

Abstract
Can ecological changes impact somatic genome development? Efforts to resolve this question could reveal a direct link between environmental changes and somatic variability, potentially illuminating our understanding of how variation can surface from a single genotype under stress. Here, we tackle this question by leveraging the biological properties of ciliates. When Paramecium tetraurelia reproduces sexually, its polyploid somatic genome regenerates from the germline genome through a developmental process that involves the removal of thousands of ORF-interrupting sequences known as internal eliminated sequences (IESs). We show that exposure to nonstandard culture temperatures impacts the efficiency of this process of programmed DNA elimination, prompting the emergence of hundreds of incompletely excised IESs in the newly developed somatic genome. These alternative DNA isoforms display a patterned genomic topography, impact gene expression, and might be inherited transgenerationally. On this basis, we conclude that environmentally induced developmental thermoplasticity contributes to genotypic diversification in Paramecium.
The DNA that is perpetuated by the germline is not always carried over to the soma in its entirety. In a broad range of eukaryotes, germline DNA sequences are reproducibly discarded at each round of sexual generation, and an abridged version of the genome is allocated to the soma (Jung et al. 2006; Wang and Davis 2014; Biederman et al. 2018; Smith et al. 2018). This process of DNA elimination is commonly regarded as highly efficient and is expected to be largely robust to environmental perturbations. However, the extent to which this expectation is well founded remains unknown. Any evidence of direct effects that ecological changes may have on somatic variability would have important implications for current understanding of evolutionary dynamics in systems for which transgenerational inheritance is at play.
In ciliates such as Paramecium tetraurelia, the process of germline-soma differentiation unfolds within a single cell and occurs at each event of sexual reproduction when P. tetraurelia undergoes meiosis and nuclear replacement (Sonneborn 1977; Prescott 1994). During sexual development, the maternal (preexisting) somatic macronucleus (MAC) is degraded, and new macronuclei are produced through the amplification (from 2n to ∼800n) and extensive rearrangement of the germline genome that is housed in mitotic copies of the zygotic micronucleus (Bétermier and Duharcourt 2015). Approximately 25% of the ∼98 Mb P. tetraurelia germline genome is eliminated during nuclear development through a highly reproducible process known as programmed DNA elimination (PDE) (Guérin et al. 2017). The characterized fraction of this germline-restricted DNA consists of some 45,000 mainly unique sequences known as internal eliminated sequences (IESs) (Arnaiz et al. 2012) and other DNA elements such as transposons and tandem repeats (Guérin et al. 2017). IESs are flanked on both sides by weak inverted terminal repeat consensus sequences with an invariable 5′-TA-3′ dinucleotide essential for their excision (Mayer and Forney 1999). IESs can be found in both noncoding and coding regions, often interrupting open reading frames (ORFs) (Arnaiz et al. 2012).
To ensure correct functioning of the somatic genome and hence production of viable sexual offspring, IESs must be precisely and reproducibly eliminated from the germline template (Arnaiz et al. 2012). Lethal developmental defects result when a domesticated PiggyBac transposase required for the excision of virtually all IESs, PiggyMac (Pgm), is silenced (Baudry et al. 2009; Dubois et al. 2012). Pgm operates within a molecular complex in conjunction with five additional Pgm-like proteins, all catalytically inactive yet also necessary for accurate IES excision (Bischerour et al. 2018). Nonetheless, two classes of developmentally specific small RNAs are also required to efficiently excise a subset of P. tetraurelia IESs: scnRNAs that mediate the cross talk between the parental and the developing nuclei (Lepère et al. 2009), and iesRNAs that relay a secondary excision signal to the developing nuclei (Sandoval et al. 2014). Besides facilitating IES excision, these sexually restricted small RNAs allow at least part of the genetic variability in the parental somatic nucleus to be inherited transgenerationally (Coyne et al. 2012; Allen and Nowacki 2017). The biogenesis of scnRNAs and iesRNAs requires, among other factors, the expression of dicer-like proteins: Dcl2/3 and Dcl5, respectively (Lepère et al. 2009; Sandoval et al. 2014; Hoehener et al. 2018). Unlike Pgm knockdown, independent DCL2, DCL3, and DCL5 silencing yields viable sexual offspring (Sandoval et al. 2014), demonstrating that inefficient IES excision can be tolerated to some degree.
In addition to variable chromosome fragmentation (Caron 1992), two types of erroneous DNA elimination under spontaneous conditions were described even before IESs were comprehensively cataloged: erroneous IES excision and “cryptic IES recognition” (Duret et al. 2008; Catania et al. 2013). The former consists largely of inefficient excision, in which IESs are excised from only a fraction of the ∼800 macronuclear copies, and to a lesser extent, of alternative or nested boundaries usage. The latter prompts the elimination of IES-like sections of somatic DNA, akin to an “off-target” effect. Therefore, inaccurate DNA elimination may take place during nuclear differentiation and prompt the production of alternatively rearranged versions of the genome. The rate of inefficient IES excision across different developmental environments has not been characterized. Equally unknown is the extent to which IES retention occurs in genes and whether it is sufficient to alter gene expression.
The somatic variability that PDE introduces to otherwise genetically identical Paramecium cells might explain at least part of the phenotypic differentiation observed in identical clones. Early studies in Paramecium have shown that exposure to different environmental conditions during nuclear differentiation, the so-called “sensitive period,” leads to phenotypic variations in genetically identical cells (Jollos 1921; Sonneborn 1977; Sonneborn and Schneller 1979). For example, the ability of homozygous clones of P. tetraurelia to discharge defensive organelles called trichocysts greatly depends on the temperature levels and food availability during the sensitive period (Sonneborn and Schneller 1979). However, it was not until recently that the mechanistic basis for developmentally regulated phenotypic differentiation in Paramecium was discovered. In P. tetraurelia and P. octaurelia, an IES-like somatic region (cryptic IES) containing the promoter and the transcription start site of the mtA gene (transmembrane protein) is variably spliced during development, resulting in complementary mating type determination (Singh et al. 2014). A similar mechanism is found in P. septaurelia, a closely related species, in which a cryptic IES is removed from the coding region of mtB, a putative transcription factor required for the expression of mtA, leading to mating type switch (Singh et al. 2014). Thus, PDE in Paramecium has been repeatedly co-opted for the regulation of gene expression through alternative DNA splicing (Orias et al. 2017). Although cryptic IES recognition has thus far received more attention as a regulated mechanism of PDE-dependent phenotypic diversification, other sources of somatic variability such as inefficient IES excision could contribute to the emergence of genetic novelties (Catania et al. 2013) or underlie adaptive phenotypic plasticity (Noto and Mochizuki 2017, 2018). DNA-splicing-controlled phenotypes may have evolved through the selection of heritable alternative DNA splicing variants, as would be expected under previously proposed models of epigenetic evolution (Coyne et al. 2012; Catania and Schmitz 2015; Allen and Nowacki 2017). The first step toward substantiating these hypothetical evolutionary scenarios is to quantify the rate of erroneous DNA elimination and establish the extent to which PDE is affected by environmental perturbations.
Here, we leverage the small somatic genome of P. tetraurelia, and the ease with which external conditions can be manipulated during development in this system, to investigate the temperature-performance response of IES elimination and its impact on genome constitution and gene expression.
Results
Temperature affects the rate of incomplete IES excision and cryptic IES recognition
After allowing isogenic lines of P. tetraurelia (strain d12) to undergo self-fertilization at three different temperatures, 18°C, 25°C, and 32°C, we inspected the independently rearranged somatic genomes of these lines (18°CF1, 25°CF1, and 32°CF1) and their progenitor (25°CF0) for incomplete IES excision. We also screened four additional somatic genomes that developed at 27°C and were previously sequenced for P. tetraurelia strain 51 (Arnaiz et al. 2012; Lhuillier-Akakpo et al. 2014; Swart et al. 2017). The ∼45,000 IES loci that we surveyed were identified in strain 51 through the knockdown of the PGM gene (Arnaiz et al. 2012) and will be hereinafter referred to as PGM-IESs. As a proxy for the level of IES retention, we used the IES retention score (IRS) (Denby Wilkes et al. 2016), a measure that ranges between zero and one, for fully excised or fully retained IESs, respectively. We arbitrarily define IES retention scores (IRSs) > 0.1 as nontrivial and label IES loci with IRS > 0.1 “somatic IESs.”
Compared to the control genomes rearranged at 25°C and 27°C, macronuclear genomes developed at 18°C and 32°C show an increase in incompletely excised IESs across a range of IRS classes (Fig. 1A). We detect an average of ∼400 somatic IESs in the line 25°CF0, its descendant 25°CF1, and the four additional strain 51 lines cultured at 27°C (Fig. 1A). In these six samples, the count of somatic IESs is comparable across the IRS thresholds applied (note that the IRS is unaffected by between-sample variation in read coverage). In contrast, the count of somatic IESs is higher in the macronuclear genomes that developed at 18°C and 32°C (nonstandard culture temperatures hereinafter also referred to as suboptimal temperatures), in which we detect up to about 800 somatic IESs. This roughly twofold increase is statistically significant when the suboptimal temperatures are grouped together (0.1 < IRS < 0.5; one-sided Wilcoxon rank-sum test, P < 0.05). The vast majority of the detected somatic IESs have scores in the range of 0.1–0.3 (Fig. 1A), potentially altering gene expression when occurring within genes (see below).
Figure 1.

The rate of incomplete IES excision and somatic DNA deletion is temperature dependent. (A) The percentage of incompletely excised IESs is larger at 18°C and 32°C in comparison with the control lines grown at 25°C–27°C for several IRS classes. The pie chart shows the relative proportions of somatic IESs (IRS > 0.1; black), the fraction of incompletely excised IESs with 0 < IRS < 0.1 (dark gray), and the fraction of completely excised IESs (light gray). IES counts (reported in percentages, log scale) for the control group (n = 6 genomes) are averages ± the standard error of the mean. (B) TA-bounded somatic deletions (Cryptic IESs) tend to be relatively more frequent (absolute numbers, log scale) when PDE occurs at suboptimal temperatures. (IESs) internal eliminated sequences; (IRS) IES retention score; (DS) deletion score.
Suboptimal temperatures also affect the rate of cryptic IES recognition (TA-bounded somatic deletions) (Fig. 1B) as measured by the deletion score (DS). Similar to the IRS, the DS is a proxy for the level of somatic DNA deletion, which ranges from zero for fully retained regions, to one for fully deleted regions. We detect up to an approximate twofold increase when comparing the number of partially excised cryptic IESs unique to 18°C and 32°C with those unique to the 25°C samples (Supplemental Fig. S1). Many TA-bounded somatic deletions lead to partial or even complete gene ablation and occasionally span multiple genes at once (Supplemental Table S1). Among the 18 somatic deletions consistently retrieved across all temperatures, we found the 195-bp DNA segment containing the promoter and transcription start site of mtA, a DNA-splicing-regulated gene (Singh et al. 2014; Orias et al. 2017). We also report a set of IES-like somatic regions that are variably spliced at different temperatures, which might represent temperature-sensitive cryptic IESs. The surge in cryptic IES deletions with temperature is less pronounced compared to that in the annotated IESs and mostly limited to DSs that tend to be smaller than 0.1. As a consequence, we decided to focus the presentation of our results on true IESs.
PDE efficiency is reduced at nonconventional culture temperatures
We asked how extensively temperature changes affect the magnitude of IES retention in the polyploid somatic genomes of the experimental lines. Supplemental Table S2 reports the full set of PGM-IESs with their corresponding retention scores (IRS) and the statistical significance of the F0-to-F1 IRS transitions.
We detect a reduction in PDE efficiency at 18°C and 32°C relative to the control temperature of 25°C (Fig. 2). Specifically, the number of IESs with a greatly intensified retention in the F1 somatic nuclei (IRSF1 > IRSF0, binomial test, Padj < 0.05) ranges from 183 to 271 at suboptimal temperatures, compared with 43 at 25°C. Further, most of the significantly retained IESs detected at 18°C and 32°C are unique to suboptimal temperatures, with a treatment-control ratio of ∼12-fold (151:13) and ∼17-fold (225:13) for 18°C and 32°C, respectively (Supplemental Fig. S2A). Conversely, the number of IESs excised with significantly increased efficiency in the F1 generation is comparable across temperatures (Fig. 2), with ∼50% overlap between each of the experimental lines and the control (Supplemental Fig. S2B). The count of significant transitions for the three temperatures tested are summarized in Figure 2D.
Figure 2.

PDE inefficiency shows a U-shaped relationship with temperature. (A–C) Bland-Altman plots displaying the Log2 fold change of IRSs from F0 to F1 for 25°C, 32°C, and 18°C. Statistically significant IRS transitions (binomial test, Padj < 0.05) are shown as filled colored circles: 25°C (dark and light blue circles); 32°C (red and orange circles); 18°C (dark and light green circles) (dark color: IRSF1 > IRSF0; light color: IRSF1 < IRSF0). Position on the x-axis reflects the log-transformed initial (F0) state of the IRSs (x-axis labels are IRSs before log transformation). (D) Counts of statistically significant IRS transitions (Padj < 0.05) after nuclear differentiation at 18°C (green), 25°C (blue), and 32°C (red). The number of somatic IESs experiencing an upward or downward IRS transition is shown above and below the horizontal dashed line, respectively.
Somatic IESs might be passed on to the sexual offspring
Regarding the potential biological significance of the PDE-mediated molecular variation, we first asked whether the somatic IESs detected in the F1 genomes were all generated anew or if they may have been obtained through transgenerational inheritance.
To gain insight into this question, we assessed how many IESs are retained simultaneously in the four independently rearranged F0 and F1 somatic nuclei. Leveraging 1000 simulated data sets based on random draws from the set of PGM-IESs, we estimated that the expected maximum number of IESs shared by chance between four genomes is about 158. We observe an overlap of 844 IESs, roughly a fivefold increase (Fig. 3A; Supplemental Table S3 contains the genes that harbor four-way-shared IESs). In addition, we found that four-way-shared incompletely excised IESs (IRS > 0) show elevated median retention scores relative to the full set of incompletely excised IESs, more so at suboptimal temperatures (Fig. 3B). This indicates that suboptimal temperatures affect the epigenetic machinery and/or the biochemical efficiency of the DNA elimination machinery.
Figure 3.

Incompletely excised IESs show elevated retention scores and might be transgenerationally inherited. (A) Venn diagram depicting sets of incompletely excised IESs (IRS > 0) shared between three F1 somatic genomes and their parental F0 genome. A large excess of incompletely excised IESs are shared across all four genomes (P < 0.001), indicating that most of the IESs in the four-way set may have been inherited transgenerationally from F0 to F1. The expected maximum number of IESs shared across all four genomes (158) (Methods) is shown under the observed value, and 229 of the observed 844 four-way-shared IESs are somatic IESs (IRS > 0.1): (blue ellipse) 25°C (F0); (green ellipse) 18°C (F1); (light blue ellipse) 25°C (F1); (red ellipse) 32°C (F1). IESs with read coverage ≥20 and length ≥26 base pairs were used. (B) Box plot showing the IRS distributions for the full set and the four-way set (4way) of incompletely excised IESs (IRS > 0) at all investigated temperatures. Pairwise comparisons between groups were performed using a Wilcoxon rank-sum test with correction for multiple testing (Benjamini–Hochberg). Statistical significance is indicated for each comparison: (****) P < 0.001; (ns) nonsignificant. Outliers are omitted for clarity.
The set of four-way-shared IESs may reflect a systematic failure to correctly excise IESs with particularly weak cis-acting recognition/excision signals. To test this hypothesis, we estimated a predictor of splicing signal quality, the Cin-score (Ferro et al. 2015). We found that four-way-shared IESs have a smaller mean Cin-score compared to the full set of PGM-IESs (IESs4-way vs. IESsPGM; 0.54 vs. 0.62; Mann–Whitney U test, P < 0.001). This difference holds true when we control for size class and genomic location and applies also to the overall set of somatic IESs (e.g., somatic IESs32°C vs. IESsPGM; 0.51 vs. 0.62; Mann–Whitney U test, P < 0.001).
A perturbation of the small RNAs guiding IES excision might also explain the observed excess of IESs shared between the F0 and the F1 genomes. We explored this hypothesis using published knockdown studies of PDE-associated epigenetic components (Dcl2/3 and Dcl5) in the P. tetraurelia strain 51 (Arnaiz and Sperling 2011; Sandoval et al. 2014). We found an excess of Dcl2/3- and Dcl5-sensitive IESs in the four-way-shared set relative to the PGM-set (46% vs. ∼10%; two-proportions Z-test, P < 0.01) under the assumption that the epigenetic regulation of IESs in the strain 51 is largely conserved in its derivative strain d12 (Rudman et al. 1991). We also found an enrichment of Dcl2/3 and Dcl5-controlled IESs in both somatic IESs (Fig. 4) and significantly retained IESs (Supplemental Fig. S3). The overrepresentation of Dcl2/3 and Dcl5-controlled IESs in the somatic sets remains largely unchanged when controlling for Cin-score and other variables such as size and expression level of somatic IES-containing genes (Supplemental Fig. S4).
Figure 4.
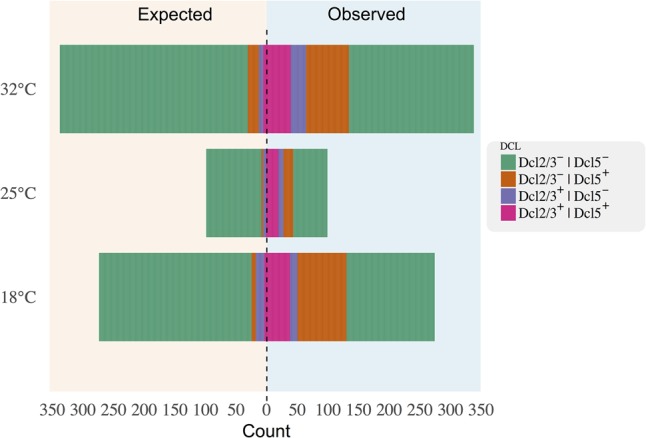
A large excess of somatic IESs is epigenetically regulated. Back-to-back stacked bar chart showing the number of intra-exonic somatic IESs for all investigated temperatures. For each temperature, IES counts are broken down into Dcl2/3-controlled IESs (Dcl2/3+ | Dcl5−, purple), Dcl5-controlled IESs (Dcl2/3− | Dcl5+, orange), Dcl2/3-Dcl5-co-controlled IESs (Dcl2/3+ | Dcl5+, fuchsia), and Dcl-independent IESs (Dcl2/3− | Dcl5−, green). Expected proportion of Dcl-dependent IESs for random samples of the same size (left) are shown back to back with the observed data (right). Two-proportions Z-test returns P < 0.01 for all expected/observed contrasts.
To complement these observations, we further investigated how extensively the transcription elongation factor TFIIS4 (Maliszewska-Olejniczak et al. 2015) and the chromatin remodeling protein Ezl1 (Lhuillier-Akakpo et al. 2014) control the excision of somatic IESs. TFIIS4 is a putative component of the small RNA-guided DNA excision machinery in P. tetraurelia, whereas Ezl1 might operate in a small-RNA-independent fashion (Swart et al. 2017). We found that a significant excess of somatic IESs is TFIIS4-sensitive (P ≤ 0.001). Conversely, we detect no or only marginal overrepresentation of Ezl1-sensitive somatic (and four-way-shared) IESs at 18°C or 32°C (P = 0.037 and P = 0.276, respectively).
Collectively, these observations indicate that perturbations of the small RNA-guided DNA excision machinery may contribute to the overrepresentation of somatic IESs at suboptimal temperatures.
Genomic distribution of somatic IESs
To deepen our understanding of the biological significance of somatic IESs, we performed a systematic analysis of their genomic distribution.
We found that somatic IESs preferentially occupy intergenic regions (Fig. 5A). Furthermore, somatic IESs are more likely to occur in genes that in the P. tetraurelia strain d12 are weakly expressed in the vegetative stage (Fig. 5B). A closer examination of the data reveals that the intergenic to intragenic ratio of somatic IESs changes with the magnitude of IES retention. The proportion of intergenic somatic IESs increases abruptly as the IRS crosses 0.1 and plateaus at ∼70% beyond an IRS threshold of ∼0.25. This is true at all the investigated temperatures (Fig. 5A). Also, the reduced efficiency of IES excision at suboptimal temperatures is not limited to intergenic IESs (Fig. 5C). When we compare the subset of IESs that are significantly retained against the set of PGM-IESs, we once again detect an excess of intergenic loci. Nevertheless, a cross-sample comparison reveals an enrichment of somatic IESs within exons at 32°C relative to 25°C (two-sided two-proportions Z-test, P < 0.05) (Fig. 5C). In particular, 83 and 158 genes show significant levels of IES retention at 18°C and 32°C compared to only 17 genes at 25°C, respectively (Supplemental Fig. S5).
Figure 5.

Purifying selection shapes the positional distribution of incompletely excised IESs. (A) IRS-dependent changes of genomic distribution for incompletely excised IESs. (B) Expression levels of genes affected by IES retention (805 genes; IRS > 0.1, red box) and the full set of macronuclear coding genes (cyan box). The expression data are from P. tetraurelia strain d12 (two replicates, cultured at 25°C; this study). Pairwise comparison was performed with a Mann–Whitney U test. Statistical significance is indicated: (***) P < 0.01. (C) Genomic distribution of significantly retained IESs at all investigated temperatures. Distribution of PGM-IESs is shown for reference (leftmost pie chart, boxed). Sample size is indicated below each set. Percentages <5% are excluded.
Overall, the pronounced bias in the distribution of somatic IESs in regions that typically evolve under reduced levels of selective constraints (Gout et al. 2010; Ferro et al. 2015) suggests that purifying selection antagonizes severe IES retention.
Genes hit by IES retention often lack a WGD1 ohnolog
P. tetraurelia has undergone a relatively recent event of whole-genome duplication (WGD1) (Aury et al. 2006). To gain further insights into the evolutionary constraints that shape the genomic distribution of IES retention, we asked whether homologous IESs in WGD1 ohnolog pairs are retained to a similar extent. We found that the excision of homologous IESs from WGD1 ohnologs is largely unlinked—most of the IESs with IRS > 0 have homologous IESs with IRS = 0 (F1 lines; 32°C = 74%, 18°C = 78%, and 25°C = 81%).
We also noticed that P. tetraurelia genes affected by IES retention in the F1 lines have significantly fewer recent WGD1 ohnologs than expected by chance (based on the total number of IES-containing genes with WGD1 ohnologs in P. tetraurelia; hypergeometric distribution, P < 0.0001). This deficit holds regardless of the selected IRS threshold or the environmental temperature at the time of nuclear development (Supplemental Table S4).
IES retention is coupled with reduced gene expression levels
To explore the potential impact of incomplete IES excision on gene expression, we collected RNA from two independent lines of P. tetraurelia strain d12 cultured at 25°C. We examined the expression levels of IES-containing WGD1 ohnolog pairs with discordant levels of IES retention. Under the assumption that the incompletely excised IESs that are shared between the 25°CF0 and 25°CF1 DNA-seq lines are also incompletely excised in the RNA-seq lines, we found that genes with incompletely excised IESs (IRS > 0) display reduced expression levels compared to their IES-free ohnologs (IRS = 0) (Fig. 6; Supplemental Table S5).
Figure 6.

The magnitude of IES retention scales negatively with the gene expression level. The average expression level of IES-containing ohnologs (derived from the most recent whole-genome duplication) varies in an IES retention score (IRS)-dependent manner. The expression levels are based on reads mapping to the somatic genome; that is, reads from IES-containing transcripts did not go into the final estimate. Average gene expression levels were compared against one another using a one-tailed Wilcoxon rank-sum test: (ns) P > 0.05; (*) P < 0.05; (**) P < 0.01. The numbers within parentheses above the bars refer to the population size of the surveyed pairs of ohnologs.
We also screened the RNA reads for the presence of IES sequences (henceforth, referred to as IES+ transcripts). In the two replicates, we found 246 and 378 IES+ transcripts (211 and 326 loci, respectively); many of the IES retention reads contain spliced-out introns, suggesting no or minimal DNA contamination. Of the detected IES+ transcripts, 87 are shared between replicates. This is a significant overlap, considering that the RNA replicates derive from two independently rearranged somatic nuclei. Furthermore, 157 IES+ transcripts, on average, have their corresponding somatic IESs in the 25°C F0 and F1 lines. This considerable overlap further strengthens our proposition that incomplete IES excision is partly nonrandom. Finally, the average IRS of these 157 somatic IESs is ∼0.29 for the 25°C F0 and F1 lines. This relatively large IRS indicates that more IES+ transcripts may occur in our RNA samples, but remain under the radar because IRS (or sequence coverage) is too low.
IES retention in exons primarily disrupts ORFs
Within genes, somatic IESs could give rise to functional alternative isoforms. To assess the impact of incomplete IES excision on protein sequence diversity, we calculated the fraction of PGM-IESs that, if retained, could prompt the introduction of premature termination codons (PTCs). We then compared such a fraction against our actual observations for the 32°C line. Approximately 21% of all PGM-IESs would lead to productive alternative DNA splicing variants through extension of the open reading frame, if retained (Fig. 7A). Roughly 16% is the counterpart in the experimental set (Fig. 7B). Thus, incomplete IES excisions may contribute rarely to protein sequence diversity, and exposure to a suboptimal temperature does not increase this tendency.
Figure 7.

IES insertion within the coding sequence (CDS) potentially diversifies protein sequences. (A) Theoretical diversification potential of PGM-IESs. Pro-IES (green): fraction of 3n, non-PTC-inducing IESs (“productive” IESs). Tail-IES (crimson red): fraction of IESs that do not introduce a premature termination codon (PTC) but lead to the ablation of the true stop codon. FS-IES (orange): fraction of IESs that disrupt the open reading frame by introducing a downstream PTC owing to frameshift (FS). PTC-IES (red): fraction of IESs that introduces a PTC within the insertion. (B) Classification of the 146 within-CDS IES insertions observed at 32°C based on the predicted transcriptional outcome. The mean excision score (1-µIRS), mean IRS (µIRS), and maximum IRS (M) are indicated beside each category. The number of IESs is indicated within parentheses. The predicted effect on the protein products is depicted schematically next to each class.
To gain some insights into the possible phenotypic consequences of IES retention, we analyzed the location of productive IESs (CDS-mapping and ORF-preserving) relative to encoded protein domains. We found that the observed fractions of productive IES retention (evidence at the DNA level) within functionally annotated domains are comparable to what is expected by chance, either before or after partitioning IESs in Dcl2/3-5-sensitive and Dcl2/3-5-insensitive (Supplemental Fig. S6).
Finally, we examined the distribution of incompletely excised IESs along genes. It was previously shown that the positional distribution of P. tetraurelia IESs is skewed toward the gene 5′ end (Ferro et al. 2015) and that incompletely excised IESs that introduce PTCs accumulate at the gene 5′ end (in lines cultured at standard temperatures) (Catania et al. 2013). We found that incompletely excised IESs accumulate at either end of genes in the 32°C line; the distribution of PTC-inducing IESs along genes becomes bimodal as the IRS increases (Supplemental Fig. S7). IES retention at the 3′ end of genes might have phenotypic consequences, because transcript degradation via nonsense-mediated mRNA decay (NMD) may be more efficient when PTC-inducing IESs are retained at the 5′ end of genes (van Hoof and Green 1996; Longman et al. 2007).
Nonrandom distribution of IESs with respect to protein families and molecular function
Finally, we examined the molecular function of genes with significant IES retention in coding sequences. The functional categorization of these genes is given in Supplemental Figure S5. The full set of genes, their expression values, as in strain d12 (this study) and 51 (Arnaiz et al. 2017), and annotations, along with parameters related to the retained IESs are presented in Supplemental Table S2.
A Gene Ontology (GO) term enrichment analysis reveals a single molecular function term, protein binding, enriched at both 18°C and 32°C (Fisher's exact test, P < 0.0001), but not at 25°C (P = 0.05). Two sets of proteins, the tetratrico peptide repeat region (TPR; IPR019734) and growth factor receptor cysteine-rich domain (GFR; IPR009030) containing proteins contribute largely to this functional enrichment. More specifically, TPR and GFR-genes together account for ∼21% and ∼24% of the genes affected at 32°C and 18°C, respectively.
Intrigued by this finding, we wondered whether P. tetraurelia genes hit by IES retention in general, and the gene families identified above in particular, are unusually IES-rich genes. We found that genes with significant IES retention at 18°C and 32°C show elevated mean IESs count (Fig. 8A) and IES density (Fig. 8B). Both TPR-motif-containing proteins and GFR cysteine-rich domain-containing proteins show significantly elevated numbers of IESs per gene, being among the most IES-rich genes in the P. tetraurelia genome (Fig. 8C). However, only TPR-genes show unusually high IES densities (Fig. 8D): With IES densities up to 10 IES/kb, the TPR-motif family of proteins alone accounts for almost 2.5% (1135 IESs) of the 44,928 PGM-IESs (an example of TPR-motif gene is in Fig. 8E).
Figure 8.

TPR-motif and GFR Cys-rich domain-containing proteins are IES-rich gene families susceptible to inefficient IES excision in response to temperature changes during autogamy. (A) Genes affected by IES retention at 18°C and 32°C have a significantly greater than average number of IESs. (B) Across all the investigated temperatures, genes hit by significant IES retention are IES dense. (C) TPR and GFR proteins show extraordinary per-gene IES counts. (D) TPR but not GFR proteins are characterized by elevated IES density (Wilcoxon rank-sum test: [ns] P > 0.05; [*] P < 0.05; [**] P < 0.01; [***] P < 0.001). (E) A TPR-motif gene (PTET.51.1.G1500001) showing a characteristic pattern of IES distribution. This gene is hit by multiple IES retention at 32°C. IESs are positioned almost invariably at the 5′ end of the TPR coding exons. According to the v2.0 annotation of macronuclear gene models, PTET.51.1.G1500001 is the gene with the greatest number of IESs to be found in the P. tetraurelia genome. (TPR) Tetratricopeptide repeat region motif (IPR019734); (GFR-Cys-rich) growth factor receptor cysteine-rich domain (IPR009030); (IES density) number of IESs per kb.
We then asked whether the pronounced representation of large gene families such as GFR- and TPR-containing proteins in our data set could be expected by chance. To address this point, we partitioned P. tetraurelia genes into three InterPro domain groups, TPR, GFR, and protein kinase-like domain (PKD) genes (with the latter group serving as a control) and performed an enrichment analysis on the experimental sets. We found that the number of GFR-genes (as well as PKD-genes) hit by significant IES retention can be expected by chance alone, both at 32°C and 18°C (binomial test, P > 0.05). Conversely, TPR-genes are highly overrepresented at either suboptimal temperature (binomial test, P < 0.0001).
Lastly, we wondered whether P. tetraurelia genes that are interrupted by IESs in the germline nucleus (IES genes) are enriched in specific molecular functions or biological processes. Indeed, we found that P. tetraurelia genes involved in ion transport, signal transduction, and microtubule-based movement, among others, are relatively more likely to be IES genes (Table 1). Conversely, IES genes are less likely to encode translation-associated factors (Table 1).
Table 1.
IES-containing genes are enriched in specific molecular functions and biological processes

Discussion
The driving question of this work was, how extensively can ecological changes affect somatic genome development? The answer may extend our view on how the cross talk between genes and environment affects organismal constitution and evolution. One possibility is that environmental changes may have generally little to no impact on this developmental process. Alternatively, environmentally induced perturbations might introduce de novo molecular variation into the freshly processed somatic DNA. Our results provide support for this latter perspective.
Studying the process of programmed DNA elimination (PDE) in the ciliate P. tetraurelia, we found that the rate of IES retention during nuclear differentiation is enhanced at nonstandard culture temperatures (18°C and 32°C) (Fig. 1A). Likewise, the efficiency of IES elimination peaks at 25°C, but is reduced at both 18°C and 32°C (Fig. 2). In as little as one sexual generation, the thermoplasticity of IES excision promotes the introduction of hundreds of IESs into the somatic genome and enhances the nuclear prevalence (to varying degrees) of IESs retained from previous sexual generations (Fig. 3B). These results suggest that IES excision, although overall highly efficient and reproducible, is to some degree sensitive to changes in the environmental temperature. This finding challenges expectations about the general robustness of somatic genome development. It may also be surprising, given that Paramecium is continuously exposed to daily and seasonal temperature fluctuations in naturally occurring conditions, and thrives along latitudinal temperature clines (Krenek et al. 2011, 2012).
Does the observed increment in somatic genetic variation have some biological relevance? The tendency of severely retained IESs to localize in intergenic regions and genes that are often under low levels of selective constraints (Fig. 5) indicates that purifying selection typically opposes IES retention. This implies that IES retention in P. tetraurelia may achieve levels that are sufficiently pronounced to impact fitness. Not only is this interpretation consistent with previous results (Gout et al. 2010; Arnaiz et al. 2012; Ferro et al. 2015), it also suggests that somatic IESs in P. tetraurelia may represent biologically relevant variation.
We show that most of this biological variation is unlikely to give rise to functional alternative protein sequence. Although our data provide direct evidence that IESs can be incorporated into transcripts, they also show that IES retention substantially disrupts ORFs (Fig. 7). Moreover, increased IES retention following environmental perturbation may result in the reduction of transcript availability (Fig. 6). These results suggest that IES retention may help fine-tune gene expression in response to environmental adversities. If maintained across sexual generations, IES retention might facilitate gene loss by disrupting expression and relaxing local selective constraints. Because the excision of homologous IES loci appears to be largely independent (under the investigated conditions), IES retention might also contribute to the diversification of gene copies in the P. tetraurelia genome. These hypothetical dynamics are expected to unfold over evolutionary time and therefore beg an important question: Can somatic IESs be inherited?
The observed overlap between IES retention profiles (Fig. 3A) cannot be explained by chance. We advance two, nonmutually exclusive hypotheses to explain the recurrence of somatic IESs in subsequent sexual generations. First, IESs flanked by weak splicing signals might be inefficiently excised at each round of PDE and thus consistently retrieved in the offspring genomes. The weak splicing signals of somatic IESs are consistent with this possibility. Second, the excision of somatic IESs might be controlled by small noncoding RNAs that facilitate IES excision from the developing macronucleus to mimic the parental genome organization. The resulting trans-somatic inheritance is consistent with previous observations (Duharcourt et al. 1995, 1998; Aronica et al. 2008; Lepère et al. 2008). It is also substantiated by the fact that four-way-shared IESs (as well as somatic IESs in general) are enriched with IESs that are cataloged as epigenetically regulated in P. tetraurelia (Fig. 4; Arnaiz and Sperling 2011). In addition, we found that the overrepresentation of epigenetically controlled somatic IESs is independent from their Cin-score. This provides support for the hypothesis that somatic IESs may be inherited. Further studies are required to strengthen this suggestion and to evaluate the stability of transgenerational epigenetic inheritance of somatic IESs alongside their effect on fitness.
Are certain genes preferentially targeted for IES retention? The analysis of the genes hit by IES retention in response to PDE's thermoplasticity reveals that TPR protein-coding genes may be particularly prone/susceptible to incomplete IES excision (Fig. 8). Although the function(s) of these proteins in Paramecium is currently unknown, in other organisms, TPR domains mediate manifold functions, mainly contributing to protein–protein interactions and the assembly of multiprotein complexes (e.g., Allan and Ratajczak 2011). Considering that TPR protein-coding genes are IES rich and yet successfully freed from IESs at 25°C, the observed high level of incomplete IES excision in these genes might simply reflect a particularly poor performance of the excision machinery in IES-rich regions at suboptimal temperatures. Alternatively, IES excision in TPR protein-coding genes may be actively modulated in suboptimal environments, for example, as a mechanism of gene expression control. This hypothetical control mechanism could be enforced through the regulation of Dcl5-sensitive IESs, because we note that TPR protein-coding genes display a twofold enrichment of Dcl5-controlled IESs and a concurrent fivefold underrepresentation of Dcl2/3-controlled IESs (Supplemental Table S7).
We found that somatic genes involved in processes such as signal transduction, cellular protein modification, and transport of ions across membranes are significantly more likely to be interrupted by IESs in the germline nucleus (Table 1). Thus, although IESs show a nearly random distribution with respect to genic and intergenic regions (Arnaiz et al. 2012), our functional enrichment analysis suggests that the roughly 45,000 IESs in P. tetraurelia are nonrandomly distributed in the coding genome in relation to molecular functions and biological processes. The functional enrichment of IES genes could be explained in terms of (1) a spatially patterned genomic invasion by transposable IES progenitors attributable to heterogeneous levels of purifying selection that antagonize gene interruption, and/or (2) differential expansion of gene families in the P. tetraurelia’s genome following the invasion. An alternative, tempting explanation is that IESs’ patterned genomic topography has been shaped by natural selection over evolutionary time to help regulate organismal responses to environmental changes.
Finally, what is the mechanism behind PDE's thermoplasticity? Suboptimal environmental conditions during nuclear differentiation might have a direct impact on the biochemical efficiency of the excision machinery. They might also alter DNA repair pathways coupled with IES elimination. Consistent with this latter hypothesis, intracellular processes coupled with DNA repair, such as meiotic recombination, are also thermoplastic, with a U-shaped temperature-performance response (Lloyd et al. 2018). Alternatively, the somatic variability introduced at high and low temperatures could result from an environmentally induced modulation or “rewiring” of the epigenetic machinery regulating IES excision. In line with this, the excision of a sizable fraction of somatic IESs in our study is known to be small RNA-dependent. Future studies investigating the temperature response of small RNAs guiding IES excision could help clarify the mechanistic basis of PDE thermoplasticity.
In sum, further research is necessary to establish the mechanisms behind PDE's thermoplasticity and its adaptive significance. The work presented here provides evidence that stressful environmental conditions experienced during nuclear differentiation can boost variability in newly developed somatic genomes. These findings offer new insights into the ways the environment can elicit plasticity in genetically identical organisms/cells.
Methods
Experimental design
To evaluate the effect of temperature change on PDE, fully homozygous isogenic Paramecium cells were cultured in daily reisolation and passed through autogamy (self-fertilization) at three different temperatures. After a first round of autogamy at 25°C to establish a parental line, one post-autogamous parental cell was isolated and allowed to divide in fresh medium. Three of the resulting isogenic cells in turn were used to start three sublines that were cultured in daily reisolation and passed through a second round of autogamy at 18°C, 25°C, or 32°C to establish filial lines. Single post-autogamous F1 cells isolated for each of the sublines as well as the remaining isogenic parental cells were expanded to mass culture for somatic DNA extraction.
Paramecium strains and culture conditions
Paramecium tetraurelia strain d12 was used in the experiment. Cells were grown in Cerophyl Medium (CM) inoculated with Enterobacter aerogenes Hormaeche and Edwards (ATCC 35028). Stocks were fed with bacterized CM every 2 wk and kept at 14°C. In preparation for the study, cells kept in stock were washed 10 times in Volvic water to ensure monoxenic growth conditions during propagation. Cells were grown in depression slides under daily reisolation regimen in 200 µL of bacterized CM. Autogamy was induced by letting approximately 25 division-old cells starve naturally for 3 d with no addition of bacterized medium. Occurrence of autogamy was confirmed by screening ≥50 Acetocarmine-stained cells for macronuclear fragmentation under a light microscope. A single ex-autogamous cell was isolated from each line, allowed to divide once, and single caryonidal progenitors brought to mass culture for somatic DNA isolation. Macronuclear fragmentation of sister caryonides was confirmed by Acetocarmine staining.
Macronuclear DNA isolation and whole-genome sequencing
The somatic nuclei of both parental (25°CF0) and filial lines (25°CF1, 32°CF1, and 18°CF1) were isolated and the macronuclear DNA (MAC) subjected to whole-genome sequencing. MACs were isolated after more than 10 vegetative divisions post-autogamy to prevent carryover of maternal MAC fragments at the time of isolation. Cells were resuspended in Volvic water for 2 h and allowed to digest their food vacuoles before MAC isolation to reduce bacterial load. MAC isolation was performed according to a described protocol (Arnaiz et al. 2012). Purified genomic DNA was subjected to pair-end Illumina sequencing (∼90–100× coverage, on average, 150-nt-long reads) on a HiSeq 4000 system.
Data preprocessing
To increase the accuracy of DNA-seq, data analysis raw reads were subjected to an initial step of quality control (QC) using FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) and preprocessed using the Joint Genome Institute (JGI) suite BBTools 37.25 (Bushnell et al. 2017). Library adapters were removed from the 3′ end of the reads with BBduk using the included reference adapter file and ensuring the same length for both reads of a pair after trimming. For the k-mer-based adapter detection, a long and short k-mer size of 23 and 11, respectively, were used and a single mismatch allowed. In addition, overlap-based adapter detection was enabled. Finally, short insert sizes in part of the library were leveraged for an overlap-based error correction with BBMerge, while keeping left and right reads separated.
Additional macronuclear genomes
We examined four additional somatic genomes, previously obtained for P. tetraurelia strain 51 cells cultured and self-fertilized at 27°C (Arnaiz et al. 2012; Lhuillier-Akakpo et al. 2014; Swart et al. 2017). These genomes were used as controls for silencing experiments and in Swart et al. (2017) are labeled as Control 1 (accession ERR1212635), Control 2 (accession ERR138450), Control 3 (accession ERR1212640), and Control 4 (accession ERR501376). In Supplemental Table S2 these “control” genomes are labeled as 27_EV, 27_AR, 27_ND7, and 27_LA, respectively.
Calculation of IES retention scores
Quality improved, trimmed reads were mapped to the P. tetraurelia strain 51 reference somatic genome available via ParameciumDB (Arnaiz and Sperling 2011) and to a pseudogermline genome containing all the known 44,928 IESs previously identified by Pgm knockdown (Arnaiz et al. 2012) that was created with the Insert module of ParTIES 1.00 (Denby Wilkes et al. 2016). Read mappings were performed with Bowtie 2.3.2 (Langmead et al. 2009) using the local alignment function for paired-end reads in very sensitive mode (--very-sensitive-local), and the resulting SAM files were manipulated with SAMtools 1.4.1 (Li et al. 2009) for downstream processing. IES retention scores were calculated with the MIRET module of ParTIES using the IES score method. Briefly, parTIES computes IRSs from DNA-seq mapping files as the ratio between IES-positive reads and the sum of IES-positive plus IES-negative reads [IES+/(IES+ + IES−)].
Genome-wide analysis of IES retention
Genome-wide analysis of IES retention was performed via statistical comparison of F0/F1 IRSs as implemented in the R script accompanying ParTIES, with the following modifications. The upper and lower bound of the 75% confidence interval (CI) constructed on the F0 retention score was taken as a reference retention score for binomial testing of upward or downward transitions, respectively. Lowly supported IESs, that is, IESs with a total support <20 reads (IES+ + IES−), were excluded from the study. The P-values were corrected for multiple testing with the Benjamini–Hochberg method, and a cutoff of 0.05 was used to designate IESs with significantly different retention levels in F1 compared to F0 samples.
Genome-wide analysis of cryptic IES excisions
To estimate the rate of cryptic IES excision in response to temperature changes during nuclear differentiation TA-bounded somatic deletions were characterized for the parental F0 genome, and the three F1 genomes were rearranged at 18°C, 25°C, and 32°C using the MILORD module implemented in ParTIES. Reads mapped on the reference macronuclear genome assembly of P. tetraurelia strain 51 were provided as input. Low coverage cryptic IESs with total read counts (support_ref + support_variant) < 20 were excluded from the analysis. Deletion scores (DSs) were calculated as the ratio of reads supporting cryptic IES excision over all reads spanning the somatic region, that is, support_var/(support_var + support_ref). Cryptic IESs alternatively excised across samples were collected by filtering deletion scores with SD > 0.3. Manual inspection with Integrative Genomics Viewer (IGV) (Robinson et al. 2011) was used to confirm putative temperature-sensitive excisions that were tagged as “Unstable.” Conversely, cryptic IESs consistently excised (DS > 0.5 in all samples) were tagged as “Stable.” The catalog of gene-mapping cryptic IESs is provided in Supplemental Table S1.
Downstream data analyses
We compiled a table (Supplemental Table S2) that reports the obtained IRSs of all samples and a plethora of additional IES-related information calculated ad hoc for this study or collected from previous studies and/or processed from various external sources, for example, the Paramecium tetraurelia strain 51's genome annotation v2 (Arnaiz et al. 2017). All external data are publicly available at ParameciumDB (Arnaiz and Sperling 2011). The following downstream analyses were conducted using in-house Python (https://www.python.org/) and R (R Core Team 2017) scripts (Supplemental Code).
IES retention profiles simulation
We simulated IES retention as a stochastic process. Four random samples were drawn without replacement from the full reference set of IESs (PGM-IESs). The size of the random samples was set equal to the number of incompletely excised IESs (IRS > 0) observed in each of the four investigated genomes. This simulation was repeated 1000 times, and the maximum number of elements shared by chance across all four drawings (theoretical four-way overlap assuming random IES retention) was used as an expectation to compare against the experimental data.
Impact of incomplete IES excision on genes
For IESs located in the coding region of protein-coding genes, we checked whether their retention promotes the induction of a premature termination codon (PTC). We calculated the IES's position with respect to the translation start codon, inserting the IES at this location and scanning this artificial CDS + IES-construct for an in-frame TGA (the only stop codon in Paramecium) upstream of the annotated one. In case a PTC was detected, we marked the distance of the PTC to the CDS start and, in case of non-3n IESs (IESs with a size that is not a multiple of three), we marked whether the PTC occurred inside the IES body or downstream. The theoretical diversification potential of PGM-IESs was calculated as the fraction of IESs whose eventual retention would neither induce PTCs nor alter the reading frame. They thus contribute to a potentially productive diversification of the coding sequence space (productive IESs). PTC-inducing IESs were further divided into PTC-IES, IESs whose insertion leads to the introduction of a PTC within the insertion itself, and FS-IES, which disrupt the open reading frame and introduce a PTC downstream from the inserted region. A small number of IESs do not introduce PTCs but lead to the ablation of the true stop codon (tail-IESs) (see also Fig. 6).
Gene families and GO-term enrichment analyses
For each investigated temperature, we tested whether specific gene families were over- or underrepresented among the genes affected by significant IES retention. We devised a sampling procedure that accounts for nonhomogeneous IES densities across P. tetraurelia’s genes. Briefly, 10,000 IES sets with size equal to the number of significantly retained IESs in the experimental samples were randomly drawn from the PGM-IESs set. A nonredundant collection of genes hit by simulated IES retention was extracted at each draw, and the proportion of proteins carrying a specific functional domain (e.g., tetratricopeptide repeat-containing genes, and protein kinase-like domain) was used to build a null distribution. The mean of this null distribution was then treated as the success probability for a two-sided binomial test with a significance cutoff of P < 0.01. Raw P-values were adjusted via the Benjamini–Hochberg procedure.
The functional enrichment analysis of IES-containing genes was performed using the statistical overrepresentation test of the PANTHER gene list analysis tool (Mi et al. 2019) and cross-validated with the topGO package (https://www.bioconductor.org/packages/release/bioc/html/topGO.html). The mRNA IDs (e.g., GSPATT00000013001) of IES-containing genes (macronuclear gene models v1) were used as supported gene identifiers for the PANTHER gene list analysis. In all cases, IES-containing genes were tested against the full set of macronuclear (coding) gene models of P. tetraurelia. The Weigh01 algorithm and F statistic (Fisher's exact test) were used for testing the GO-terms overrepresentation with the topGO package. Raw P-values provided by topGO were adjusted with the P.adjust function (method = “hochberg”) implemented in the R package stats (version 3.4.0). A critical value of 0.05 was adopted as significance threshold in all tests.
mRNA samples and transcript quantification
Transcriptomic data were obtained from two lines of P. tetraurelia strain d12, each expanded from a distinct post-autogamous cell and exposed to 25°C during vegetative growth. After the extraction with TRIzol, the total RNA samples were used for library construction, a protocol that includes mRNA isolation by poly(A)-capture, fragmentation, cDNA synthesis, adapter ligation, amplification by PCR, and fragment length assessment. The prepared RNA was sequenced on an Illumina HiSeq 4000 platform yielding approximately 20 million 150-bp paired-end reads per sample. The raw reads were subjected to a quality control with FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) and trimmed with Atropos (Didion et al. 2017). The processed reads were aligned to the P. tetraurelia reference genome (Aury et al. 2006; Arnaiz et al. 2017) with STAR (Dobin et al. 2013). The RSEM software package (Li and Dewey 2011) was used for transcript quantification.
Data access
High-throughput sequencing data generated in this study have been submitted to the European Nucleotide Archive (https://www.ebi.ac.uk/ena) under the accession number PRJEB28697 for the whole-genome resequencing experiment (DNA-seq for control and temperature-exposed samples) and ERS3357003–ERS3357004, for the mRNA experiment (two biological replicates cultured at 25°C). Custom analysis scripts are available as Supplemental Code.
Supplementary Material
Acknowledgments
We thank Gennady Churakov, Franz Goller, and Hans-Dieter Görtz for their comments on a draft of the manuscript. Kathrin Brüggemann is gratefully acknowledged for her technical assistance. We thank the editor of Genome Research and three anonymous reviewers for suggesting additional analyses that improved the manuscript. This work was supported by a Deutsche Forschungsgemeinschaft (DFG) research grant to F.C. (CA1416/ 1-1) and carried out within the DFG Research Training Group 2220 “Evolutionary Processes in Adaptation and Disease” at the University of Münster.
Author contributions: V.V. was responsible for investigation, formal analysis, software, methodology, visualization, writing the original draft, and review and editing. R.H. was responsible for data curation, software, and review and editing of the manuscript. F.C. was responsible for funding acquisition, conceptualization, project administration, supervision, visualization, writing the original draft, and review and editing.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.245332.118.
References
- Allan RK, Ratajczak T. 2011. Versatile TPR domains accommodate different modes of target protein recognition and function. Cell Stress Chaperones 16: 353–367. 10.1007/s12192-010-0248-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen SE, Nowacki M. 2017. Necessity is the mother of invention: ciliates, transposons, and transgenerational inheritance. Trends Genet 33: 197–207. 10.1016/j.tig.2017.01.005 [DOI] [PubMed] [Google Scholar]
- Arnaiz O, Sperling L. 2011. ParameciumDB in 2011: new tools and new data for functional and comparative genomics of the model ciliate Paramecium tetraurelia. Nucleic Acids Res 39: D632–D636. 10.1093/nar/gkq918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnaiz O, Mathy N, Baudry C, Malinsky S, Aury JM, Denby Wilkes C, Garnier O, Labadie K, Lauderdale BE, Le Mouël A, et al. 2012. The Paramecium germline genome provides a niche for intragenic parasitic DNA: evolutionary dynamics of internal eliminated sequences. PLoS Genet 8: e1002984 10.1371/journal.pgen.1002984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnaiz O, Van Dijk E, Bétermier M, Lhuillier-Akakpo M, de Vanssay A, Duharcourt S, Sallet E, Gouzy J, Sperling L. 2017. Improved methods and resources for paramecium genomics: transcription units, gene annotation and gene expression. BMC Genomics 18: 483 10.1186/s12864-017-3887-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aronica L, Bednenko J, Noto T, DeSouza LV, Siu KW, Loidl J, Pearlman RE, Gorovsky MA, Mochizuki K. 2008. Study of an RNA helicase implicates small RNA–noncoding RNA interactions in programmed DNA elimination in Tetrahymena. Genes Dev 22: 2228–2241. 10.1101/gad.481908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aury JM, Jaillon O, Duret L, Noel B, Jubin C, Porcel BM, Ségurens B, Daubin V, Anthouard V, Aiach N, et al. 2006. Global trends of whole-genome duplications revealed by the ciliate Paramecium tetraurelia. Nature 444: 171–178. 10.1038/nature05230 [DOI] [PubMed] [Google Scholar]
- Baudry C, Malinsky S, Restituito M, Kapusta A, Rosa S, Meyer E, Bétermier M. 2009. PiggyMac, a domesticated piggyBac transposase involved in programmed genome rearrangements in the ciliate Paramecium tetraurelia. Genes Dev 23: 2478–2483. 10.1101/gad.547309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bétermier M, Duharcourt S. 2015. Programmed rearrangement in ciliates: Paramecium. In Mobile DNA III (ed. Craig N, et al. ), pp. 369–388. ASM Press, Washington, DC: 10.1128/microbiolspec.MDNA3-0035-2014 [DOI] [PubMed] [Google Scholar]
- Biederman MK, Nelson MM, Asalone KC, Pedersen AL, Saldanha CJ, Bracht JR. 2018. Discovery of the first germline-restricted gene by subtractive transcriptomic analysis in the Zebra finch, Taeniopygia guttata. Curr Biol 28: 1620–1627.e5. 10.1016/j.cub.2018.03.067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischerour J, Bhullar S, Denby Wilkes C, Régnier V, Mathy N, Dubois E, Singh A, Swart E, Arnaiz O, Sperling L, et al. 2018. Six domesticated PiggyBac transposases together carry out programmed DNA elimination in Paramecium. eLife 7: e37927 10.7554/eLife.37927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushnell B, Rood J, Singer E. 2017. BBMerge—accurate paired shotgun read merging via overlap. PLoS One 12: e0185056 10.1371/journal.pone.0185056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caron F. 1992. A high degree of macronuclear chromosome polymorphism is generated by variable DNA rearrangements in Paramecium primaurelia during macronuclear differentiation. J Mol Biol 225: 661–678. 10.1016/0022-2836(92)90393-X [DOI] [PubMed] [Google Scholar]
- Catania F, Schmitz J. 2015. On the path to genetic novelties: insights from programmed DNA elimination and RNA splicing. Wiley Interdiscip Rev RNA 6: 547–561. 10.1002/wrna.1293 [DOI] [PubMed] [Google Scholar]
- Catania F, McGrath CL, Doak TG, Lynch M. 2013. Spliced DNA sequences in the Paramecium germline: their properties and evolutionary potential. Genome Biol Evol 5: 1200–1211. 10.1093/gbe/evt087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyne RS, Lhuillier-Akakpo M, Duharcourt S. 2012. RNA-guided DNA rearrangements in ciliates: Is the best genome defence a good offence? Biol Cell 104: 309–325. 10.1111/boc.201100057 [DOI] [PubMed] [Google Scholar]
- Denby Wilkes C, Arnaiz O, Sperling L. 2016. ParTIES: a toolbox for Paramecium interspersed DNA elimination studies. Bioinformatics 32: 599–601. 10.1093/bioinformatics/btv691 [DOI] [PubMed] [Google Scholar]
- Didion JP, Martin M, Collins FS. 2017. Atropos: specific, sensitive, and speedy trimming of sequencing reads. PeerJ 5: e3720 10.7717/peerj.3720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29: 15–21. 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubois E, Bischerour J, Marmignon A, Mathy N, Régnier V, Bétermier M. 2012. Transposon invasion of the Paramecium germline genome countered by a domesticated PiggyBac transposase and the NHEJ pathway. Int J Evol Biol 2012: 436196 10.1155/2012/436196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duharcourt S, Butler A, Meyer E. 1995. Epigenetic self-regulation of developmental excision of an internal eliminated sequence on Paramecium tetraurelia. Genes Dev 9: 2065–2077. 10.1101/gad.9.16.2065 [DOI] [PubMed] [Google Scholar]
- Duharcourt S, Keller AM, Meyer E. 1998. Homology-dependent maternal inhibition of developmental excision of internal eliminated sequences in Paramecium tetraurelia. Mol Cell Biol 18: 7075–7085. 10.1128/MCB.18.12.7075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duret L, Cohen J, Jubin C, Dessen P, Gout JF, Mousset S, Aury JM, Jaillon O, Noel B, Arnaiz O, et al. 2008. Analysis of sequence variability in the macronuclear DNA of Paramecium tetraurelia: a somatic view of the germline. Genome Res 18: 585–596. 10.1101/gr.074534.107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferro D, Lepennetier G, Catania F. 2015. Cis-acting signals modulate the efficiency of programmed DNA elimination in Paramecium tetraurelia. Nucleic Acids Res 43: 8157–8168. 10.1093/nar/gkv843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gout JF, Kahn D, Duret L. 2010. The relationship among gene expression, the evolution of gene dosage, and the rate of protein evolution. PLoS Genet 6: e1000944 10.1371/journal.pgen.1000944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guérin F, Arnaiz O, Boggetto N, Denby Wilkes C, Meyer E, Sperling L, Duharcourt S. 2017. Flow cytometry sorting of nuclei enables the first global characterization of Paramecium germline DNA and transposable elements. BMC Genomics 18: 327 10.1186/s12864-017-3713-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoehener C, Hug I, Nowacki M. 2018. Dicer-like enzymes with sequence cleavage preferences. Cell 173: 234–247.e7. 10.1016/j.cell.2018.02.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jollos V. 1921. Experimentelle Protistenstudien: Untersuchungen über Variabilität und Vererbung bei Infusorien. (Experimental studies on protists: studies on variability and inheritance in infusoria). G. Fischer, Jena, Germany. [Google Scholar]
- Jung D, Giallourakis C, Mostoslavsky R, Alt FW. 2006. Mechanism and control of V(D)J recombination at the immunoglobulin heavy chain locus. Annu Rev Immunol 24: 541–570. 10.1146/annurev.immunol.23.021704.115830 [DOI] [PubMed] [Google Scholar]
- Krenek S, Berendonk TU, Petzoldt T. 2011. Thermal performance curves of Paramecium caudatum: a model selection approach. Eur J Protistol 47: 124–137. 10.1016/j.ejop.2010.12.001 [DOI] [PubMed] [Google Scholar]
- Krenek S, Petzoldt T, Berendonk TU. 2012. Coping with temperature at the warm edge—patterns of thermal adaptation in the microbial eukaryote Paramecium caudatum. PLoS One 7: e30598 10.1371/journal.pone.0030598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25 10.1186/gb-2009-10-3-r25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepère G, Bétermier M, Meyer E, Duharcourt S. 2008. Maternal noncoding transcripts antagonize the targeting of DNA elimination by scanRNAs in Paramecium tetraurelia. Genes Dev 22: 1501–1512. 10.1101/gad.473008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepère G, Nowacki M, Serrano V, Gout JF, Guglielmi G, Duharcourt S, Meyer E. 2009. Silencing-associated and meiosis-specific small RNA pathways in Paramecium tetraurelia. Nucleic Acids Res 37: 903–915. 10.1093/nar/gkn1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lhuillier-Akakpo M, Frapporti A, Denby Wilkes C, Matelot M, Vervoort M, Sperling L, Duharcourt S. 2014. Local effect of enhancer of zeste-like reveals cooperation of epigenetic and cis-acting determinants for zygotic genome rearrangements. PLoS Genet 10: e1004665 10.1371/journal.pgen.1004665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Dewey CN. 2011. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12: 323 10.1186/1471-2105-12-323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R; 1000 Genome Project Data Processing Subgroup. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd A, Morgan C, H. Franklin FC, Bomblies K. 2018. Plasticity of meiotic recombination rates in response to temperature in Arabidopsis. Genetics 208: 1409–1420. 10.1534/genetics.117.300588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longman D, Plasterk RH, Johnstone IL, Caceres JF. 2007. Mechanistic insights and identification of two novel factors in the C. elegans NMD pathway. Genes Dev 21: 1075–1085. 10.1101/gad.417707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maliszewska-Olejniczak K, Gruchota J, Gromadka R, Denby Wilkes C, Arnaiz O, Mathy N, Duharcourt S, Bétermier M, Nowak JK. 2015. TFIIS-dependent non-coding transcription regulates developmental genome rearrangements. PLoS Genet 11: e1005383 10.1371/journal.pgen.1005383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer KM, Forney JD. 1999. A mutation in the flanking 5′-TA-3′ dinucleotide prevents excision of an internal eliminated sequence from the Paramecium tetraurelia genome. Genetics 151: 597–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi H, Muruganujan A, Ebert D, Huang X, Thomas PD. 2019. PANTHER version 14: more genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res 47: D419–D426. 10.1093/nar/gky1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noto T, Mochizuki K. 2017. Whats, hows and whys of programmed DNA elimination in Tetrahymena. Open Biol 7: 170172 10.1098/rsob.170172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noto T, Mochizuki K. 2018. Small RNA-mediated trans-nuclear and trans-element communications in Tetrahymena DNA elimination. Curr Biol 28: 1938–1949.e5. 10.1016/j.cub.2018.04.071 [DOI] [PubMed] [Google Scholar]
- Orias E, Singh DP, Meyer E. 2017. Genetics and epigenetics of mating type determination in Paramecium and Tetrahymena. Annu Rev Microbiol 71: 133–156. 10.1146/annurev-micro-090816-093342 [DOI] [PubMed] [Google Scholar]
- Prescott DM. 1994. The DNA of ciliated protozoa. Microbiol Rev 58: 233–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. 2017. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna: https://www.R-project.org/. [Google Scholar]
- Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. 2011. Integrative genomics viewer. Nat Biotechnol 29: 24–26. 10.1038/nbt.1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudman B, Preer LB, Polisky B, Preer JR. 1991. Mutants affecting processing of DNA in macronuclear development in Paramecium. Genetics 129: 47–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandoval PY, Swart EC, Arambasic M, Nowacki M. 2014. Functional diversification of Dicer-like proteins and small RNAs required for genome sculpting. Dev Cell 28: 174–188. 10.1016/j.devcel.2013.12.010 [DOI] [PubMed] [Google Scholar]
- Singh DP, Saudemont B, Guglielmi G, Arnaiz O, Goût JF, Prajer M, Potekhin A, Przybòs E, Aubusson-Fleury A, Bhullar S, et al. 2014. Genome-defence small RNAs exapted for epigenetic mating-type inheritance. Nature 509: 447–452. 10.1038/nature13318 [DOI] [PubMed] [Google Scholar]
- Smith JJ, Timoshevskaya N, Ye C, Holt C, Keinath MC, Parker HJ, Cook ME, Hess JE, Narum SR, Lamanna F, et al. 2018. The sea lamprey germline genome provides insights into programmed genome rearrangement and vertebrate evolution. Nat Genet 50: 270–277. 10.1038/s41588-017-0036-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonneborn TM. 1977. Genetics of cellular differentiation: stable nuclear differentiation in eucaryotic unicells. Annu Rev Genet 11: 349–367. 10.1146/annurev.ge.11.120177.002025 [DOI] [PubMed] [Google Scholar]
- Sonneborn TM, Schneller MV. 1979. A genetic system for alternative stable characteristics in genomically identical homozygous clones. Dev Genet 1: 21–46. 10.1002/dvg.1020010105 [DOI] [Google Scholar]
- Swart EC, Denby Wilkes C, Sandoval PY, Hoehener C, Singh A, Furrer DI, Arambasic M, Ignarski M, Nowacki M. 2017. Identification and analysis of functional associations among natural eukaryotic genome editing components. F1000Res 6: 1374 10.12688/f1000research.12121.1 [DOI] [Google Scholar]
- van Hoof A, Green PJ. 1996. Premature nonsense codons decrease the stability of phytohemagglutinin mRNA in a position-dependent manner. Plant J 10: 415–424. 10.1046/j.1365-313x.1996.10030415.x [DOI] [PubMed] [Google Scholar]
- Wang J, Davis RE. 2014. Programmed DNA elimination in multicellular organisms. Curr Opin Genet Dev 27: 26–34. 10.1016/j.gde.2014.03.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.