

Abstract
Malignant tumors of the central nervous system (CNS) continue to be a leading cause of cancer-related mortality in both children and adults. Traditional therapies for malignant brain tumors consist of surgical resection and adjuvant chemoradiation; such approaches are often associated with extreme morbidity. Accordingly, novel, targeted therapeutics for neoplasms of the CNS, such as immunotherapy with oncolytic engineered herpes simplex virus (HSV) therapy, are urgently warranted. Herein, we discuss treatment challenges related to HSV virotherapy delivery, entry, replication, and spread, and in so doing focus on host antiviral immune responses and the immune microenvironment. Strategies to overcome such challenges including viral re-engineering, modulation of the immunoregulatory microenvironment and combinatorial therapies with virotherapy, such as checkpoint inhibitors, radiation, and vaccination are also examined in detail.
Keywords: HSV, virotherapy, oncolytic, brain tumors, experimental therapeutics, immunotherapy, cancer vaccination
Introduction:
Cancers affecting the central nervous system (CNS) result in severe morbidity/mortality in both pediatric and adult populations (1, 2). As such cancer survivors often sustain long-term disabilities such as neurosensory and neurocognitive impairments, resulting from the current standards of care (3). Such long-term complications as well as tumor resistance to current therapies have increased the need for innovative approaches to treat malignant brain tumors, especially within pediatric populations. In line with such needs, oncolytic viral therapy has emerged as a promising approach.
Among viral therapeutics, genetically engineered oncolytic herpes simplex viruses (oHSV) offer several advantages. HSV-1 has been extensively studied and its essential and non-essential genes have been defined. It is a large, enveloped neurotropic DNA virus that does not integrate into host DNA (4). Accordingly, HSV has proven ideal for targeting cancers of neural origin due to its intrinsic neurotropism (5). Of note, the γ134.5 neurovirulence diploid gene, which is necessary for viral replication in normal cells but not tumor cells, may be deleted in oHSV, thereby allowing normal cells to be spared while tumor cells are selectively targeted (6). The virus is highly immunogenic and can stimulate a robust anti-tumor immune response. In addition to deleting the neurovirulence gene, foreign genes can be inserted into the virus genome to augment the oncolytic effects and enhance anti-tumor immunity while not interfering with other dispensable and indispensable gene functions. Lastly, oHSV is sensitive to anti-viral medications such as acyclic guanosine analogs (acyclovir, ganciclovir, valganciclovir) in the unlikely event that it causes disease.
The first genetically engineered oHSV dlsptk (see Table 1 for a summary of oHSVs discussed in the text) which is thymidine-kinase deficient, effectively killed human glioma cells in vitro and led to prolonged survival when used in an in vivo model (7). While promising, such preclinical testing of first-generation viral vectors demonstrated the need for increased efficacy and safety testing due to poor replication and resistance to antiviral drugs. Subsequently, other first-generation mutant HSV vectors were developed such as G207, which has an ICP34.5 deletion at both γ134.5 loci and a lacZ insertion inactivating the ICP6 gene (UL39), helping inhibit late viral protein synthesis (6). Of note, the multi-mutated G207 virus is sensitive to anti-HSV therapies thereby increasing its safety profile. Preclinical evaluation of G207 in mice and non-human primates led to its use as the first oHSV in clinical trials in the United States (8). Such work led to clinical trials with other first-generation oHSV such as HSV1716 (9), which contains deletions of both ICP34.5 encoding genes. HSV1716 was evaluated in the United Kingdom in three clinical trials at the same time G207 was studied in the United States (10–12). These trials ultimately paved the way for talimogene laherparepvec (T-Vec), a second-generation oHSV which has become the first oncolytic virus approved by the US Food and Drug Administration (FDA) for the treatment of any cancer, melanoma in this case (13); T-Vec has two deletions, namely γ134.5 and α47 genes, and contains an insertion of human granulocyte-macrophage colony-stimulating factor into the γ134.5 loci. Interestingly, third-generation agents containing therapeutic mutations in the genome are now being investigated; G47∆, a triple-mutated vector with deletions in both copies of the γ134.5 gene, a deletion in the α47 gene, and a lacZ insertion in the ICP6 locus became the first third-generation HSV-1 to be tested in humans with glioblastoma (GBM) in Japan (14). Multiple clinical trials using oHSV to treat malignant tumors of the CNS in children and adults are underway or have been completed (Table 2).
Table 1:
Summary of oHSVs
| Virus | Deletion/Mutation | Foreign gene/promoter insertion | Reference |
|---|---|---|---|
| C130 | Deletion in both copies of γ134.5 gene | Expresses the HCMV TRS1 gene product | (43) |
| C134 | Deletion in both copies of γ134.5 gene | IRS1 gene under control of an HCMV immediate early promoter | (43, 44, 60) |
| dlsptk | Thymidine-kinase deficient | None | (7) |
| G207 | Deletion in both copies of γ134.5 gene and disabling lacZ insertion in UL39 | None | (6, 8, 16, 17, 24, 30, 37, 51, 89) |
| G47∆ | Deletion of the γ134.5 and α47 genes and a disabling lacZ insertion within ICP6 | Murine angiostatin | (14, 20, 75) |
| G47∆-IL12 | Deletion of the γ134.5 and α47 genes and a disabling lacZ insertion within ICP6 | Murine IL-12 gene insert | (72, 73, 85) |
| HSV1716 | Deletion in both copies of γ134.5 gene | None | (9–12, 23) |
| M002 | Deletion in both copies of γ134.5 gene | Murine IL-12 gene insert | (30, 67, 68, 70) |
| M032 | Deletion in both copies of γ134.5 gene | Human IL-12 gene insert | (71) |
| NG34 | Deletion in ICP6 and ICP34.5 genes | Human GADD34 gene transcriptionally controlled by the Nestin promoter | (41) |
| R-115 | None | Murine IL-12; CMV promoter | (36) |
| R5141 | Deletion of polylysine tract in gB | Insertion of IL-13 between amino acids 24 and 25 of gD | (38) |
| R5181 | Deletion of polylysine tract in gB | Insertion of urokinase plasminogen activator between amino acids 24 and 25 of gD | (39) |
| RAMBO | Deletion in both copies of γ134.5 gene and in-frame gene-disrupting insertion of GFP within ICP6 gene | Vasculostatin (Vstat120) under control of the HSV IE 4/5 promoter | (74) |
| rHSVQ1 | Deletion in UL39 and γ134.5 genes | None | (40, 45) |
| rQNestin34.5v.2 | Deletion in γ134.5 gene and UL39 | ICP-34.5 under control of synthetic nestin promoter | (40, 45) |
| talimogene laherparepvec (T-Vec) | Deletions of the ICP34.5 and ICP47 genes | Granulocyte-macrophage colony-stimulating factor, CMV promoter | (13, 86) |
Table 2:
Summary of past and ongoing oHSV brain tumor clinical trials
| Tumor Typea | Virus Type | Clinical Trial Identification | Pediatric or Adult | Status | Reference |
|---|---|---|---|---|---|
| HGG | rQNestin 34.5v.2 | Adult | Recruiting | (40) | |
| M032-HSV-1 | Adult | Recruiting | (71) | ||
| HSV G207 | Adult | Completed | (8, 17, 89) | ||
| HGG | C134-HSV-1 | Adult | Not Yet Recruiting | (57) | |
| Supratentorial Neoplasm | HSV G207 | Pediatric | Recruiting | (16, 90) | |
| HGG | HSV-1716 | Pediatric | Terminated | ||
| HGG | HSV-1716 | Adult | Completed | (10–12) | |
HGG = high grade glioma
While past and present clinical trials have demonstrated safety and some evidence of efficacy, it has become apparent that mechanisms of therapeutic resistance to oHSV exist and strategies for overcoming these mechanisms are, therefore, needed. Herein, we review the treatment challenges and mechanisms of therapeutic resistance to oHSV, and we discuss approaches to overcoming these challenges and maximizing oHSV immunotherapy (Figure 1). Failure of oHSV treatment may result from several causes. These can be broadly grouped into three categories that include those related to the viral vector, the tumor/microenvironment, and/or failure of the host immune response.
Figure 1.
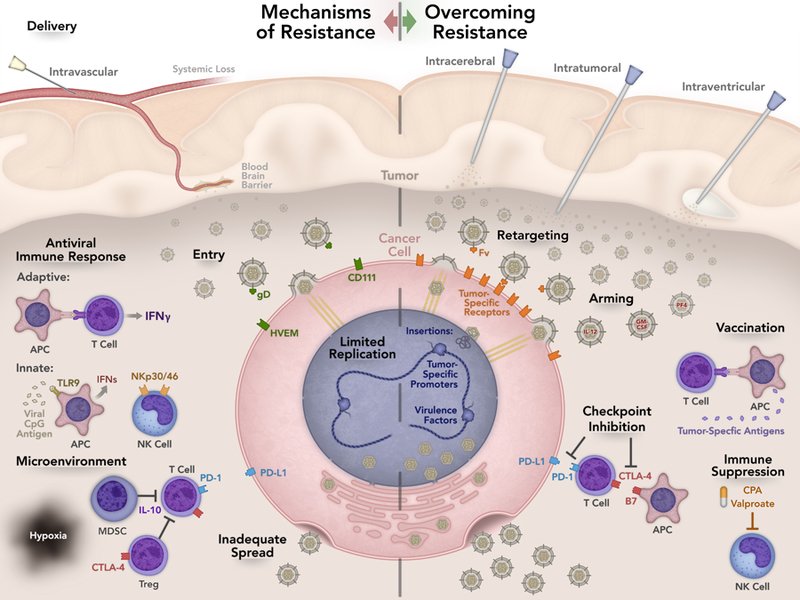
Summary of mechanisms of resistance and strategies to overcome resistance. Suboptimal delivery is one possible mechanism of treatment failure. Intratumoral injection is the most common route of delivery but other routes have been tested. Intravascular and intraventricular delivery are currently being investigated and may allow access to otherwise inaccessible tumors. Efficient entry is imperative for oHSV action. CD111 is the most efficient entry receptor but expression varies among tumors. To enhance targeting of oHSV, viral retargeting techniques to tumor specific antigens are being investigated. Viral replication and spread is necessary to prolong viral infection and effectively maximize the anti-tumor response. Host recognition of the virus and degradation within vesicles prevents viral infection. Chimeric HSVs, HDAC6 inhibition, and STAT3 expression can alter oHSV replication. The tumor microenvironment contains immune and endothelial cells, fibroblasts, pericyte cells, and neurons, astrocytes, and microglia. The extracellular matrix created by these cells aids in tumor growth, progression, invasion, and metastasis. Altering the tumor microenvironment can help modulate the anti-tumor response. The host anti-viral immune response may decrease viral efficacy and development of an anti-tumor response. Antigen presenting cells (APCs) recognize viral antigen, which leads to interferon (IFN) production resulting in less viral replication. Suppressing the innate immune response to the virus decreases IFN production resulting in greater viral efficacy. Vaccination with tumor antigens can augment the immune response to the tumor. Arming oHSVs with various cytokines in addition to checkpoint inhibition allows sustained T cell activation contributing to a robust anti-tumor response.
Viral Delivery
Delivery of the virus to the tumor is critical; suboptimal delivery is one potential mechanism of treatment failure. Various routes of delivery to CNS tumors have been tested preclinically including intravascular, intrathecal, intracerebral, and intratumoral (15). Clinical trials are investigating administration of oHSVs for CNS tumors via catheters designed to deliver virus into the region of the tumor (16) (, ), by injecting intracerebrally into the surrounding tumor resection cavity (17), and by intratumoral/peritumoral injection (, ). Systemic administration is less reliable due to leaky vasculature and limited interstitial transportation in addition to the loss of virus during first-pass elimination through the liver (18).
The blood-brain barrier (BBB) creates a unique challenge for systemic delivery of oHSV in treating CNS tumors. While the BBB may be disrupted in certain metastatic and high-grade brain tumors (19), overcoming an intact BBB via targeted disruption may allow for greater viral entry and replication within the tumor following systemic delivery. This method was effective in preclinical studies of metastatic breast cancer in the brain when combined with the systemic injection of G47∆ oHSV (20). Even if the BBB is disrupted, it is prudent to note that intravenous delivery risks neutralization of the virus through host immune defense. In the case of HSV-1, most adults are seropositive, possibly leading to greater clearance when injected systemically. However, a study performed in HSV-1 immunized mice revealed that the ability of the virus to kill tumor cells was not hindered by seropositivity (21). While this study suggests that systemically delivered viral therapies should be effective in HSV seropositive humans, the clinical implications of these findings remain to be fully elucidated, as there may be a differential effect of pre-existing antibodies on viral oncolysis and subsequent development of an anti-tumor immune response.
Lastly, off target viral replication is a potential risk. Virus that has been poorly targeted or virus that has evolved may lead to infection of host tissue. As virus replicates, the probability for mutant progeny increases. Although this has not been reported to occur clinically, it is a theoretical concern as viruses continue to be advanced to human trials (22). This concern has resulted in direct inoculation of the tumor as the favored technique. Limitations related to intratumoral injection also exist such as the inability to specifically target difficult-to-access tumors, tumors located within eloquent areas of the brain, and metastatic disease. Additionally, the risks and expense associated with complex neurosurgical procedures involved in intratumoral delivery make repeat dosing difficult. Given such limitations, different methods of delivery are being explored. Intraventricular injection may ultimately provide access to tumors that are otherwise inaccessible without the need for repeated invasive cranial procedures, but potential toxicity from this delivery approach requires additional studies (8). Preclinically, HSV 1716 delivered intracerebroventricularly into BALB/c mice led to the loss of ependymal cells and hydrocephalus, without adverse effects on mortality (23). Conversely, an earlier study of G207 injected intracerebrally or intracerebroventricularly did not lead to adverse effects on the ependymal cells (24). These divergent findings may be related to the difference in virulence between the two viral constructs and/or due to differences in HSV sensitivity of the mouse strains used in the studies.
Viral entry, replication and spread
Following delivery, the next stage that may impede action of oHSV is efficient entry of the virus into the tumor. The primary mode of entry for HSV-1 in the normal replicative process is via fusion of the viral envelope to the target cell membrane utilizing interaction of glycoprotein gD, G, B, gH and gL to cell attachment receptors with subsequent transport of genetic material to the nucleus through microtubular apparatus (25). However, another, less common, mode of entry is via endocytic vesicles that fuse with viral envelopes (26). In this type of entry, it is possible for the endosome to recognize the virus before it travels to the nucleus (27), thereby preventing lysis and resultant viral spread. Additionally, there is evidence that virus particles undergo degradation within the pre-lysosomal vesicles, resulting in a failure of viral infection (28).
For the membrane to begin fusion, a receptor binding protein must be activated by the virus. The three classes of membrane receptors that have proven important for HSV-1 entry are heparin sulfate proteoglycan, herpes virus entry mediator (HVEM, HveA, TNFRSF14, ATAR and CD270), and nectin-1 (CD111, poliovirus receptor-related-1) (26). One of the most abundant heparin sulfate proteoglycans is the syndecan family. Syndecans act as co-receptors on the cell surface and attach to glycoproteins that have been enveloped by HSV-1 (29) acting to stabilize the virus and allow for interactions of its glycoproteins with the receptors directly involved in fusion. HVEM is a member of the tumor necrosis factor receptor family and is mainly expressed on T and B lymphocytes. CD111, the most efficient HSV entry receptor, is expressed in many cell types, including neurons and brain tumors including glioma, ependymoma, medulloblastoma, and supratentorial primitive neuroectodermal tumor (30). Further, in both adult glioblastoma and pediatric brain tumor xenografts, CD111 expression was inversely correlated with the half maximal lethal dose (LD50) of oHSV (30). These findings suggest that expression levels of CD111 may predict tumor cell killing by oHSV and that CD111 expression may be a useful biomarker to identify individuals most likely to benefit from the therapy. While CD111 expression similarly predicted sensitivity to oHSV in several extracranial solid tumor models, effective cell infection and killing by oHSV in neuroblastoma was seen even at low levels of CD111 (31). Another study showed that induction of CD111 on malignant peripheral nerve sheath tumors that initially expressed low CD111 did not improve cell killing (32). Therefore, CD111 expression may only predict viral infectivity in certain tumor types. Possibly, other receptors or entry mechanisms described above may ultimately be involved.
Retargeting oHSV
To enhance tumor cell specific targeting and to reduce the risk of adversely harming normal cells, a myriad of viral retargeting techniques are being tested. The two-primary methods for retargeting oHSV have thus far focused on altering the glycoprotein receptor specificity or modifying transcription through the insertion of tumor-specific promoters. One example of inserting a gene coding for a sequence that is tumor-specific has been to replace the capsid epitope for HVEM with single-chain Fv fragments that target cell receptors only on the tumor, which increases infection selectivity (33). Human epidermal growth factor receptor 2 (HER2) has been studied as a target receptor and achieved specificity by deleting the gD sequences involved in binding to HVEM and CD111 and replacing them with single-chain variable-fragment antibody (34). In an orthotopic mouse model of HER2-engineered human glioma, epidermal growth factor receptor-retargeted virus led to a complete response in 73% of mice (35). Recently, Campadelli-Fiume and colleagues (36) developed an IL-12-armed, HER2-retargeted, fully-virulent oHSV that carries no deletion/mutation (R-115). The virus was safe and effective at inhibiting the growth of primary HER2+ lung carcinoma in mice and induced a systemic immunotherapeutic vaccine response in survivors. The safety of such an approach will have to be tested in brain tumor models; nevertheless, this study highlights the potential of fully virulent retargeted oHSVs.
Other retargeted, mutant oHSVs have been developed. Retargeting G207 through regulation of the virulence gene by a Musashi1 promoter in an in vivo glioma model resulted in increased therapeutic efficacy without altering the safety profile (37). R5141 was developed to only enter and replicate in cells that express IL-13Rα2 and not through CD111 (38); in addition, R5181, containing a urokinase plasminogen activator insertion, was capable of entering cells expressing the non-viral, urokinase plasminogen activator receptor (39). These results provide evidence that viruses may be consistently targeted to enter select populations within tumors. While promising, advanced combinations of targets will ultimately be required to deal with inter and intratumoral heterogeneity.
An example of modifying transcription through the insertion of tumor-specific promoter is oHSV rQNestin34.5v.2 (rQNestin), which contains a reinsertion of one of the two deleted neurovirulence genes (γ134.5) under transcriptional control of a nestin gene enhancer/promoter instead of the constitutive viral promoter (40). Thus, transcription of these viral replication promotors increases in the targeted tumor tissue. This oHSV is being tested in a Phase 1 clinical trial in GBM patients (). rQNestin is the only ICP34.5-positive oHSV to be used in brain tumor patients, and safety of this approach will need to be confirmed. Recently, rQNestin was re-engineered to possess GADD34, the human version of ICP34.5 with expression selective to GBM. This oHSV, termed NG34, resulted in similar replication and efficacy in mice as compared to its parent oHSV, but with less neurovirulence (41). The above mentioned studies demonstrate the capability of oHSV to be transcriptionally retargeted to specifically infect tumor cells.
Improving replication
Viral replication is necessary to attain appropriate viral titers and prolong viral infection to maximize the anti-tumor response. Multiple modifications and deletions attenuate viral replication of oHSV (42), and an unfavorable microenvironment including the extracellular matrix (ECM), hypoxia, and innate immune cells may contribute to decreased replication. To improve replication without increasing neurovirulence, Cassady, et al. developed chimeric HSVs that express the human cytomegalovirus (HCMV) PKR-evasion genes, TRS1 (C130) and IRS1 (C134) (43). Transfer of these genes from HCMV to the ∆γ134.5 HSV (HCMV/HSV-1) resulted in replication and virus protein production similar to that of wild-type HSV (44). They also noted that introduction of the IRS1 gene did not restore neurovirulence and led to increased survival in an in vivo xenograft model of GBM. Finally, they found that some mice with TRS1 insertion died compared to IRS1, suggesting limited neurovirulence. A clinical trial employing C134 with the IRS1 insertion is forthcoming (Table 2) and has received FDA approval.
In a similar effort to enhance replication, Nakashima et al. (45) inhibited histone deacetylase 6 (HDAC6), which may have antiviral functions leading to viral autophagy instead of viral shuttling to the nucleus for replication. Inhibition of HDAC6 resulted in increased replication of oHSV rQNestin and rHSVQ1 in glioma cell lines and increased viral shuttling to the nucleus. Inhibition of HDAC6 also led to increased viral titers in vivo, again suggesting improved replication (45). Beyond HDAC6 inhibition, recent evidence has emerged to suggest that the signal transducer and activator of transcription 3 (STAT3) activation may enhance replication (46). Over-expression of STAT3 increased oHSV replication while decreased levels of STAT3 impaired replication of oHSV (46). More studies will ultimately be needed to clarify the clinical relevance of such therapeutic approaches.
Tumor Features
Brain tumor genotypic/phenotypic heterogeneity poses an innate clinical problem. Tumors may differ in their expression of HSV entry molecules or foster a local microenvironment that is not conducive to viral replication and spread. Intact innate responses of tumor cells, normal neuroglia, or immune cells within the tumor microenvironment, pose a barrier to viral replication. The microenvironment contains a variety of cells including immune and endothelial cells, host fibroblasts, and pericyte cells as well as neurons, astrocytes, and microglia recruited to the growing tumor. These cells secrete an ECM which provides structural support to cancer cells. In addition to structural support, the matrix is also responsible for aiding tumor growth, progression, neovasculature, invasion, and metastasis (reviewed in (47)). Given such a complicated milieu, it is not surprising that numerous factors within the microenvironment can both aid and modulate the anti-tumor response. As a clever approach to improve spread of oHSV in brain tissue, Dmitrieva et al. (48) generated an oHSV (OV-Chase) that expressed Chondroitinase-ABC (Chase-ABC), which is a bacterial enzyme that can remove chondroitin sulfate glycoso-aminogylcans from proteoglycans. Secreted and membrane-bond forms of chondroitin sulfate glycosoaminoglycans link to hyaluronan, forming the major ECM in the brain. By degrading the ECM, they demonstrated increased virus spread throughout intracranial tumors (48). Therefore, the microenvironment must be considered when developing novel oHSV.
Among this myriad of microenvironmental factors, tissue hypoxia plays a crucial role in the development and progression of malignant gliomas having been associated with tumor growth, aggressiveness, invasion and spread (49). Friedman, et al. reported that GBM xenograft lines exposed to hypoxia led to a decrease in oHSV infectivity, replication, and cytotoxicity compared to wild-type virus (50); however, they were able to successfully employ the chimeric HCMV/HSV-1 virus, which has improved late viral protein synthesis during states of hypoxia (44). This second-generation virus demonstrated similar efficacy to wild-type HSV-1 in targeting glioma cells and glioma stem-like cells during hypoxia. This effect was at least partially due to p38 mitogen-activated protein kinase signaling pathway, which enhances late viral gene expression. These findings differ from those reported previously by Aghi et al. (51), who found that G207 replication increased in U87 glioma cells under hypoxia. One explanation for this difference may be the use of a single, long-term established tumor cell line in the latter study. Further, when the viral ICP4 gene in glioma cells was placed under the control of an HIF-responsive promoter of the HSV-1 d120, which contains partial ICP4 gene deletions (52), HIF-HSV resulted in similar replication in hypoxia and normoxia. Importantly, wild-type ICP4 was restored under the control of the HIF promoter creating potential toxicity in patients that could result in encephalitis. Together, these results suggest that oxygen tension should be considered when developing next-generation viral vectors for clinical therapy.
Host antiviral immune response; improving the immune microenvironment
The host immune response poses another obstacle to effective oHSV therapy. In order to maximize the efficacy and durability of clinical responses to oHSV, the development of an anti-tumor immune response is likely critical; while an antiviral immune response may decrease efficacy. Antigen-presenting cells (APCs) of the innate immune system, particularly dendritic cells, first recognize viral nucleotides and respond to viral infection by producing interferons (IFNs) in order to create a less permissive environment for viral replication (53). Indeed, early HSV replication can be inhibited by treatment with IFN. Virus is recognized by APCs through the binding of pattern recognition receptor toll-like receptor 9 (TLR9) with unmethylated CpG motifs present in the HSV genome, which leads to a downstream signaling cascade resulting in IFN production (54). In addition to IFNs, virus recognition also leads to the production and release of TNFα, IL-1 beta (IL-1β), IL-6, IL-12, chemokine (C-C motif) ligand 2 (CCL2), CCL7, CCL8, CCL9, chemokine (C-X-C motif) ligand 1 (CXCL1), CXCL4, and CXCL5 (55), which further augments the anti-viral response. When the innate immune system targets and removes the virus, it limits the oncolytic effects of the virus. Viral replication and spread within the tumor is also compromised due to the destruction of virally infected cells by infiltrating inflammatory cells (56). Specifically, macrophages can infiltrate and engulf the virus (57), whereas NK cells may kill infected cells prior to the release of functional progeny virions leading to ineffective viral propagation and a blunted anti-tumor response.
In conjunction with innate immunity, adaptive immunity plays a role in oncolytic viral infection. Following infection, viral antigens may be presented to cytotoxic T cells via APCs prior to tumor associated antigens (58). In this way, activation of virus-specific T cells contributes to premature viral clearance. These T cells also contribute to the production of IFN-γ, which has been shown to limit viral spread and protect against viral challenge. B cells have also been shown to play a role in anti-viral immunity to HSV. For example, in mice immunocompromised as a result of cyclophosphamide (CPA) treatment, HSV-1 antiserum transfer resulted in 100% survival in mice compared to mice that received a control serum (59).
Recent studies suggest the adaptive immune system also plays a critical role in the anti-tumor response, independent of replication (60). C134 and its control ∆γ134.5 oHSV prolonged survival in immunocompetent mice bearing a murine malignant glioma, but not immunocompromised mice. The authors suggest this effect was due to the lack of a T cell response in immunodeficient mice, showing that adaptive immunity was integral for the anti-tumor response.
To overcome some of the aforementioned challenges, investigators have employed drugs to block the migration or activity of pro-inflammatory cells. One example is pretreatment with CPA, a chemotherapy and immunomodulatory drug. Pretreatment with CPA decreased HSV clearance and increased replication in vivo by inhibiting innate immunity (61). Currently, a clinical trial is recruiting patients with recurrent glioma to investigate the effectiveness of combining rQNestin with CPA (). Another example is to use anti-inflammatory cytokines to dampen the anti-viral response. Pretreatment with TGF-β, an immunosuppressant, led to a decrease in NK cytolytic activity and increased viral titers in an in vitro model of GBM (62). Further, TGF-β treatment prior to oncolytic HSV inhibited tumor growth and led to prolonged survival in a mouse model of GBM. Finally, inhibiting angiogenesis by inhibiting vascular endothelial growth factor (VEGF) enhanced the anti-tumor effects of oHSV (63).
Clearly, the tumor immune microenvironment greatly influences therapeutic responses to oHSV. For example, modulation of NK cells can both positively and negatively affect viral therapy. NK cells promote viral clearance but are also involved in anti-tumor immunity. In preclinical studies, following oHSV infection, NK cells rapidly infiltrated the tumor and led to premature clearance of the virus in a GBM model (64). Depletion of NK cells in mice bearing GBM resulted in higher viral titers and increased survival compared to control mice. The anti-viral effects were dependent on the NK cytotoxicity receptors NKp30 and NKp46. Importantly, human GBMs up-regulate NK cell receptor ligands, suggesting that in this tumor type, NK cells are detrimental to oHSV therapy (64). Similarly, inhibition of NK cells by valproic acid, a HDAC inhibitor, led to a decrease in both NK and macrophage recruitment in a murine GBM tumor model, thereby suppressing the killing of oHSV-infected tumor cells (65). Pretreatment with low-dose CPA increased viral replication and decreased HSV-related immune cell infiltration into the tumor (61). These results suggest that pharmacologically altering the tumor microenvironment, specifically making it temporarily immunosuppressive, may enhance viral oncolysis. However, making the tumor microenvironment more immunosuppressive may be detrimental to the development of an anti-tumor immune response. The challenge for researchers is discovering the ideal balance to maximize the therapeutic effect.
Myeloid-derived suppressor cells also contribute to the immunosuppressive tumor microenvironment through inhibition of T cell activity. These cells infiltrate the tumor microenvironment and lead to an unfavorable microenvironment and inhibit an anti-tumor immune cell response likely through production of anti-inflammatory cytokines such as IL-10 (66). In support of this concept, M002, which produces murine IL-12 (67), led to a decrease in myeloid-derived suppressor cells in a sarcoma model in immunocompetent mice leading to a more favorable environment for tumor killing (68).
Arming oHSV – the addition of therapeutic transgenes
HSV has a large genome (152 kilobase [kb]) with approximately 30kb of genes that are non-essential for productive replication in tumor cells (69). These genes can be removed to provide room for packaging genetically engineered viral DNA (containing foreign genes designed to augment anti-tumor effects) into infectious capsids. The different types of genes that have been utilized include genes that can influence the tumor microenvironment, induce apoptosis, enzymes that activate pro-drugs, and reporter genes (47).
Cytokine insertions have been investigated due to their ability to modulate the anti-tumor response. Cytokines enhance the activity of cytotoxic T cells, leading to greater anti-tumor efficacy. The first clinically approved oHSV, T-Vec, contains an insertion of granulocyte-macrophage colony-stimulating factor, which has been shown to induce myeloid precursor cells and recruit dendritic cells (13). Preclinically, murine IL-12 producing M002 virus (67) improved survival in mice with intracranial gliomas and M002 outperformed first-generation oHSV in vivo in patient-derived xenograft high-grade brain tumors (30, 70). Following the preclinical evaluation of the safety of M032, which encodes a functional human IL-12, a Phase 1 clinical trial was designed and is ongoing in adults with recurrent GBM () (71). Another murine IL-12 expressing virus, G47Δ-mIL12, was effective at targeting GBM stem cells and the tumor microenvironment by increasing IFN-γ release, inhibiting angiogenesis, and reducing Tregs (72). When G47Δ-mIL12 was combined with axitinib, a VEGF receptor tyrosine kinase inhibitor, the combination therapy enhanced anti-tumor efficacy in immunodeficient and immunocompetent orthotopic GBM murine models compared to either therapy alone (73).
Another arming strategy involves inserting anti-angiogenic transgenes into the oHSV vector as increased vascularity and microvascular proliferation are hallmarks of malignant gliomas (74). G47∆ armed with platelet factor-4, a member of the CXC chemokine family has been shown to inhibit angiogenesis (75). Specifically, expression of platelet factor-4 resulted in inhibition of tumor growth and increased survival without altering viral replication in two neural tumor models in vivo. Similarly, vasculostatin-expressing Rapid Anti-angiogenesis Mediated By Oncolytic (RAMBO) virus resulted in a significant reduction in angiogenesis compared to control virus in vivo (74). Mice with intracranial gliomas treated with RAMBO had increased survival compared to control mice, and RAMBO led to a significant reduction in microvascular density, suggesting the virus hindered recruitment of the vascular supply to the tumor. These data demonstrate that modulating the tumor vasculature through armed oncolytic viruses can aid in reducing tumor growth and progression.
Recently, a novel oHSV was developed to express a single-chain antibody against programmed cell death protein 1 (PD-1) (76). When injected into two preclinical mouse models of GBM, the virus led to an effective anti-tumor response. Importantly, when immunocompetent mice were re-challenged with GBM, they were able to reject the challenge, suggesting the presence of anti-tumor memory. These data support the use of oHSV armed with checkpoint inhibitors.
Checkpoint inhibition
Immune checkpoints are self-regulatory pathways that ensure the immune system is regulated and can discriminate between self and non-self (77). T cells, which play a vital role in immune function, express a number of immune checkpoint receptors, and, thus, have become the focus of checkpoint inhibition therapy (77). These receptors regulate T cell function in response to various stimuli including infection and tumor antigens (77). When an antigen is recognized by a T-cell in the correct inflammatory context, the T cell performs effector functions that amplify the immune response, including direct cytolysis and production of IFNs that inhibit tumor cell replication. The IFNs, however, also increase checkpoint molecule expression (78), which may downregulate the T cell response and activate T regulatory cells (Tregs). Clinically, increased Tregs within the tumor are associated with a poor prognosis (79). Of note, tumor cells infected with virus also produce IFNs and thus, have increased checkpoint expression, providing a rational basis for the combination of oHSV and checkpoint inhibitor therapy.
Tumor cells have the ability to evade immune recognition through checkpoint pathway activation (80). When the tumor cell is recognized by T cells, checkpoint protein expression (e.g. Cytotoxic T Lymphocyte Antigen-4 [CTLA-4], Programmed Cell Death protein-1 and Programmed Death Ligand 1 [PD-1/PD-L1]) may be upregulated and can competitively engage specific signaling receptors expressed by APCs, activating the checkpoint pathway and essentially inactivating the T-cell response against the tumor (80). When these proteins are present on the tumor or within the microenvironment, they allow tumor cells to evade immune cell recognition. Members of the indoleamine 2,3-dioxygenase (IDO) family are able to convert tryptophan to kynurenine required to maintain the physiologic differentiation and maturation of Tregs (81). Specific inhibitors that block the actions of the IDO family members diminish the differentiation of T cells into Tregs, thereby minimizing their negative regulatory effect on ongoing immune responses. Therefore, a strategy to increase the virus-induced T-cell anti-tumor response can be designed via the rational application of checkpoint inhibitors. Currently, there are a number of checkpoint inhibitors that have advanced to clinical trials including those for CTLA-4, PD-1/PD-L1, and IDO (82).
Due to the heterogeneous and ever-evolving tumor environment, it is possible for tumor cells to adapt to checkpoint inhibitor therapy when used alone. These adaptations may include down-regulation of checkpoint protein ligands, changes in cytokine signaling, and utilization of alternative pathways (83). Brain tumors pose additional challenges due to systemic immune dysfunction, reduced levels of circulating lymphocytes, and/or decreased penetration of drugs through the BBB (84). In the first randomized phase 3 clinical trial of PD-1/PD-L1 pathway inhibition, nivolumab, an anti-PD-1 monoclonal antibody, failed to prolong overall survival in recurrent GBM patients when used alone (). Combining checkpoint inhibition with oHSV may lead to a sustained anti-tumor immune response due to the release of tumor antigens following virus-mediated tumor cell lysis (82). In a syngeneic mouse glioma model, the triple combination of oHSV G47Δ-mIL12 with anti-PD-1 and anti-CTLA-4 checkpoint inhibitors cured most mice, and surviving mice that were re-challenged with tumor cells did not succumb to tumors demonstrating the effectiveness of the treatment on immunologic memory (85). Recently, clinical trials have sought to investigate the impact of combined oncolytic virotherapy with checkpoint inhibition. In the first randomized open-label Phase 2 study combining oHSV T-Vec and CTLA-4 inhibitor ipilimumab, the combination improved the objective response rate in patients with advanced melanoma to 39% from 18% for those that received ipilimumab alone (86). The combination therapy led to greater anti-tumor activity without additional adverse events and provides evidence for continued investigation of oHSV in parallel with checkpoint inhibitors.
Radiation
Combination therapies may increase the therapeutic effects of treatment by altering the microenvironment in a way that amplifies the anti-tumor immune response. One clinically relevant example is via the addition of ionizing radiation. Radiation may synergize with oHSV by increasing the innate and adaptive anti-tumor immune response. Tumor antigens are released following radiation leading to an increase in their presentation and resulting in chemokine induction and subsequent effector T cell recruitment thereby priming cytotoxic T cells specific to the tumor and increasing T cell function in tumors (87). Preclinical studies demonstrated that oHSV-1 treatment followed by radiation increased viral replication and efficacy and prolonged survival in a malignant glioma model (88). Furthermore, combining radiation with oHSV may be a way to induce an anti-tumor immune response to distant sites of metastatic disease not specifically targeted by the virus. G207 combined with a single 5 Gray dose of radiation was tested in adults with recurrent high-grade gliomas (89). The combination treatment was safe and six of nine patients had stable disease or partial response. The concept of combining oHSV with radiation is being further tested in an ongoing Phase 1 clinical trial of G207 with a single dose of radiation in children with recurrent supratentorial brain tumors (Table 2) (90).
Vaccination
Viral replication resulting in tumor lysis provides a tumor debris field that makes tumor antigens accessible to dendritic cells and other APCs. These cells are activated by the presence of viral nucleic acids that bind to TLR9 and induce cytokines that promote the priming of cytotoxic T cells directed against the tumor. This stimulatory environment may also prime an anti-viral immune response that results in viral clearance. Therefore, strategies for directing the immune response to specific tumor antigens to enhance the anti-tumor effect while mitigating viral clearance should be explored.
A promising approach is the use of cancer vaccines to prime cytotoxic T cells against specific tumor antigens (91), which can be further augmented by virotherapy. Cancer vaccines can be grouped into different categories including cell vaccines, genetic vaccines, and protein/peptide vaccines (92). Cell vaccines are prepared using either patient-derived tumor cells or dendritic cells. In the case of dendritic cells, patients are treated with dendritic cells that have been loaded with tumor-associated antigens in order to provoke an anti-tumor immune response (93). Another strategy is to use nucleic acids encoding multiple tumor antigens as DNA or RNA, which may be delivered as either the naked nucleic acid sequence or packaged within synthetic- or viral- vectors (94, 95). Nucleic acid-based vaccines can be programmed to have innate stimulatory capacity (96), or may even encode adjuvants, such as IL-12, that can be produced in situ (97). Protein/peptide vaccines provide the advantage that the antigen does not require production in situ but can be processed and presented by dendritic cells for T-cell priming. However, protein and peptides alone are weakly immunogenic and require packaging into particles, such as lipid or polymer-based systems, with specific adjuvants (e.g. TLR agonists) that promote dendritic cell uptake and induce the production of IFNs needed to prime T-cell immunity (98). In this regard, polymer carriers of TLR-7 and −8 agonists linked to protein/peptide antigens that self-assemble into nanoparticles have been developed as an effective strategy for delivering antigens to dendritic cells to induce high magnitude and quality T cell immunity (99). Such a strategy may be used to safely prime cytotoxic T cell responses prior to oncolytic virotherapy treatment thereby maximizing the tumor-directed immune response.
A central challenge for vaccination approaches is selecting appropriate antigens that direct the T-cell response to tumors but avoid off-target effects. Suitable antigens include tumor-associated self-antigens, which are germ-line encoded and preferentially expressed on tumor cells; and “neoantigens,” which are mutant self-proteins that are necessarily tumor-specific (100). A major focus of ongoing research and development activities is to develop optimal strategies for maximizing the breadth and magnitude of anticancer T-cell immunity using such technologies that can be scaled as patient-specific, on-demand therapies and determining ideal combination therapies to use with tumor vaccination such as virotherapy.
Conclusions:
Therapeutic resistance and treatment challenges related to viral delivery, entry, and replication must be overcome before the promise of oHSV can be fully realized. Oncolytic HSV have many advantages as compared to other viral vectors and may ultimately be engineered and/or used in combination with other treatment strategies in an effort to overcome resistance mechanisms and maximize clinical response. The various approaches described above hold great promise, and if successful, these treatments may be extended to all solid tumors, both within and outside the CNS.
Acknowledgements/Funding:
This research was supported in part by grants from the Food and Drug Administration Office of Orphan Products Development (R01FD005379), the Department of Defense (W81XWH-15–1-0108) Rally Foundation for Childhood Cancer Research, Hyundai Hope on Wheels, and the Kaul Pediatric Research Institute to GKF and R01CA217179 to JMM and GYG. JDB was supported by the UAB Medical Scientist Training Program (MSTP) and an American Brain Tumor Association summer fellowship. SKT was supported by the National Cancer Institute of the National Institutes of Health under Award Number T32CA183926. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health, the U.S. FDA or the Department of Defense. The authors apologize to colleagues we couldn’t cite given limitations on the number of references.
Footnotes
Competing Interests:
Drs. Markert, Whitley and Gillespie are founders of and own stock and stock options (<8% interest) in Aettis, Inc., a biotech company that holds intellectual property surrounding oncolytic HSV. Dr. Gillespie currently serves as one of five unpaid members of the Board of Directors for Aettis, Inc. Drs Markert, Whitley, and Gillespie were also founders of and owned stock and stock options (<8%) in Catherex Inc., a biotechnology company that had licensed additional intellectual property related to oHSV. Catherex, Inc., was sold to Amgen, Inc., on December 18, 2015, and they no longer participate in any decision making or have any control of any aspect of Catherex or Amgen, although they did receive proceeds from the sale of the company. Dr. Gillespie has served as a paid advisor to a Program Project at the Ohio State University that seeks to find improved methods for application of distinct oHSV to treat localized and metastatic cancers. This is generally, but not specifically, related to the subject matter of this investigation. Dr. Bernstock has positions/equity in CITC Ltd. Drs. Bernstock, Ishizuka, and Lynn have positions/equity Avidea Technologies. Dr.Whitley is a member of the Board of Directors of Gilead Sciences. The remaining authors declare that they have no conflict of interest.
References:
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin 2016. Jan-Feb;66(1):7–30. [DOI] [PubMed] [Google Scholar]
- 2.Ostrom QT, Gittleman H, Liao P, Vecchione-Koval T, Wolinsky Y, Kruchko C, et al. CBTRUS Statistical Report: Primary brain and other central nervous system tumors diagnosed in the United States in 2010–2014. Neuro Oncol 2017. November 6;19(suppl_5):v1–v88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Krull KR, Hardy KK, Kahalley LS, Schuitema I, Kesler SR. Neurocognitive Outcomes and Interventions in Long-Term Survivors of Childhood Cancer. J Clin Oncol 2018. July 20;36(21):2181–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shah AC, Benos D, Gillespie GY, Markert JM. Oncolytic viruses: clinical applications as vectors for the treatment of malignant gliomas. J Neurooncol 2003. December;65(3):203–26. [DOI] [PubMed] [Google Scholar]
- 5.Friedman GK, Pressey JG, Reddy AT, Markert JM, Gillespie GY. Herpes simplex virus oncolytic therapy for pediatric malignancies. Mol Ther 2009. July;17(7):1125–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mineta T, Rabkin SD, Yazaki T, Hunter WD, Martuza RL. Attenuated multi-mutated herpes simplex virus-1 for the treatment of malignant gliomas. Nat Med 1995. September;1(9):938–43. [DOI] [PubMed] [Google Scholar]
- 7.Martuza RL, Malick A, Markert JM, Ruffner KL, Coen DM. Experimental therapy of human glioma by means of a genetically engineered virus mutant. Science (New York, NY) 1991. May 10;252(5007):854–6. [DOI] [PubMed] [Google Scholar]
- 8.Markert JM, Medlock MD, Rabkin SD, Gillespie GY, Todo T, Hunter WD, et al. Conditionally replicating herpes simplex virus mutant, G207 for the treatment of malignant glioma: results of a phase I trial. Gene Ther 2000. May;7(10):867–74. [DOI] [PubMed] [Google Scholar]
- 9.Streby KA, Geller JI, Currier MA, Warren PS, Racadio JM, Towbin AJ, et al. Intratumoral Injection of HSV1716, an Oncolytic Herpes Virus, Is Safe and Shows Evidence of Immune Response and Viral Replication in Young Cancer Patients. Clin Cancer Res 2017. July 15;23(14):3566–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rampling R, Cruickshank G, Papanastassiou V, Nicoll J, Hadley D, Brennan D, et al. Toxicity evaluation of replication-competent herpes simplex virus (ICP 34.5 null mutant 1716) in patients with recurrent malignant glioma. Gene Ther 2000. May;7(10):859–66. [DOI] [PubMed] [Google Scholar]
- 11.Papanastassiou V, Rampling R, Fraser M, Petty R, Hadley D, Nicoll J, et al. The potential for efficacy of the modified (ICP 34.5(−)) herpes simplex virus HSV1716 following intratumoural injection into human malignant glioma: a proof of principle study. Gene Ther 2002. March;9(6):398–406. [DOI] [PubMed] [Google Scholar]
- 12.Harrow S, Papanastassiou V, Harland J, Mabbs R, Petty R, Fraser M, et al. HSV1716 injection into the brain adjacent to tumour following surgical resection of high-grade glioma: safety data and long-term survival. Gene Ther 2004. November;11(22):1648–58. [DOI] [PubMed] [Google Scholar]
- 13.Andtbacka RH, Kaufman HL, Collichio F, Amatruda T, Senzer N, Chesney J, et al. Talimogene Laherparepvec Improves Durable Response Rate in Patients With Advanced Melanoma. J Clin Oncol 2015. September 1;33(25):2780–8. [DOI] [PubMed] [Google Scholar]
- 14.Fukuhara H, Ino Y, Todo T. Oncolytic virus therapy: A new era of cancer treatment at dawn. Cancer Sci 2016. October;107(10):1373–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Foreman PM, Friedman GK, Cassady KA, Markert JM. Oncolytic Virotherapy for the Treatment of Malignant Glioma. Neurotherapeutics 2017. April;14(2):333–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bernstock JD, Wright Z, Bag AK, Gessler F, Gillespie GY, Markert JM, et al. Stereotactic Placement of Intratumoral Catheters for Continuous Infusion Delivery of Herpes Simplex Virus −1 G207 in Pediatric Malignant Supratentorial Brain Tumors. World Neurosurg 2018 November 24 2019;122:e1592–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Markert JM, Liechty PG, Wang W, Gaston S, Braz E, Karrasch M, et al. Phase Ib trial of mutant herpes simplex virus G207 inoculated pre-and post-tumor resection for recurrent GBM. Mol Ther 2009. January;17(1):199–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pond SM, Tozer TN. First-pass elimination. Basic concepts and clinical consequences. Clin Pharmacokinet 1984. Jan-Feb;9(1):1–25. [DOI] [PubMed] [Google Scholar]
- 19.Sarkaria JN, Hu LS, Parney IF, Pafundi DH, Brinkmann DH, Laack NN, et al. Is the blood-brain barrier really disrupted in all glioblastomas? A critical assessment of existing clinical data. Neuro Oncol 2018. January 22;20(2):184–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu R, Martuza RL, Rabkin SD. Intracarotid delivery of oncolytic HSV vector G47Delta to metastatic breast cancer in the brain. Gene Ther 2005. April;12(8):647–54. [DOI] [PubMed] [Google Scholar]
- 21.Zhu H, Su Y, Zhou S, Xiao W, Ling W, Hu B, et al. Immune analysis on mtHSV mediated tumor therapy in HSV-1 seropositive mice. Cancer Biol Ther 2007. May;6(5):724–31. [DOI] [PubMed] [Google Scholar]
- 22.Maroun J, Munoz-Alia M, Ammayappan A, Schulze A, Peng KW, Russell S. Designing and building oncolytic viruses. Future Virol 2017. April;12(4):193–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kesari S, Lasner TM, Balsara KR, Randazzo BP, Lee VM, Trojanowski JQ, et al. A neuroattenuated ICP34.5-deficient herpes simplex virus type 1 replicates in ependymal cells of the murine central nervous system. J Gen Virol 1998. March;79 ( Pt 3):525–36. [DOI] [PubMed] [Google Scholar]
- 24.Sundaresan P, Hunter WD, Martuza RL, Rabkin SD. Attenuated, replication-competent herpes simplex virus type 1 mutant G207: safety evaluation in mice. J Virol 2000. April;74(8):3832–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou G, Avitabile E, Campadelli-Fiume G, Roizman B. The domains of glycoprotein D required to block apoptosis induced by herpes simplex virus 1 are largely distinct from those involved in cell-cell fusion and binding to nectin1. J Virol 2003. March;77(6):3759–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Spear PG. Herpes simplex virus: receptors and ligands for cell entry. Cellular microbiology 2004. May;6(5):401–10. [DOI] [PubMed] [Google Scholar]
- 27.Brencicova E, Diebold SS. Nucleic acids and endosomal pattern recognition: how to tell friend from foe? Front Cell Infect Microbiol 2013;3:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Laquerre S, Anderson DB, Stolz DB, Glorioso JC. Recombinant herpes simplex virus type 1 engineered for targeted binding to erythropoietin receptor-bearing cells. J Virol 1998. December;72(12):9683–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Trybala E, Liljeqvist JA, Svennerholm B, Bergstrom T. Herpes simplex virus types 1 and 2 differ in their interaction with heparan sulfate. J Virol 2000. October;74(19):9106–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Friedman GK, Bernstock JD, Chen D, Nan L, Moore BP, Kelly VM, et al. Enhanced Sensitivity of Patient-Derived Pediatric High-Grade Brain Tumor Xenografts to Oncolytic HSV-1 Virotherapy Correlates with Nectin-1 Expression. Sci Rep 2018. September 17;8(1):13930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang PY, Swain HM, Kunkler AL, Chen CY, Hutzen BJ, Arnold MA, et al. Neuroblastomas vary widely in their sensitivities to herpes simplex virotherapy unrelated to virus receptors and susceptibility. Gene Ther 2016. February;23(2):135–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jackson JD, McMorris AM, Roth JC, Coleman JM, Whitley RJ, Gillespie GY, et al. Assessment of oncolytic HSV efficacy following increased entry-receptor expression in malignant peripheral nerve sheath tumor cell lines. Gene Ther 2014. November;21(11):984–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Miest TS, Cattaneo R. New viruses for cancer therapy: meeting clinical needs. Nat Rev Microbiol 2014. January;12(1):23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Menotti L, Cerretani A, Hengel H, Campadelli-Fiume G. Construction of a fully retargeted herpes simplex virus 1 recombinant capable of entering cells solely via human epidermal growth factor receptor 2. J Virol 2008. October;82(20):10153–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Uchida H, Marzulli M, Nakano K, Goins WF, Chan J, Hong CS, et al. Effective treatment of an orthotopic xenograft model of human glioblastoma using an EGFR-retargeted oncolytic herpes simplex virus. Mol Ther 2013. March;21(3):561–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Leoni V, Vannini A, Gatta V, Rambaldi J, Sanapo M, Barboni C, et al. A fully-virulent retargeted oncolytic HSV armed with IL-12 elicits local immunity and vaccine therapy towards distant tumors. PLoS Pathog 2018. August;14(8):e1007209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kanai R, Tomita H, Shinoda A, Takahashi M, Goldman S, Okano H, et al. Enhanced therapeutic efficacy of G207 for the treatment of glioma through Musashi1 promoter retargeting of gamma34.5-mediated virulence. Gene Ther 2006. January;13(2):106–16. [DOI] [PubMed] [Google Scholar]
- 38.Zhou G, Roizman B. Construction and properties of a herpes simplex virus 1 designed to enter cells solely via the IL-13alpha2 receptor. Proc Natl Acad Sci U S A 2006. April 4;103(14):5508–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kamiyama H, Zhou G, Roizman B. Herpes simplex virus 1 recombinant virions exhibiting the amino terminal fragment of urokinase-type plasminogen activator can enter cells via the cognate receptor. Gene Ther 2006. April;13(7):621–9. [DOI] [PubMed] [Google Scholar]
- 40.Kambara H, Okano H, Chiocca EA, Saeki Y. An oncolytic HSV-1 mutant expressing ICP34.5 under control of a nestin promoter increases survival of animals even when symptomatic from a brain tumor. Cancer Res 2005. April 1;65(7):2832–9. [DOI] [PubMed] [Google Scholar]
- 41.Nakashima H, Nguyen T, Kasai K, Passaro C, Ito H, Goins WF, et al. Toxicity and Efficacy of a Novel GADD34-expressing Oncolytic HSV-1 for the Treatment of Experimental Glioblastoma. Clin Cancer Res 2018. June 1;24(11):2574–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wakimoto H, Kesari S, Farrell CJ, Curry WT Jr., Zaupa C, Aghi M, et al. Human glioblastoma-derived cancer stem cells: establishment of invasive glioma models and treatment with oncolytic herpes simplex virus vectors. Cancer research 2009. April 15;69(8):3472–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cassady KA. Human cytomegalovirus TRS1 and IRS1 gene products block the double-stranded-RNA-activated host protein shutoff response induced by herpes simplex virus type 1 infection. J Virol 2005. July;79(14):8707–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Friedman GK, Nan L, Haas MC, Kelly VM, Moore BP, Langford CP, et al. gamma(1)34.5-deleted HSV-1-expressing human cytomegalovirus IRS1 gene kills human glioblastoma cells as efficiently as wild-type HSV-1 in normoxia or hypoxia. Gene Ther 2015. April;22(4):348–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nakashima H, Kaufmann JK, Wang PY, Nguyen T, Speranza MC, Kasai K, et al. Histone deacetylase 6 inhibition enhances oncolytic viral replication in glioma. J Clin Invest 2015. November 2;125(11):4269–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Okemoto K, Wagner B, Meisen H, Haseley A, Kaur B, Chiocca EA. STAT3 activation promotes oncolytic HSV1 replication in glioma cells. PLoS One 2013;8(8):e71932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kaur B, Cripe TP, Chiocca EA. “Buy one get one free”: armed viruses for the treatment of cancer cells and their microenvironment. Curr Gene Ther 2009. October;9(5):341–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dmitrieva N, Yu L, Viapiano M, Cripe TP, Chiocca EA, Glorioso JC, et al. Chondroitinase ABC I-mediated enhancement of oncolytic virus spread and antitumor efficacy. Clin Cancer Res 2011. March 15;17(6):1362–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Evans SM, Judy KD, Dunphy I, Jenkins WT, Hwang WT, Nelson PT, et al. Hypoxia is important in the biology and aggression of human glial brain tumors. Clin Cancer Res 2004. December 15;10(24):8177–84. [DOI] [PubMed] [Google Scholar]
- 50.Friedman GK, Haas MC, Kelly VM, Markert JM, Gillespie GY, Cassady KA. Hypoxia Moderates gamma(1)34.5-Deleted Herpes Simplex Virus Oncolytic Activity in Human Glioma Xenoline Primary Cultures. Transl Oncol 2012. June;5(3):200–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Aghi MK, Liu TC, Rabkin S, Martuza RL. Hypoxia enhances the replication of oncolytic herpes simplex virus. Mol Ther 2009. January;17(1):51–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Longo SL, Griffith C, Glass A, Shillitoe EJ, Post DE. Development of an oncolytic herpes simplex virus using a tumor-specific HIF-responsive promoter. Cancer Gene Ther 2011. February;18(2):123–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mittnacht S, Straub P, Kirchner H, Jacobsen H. Interferon treatment inhibits onset of herpes simplex virus immediate-early transcription. Virology 1988. May;164(1):201–10. [DOI] [PubMed] [Google Scholar]
- 54.Rasmussen SB, Sorensen LN, Malmgaard L, Ank N, Baines JD, Chen ZJ, et al. Type I interferon production during herpes simplex virus infection is controlled by cell-type-specific viral recognition through Toll-like receptor 9, the mitochondrial antiviral signaling protein pathway, and novel recognition systems. J Virol 2007. December;81(24):13315–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Davis BK, Wen H, Ting JP. The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu Rev Immunol 2011;29:707–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fulci G, Dmitrieva N, Gianni D, Fontana EJ, Pan X, Lu Y, et al. Depletion of peripheral macrophages and brain microglia increases brain tumor titers of oncolytic viruses. Cancer Res 2007. October 1;67(19):9398–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Thorne AH, Meisen WH, Russell L, Yoo JY, Bolyard CM, Lathia JD, et al. Role of cysteine-rich 61 protein (CCN1) in macrophage-mediated oncolytic herpes simplex virus clearance. Mol Ther 2014. September;22(9):1678–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Filley AC, Dey M. Immune System, Friend or Foe of Oncolytic Virotherapy? Front Oncol 2017;7:106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mitchell BM, Stevens JG. Neuroinvasive properties of herpes simplex virus type 1 glycoprotein variants are controlled by the immune response. J Immunol 1996. January 1;156(1):246–55. [PubMed] [Google Scholar]
- 60.Ghonime MG, Jackson J, Shah A, Roth J, Li M, Saunders U, et al. Chimeric HCMV/HSV-1 and Deltagamma134.5 oncolytic herpes simplex virus elicit immune mediated antigliomal effect and antitumor memory. Transl Oncol 2018. February;11(1):86–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fulci G, Breymann L, Gianni D, Kurozomi K, Rhee SS, Yu J, et al. Cyclophosphamide enhances glioma virotherapy by inhibiting innate immune responses. Proc Natl Acad Sci U S A 2006. August 22;103(34):12873–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Han J, Chen X, Chu J, Xu B, Meisen WH, Chen L, et al. TGFbeta Treatment Enhances Glioblastoma Virotherapy by Inhibiting the Innate Immune Response. Cancer Res 2015. December 15;75(24):5273–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Currier MA, Eshun FK, Sholl A, Chernoguz A, Crawford K, Divanovic S, et al. VEGF blockade enables oncolytic cancer virotherapy in part by modulating intratumoral myeloid cells. Mol Ther 2013. May;21(5):1014–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Alvarez-Breckenridge CA, Yu J, Price R, Wojton J, Pradarelli J, Mao H, et al. NK cells impede glioblastoma virotherapy through NKp30 and NKp46 natural cytotoxicity receptors. Nat Med 2012. December;18(12):1827–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Alvarez-Breckenridge CA, Yu J, Price R, Wei M, Wang Y, Nowicki MO, et al. The histone deacetylase inhibitor valproic acid lessens NK cell action against oncolytic virus-infected glioblastoma cells by inhibition of STAT5/T-BET signaling and generation of gamma interferon. J Virol 2012. April;86(8):4566–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Leddon JL, Chen CY, Currier MA, Wang PY, Jung FA, Denton NL, et al. Oncolytic HSV virotherapy in murine sarcomas differentially triggers an antitumor T-cell response in the absence of virus permissivity. Mol Ther Oncolytics 2015;1:14010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Parker JN, Gillespie GY, Love CE, Randall S, Whitley RJ, Markert JM. Engineered herpes simplex virus expressing IL-12 in the treatment of experimental murine brain tumors. Proc Natl Acad Sci U S A 2000. February 29;97(5):2208–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ring EK, Li R, Moore BP, Nan L, Kelly VM, Han X, et al. Newly Characterized Murine Undifferentiated Sarcoma Models Sensitive to Virotherapy with Oncolytic HSV-1 M002. Mol Ther Oncolytics 2017. December 15;7:27–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Roizman B The function of herpes simplex virus genes: a primer for genetic engineering of novel vectors. Proc Natl Acad Sci U S A 1996. October 15;93(21):11307–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Markert JM, Cody JJ, Parker JN, Coleman JM, Price KH, Kern ER, et al. Preclinical evaluation of a genetically engineered herpes simplex virus expressing interleukin-12. J Virol 2012. May;86(9):5304–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Patel DM, Foreman PM, Nabors LB, Riley KO, Gillespie GY, Markert JM. Design of a Phase I Clinical Trial to Evaluate M032, a Genetically Engineered HSV-1 Expressing IL-12, in Patients with Recurrent/Progressive Glioblastoma Multiforme, Anaplastic Astrocytoma, or Gliosarcoma. Hum Gene Ther Clin Dev 2016. June;27(2):69–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cheema TA, Wakimoto H, Fecci PE, Ning J, Kuroda T, Jeyaretna DS, et al. Multifaceted oncolytic virus therapy for glioblastoma in an immunocompetent cancer stem cell model. Proc Natl Acad Sci U S A 2013. July 16;110(29):12006–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Saha D, Wakimoto H, Peters CW, Antoszczyk SJ, Rabkin SD, Martuza RL. Combinatorial Effects of VEGFR Kinase Inhibitor Axitinib and Oncolytic Virotherapy in Mouse and Human Glioblastoma Stem-Like Cell Models. Clin Cancer Res 2018. July 15;24(14):3409–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hardcastle J, Kurozumi K, Dmitrieva N, Sayers MP, Ahmad S, Waterman P, et al. Enhanced antitumor efficacy of vasculostatin (Vstat120) expressing oncolytic HSV-1. Mol Ther 2010. February;18(2):285–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tanaka T, Manome Y, Wen P, Kufe DW, Fine HA. Viral vector-mediated transduction of a modified platelet factor 4 cDNA inhibits angiogenesis and tumor growth. Nat Med 1997. April;3(4):437–42. [DOI] [PubMed] [Google Scholar]
- 76.Passaro C, Alayo Q, DeLaura I, McNulty JJ, Grauwet K, Ito H, et al. Arming an oncolytic herpes simplex virus Type 1 with a single chain fragment variable antibody against PD-1 for experimental glioblastoma therapy. Clin Cancer Res 2018 October 2 2019;25:290–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 2012. March 22;12(4):252–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Liu Z, Ravindranathan R, Kalinski P, Guo ZS, Bartlett DL. Rational combination of oncolytic vaccinia virus and PD-L1 blockade works synergistically to enhance therapeutic efficacy. Nat Commun 2017. March 27;8:14754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Facciabene A, Motz GT, Coukos G. T-regulatory cells: key players in tumor immune escape and angiogenesis. Cancer Res 2012. May 1;72(9):2162–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ricklefs FL, Alayo Q, Krenzlin H, Mahmoud AB, Speranza MC, Nakashima H, et al. Immune evasion mediated by PD-L1 on glioblastoma-derived extracellular vesicles. Sci Adv 2018. March;4(3):eaar2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhai L, Ladomersky E, Lenzen A, Nguyen B, Patel R, Lauing KL, et al. IDO1 in cancer: a Gemini of immune checkpoints. Cell Mol Immunol 2018. May;15(5):447–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ring EK, Markert JM, Gillespie GY, Friedman GK. Checkpoint Proteins in Pediatric Brain and Extracranial Solid Tumors: Opportunities for Immunotherapy. Clin Cancer Res 2017. January 15;23(2):342–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Pulluri B, Kumar A, Shaheen M, Jeter J, Sundararajan S. Tumor microenvironment changes leading to resistance of immune checkpoint inhibitors in metastatic melanoma and strategies to overcome resistance. Pharmacol Res 2017. September;123:95–102. [DOI] [PubMed] [Google Scholar]
- 84.Aldape K, Brindle KM, Chesler L, Chopra R, Gajjar A, Gilbert MR, et al. Challenges to curing primary brain tumours. Nat Rev Clin Oncol 2019. February 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Saha D, Martuza RL, Rabkin SD. Macrophage Polarization Contributes to Glioblastoma Eradication by Combination Immunovirotherapy and Immune Checkpoint Blockade. Cancer Cell 2017. August 14;32(2):253–67 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chesney J, Puzanov I, Collichio F, Singh P, Milhem MM, Glaspy J, et al. Randomized, Open-Label Phase II Study Evaluating the Efficacy and Safety of Talimogene Laherparepvec in Combination With Ipilimumab Versus Ipilimumab Alone in Patients With Advanced, Unresectable Melanoma. J Clin Oncol 2018. June 10;36(17):1658–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Herrera FG, Bourhis J, Coukos G. Radiotherapy combination opportunities leveraging immunity for the next oncology practice. CA Cancer J Clin 2017. January;67(1):65–85. [DOI] [PubMed] [Google Scholar]
- 88.Bradley JD, Kataoka Y, Advani S, Chung SM, Arani RB, Gillespie GY, et al. Ionizing radiation improves survival in mice bearing intracranial high-grade gliomas injected with genetically modified herpes simplex virus. Clin Cancer Res 1999. June;5(6):1517–22. [PubMed] [Google Scholar]
- 89.Markert JM, Razdan SN, Kuo HC, Cantor A, Knoll A, Karrasch M, et al. A phase 1 trial of oncolytic HSV-1, G207, given in combination with radiation for recurrent GBM demonstrates safety and radiographic responses. Mol Ther 2014. May;22(5):1048–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Waters AM, Johnston JM, Reddy AT, Fiveash J, Madan-Swain A, Kachurak K, et al. Rationale and Design of a Phase 1 Clinical Trial to Evaluate HSV G207 Alone or with a Single Radiation Dose in Children with Progressive or Recurrent Malignant Supratentorial Brain Tumors. Hum Gene Ther Clin Dev 2017. March;28(1):7–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Banchereau J, Palucka K. Immunotherapy: Cancer vaccines on the move. Nat Rev Clin Oncol 2018. January;15(1):9–10. [DOI] [PubMed] [Google Scholar]
- 92.Guo C, Manjili MH, Subjeck JR, Sarkar D, Fisher PB, Wang XY. Therapeutic cancer vaccines: past, present, and future. Adv Cancer Res 2013;119:421–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Stronen E, Toebes M, Kelderman S, van Buuren MM, Yang W, van Rooij N, et al. Targeting of cancer neoantigens with donor-derived T cell receptor repertoires. Science 2016. June 10;352(6291):1337–41. [DOI] [PubMed] [Google Scholar]
- 94.Draper SJ, Heeney JL. Viruses as vaccine vectors for infectious diseases and cancer. Nat Rev Microbiol 2010. January;8(1):62–73. [DOI] [PubMed] [Google Scholar]
- 95.Suschak JJ, Williams JA, Schmaljohn CS. Advancements in DNA vaccine vectors, non-mechanical delivery methods, and molecular adjuvants to increase immunogenicity. Hum Vaccin Immunother 2017. December 2;13(12):2837–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kranz LM, Diken M, Haas H, Kreiter S, Loquai C, Reuter KC, et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature 2016. June 16;534(7607):396–401. [DOI] [PubMed] [Google Scholar]
- 97.Flingai S, Czerwonko M, Goodman J, Kudchodkar SB, Muthumani K, Weiner DB. Synthetic DNA vaccines: improved vaccine potency by electroporation and co-delivered genetic adjuvants. Frontiers in immunology 2013. November 4;4:354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Bookstaver ML, Tsai SJ, Bromberg JS, Jewell CM. Improving Vaccine and Immunotherapy Design Using Biomaterials. Trends Immunol 2018. February;39(2):135–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lynn GM, Laga R, Darrah PA, Ishizuka AS, Balaci AJ, Dulcey AE, et al. In vivo characterization of the physicochemical properties of polymer-linked TLR agonists that enhance vaccine immunogenicity. Nat Biotechnol 2015. November;33(11):1201–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Yarchoan M, Johnson BA 3rd, Lutz ER, Laheru DA, Jaffee EM. Targeting neoantigens to augment antitumour immunity. Nat Rev Cancer 2017. April;17(4):209–22. [DOI] [PMC free article] [PubMed] [Google Scholar]