR screens identify genomic ribonucleotides as a source of PARP-trapping lesions. Nature 559: 285–289. 10.1038/s41586-018-0291-z [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.