

Abstract
The global obesity epidemic is driving the concomitant rise in nonalcoholic fatty liver disease (NAFLD). To identify new genes involved in central liver functions, we examined liver RNA‐sequence data from 259 patients who underwent morbidly obese bariatric surgery. Of these patients, 84 had normal liver histology, 40 simple steatosis, 43 nonalcoholic steatohepatitis, and the remaining 92 patients had varying degrees of NAFLD based on liver histology. We discovered oligodendrocyte maturation‐associated long intergenic noncoding RNA (OLMALINC), a long intervening noncoding RNA (lincRNA) in a human liver co‐expression network (n = 75 genes) that was strongly associated with statin use and serum triglycerides (TGs). OLMALINC liver expression was highly correlated with the expression of known cholesterol biosynthesis genes and stearoyl‐coenzyme A desaturase (SCD). SCD is the rate‐limiting enzyme in monounsaturated fatty acids and a key TG gene that is known to be up‐regulated in liver steatosis and NAFLD and resides adjacent to OLMALINC on the human chromosome 10q24.31. Next, we functionally demonstrated that OLMALINC regulates SCD as an enhancer‐RNA (eRNA), thus describing the first lincRNA that functions as an eRNA to regulate lipid metabolism. Specifically, we show that OLMALINC promotes liver expression of SCD in cis through regional chromosomal DNA–DNA looping interactions. Conclusion: The primate‐specific lincRNA OLMALINC is a novel epigenetic regulator of the key TG and NAFLD gene SCD.
Abbreviations
- aCRISPR
activating clustered regularly interspaced short palindromic repeats
- ASO
antistreptolysin O
- cDNA
complementary DNA
- ChIP‐seq
chromatin immunoprecipitation sequencing
- D1
aggregated meta‐liver trait
- dCas9
dead Cas9
- ENCODE
Encyclopedia of DNA Elements
- ERCC
External RNA Controls Consortium
- eRNA
enhancer RNA
- FBS
fetal bovine serum
- FDR
false discovery rate
- gRNA
guide RNA
- GRO‐seq
global run‐on sequencing
- GTEx
genotype‐tissue expression
- IDT
Integrated DNA Technologies, Inc.
- kb
kilobase
- KOBS
Kuopio obesity surgery
- LincRNA
long intervening noncoding RNA
- LncRNA
long noncoding RNA
- LXR
liver X receptor
- LXRE
liver X receptor responsive element
- MetS
metabolic syndrome
- MEM
minimum essential medium
- MUFA
monounsaturated fatty acid
- NAFLD
nonalcoholic fatty liver disease
- NASH
nonalcoholic steatohepatitis
- OLMALINC
oligodendrocyte maturation‐associated long intergenic noncoding RNA
- RNA‐seq
RNA sequencing
- RT‐qPCR
real‐time quantitative polymerase chain reaction
- SCD
stearoyl‐coenzyme A desaturase
- siRNA
small interfering RNA
- SREBP
sterol regulatory element binding protein
- sSVA
supervised surrogate variable analysis
- TG
triglyceride
- TPM
transcript per million
- TSS
transcription start site
- UCLA
University of California Los Angeles
- WGCNA
weighted gene co‐expression network analysis
- WNT8B
wingless‐type MMTV integration site family, member 8B
Metabolic syndrome (MetS), as defined by the clustering of phenotypic, biochemical, and clinical factors, has reached epidemic proportions in the United States.1 Nonalcoholic fatty liver disease (NAFLD), the liver manifestation of MetS, has also increased in parallel with other determinants of MetS.2 NAFLD ranges from simple steatosis to inflammatory nonalcoholic steatohepatitis (NASH), which can lead to fibrosis, cirrhosis, and hepatocellular carcinoma.3 The pathophysiology and interplay of MetS and NAFLD are complex, multifactorial, and include both genetic and environmental contributions.
Intrahepatic lipid accumulation, i.e., steatosis, is the hallmark of NAFLD.4, 5 Although the pathogenic pathways that cause progression from steatosis to steatohepatitis and fibrosis remain elusive, human and murine models have demonstrated that lipid dysregulation plays an important role in NAFLD pathogenesis.5, 6, 7, 8 Blood lipidomics data in patients with NAFLD6, 9, 10 and murine knockout models11 have also shown the importance of the monounsaturated fatty acid (MUFA) rate‐limiting enzyme stearoyl‐coenzyme A desaturase (SCD [also known as SCD‐1]), in MetS, steatosis, and NAFLD.6, 9, 10 Targeting SCD in murine NASH models has shown promising results12 and has recently led to human clinical trials with early phase data demonstrating reversal of hepatic steatosis using Aramchol, an SCD activity inhibitor.13
As advances in deep and high‐throughput sequencing have emerged, novel players have been identified in lipid biology, including the identification of a unique group of noncoding genes called long noncoding RNAs (lncRNAs).14 LncRNAs are >200 nucleotides long, show tissue and cell‐type specificity, and can differentially regulate signaling pathways.15 Understanding their biology has provided insight into new ways in which known key metabolic genes and proteins are regulated beyond previously described mechanisms; such novel ways include acting as scaffolds to complex proteins and enhancer RNAs (eRNAs) and modifying chromatin states.14, 16 This has included the role of lncRNAs in the regulation of cholesterol and lipid pathways.17 However, to the best of our knowledge, no eRNA lncRNAs have been discovered to regulate lipid metabolism.
In the present study, we identified the long intervening noncoding RNA (lincRNA) oligodendrocyte maturation‐associated long intergenic noncoding RNA (OLMALINC) in a statin‐ and triglyceride (TG)‐associated liver co‐expression network using liver RNA‐sequencing (RNA‐seq) from 259 Finnish patients who had undergone bariatric surgery. These patients were from the Kuopio Obesity Surgery (KOBS) cohort and had refined clinical phenotypic and liver histology data. We demonstrate that OLMALINC liver expression is highly correlated with the key lipid and TG pathway genes, including SCD, in the liver RNA‐seq data. We further functionally show that OLMALINC regulates this central TG metabolism gene, SCD, as a regional eRNA. Taken together, these novel data indicate that SCD is regulated by the adjacent lincRNA OLMALINC, which likely contributes to the central function of SCD in TG metabolism and liver steatosis.
Participants and Methods
Study Cohorts
The KOBS cohort was recruited at the University of Eastern Finland and Kuopio University Hospital, Kuopio, Finland.18 All participants provided informed consent, and the study was approved by the Ethics Committee of the Kuopio University Hospital, Kuopio, Finland. The liver RNA‐seq cohort comprises 259 Finnish KOBS participants who underwent bariatric surgery during which liver biopsies were obtained. Clinical measurements were performed as described.18 We also analyzed liver RNA‐seq data on 96 genotype‐tissue expression (GTEx) samples.19 We obtained the GTEx data used for the analyses in this manuscript from the GTEx Portal on March 23, 2017.
Histologic Assessment of the Liver Biopsy and Meta‐Liver Trait D1
NASH Clinical Research Network criteria were used to evaluate the liver histologic data.20 The following attributes were used: steatosis grade (0‐3), lobular inflammation (0‐2), ballooning (0‐2), and fibrosis stage (0‐4). The diagnosis for NASH was also determined by the pathologist following the standard guidelines.21, 22 To determine NAFLD status with liver RNA‐seq data, we performed a nonlinear principal component analysis using the homals R package23 on the four Clinical Research Network liver histologic phenotypes and used the first principal component as the aggregated meta‐liver trait (D1) for NAFLD (Fig. 1A). D1 is negatively correlated with the histologic parameters, i.e., a higher D1 represents a healthier liver (Fig. 1A).
Figure 1.
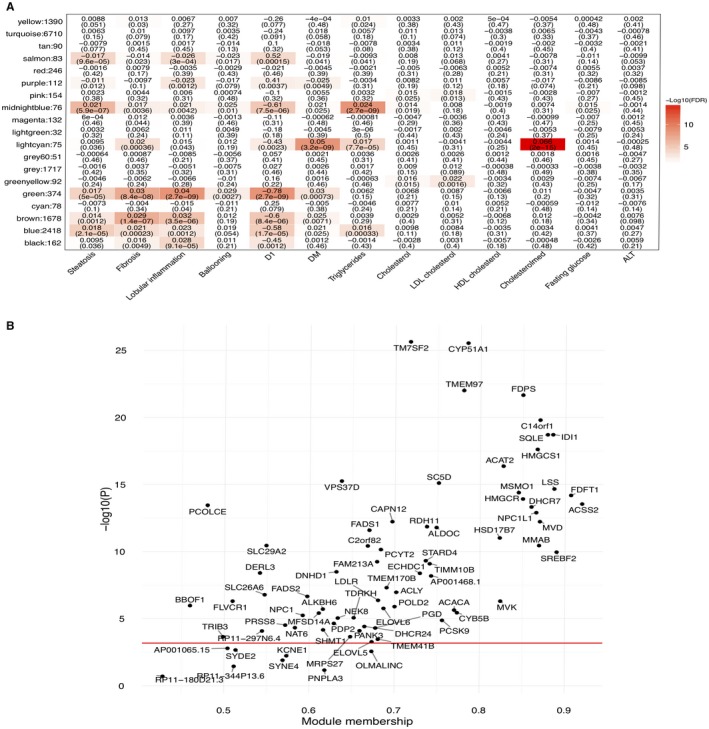
Liver weighted gene co‐expression network analyses (WGCNA) identify a statin‐associated network module (i.e. the light cyan module). (A) The association results between the liver WGCNA modules and statin use, metabolic traits, and histologic liver phenotypes in the Finnish KOBS cohort. D1 indicates the aggregated meta‐liver trait for NAFLD (see Participants and Methods). Numbers in the cells and parentheses indicate effect sizes and FDRs, respectively. (B) Genes in the light cyan module (n = 75) are strongly associated with statin medication and involved in cholesterol synthesis. The strength of association with statin medication is highly correlated with the module membership of the light cyan module. The red line indicates the threshold for the Bonferroni‐corrected P value of 0.05. Abbreviations: ALT, alanine aminotransferase; DM, diabetes mellitus; HDL, high‐density lipoprotein; LDL, low‐density lipoprotein; WGCNA, weighted gene co‐expression network analysis.
Liver RNA‐seq and Expression Quantification
RNA samples were isolated using the miRNeasy (Qiagen) kit, and sequencing libraries were prepared using the Ribo‐Zero Gold (Illumina) kit to remove ribosomal RNAs. External RNA Controls Consortium (ERCC) spike‐ins (Thermo Fisher Scientific) were added as controls. We quantified the transcript abundance as read counts and transcript per million (TPM) using Kallisto24 based on GENCODE version 25 liftover to hg19 gene annotation. Gene‐level quantification was estimated as the sums of read counts and TPM of all transcripts of a gene. To remove lowly expressed genes, a gene had to have >10 reads in 80% of samples, resulting in 15,670 genes in the final analysis.
Hidden Covariate Estimation for RNA‐seq
We performed a supervised surrogate variable analysis (sSVA)25 on TPMs and used the 92 ERCC spike‐in transcripts as invariable controls to estimate hidden confounders in the liver RNA‐seq data. The following covariates were included in the sSVA analysis: uniquely aligned reads %, mitochondrial reads %, 3′ bias, body mass index, sex, and age. Overall, 25 latent factors were estimated, and we included all sSVA factors and known covariates in downstream analyses. GTEx data do not contain ERCC spike‐ins so we did not carry out sSVA analysis but adjusted for the same covariates as in KOBS.
Statistical Analysis for Weighted Gene Co‐Expression Network Analysis, Gene Correlations, and Expression‐Trait Associations
Statistical analyses were performed in R. We transformed raw TPM to log2(TPM + 1) and then performed empirical Bayes‐moderated linear regression implemented in the weighted gene co‐expression network analysis (WGCNA) package26 (function empiricalBayesLM) to correct for covariates while retaining the variation due to the trait of interest. We calculated pairwise gene correlation using biweight correlation allowing a maximum of 5% outliers and subsequently built a signed network using the soft threshold power of 12. The eigen‐gene of each module was calculated and used for trait association tests. To test the module preservation in GTEx, we reprocessed the RNA‐seq raw reads using our pipeline, the same quality control, and genes expressed in both KOBS and GTEx. A module with a preservation summary Z statistic >10 was considered as strongly preserved.27 Pair‐wise gene expression correlation between OLMALINC and all other genes was calculated using biweight correlation and the adjusted TPMs. We used linear and logistic regression in all trait association tests, where the adjusted gene expression level and trait were treated as dependent and independent variables, respectively. Quantitative traits were adjusted for age and sex and were inverse normal transformed to avoid outlier effects.
Cell Culture
We maintained HepG2 (American Type Culture Collection, Manassas, VA) and Fa2N4 (XenoTech, Kansas City, Kansas) cells in a monolayer culture at 37°C with 5% CO2. The base medium was Eagle’s minimum essential medium (MEM) (Corning) for HepG2 and recommended media (XenoTech) containing 100 U/mL penicillin and 100 μg/mL streptomycin sulfate (GE Healthcare Sciences) for Fa2N4. We tested the cells for mycoplasma contamination using the SouthernBiotech Mycoplasma Detection kit.
Reagents and Transfections
For antistreptolysin O (ASO) treatment, 0.5 million cells were grown to ~70% confluency in six‐well plates in triplicates (in 10% fetal bovine serum [FBS] containing 1 g/L glucose with penicillin/ampicillin). Cells were treated with Opti‐MEM (Gibco), lipofectamine RNAiMax (13778100; Invitrogen) and ASO (Integrated DNA Technologies, Inc. [IDT]) at a final concentration of 50‐100 nM. The control ASO was designed to have similar modifications to the OLMALINC ASO. Cells were transfected at a final concentration to 30 pM for small interfering RNAs (siRNAs). ASO and siRNA sequences are provided in Supporting Tables S3 and S4. For plasmid transfections, we used Lipofectamine 3000 (Invitrogen) with 2 μg DNA. For the time‐point experiments, cells were incubated overnight in 0.25% bovine serum albumin (Sigma), followed by treatment in corresponding conditions outlined in the figures.28 We obtained lipoprotein‐deficient medium from Kalen Biomedical, LLC; simvastatin sodium salt from Calbiochem dissolved in dimethyl sulfoxide; and GW 3965 and mavelonic acid were kindly provided by Thomas Q. de Aguilar Vallim, Department of Biological Chemistry, University of California, Los Angeles, CA. Oleic acid was purchased from Sigma Aldrich. For cellular localization experiments, we used the PARIS kit (Invitrogen). Green fluorescent protein control and OLMALINC complementary DNA (cDNA) plasmids were obtained from GeneCopoeia.
Western Blots
Cells were washed and lysed in 1X Laemmli sodium dodecyl sulfate (SDS) sample buffer (Alfa Aesar). Lysates were separated by SDS–polyacrylamide gel electrophoresis (4%‐15% polyacrylamide) precast gels (Bio‐Rad Laboratories) overnight, transferred to a polyvinylidene difluoride membrane (Immobilon, Millipore Corp.), and blocked for 1 hour in 5% blocking solution (Bio‐Rad Laboratories). The membrane was incubated in 1:1,000 primary SCD antibody (Thermo Fisher Scientific) overnight at 4°C, followed by washes in 1:1,000 secondary mouse antibody for 45 minutes. The membrane was washed, after which immunoreactive proteins were detected using chemiluminescence (Bio‐Rad Laboratories). Beta‐actin (used for the loading control) and secondary mouse antibodies were kindly provided by Dr. Enrique Rozengurt’s laboratory (CURE: Digestive Diseases Research Institute, University of California Los Angeles [UCLA], Los Angeles, CA).
RNA Purification, cDNA Synthesis, and Real‐Time Quantitative Polymerase Chain Reaction
We harvested cells in TRIzol (Invitrogen) and extracted their RNA using Direct‐Zol (Zymo Research) according to the manufacturer’s protocol. We synthesized cDNA using the Maxima First Strand cDNA Synthesis kit (Thermo Scientific). Real‐time quantitative polymerase chain reaction (RT‐qPCR) was performed using SYBR Green reaction mix (Applied Biosystems) and the Studio 5 detection system (Applied Biosystems). We used 36B4 as an internal control to normalize the data. The primer list is provided in Supporting Table S2.
Conservation and Synteny of OLMALINC
To study the conservation of the OLMALINC locus, we used the National Center for Biotechnology Information HomoloGene and mouse and human Ensembl data. We evaluated the conservation of OLMALINC between human and mouse by aligning DNA segments sequentially between mouse and human using blast (GRCh37/hg19) with the blastn function word size 11, expected threshold 10, match score 2, and mismatch score –3. We also used the mouse Encyclopedia of DNA Elements (ENCODE) data (Mouse mm10) to identify RNA polymerase II and histone methylation markers.
Promoter Capture HI‐C
We performed promoter Capture Hi‐C in two biological replicates of 10 million HepG2 cells.29 The libraries were sequenced on an Illumina HiSeq 4000 to obtain ~114 million paired‐end reads. The reads were processed as described30 using HiCUP31 version 0.7.2 software and aligning to GRCh37/hg19.31 Significant interactions were identified using CHiCAGO software32 version 1.1.1.
Global Run‐On Sequencing
Global run‐on sequencing (GRO‐seq) libraries were prepared according to described protocols in HepG2 cells (10% FBS).33, 34 The Illumina HiSeq 2000 platform was used to sequence the libraries after size selection (180‐350 base pairs). After quality control, the data were aligned using GRCh37/hg19. GRO‐seq data are accessible under Gene Expression Omnibus accession GSE92375.
Activating CRISPR Dead Cas9 Stable Cell Lines
To generate the activating clustered regularly interspaced short palindromic repeats (aCRISPR) dead Cas9‐VP64 (aCRISPR‐dCas9) stable cell lines, we used the pHAGE EF10apha dCas9‐VP64 (#50918l; Addgene) plasmid. Cells were transduced with polybrene (1 μg/mL) for 2‐3 days, followed by selection with 4 μg/mL of puromycin for 7 days. Single‐clone isolation was obtained following serial dilutions. Clones expressing dCas9 were confirmed by RT‐qPCR of the dCas9 gene. We used two OLMALINC guide RNAs (gRNAs) targeting the promoter region of OLMALINC. 35 gRNAs were obtained from VectorBuilder (Shenandoah, TX).
CRISPR‐Cas9 of the OLMALINC Enhancer/Promoter Region
Using IDT Alt‐R CRISPR‐Cas9 genome editing tools, gRNAs were designed to flank the enhancer/promoter region of OLMALINC, which was identified using ENCODE, GRO‐seq, and promoter Capture Hi‐C. Four gRNAs were used to identify the most efficient gRNAs (Supporting Table S3). RNA protein complexes were prepared using Alt‐R S.p. Cas9 Nuclease V3 (IDT) with the OLMALINC gRNAs. HepG2 cells were transfected with Opti‐MEM (Thermo Fisher Scientific) and Lipofectamine RNAiMax (Invitrogen) for 48 hours. Transfection efficiency was evaluated using light and fluorescent microscopy (Texas Red‐X) using the BZ‐X710 fluorescent microscope. A FACAriaII cytometer was used to quantify the efficiency of transfection using FACDiva version 8.0.2. We extracted HepG2 genomic DNA using the PureLink Genomic DNA extraction kit (Thermo Fisher Scientific). PCR of the genomic DNA was conducted using primers flanking the gRNA cut sites to detect efficiency of all clones as well as to amplify regions within the OLMALINC wild type. These were confirmed using RT‐qPCR.
Statistical Methods of the Cellular Data
For the in vitro HepG2 experiments, numeric outcomes are summarized as means ± SD or SEM. All relative expression values were measured using ΔΔCt. Experimental groups were compared using the unpaired Student t test (for two groups). Analyses were performed using GraphPad Prism version 7.0c. Statistical significance was defined as P < 0.05. Graphs were made in GraphPad Prism and assembled in Inkspace.
Results
Identification of OLMALINC in the Statin‐ and TG‐Associated Liver Co‐Expression Network
To identify new genes involved in central liver functions, we performed a WGCNA on the liver transcriptomes from 259 participants (40% statin users) in the KOBS surgery cohort and tested the association of co‐expression modules with statin use, serum TGs, and other metabolic and liver histology phenotypes measured in this cohort. Thirteen of the 19 co‐expression modules were significantly associated (false discovery rate [FDR], <0.05) with at least one of the clinical or histologic traits (Fig. 1A), including the light cyan module (75 genes) that was significantly associated with statin use (FDR, 2.0 × 10–15) and serum TGs (FDR, 7.7 × 10–5), among other traits (Fig. 1A). We validated the module preservation in an independent human liver RNA‐seq cohort, GTEx, by investigating the GTEx subjects whose causes of death were not liver diseases (n = 96). Most trait‐associated liver modules, such as the statin‐ and TG‐associated light cyan module, were either preserved (Z score, >3) or highly preserved (Z score, >10) in the GTEx livers (Supporting Fig. S1), respectively, suggesting that gene coregulation related to main liver functions is robust and consistent across human cohorts. Notably, we observed that the 75 genes in the statin‐ and TG‐associated light cyan network module (Fig. 1A,B) comprise 19 known cholesterol pathway genes, 33 fatty acid and metabolic pathway genes, and several potentially novel statin response and TG genes, including the lincRNA OLMALINC. In line with its statin and TG associations, this light cyan module was enriched for the steroid biosynthesis pathway, fatty acid metabolism, and other metabolic pathways (FDR, <0.05) (Supporting Fig. S2), using the Kyoto Encyclopedia of Genes and Genomes pathway database.
Because the lincRNA OLMALINC identified in the light cyan module resides immediately downstream from the main TG metabolism gene SCD on human chromosome 10 and given that lincRNAs often regulate adjacent coding genes,18 we next individually tested the correlation of OLMALINC liver expression with SCD and detected a significant correlation (ß = 0.44; FDR, 4.57 × 10–11) (Supporting Table S1). We observed that SCD, in turn, resides in another WGCNA network, the midnight blue module, that is strongly associated with serum TGs (FDR, 2.7 × 10–9) and liver steatosis (FDR, 5.9 × 10–7) (Fig. 1A).
Next, we followed up the OLMALINC and SCD co‐expression findings and their mutual associations. We first tested if the liver expression of OLMALINC is individually associated with statin usage. When counting for multiple testing of the 75 genes in the light cyan module using Bonferroni (which is a conservative approach because these co‐expressed module genes are not entirely independent), OLMALINC was nominally associated with statin use (P = 0.0035; Fig. 1B). Thus, the statin users appear to have a higher OLMALINC liver expression than the non‐users in the KOBS cohort; this finding was fully supported by our in vitro statin response results in HepG2 cells (see below). Similarly, SCD liver expression was also higher in the statin users of the KOBS cohort (P = 0.0027), which was again in line with our in vitro HepG2 results (see below). We also detected a significant association between OLMALINC liver expression and fasting serum TGs in the KOBS cohort (ß = 0.27; P = 0.001), passing the Bonferroni correction for six traits (Supporting Table S2). In line with this observation, SCD liver expression was significantly associated with serum TGs (ß = 0.48; P = 0.13 × 10–7) in the KOBS cohort as well. Finally, although OLMALINC was not associated with steatosis or other liver histology traits (Supporting Table [Link], [Link]), SCD liver expression was associated with liver steatosis (ß = 0.35; P = 0.0054) but not with NASH (ß = 0.27; P = 0.107). Taken together, these novel data suggest the possibility that OLMALINC regulates its adjacent regional protein coding gene SCD, which is likely the driver in liver steatosis among the two, while both genes are associated with serum TG levels and respond to statin use. To further investigate this new hypothesis that OLMALINC regulates SCD, we performed functional genomics studies, as described below.
To assess OLMALINC gene expression in other human tissues, we analyzed the RNA‐seq data from the GTEx project and found that OLMALINC is ubiquitously lowly expressed as expected from a lincRNA. After the brain, the most abundant OLMALINC expression can be seen in the liver and other endocrine/hormone‐regulated organs (Supporting Fig. S3).
Overview of Our Functional Genomic Approaches to Study OLMALINC in Lipid Metabolism
We aimed to study the function of OLMALINC by using molecular genomics approaches (Supporting Fig. S4). Because the chromosomal location of OLMALINC is directly downstream of SCD (see below), we first demonstrated that OLMALINC is an enhancer of SCD transcription by forming a DNA–DNA looping interaction (Supporting Fig. S4A). This was confirmed by CRISPR‐Cas9 genetic deletion of this region (Supporting Fig. S4B) and endogenous transcriptional overexpression using the aCRISPR‐dCas9 gene editing system (Supporting Fig. S4D). To complement our CRISPR‐Cas9 gene editing, we confirmed that OLMALINC positively regulates SCD expression (Supporting Fig. S4C) by using an ASO that preferentially localizes to the nucleus. We further showed that OLMALINC expression increases with SCD siRNA (Supporting Fig. S4E) but decreases with oleic acid treatment, a by‐product of SCD enzyme activity.
OLMALINC Is Statin, Sterol, and Liver X Receptor Responsive
Using data from the ENCODE project and chromatin immunoprecipitation sequencing (ChIP‐seq) from HepG2 cells, we found two active transcription start sites (TSSs) characterized by an RNA polymerase II binding site, a 5′ capped analysis of gene expression (CAGE) peak, and active histone modification markers (characteristic of enhancer and promoter elements) in the OLMALINC–SCD region (Fig. 2A). GRO‐seq data in HepG2 cells, used to assess nascent RNA, not only confirmed two active TSSs in the enhancer and promoter of OLMALINC but also demonstrated bidirectional transcription, suggesting that OLMALINC could function as an enhancer to SCD (Fig. 2A). Using ENCODE project data, we identified sterol regulatory element binding protein (SREBP)1 and SREBP2 ChIP‐seq sites at the OLMALINC TSSs (Fig. 2B). We hypothesized that OLMALINC expression would be statin and sterol responsive, based on our correlative results from the liver RNA‐seq data in the KOBS cohort. Using RT‐qPCR, we showed that OLMALINC expression increases with statin and sterol treatments in a time‐dependent manner, demonstrating that it is both a sterol‐ and statin‐responsive gene (Fig. 3A,B). These data were consistent with the human liver RNA‐seq results in the KOBS cohort and demonstrated a positive correlation of OLMALINC with liver cholesterol gene expression and a membership in the statin module of our WGCNA analysis (Fig. 1; Supporting Table S1). We also showed that OLMALINC expression is liver X receptor (LXR) responsive because cells treated with the synthetic liver LXRα and LXRβ agonist GW3965 increase OLMALINC expression (Fig. 3C). We identified an LXR responsive element (LXRE‐DR4) T(G/A) A(C/A) C(T/C) XXXXT(G/A) A(C/A) C(T/C) in the OLMALINC promoter (Supporting Fig. S5). This is consistent with OLMALINC having a retinoid X receptor alpha ChIP‐seq binding site, which forms a heterodimer with LXRα and LXRβ to activate transcription (Fig. 2B), suggesting a direct role of LXR in regulating OLMALINC liver expression. We observed similar data in the immortalized human hepatocyte cell line Fa2N4 when treated with statins and GW3965, thereby corroborating our findings in the HepG2 cell line (Supporting Fig. S6).
Figure 2.
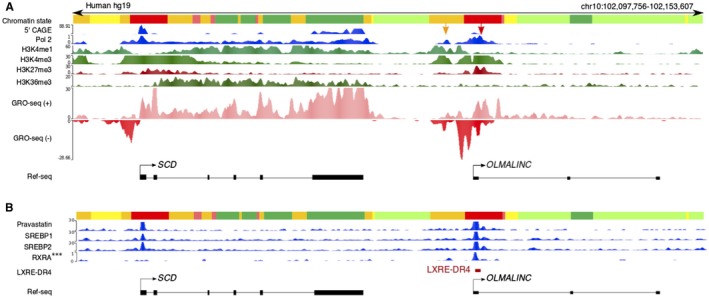
OLMALINC resides downstream of SCD and demonstrates similar regulatory regions. (A) The annotated OLMALINC promoter (red) and enhancer (orange) demonstrate histone methylation marks, 5′ CAGE, and polymerase II ChIP‐seq binding sites using ENCODE data. There are two TSSs: the orange arrow denotes the enhancer‐TSS, while the red arrow highlights the promoter‐TSS. Our GRO‐seq data in HepG2 cells show active transcription and nascent OLMALINC RNA expression bidirectionally. (B) OLMALINC has SREBP1/2, pravastatin (pravastatin‐treated HepG2 cells with SREBP1/2 peaks), and RXRA binding sites where an LXRE (LXRE‐DR4) is identified using sequence comparisons. Abbreviations: CAGE, capped analysis of gene expression; RXRA, retinoid X receptor alpha.
Figure 3.
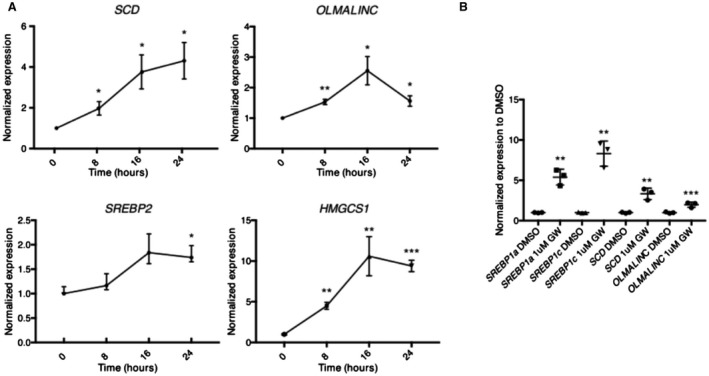
OLMALINC expression is responsive to sterols, statins, and LXR agonists in HepG2 cells. (A) OLMALINC and SCD increase expression by RT‐qPCR in a time‐dependent manner under sterol‐depleted conditions supplemented with statin treatment (5% lipoprotein‐deficient media with 5 µM simvastatin and 50 µM mavelonic acid) when compared to sterol‐rich conditions (10% FBS) supplemented with DMSO vehicle control, similarly to SREBP2 and its downstream gene HMGCS1. Each time point was normalized to its DMSO 10% FBS‐treated time point. (B) OLMALINC gene expression increases after 24‐hour treatment of GW3695 (an LXRα and LXRβ agonist) when compared to the DMSO vehicle control in 5% LPDS with 5 µM simvastatin and 50 µM mavelonic acid, as measured by RT‐qPCR. Values are mean ± SD (n = 3) for A and C or mean ± SEM for B (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001 (unpaired Student t test was used for two groups). Abbreviations: DMSO, dimethyl sulfoxide; HMGCS1, 3‐hydroxy‐3‐methylglutaryl‐coenzyme A synthase 1.
OLMALINC Function
To study OLMALINC function, we analyzed its cellular localization, which did not demonstrate a significant difference between the cytoplasmic and nuclear extracts for exons 1‐2 (RT‐qPCR of exons 2‐3 demonstrated a preferential cytoplasmic expression of the stable transcript) (Supporting Fig. S7B). All subsequent RT‐qPCR data that we present were conducted by measuring exons 1‐2 (shared between the identified isoforms). A ~50% knockdown of OLMALINC by ASO (of exon 2) resulted in a decrease in SCD expression (Fig. 4A; Supporting Fig. S4C). Conversely, when SCD was knocked down, we observed an increase in OLMALINC expression (Fig. 4B,C). These data suggest that OLMALINC expression is responsive to SCD expression, its protein level, or the MUFA by‐products. Given that SCD resides upstream of OLMALINC as well as previous observations that lincRNAs can regulate genes in cis, we hypothesized that SCD is regulated locally by OLMALINC in cis.
Figure 4.
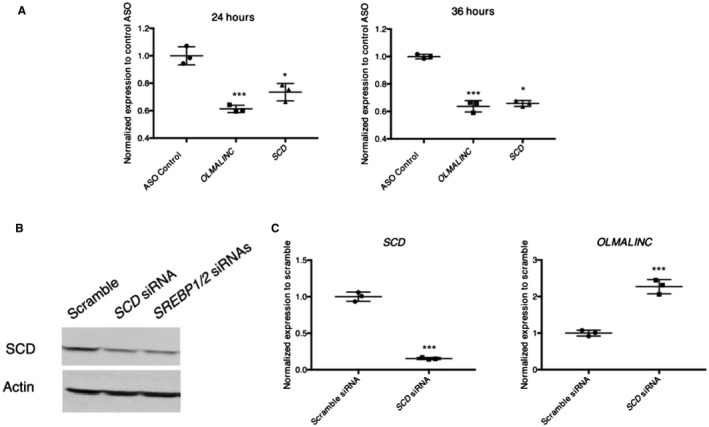
OLMALINC ASO introduced to HepG2 cells causes a decrease in expression of OLMALINC and target genes. (A) OLMALINC and target gene expression, measured by RT‐qPCR, decrease after 24‐hour and 36‐hour treatment with ASO targeting exon 2 of the OLMALINC gene. (B) Validation of SCD protein antibody (38 kDa) after treatment with scramble, SCD, and SREBP1 with SREBP2 siRNAs after 96 hours. (C) OLMALINC gene expression increases after 48‐hour treatment with an SCD siRNA compared to the scramble control. Values are mean ± SD (n = 3). *P < 0.05, ***P < 0.001 (unpaired Student t test was used for two groups).
The cis Effects of OLMALINC on SCD Expression
OLMALINC resides directly downstream of SCD, the microsomal enzyme that converts polyunsaturated fatty acids into MUFAs. OLMALINC liver expression is significantly correlated with SCD expression (ß = 0.44; FDR, 4.57E–11; Supporting Table S1) and serum TGs (Supporting Table [Link], [Link]), suggesting a role for OLMALINC in TG regulation. The chromosome 10 region of OLMALINC and SCD in humans has synteny with chromosome 19 of the mouse genome where wingless‐type MMTV integration site family, member 8B (WNT8B), SCD1, SCD2, SCD3, and SCD4 are localized in a ~330‐kilobase (kb) region (Supporting Fig. S8). However, no orthologues of OLMALINC were identified in the mouse. Consistent with these findings, no histone methylation markers or RNA polymerase II ChIP‐seq sites were found in the mouse genome between WNT8B and SCD1 to suggest a TSS (Supporting Fig. S9). Similar to other lincRNAs, OLMALINC only shows a high homology in primates.37
Since lincRNAs often exert their function by affecting adjacent genes, we hypothesized that OLMALINC may regulate SCD expression in cis by acting as an enhancer. To further investigate this, we performed promoter Capture Hi‐C in liver HepG2 cells (in 10% FBS) and identified a DNA–DNA looping interaction between the promoter of SCD and the annotated promoter/enhancer of OLMALINC (Fig. 5A; Supporting Fig. S4A). This interaction is cell‐type specific given that no interaction was identified between SCD and OLMALINC in human adipocytes despite the high SCD adipocyte expression.29 These promoter Capture Hi‐C interaction data suggest that OLMALINC acts by looping in cis to affect transcription of SCD. It is worth noting that because OLMALINC and SCD have a bidirectional promoter (Fig. 2B), it is possible that the looping interaction is strand specific; however, only the positive strand was interrogated when targeting the promoter for CRISPR‐Cas9 (see below).
Figure 5.
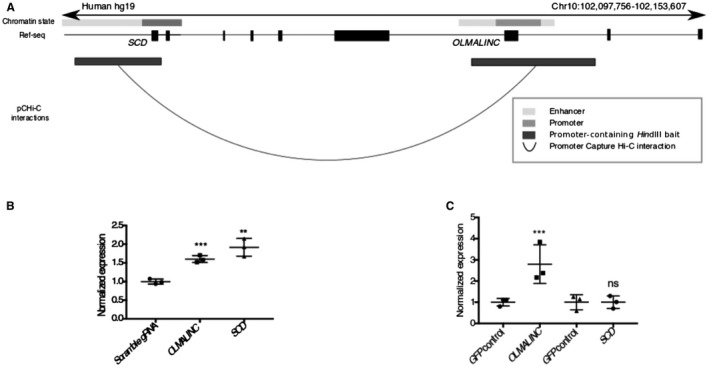
OLMALINC regulates SCD gene expression in cis by forming DNA–DNA looping interactions. (A) Promoter Capture Hi‐C data in HepG2 cells demonstrate DNA–DNA looping interactions between the OLMALINC enhancer/promoter and the SCD promoter/enhancer regions. (B) Endogenous OLMALINC overexpression using aCRISPR‐dCa9 gene editing increases expression of SCD. (C) Overexpression of the spliced OLMALINC stable transcript (exons 1‐3) for 48 hours does not affect SCD gene expression. Expression data are normalized to a GFP negative control. Values are mean ± SD (n = 3). **P < 0.01, ***P < 0.001 (unpaired Student t test was used for two groups). Abbreviations: GFP, green fluorescent protein; ns, not significant.
To further investigate the cis local regulatory effects, we used aCRISPR‐dCas9‐VP64 to overexpress OLMALINC endogenously using previously validated gRNAs in a constitutively expressing dCas9 cell line.35, 38 By RT‐qPCR, we demonstrated that a ~1.8‐fold increase in OLMALINC expression resulted in a 2‐fold increase in SCD expression (Fig. 5B; Supporting Fig. S4D).
To further tease out the local transcriptional versus posttranscriptional effects of OLMALINC regulation, we investigated the effects of its transcript on SCD expression. OLMALINC is annotated to have several transcripts (data not shown). Expression of a stable transcript with three exons was confirmed by Sanger sequencing of the PCR products (Supporting Fig. S7A) and alignment analysis of the liver RNA‐seq (data not shown). When the mature OLMALINC transcript is overexpressed using a cDNA construct (exons 1‐3), we observed no downstream effects on SCD gene expression (Fig. 5C). In conjunction with the endogenous overexpression data (aCRISPR‐dCas9), our results confirm that SCD regulation by OLMALINC occurs at the transcriptional level, likely through the cis effects.
To target the cis effects of OLMALINC on SCD, we used CRISPR‐Cas9 gene editing to delete the ~3.5‐kb region of OLMALINC, which encompasses the SREBP1/2 binding sites, TSSs, LXRE, and the Capture Hi‐C looping interactions (Fig. 6A‐C). Using a fluorescently labeled, trans‐activating CRISPR RNA (tracrRNA), we determined that our transfection efficiency of the HepG2 cells was 84% (Supporting Fig. S10), thus showing success in targeting the majority of the cells. The cells demonstrate ~50% decrease in OLMALINC expression, which causes a decrease in SCD expression (Fig. 6D; Supporting Fig. S4B). Whether the SCD expression effects are specific to disruption of DNA–DNA interactions between SCD and OLMALINC encompassing the promoter/enhancer region or are a by‐product of large DNA deletions remains to be tested. Wnt8B, the gene downstream of OLMALINC, is not expressed in human liver, as confirmed by the GTEx cohort and our RT‐qPCR data in HepG2 cells (data not shown), thus ruling out a Wnt8B‐specific effect. Taken together, our detailed functional genomic manipulation of OLMALINC expression (overexpression at the transcriptional level using aCRISPR‐dCas9, overexpression posttranscriptionally using the mature cDNA transcript, and knocking down OLMALINC RNA by CRISPR‐Cas9 and ASO) showed that OLMALINC regulates SCD expression in cis as an enhancer, likely through looping interactions.
Figure 6.
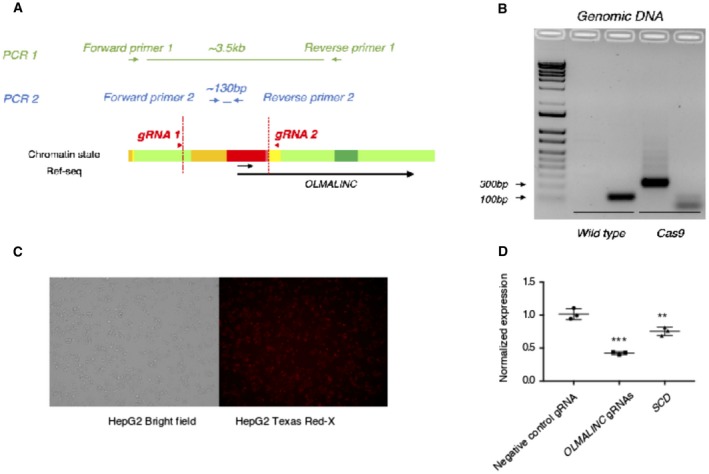
OLMALINC enhancer/promoter deletion using CRISPR‐Cas9 gene editing decreases SCD gene expression. (A) Schematic of primer designs for genomic PCR amplification of wild type versus CRISPR‐Cas9‐mediated OLMALINC promoter/enhancer deletion. Per ENCODE HepG2 chromatin state data, red highlights OLMALINC promoter while yellow highlights the enhancer. (B) Gel electrophoresis of PCR products from amplification of the wild type and CRISPR‐Cas9 OLMALINC enhancer/promoter deletions from the genomic DNA from HepG2 cells. (C) Evaluation of transfection efficiency of HepG2 with fluorescently labeled tracRNA with ATTO‐550 after 24 hours; left panel demonstrating bright field cells and right panel the corresponding labeled cells. (D) OLMALINC and SCD gene expression by RT‐qPCR after 48‐hour transfection with the Cas9 enzyme and OLMALINC gRNAs flanking the enhancer/promoter region. Values are mean ± SD (n = 3). **P < 0.01, ***P < 0.001 (unpaired Student t test was used for two groups). Abbreviations: bp, base pair; tracRNA, trans‐activating RNA.
OLMALINC Regulation
In conjunction with the ENCODE data, we demonstrated that OLMALINC is sterol, statin, and LXR responsive (Figs. 2 and 3). Given the cis effect of OLMALINC on SCD and the known regulation of SCD by the SREBP1 pathway,39 we sought to further understand OLMALINC regulation by these transcription factors. To accomplish this, we knocked down SREBP1 and SREBP2 using siRNAs to study those effects on OLMALINC expression. We observed that the knockdown of SREBP2 or SREBP1 alone does not affect OLMALINC expression or SREBP1/2‐dependent genes, likely from compensatory effects of the SREBPs (data not shown). However, when both SREBP1 and SREBP2 siRNAs are used in conjunction, their target genes, including SCD, are decreased while OLMALINC expression does not decrease (Fig. 7A). We therefore hypothesized that OLMALINC expression is regulated by SCD byproducts, which are MUFAs, possibly through a feedback mechanism. To test this hypothesis, we treated HepG2 cells with the MUFA oleic acid at different time points and demonstrated that OLMALINC expression decreases with oleic acid treatment (Fig. 7B), which is consistent with the observed increase in OLMALINC expression when knocking down SCD (Fig. 4B). We observed that OLMALINC gene expression decreases early (18 hours) before seeing an effect on SCD gene expression; SCD gene expression occurs later at 24 and 48 hours of treatment (Fig. 4B) when we also see a decrease in SREBP1a and SREBP1c. These data suggest that OLMALINC senses and mediates SCD gene expression locally before SREBP1 transcription factor proteins can regulate SCD expression. This is in line with our finding that the OLMALINC expression is positively correlated only with serum TGs and not with the other phenotypes in the KOBS cohort (Supporting Table [Link], [Link]).
Figure 7.
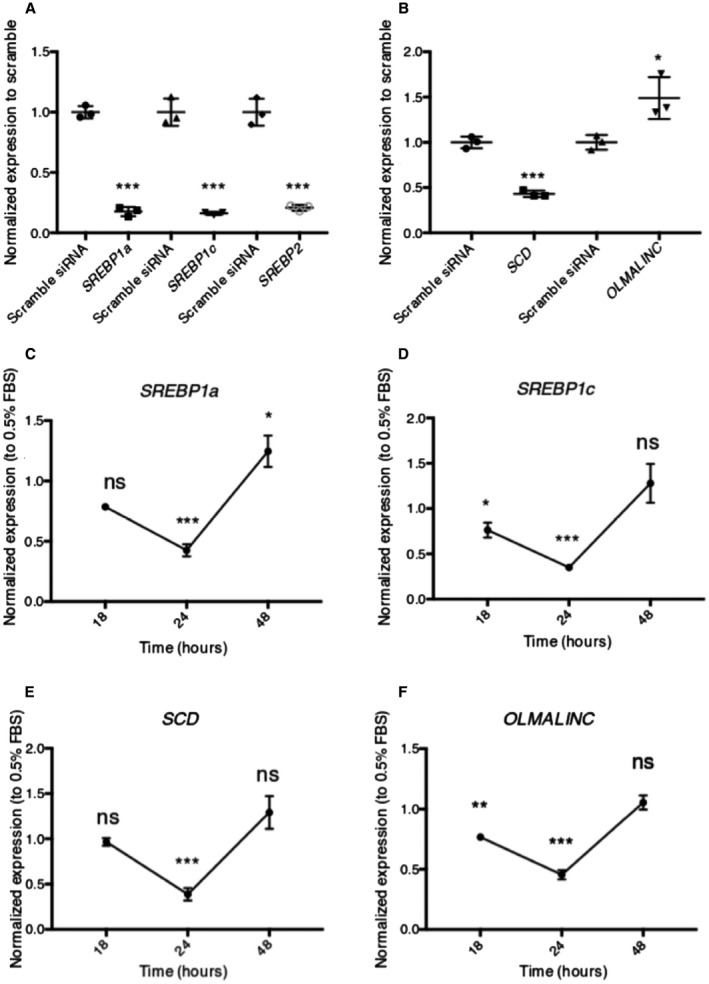
OLMALINC is regulated by MUFAs but not by SREBP1/2. (A) SREBP1a, SREBP1c, and SREBP2 gene expression after SREBP1 and SREBP2 siRNA cotransfection for 48 hours, relative to scramble siRNA control. (B) OLMALINC expression does not decrease after a 48‐hour cotransfection with SREBP1 and SREBP2 siRNAs; SCD decreases. (C‐E) SREBP1a, SREBP1c, and SCD expression decreases after lipid loading with MUFAs (200 µM oleic acid) 24‐hour treatment only, following 8 hours of starvation in 0.5% FBS. (F) OLMALINC decreases its expression after lipid loading with MUFAs (200 µM oleic acid) after 18‐hour and 24‐hour treatment, following 8 hours of starvation in 0.5% FBS. All expression time points are normalized to the corresponding gene expression in 0.5% FBS. Values are mean ± SD (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001 (unpaired Student t test was used for two groups).
Discussion
In the present study, we combined human liver transcriptomic and in vitro experimental data to identify and characterize the lincRNA OLMALINC in lipid metabolism. We first detected OLMALINC in tight correlation with known lipid genes in human liver RNA‐seq data and then demonstrated that our human correlative expression data translate to important effects of OLMALINC on a key TG gene, SCD. Our study also describes the first eRNA in lipid metabolism as our data showed that OLMALINC regulates the SCD gene in cis. Specifically, we observed that OLMALINC regulates SCD at the transcriptional level in cis by forming a looping interaction with the SCD enhancer/promoter region at important DNA elements where transcription factors and enhancers can interact and activate gene transcription. Furthermore, as SCD encodes an enzyme involved in fatty acid biosynthesis, including the synthesis of the MUFA oleic acid,8 it is noteworthy that in our context‐specific lipid‐loading experiments, OLMALINC expression is responsive to the SCD by‐product oleic acid early, independently of SREBP1, before seeing changes in SREBP1a/c, which occurs later. This suggests that OLMALINC may have evolved through an independent mechanism to sense and fine tune SCD gene expression early through feedback regulation given its proximity to the gene, perhaps to maintain the important MUFA homeostasis. The underlying molecular mechanism by which oleic acid directly or indirectly regulates OLMALINC gene expression warrants further investigation.
Cellular cholesterol and lipid homeostasis are tightly regulated to maintain essential lipid‐related processes in the human membrane.40 Important feedback mechanisms are in place to preserve homeostasis at the transcriptional, posttranscriptional, and protein level. This is partly through the SREBP transcription factors, which are the master regulators of cellular lipid and cholesterol processes, with SREBP1c preferentially activating the fatty acid synthesis pathway.40, 41, 42 Recent studies have demonstrated the role of lincRNAs in regulating and helping regulate SREBPs in their functions.14 For instance, metastasis‐associated lung adenocarcinoma transcript 1 (MALAT1), the nucleus‐specific lincRNA, inhibits degradation of SREBP1c protein by preventing its ubiquitination in the nucleus.43 Similarly, the lncRNA H19 stabilizes SREBP1c both at the transcript and protein levels depending if it exerts its function in the cytoplasm or nucleus, respectively.44 In this study, we demonstrate that OLMALINC acts as an enhancer for SCD and regulates SCD expression through sensing of its by‐products before SREBP1‐dependent effects.
Patients with NASH and NAFLD have previously been shown to exhibit altered cholesterol and TG metabolism.6, 9 Because the majority of the participants in the KOBS cohort have some form of NAFLD, it is possible that the statin‐associated co‐expression module we identified in the WGCNA analysis may also reflect the primary effect that NAFLD and NASH have on cholesterol metabolism. However, the correlative WGCNA data cannot alone separate these two possibilities. As SCD has been shown to be dysregulated in NAFLD and NASH,5, 6, 9 future studies are warranted to elucidate the role of OLMALINC in cholesterol metabolism. Because OLMALINC expression was not correlated with the liver phenotypes of steatosis and NASH in the KOBS cohort, it is unlikely to play a direct role in the pathophysiology of NAFLD and/or NASH. Future liver lipidomic studies could potentially provide further evidence of how this novel lincRNA affects or is affected by liver‐specific lipids. However, some of its specific lincRNA characteristics would have to be taken into account in future studies of OLMALINC. We demonstrate that OLMALINC regulates SCD as an enhancer RNA locally through chromosomal looping interactions (Fig. 5) and early by responding to oleic acid (Fig. 7C‐F); given these local and early changes, it may be challenging to measure lipid changes using the current ASO and CRISPR methods because transfection protocols require longer time courses for sufficient knockdown and knockout, respectively. As OLMALINC is a primate‐specific lincRNA35 (Supporting Figs. S7 and S8), the value of in vivo rodent models will also likely be somewhat limited.
Recent studies have demonstrated that lncRNAs affect nearby coding gene expression similarly to the effects of OLMALINC on SCD expression.45 Through detailed transcriptional analyses, it has also been elucidated that the effects on the nearby genes by lncRNAs are not necessarily mediated through the transcript but rather by transcriptional regulation (through enhancers and promoters) and/or splicing machinery.16 In addition to the important enhancer/promoter region through which OLMALINC affects SCD, we show that OLMALINC has a stable, spliced, and polyadenylated transcript. Given that enhancers generally produce unstable transcripts without a poly‐A tail or splicing,46 OLMALINC likely has a secondary function on other targets independently of its cis effects on SCD expression; this function remains to be elucidated.
Consistent with the importance of SCD in metabolic disorders, patients with NASH demonstrate increased SCD expression in the liver.9 Plasma oleate to stearate (18:1/18:0) and palmitoleate to palmitate (16:1/16:0) ratios, which are used as surrogates for systemic SCD activity, are also increased in patients with MetS and NASH, supporting an increase in SCD activity.10 These data are corroborated by recent clinical trials targeting SCD protein in patients with NASH (n = 58) and human immunodeficiency virus (who also develop hepatic steatosis; n = 25), which demonstrates reversal of hepatic steatosis with treatment.13, 47 In agreement with the human data, SCD –/– mouse models are protected from adiposity, have decreased de novo lipogenesis, and have increased fatty acid oxidation.11 It has also been shown that repletion of oleate through dietary supplementation in global and liver‐specific SCD knockout murine models prevents hepatic endoplasmic reticulum stress and inflammation.48 Given these findings, it would not be surprising for a lincRNA to have evolved to maintain MUFA homeostasis and provide another layer of early regional regulation to SCD gene expression epigenetically through chromosomal looping of this adjacent coding gene. Although far from therapeutic considerations, further understanding of OLMALINC function opens up unexplored avenues for gene modification and treatment considering its cell and tissue specificity.
The present study highlights a novel lincRNA, OLMALINC, that affects a key TG gene by affecting SCD expression in cis as a regional eRNA. OLMALINC joins a group of lipid lincRNAs that have been described and continue to emerge in lipid homeostasis and pathology.17 In addition to their role in regulating important coding genes, they could be one of many factors that explain the cross‐species differences in lipid metabolism. Further unraveling of their biology will provide insight into new cellular mechanisms and may pave the way for better understanding of complex cardiometabolic disorders in humans.
Supporting information
Acknowledgment
We thank the participants of the KOBS and GTEx cohorts. We also thank the sequencing core at UCLA for performing the liver RNA‐seq; the EMBL GeneCore Sequencing Facility (https://www.embl.de/services/core_facilities/genecore/index.html) for GRO‐seq; and the UCLA flow cytometry core for cell sorting. We thank the UCLA Integrated Molecular Technologies Core (CURE/P30 DK041301), Dr. Enrique Rozengurt and Jim Sinnet‐Smith for their help with western blotting, and Dr. Gregory Brent and Dr. Thomas Q. de Aguinar Vallim for their feedback. GTEx was supported by the Common Fund of the Office of the Director of the National Institutes of Health and by the National Cancer Institute, National Human Genome Research Institute, National Heart, Lung, and Blood Institute, National Institute on Drug Abuse, National Institute of Mental Health, and National Institute of Neurological Disorders and Stroke.
Supported by the National Institutes of Health (NIH) (grants HL‐095056, HL‐28481, and DKP3041301 to J.R.P. and P.P.; U01 DK105561 to P.P.; training grant DK007180 to J.N.B.); University of California Los Angeles Specialty Training and Advanced Research Program (to J.N.B.); Howard Hughes Medical Institute Gilliam Fellowship (to M.A.); NIH‐National Heart, Lung, and Blood Institute (grant 1F31HL142180 to K.M.G.); Academy of Finland (grants 287478 and 294073 to M.U.K.); Sigrid Jusélius Foundation (to M.U.K.), Finnish Foundation for Cardiovascular Research and European Research Council (Starting Grant 802825 to M.U.K.) and Academy of Finland (grant 316458 to D.K.).
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the article.
Potential conflict of interest: Nothing to report.
This article was originally published in International Journal of Obesity 2019. doi:10.1038/s41366-019-0424-y
References
Author names in bold designate shared co‐first authorship.
- 1. Vernon G, Baranova A, Younossi ZM. Systematic review: the epidemiology and natural history of non‐alcoholic fatty liver disease and non‐alcoholic steatohepatitis in adults. Aliment Pharmacol Ther 2011;34:274‐285. [DOI] [PubMed] [Google Scholar]
- 2. Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease‐meta‐analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016;64:73‐84. [DOI] [PubMed] [Google Scholar]
- 3. Brunt EM, Tiniakos DG. Histopathology of nonalcoholic fatty liver disease. World J Gastroenterol 2010;16:5286‐5296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Browning JD, Horton JD. Molecular mediators of hepatic steatosis and liver injury. J Clin Invest 2004;114:147‐152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Min HK, Kapoor A, Fuchs M, Mirshahi F, Zhou H, Maher J, et al. Increased hepatic synthesis and dysregulation of cholesterol metabolism is associated with the severity of nonalcoholic fatty liver disease. Cell Metab 2012;15:665‐674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chiappini F, Coilly A, Kadar H, Gual P, Tran A, Desterke C, et al. Metabolism dysregulation induces a specific lipid signature of nonalcoholic steatohepatitis in patients. Sci Rep 2017;7:46658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mannisto VT, Simonen M, Soininen P, Tiainen M, Kangas AJ, Kaminska D, et al. Lipoprotein subclass metabolism in nonalcoholic steatohepatitis. J Lipid Res 2014;55:2676‐2684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ntambi JM. Regulation of stearoyl‐CoA desaturase by polyunsaturated fatty acids and cholesterol. J Lipid Res 1999;40:1549‐1558. [PubMed] [Google Scholar]
- 9. Walle P, Takkunen M, Mannisto V, Vaittinen M, Lankinen M, Kärjä V, et al. Fatty acid metabolism is altered in non‐alcoholic steatohepatitis independent of obesity. Metabolism 2016;65:655‐666. [DOI] [PubMed] [Google Scholar]
- 10. Puri P, Wiest MM, Cheung O, Mirshahi F, Sargeant C, Min HK, et al. The plasma lipidomic signature of nonalcoholic steatohepatitis. Hepatology 2009;50:1827‐1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ntambi JM, Miyazaki M, Stoehr JP, Lan H, Kendziorski CM, Yandell BS, et al. Loss of stearoyl‐CoA desaturase‐1 function protects mice against adiposity. Proc Natl Acad Sci U S A 2002;99:11482‐11486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Iruarrizaga‐Lejarreta M, Varela‐Rey M, Fernandez‐Ramos D, Martínez‐Arranz I, Delgado TC, Simon J, et al. Role of Aramchol in steatohepatitis and fibrosis in mice. Hepatol Commun 2017;1:911‐927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Safadi R, Konikoff FM, Mahamid M, Zelber‐Sagi S, Halpern M, Gilat T, et al.; FLORA Group . The fatty acid‐bile acid conjugate Aramchol reduces liver fat content in patients with nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol 2014;12:2085‐2091.e2081. [DOI] [PubMed] [Google Scholar]
- 14. van Solingen C, Scacalossi KR, Moore KJ. Long noncoding RNAs in lipid metabolism. Curr Opin Lipidol 2018;29:224‐232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kopp F, Mendell JT. Functional classification and experimental dissection of long noncoding RNAs. Cell 2018;172:393‐407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kaikkonen MU, Adelman K. Emerging roles of non‐coding RNA transcription. Trends Biochem Sci 2018;43:654‐667. [DOI] [PubMed] [Google Scholar]
- 17. Sallam T, Jones MC, Gilliland T, Zhang L, Wu X, Eskin A, et al. Feedback modulation of cholesterol metabolism by the lipid‐responsive non‐coding RNA LeXis. Nature 2016;534:124‐128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mannisto VT, Simonen M, Hyysalo J, Soininen P, Kangas AJ, Kaminska D, et al. Ketone body production is differentially altered in steatosis and non‐alcoholic steatohepatitis in obese humans. Liver Int 2015;35:1853‐1861. [DOI] [PubMed] [Google Scholar]
- 19. GTEx Consortium . The Genotype‐Tissue Expression (GTEx) project. Nat Genet 2013;45:580‐585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, et al.; Nonalcoholic Steatohepatitis Clinical Research Network . Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005;41:1313‐1321. [DOI] [PubMed] [Google Scholar]
- 21. Brunt EM. Histopathology of non‐alcoholic fatty liver disease. Clin Liver Dis 2009;13:533‐544. [DOI] [PubMed] [Google Scholar]
- 22. Simonen M, Mannisto V, Leppanen J, Kaminska D, Kärjä V, Venesmaa S, et al. Desmosterol in human nonalcoholic steatohepatitis. Hepatology 2013;58:976‐982. [DOI] [PubMed] [Google Scholar]
- 23. de Leeuw J, Mair P. Gifi methods for optimal scaling in R: the package homals. J Stat Softw 2009;31:1–21. [Google Scholar]
- 24. Bray NL, Pimentel H, Melsted P, Pachter L. Near‐optimal probabilistic RNA‐seq quantification. Nat Biotechnol 2016;34:525‐527. Erratum in: Nat Biotechnol 2016;34:888. [DOI] [PubMed] [Google Scholar]
- 25. Leek JT. svaseq: removing batch effects and other unwanted noise from sequencing data. Nucleic Acids Res 2014;42:e161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 2008;9:559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Langfelder P, Luo R, Oldham MC, Horvath S. Is my network module preserved and reproducible? PLoS Comput Biol 2011;7:e1001057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tarling EJ, Edwards PA. ATP binding cassette transporter G1 (ABCG1) is an intracellular sterol transporter. Proc Natl Acad Sci U S A 2011;108:19719‐19724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mifsud B, Tavares‐Cadete F, Young AN, Sugar R, Schoenfelder S, Ferreira L, et al. Mapping long‐range promoter contacts in human cells with high‐resolution Capture Hi‐C. Nat Genet 2015;47:598‐606. [DOI] [PubMed] [Google Scholar]
- 30. Wingett S, Ewels P, Furlan‐Magaril M, Nagano T, Schoenfelder S, Fraser P, et al. HiCUP: pipeline for mapping and processing Hi‐C data. F1000Res 2015;4:1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cairns J, Freire‐Pritchett P, Wingett SW, Várnai C, Dimond A, Plagnol V, et al. CHiCAGO: robust detection of DNA looping interactions in Capture Hi‐C data. Genome Biol 2016;17:127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang D, Garcia‐Bassets I, Benner C, Li W, Su X, Zhou Y, et al. Reprogramming transcription by distinct classes of enhancers functionally defined by eRNA. Nature 2011;474:390‐394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kaikkonen MU, Niskanen H, Romanoski CE, Kansanen E, Kivelä AM, Laitalainen J, et al. Control of VEGF‐A transcriptional programs by pausing and genomic compartmentalization. Nucleic Acids Res 2014;42:12570‐12584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Engreitz JM, Haines JE, Perez EM, Munson G, Chen J, Kane M, et al. Local regulation of gene expression by lncRNA promoters, transcription and splicing. Nature 2016;539:452‐455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mills JD, Kavanagh T, Kim WS, Chen BJ, Waters PD, Halliday GM, et al. High expression of long intervening non‐coding RNA OLMALINC in the human cortical white matter is associated with regulation of oligodendrocyte maturation. Mol Brain 2015;8:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pan DZ, Garske KM, Alvarez M, Bhagat YV, Boocock J, Nikkola E, et al. Integration of human adipocyte chromosomal interactions with adipose gene expression prioritizes obesity‐related genes from GWAS. Nat Commun 2018;9:1512. Erratum in: Nat Commun 2018;9:3472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Konermann S, Brigham MD, Trevino AE, Joung J, Abudayyeh OO, Barcena C, et al. Genome‐scale transcriptional activation by an engineered CRISPR‐Cas9 complex. Nature 2015;517:583‐588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Liu SJ, Horlbeck MA, Cho SW, Birk HS, Malatesta M, He D, et al. CRISPRi‐based genome‐scale identification of functional long noncoding RNA loci in human cells. Science 2017;355:1‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shimano H, Horton JD, Shimomura I, Hammer RE, Brown MS, Goldstein JL. Isoform 1c of sterol regulatory element binding protein is less active than isoform 1a in livers of transgenic mice and in cultured cells. J Clin Invest 1997;99:846‐854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Shimano H, Shimomura I, Hammer RE, Herz J, Goldstein JL, Brown MS, et al. Elevated levels of SREBP‐2 and cholesterol synthesis in livers of mice homozygous for a targeted disruption of the SREBP‐1 gene. J Clin Invest 1997;100:2115‐2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Brown MS, Goldstein JL. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane‐bound transcription factor. Cell 1997;89:331‐340. [DOI] [PubMed] [Google Scholar]
- 42. Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest 2002;109:1125‐1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yan C, Chen J, Chen N. Long noncoding RNA MALAT1 promotes hepatic steatosis and insulin resistance by increasing nuclear SREBP‐1c protein stability. Sci Rep 2016;6:22640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Liu C, Yang Z, Wu J, Zhang L, Lee S, Shin DJ, et al. Long noncoding RNA H19 interacts with polypyrimidine tract‐binding protein 1 to reprogram hepatic lipid homeostasis. Hepatology 2018;67:1768‐1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ebisuya M, Yamamoto T, Nakajima M, Nishida E. Ripples from neighbouring transcription. Nat Cell Biol 2008;10:1106‐1113. [DOI] [PubMed] [Google Scholar]
- 46. Kim TK, Shiekhattar R. Architectural and functional commonalities between enhancers and promoters. Cell 2015;162:948‐959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ajmera VH, Cachay E, Ramers C, Vodkin I, Bassirian S, Singh S, et al. MRI assessment of treatment response in HIV‐associated NAFLD: a randomized trial of a stearoyl‐coenzyme‐A‐desaturase‐1 inhibitor inhibitor (ARRIVE Trial). Hepatology 2019. 10.1002/hep.30674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Liu X, Burhans MS, Flowers MT, Ntambi JM. Hepatic oleate regulates liver stress response partially through PGC‐1alpha during high‐carbohydrate feeding. J Hepatol 2016;65:103‐112. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials