

Abstract
This study was designed to investigate mechanisms of lipid metabolic inflexibility in human obesity and the ability of fenofibrate (FENO) to increase skeletal muscle fatty acid oxidation (FAO) in primary human skeletal muscle cell cultures (HSkMC) exhibiting metabolic inflexibility. HSkMC from 10 lean and 10 obese, insulin resistant subjects were treated with excess fatty acid for 24 hours (24hFA) to gauge lipid-related metabolic flexibility. Metabolically inflexible HSkMC from obese individuals were then treated with 24hFA in combination with FENO to determine effectiveness for increasing FAO. Mitochondrial enzyme activity and FAO were measured in skeletal muscle from subjects with pre-diabetes (n=11) before and after 10 weeks of fenofibrate in vivo. 24hFA increased FAO to a greater extent in HSkMC from lean vs. obese subjects (+49% vs. +9%, for lean vs. obese, respectively; p<0.05) indicating metabolic inflexibility with obesity. Metabolic inflexibility was not observed for measures of cellular resporiration in permeabilized cells using carbohydrate substrate. Fenofibrate co-incubation with 24hFA, increased FAO in a subset of HSkMC from metabolically inflexible, obese subjects (p<0.05), which was eliminated by PPARα antagonist. In vivo, fenofibrate treatment increased skeletal muscle FAO in a subset of subjects with pre-diabetes but did not affect gene transcription or mitochondrial enzyme activity. Lipid metabolic inflexibility observed in HSkMC from obese subjects is not due to differences in electron transport flux, but rather upstream decrements in lipid metabolism. Fenofibrate increases the capacity for FAO in human skeletal muscle cells, though its role in skeletal muscle metabolism in vivo remains unclear.
Keywords: mitochondria, fat oxidation, insulin resistance
Introduction
The capacity to modify substrate oxidation in response to changes in macronutrient availability is a hallmark of healthy metabolic tissues [1]. While many studies have investigated this metabolic flexibility in response to changes in glucose supply [2–4], fewer have examined the ability to adjust fatty acid oxidation (FAO) in response to fatty acid exposure. There is evidence of reduced mitochondrial capacity and FAO in the skeletal muscle of obese and/or diabetic humans 5–8], however, the underlying mechanisms linked with this metabolic inflexibility and interventions aimed increasing skeletal muscle FAO are not clearly evident.
We have previously reported a dampened ability to increase FAO in response to lipid exposure in skeletal muscle and primary human skeletal muscle cells (HSkMC) from obese, insulin resistant humans [6,9]. In addition, we observed an inability to upregulate peroxisome proliferator-activated receptor (PPAR)α and PPARα-responsive genes in response to high fat feeding in skeletal muscle of obese, insulin resistant humans [10]. PPARα is a global transcriptional regulator of FAO, activated by endogenous fatty acids and fatty acid derivatives [11]. Thus, PPARα is a prominent candidate for interventions aimed at increasing FAO and improving metabolic flexibility in obese, insulin resistant individuals who exhibit a reduced capacity for appropriately modulating FAO in response to nutrient availability.
Fibrates such as fenofibrate (FENO) are PPARα activators commonly used for treatment of hypertriglyceridemia [12]. These PPARα activators are known to induce the expression of genes involved in lipid oxidation, including pyruvate dehydrogenase kinase (PDK)4 and carnitine palmitoyl transferase (CPT)1 [13] in vitro or in animal models; however, there are limited data on whether FENO can increase FAO in metabolically inflexible, obese insulin-resistant humans. In addition, there are conflicting reports on effects of fibrates on insulin resistance [14–20]. Thus, as a regulator of PPARα, and potentially FAO, this series of experiments was designed to investigate the mechanisms linked with the lipid metabolic inflexibility of human obesity and the ability of FENO to increase FAO in human skeletal muscle.
Methods
HSkMC were derived from muscle biopsy specimens of lean and obese, insulin resistant humans and used to examine metabolid inflexibilty, FAO, and the effects of FENO. In vivo effects of FENO were determined in skeletal muscle samples collected before and after 10 weeks of FENO treatment in pre-diabetic individuals.
HSkMC
The East Carolina University Policy and Review Committee on Human Research approved this protocol and written, informed consent was obtained prior to the muscle biopsy procedure. For the HSkMC studies, skeletal muscle biopsies were performed as described [10,21] in the vastus lateralis of 10 lean (BMI<25 kg/m2) and 10 obese (BMI>30 kg/m2) Caucasian males (ages 18–31 y). Participants were free from disease, nonsmokers, and not taking medications known to alter lipid or carbohydrate metabolism. Subjects were excluded if they were performing more than 60 min/week of organized physical activity, or if they experienced a weight change greater than 2 kg in the previous 3 months.
Skeletal muscle satellite cells were isolated and cultured from fresh skeletal muscle tissue and differentiated to myotubes for 7 days [9]. Metabolic flexibility was tested by incubating cells for 24 h in differentiation medium supplemented with either 0.1% BSA (control; CTRL) or 200 μM oleate:palmitate (1:1 ratio) bound to BSA (2.5:1 molar ratio) plus 2 mM carnitine (24hFA), after which metabolic measures were made to determine: 1) FAO of intact HSkMC using 14C-labeled fatty acid incorporation to 14CO2; or 2) mitochondrial oxygen consumption of permeabilized HSkMC (Seahorse XF-24, Seahorse Bioscience, Billerica, MA).
Experiments were performed to determine the optimal FENO concentration and duration of incubation considering cell viability and capacity for increasing FAO (2h, 6 μM). In subsequent experiments, HSkMCs were exposed to 24hFA and 2h, 6 μM FENO in combination with 10 μM GW6471 (PPARα inhibitor), 20 μM Compound C (AMPK inhibitor), or 0.5 M AICAR (AMPK activator) to discern the mechanism of FENO action on HSkMC FAO.
HSkMC Fatty Acid Oxidation Assays
FAO assays were performed as described [22]. Briefly, myotubes were incubated at 37 °C in sealed 24-well plates containing differentiation media containing 0.25 μCi/ml [14C]oleate and 0.25 μCi/ml [14C]palmitate (PerkinElmer Life Sciences, Waltham, MA), with 200 μM, 1:1 ‘cold’ oleate:palmitate. After a 2h incubation, the rate of FAO was determined by measuring the 14CO2 released from the media following acidification. Measures were performed in triplicate and data were corrected for total protein content, as measured by BCA method (bicinchoninic acid assay; Pierce Biotechnology, Inc.)
HSkMC Respiration Experiments
For permeabilized cell respiration, media was removed and 200 μl of sucrose-based respiration buffer (MiR05), supplemented with 2.5 μg/ml digitonin, were added. After 5 min, an additional 300 μl MiR05 respiration buffer containing basal substrate conditions (5 μM palmitoyl carnitine, 1 mM malate) but without digitonin supplement, were added. Cells were then subjected to the following respiration experiment: basal state 4 respiration (palmitoyl carnitine + malate, PCM4), state 3 respiration supported by palmitoyl carnitine (+2 mM ADP; PCM3), state 3 respiration supported by succinate (+3 mM succinate +1 μM rotenone; SR3), state 2 respiration (+2 μM oligomycin; Olig.), and maximal uncoupled respiration (+4 μM FCCP). Respiratory control ratios (RCR)s were calculated as the state 3 respiration rate (succinate + rotenone) / state 4 respiration rates and were used to assess mitochondrial integrity. Following experiments, cells were rinsed with PBS, and lysed with protein lysis buffer, and total protein content was measured by BCA assay. All respiration data were normalized to cell-free wells and total protein content.
In Cell ELISA
HSkMC were rinsed twice in PBS then fixed to culture plates with 4% formalin for 5 min. Cells were permeabilized using permeabilization/blocking buffer (0.1% Fraction V BSA, 5% goat serum, 0.3% Triton X-100, 0.2% sodium azide in PBS) with rotation for 1 hr. After rinsing, 1:250 dilution of rabbit anti-AMPK or rabbit anti-phosphorylated AMPK [Thr172] in antibody buffer (0.1% Fraction V BSA, 5% goat serum in PBS) were added, then incubated overnight at 4°C with rotation. After rinsing. goat anti-rabbit horseradish peroxidase (HRP)-conjugated antibody diluted 1:5,000 in antibody buffer was added, then incubated at room temperature for 1 hr. Rinsed wells were emptied and blotted dry, HRP developing solution was added, and color change was measured at 1 min intervals for 10 min at 650 nM. Stable slopes for 5 consecutive minutes were used to quantify the content of phosphorylated AMPK relative to total AMPK (phosphorylated AMPK/total AMPK) protein.
Pre-diabetic Subjects and Study Design
To study the effects of FENO on in vivo skeletal muscle metabolism, muscle biopsies were obtained from individuals (age 18–65 y, BMI 28–38 kg/m2) with pre-diabetes (defined as impaired glucose tolerance [IGT; plasma glucose of 140–199 mg/dl 2 h after a 75 g oral glucose tolerance test] or impaired fasting glucose [IFG; fasting plasma glucose of 100–126 mg/dl]) recruited as part of a randomized clinical trial () [21]. All subjects provided written, informed consent under a protocol that was approved by the local Institutional Review Board. Studies were conducted at the Clinical Research Center at the University of Colorado Anschutz Medical Campus.
Subjects with a history of diabetes, renal insufficiency (creatinine >1.4), liver disease (AST or ALT> 2x normal), congestive heart failure, or coronary artery disease were excluded, as were subjects who were taking medications known to affect inflammation or insulin resistance, such as steroids or NSAIDS. Baseline measures included insulin resistance by insulin-modified, frequently sampled intravenous glucose tolerance tests (FSIGT) and body composition by dual-energy X-ray absorptiometry (DXA) followed by vastus lateralis skeletal muscle biopsies (n=11). Subjects then received FENO (145 mg/day) for 10 weeks and FSIGT, DXA, and muscle biopsy repeated.
Fasting insulin was measured using an immunochemiluminescent assay (MLT Assay, Wales, UK). Plasma glucose was measured in duplicate by a glucose oxidase assay. Insulin sensitivity was calculated from the insulin and glucose data using the MinMod Millennium program [23].
Skeletal Muscle Fatty Acid Oxidation
Skeletal muscle FAO was measured as for HSkMC with modification for tissue homogenates as previously described [24] (n=3). Tissue homogenates were assayed for total protein content by BCA assay for data normalization. All measures were performed in quintuplet.
Mitochondrial Enzyme Activity
Mitochondrial-enriched supernatants (post 600 g) were prepared from frozen skeletal muscle samples [25]. Supernatants were used to assay activity of respiratory chain enzyme complexes I and II+III, and citrate synthase (CS) spectrophotometrically on a Synergy H1 microplate reader (Biotek, Winooski, VT). For complexes I, II+III, and CS enzyme activities were calculated as initial rates (nmol/min). For complex III, enzyme activity was calculated as the first-order rate constants derived within 2–3 min of reaction initiation. The protein content of each sample was determined by BCA assay. All enzyme activities were normalized to the total protein content of each sample and results are expressed relative to CS activity.
Quantitative PCR
Total RNA was isolated from skeletal muscle tissue using the Ultraspec RNA kit (Biotecx Laboratories, Inc, Houston, TX). cDNA was transcribed from 200 ng total RNA using iScript cDNA Synthesis kit (Bio-Rad). qPCR was performed using primer sets for genes of interest and RPL13a and ubiquitin C as reference genes and iQ SYBR Supermix (Bio-Rad, Hercules, CA) as described [25]. Reactions were run in duplicate on an iQ5 Real-Time PCR Detection System (Bio-Rad) along with a no-template control per gene. RNA expression data was normalized to reference genes using the comparative threshold cycle method.
Mitochondrial DNA Copy Number
Total cellular DNA was isolated by phenol/chloroform extraction and quantified using the PicoGreen DNA quantification kit (Thermo Fisher, Waltham, MA). mtDNA content was measured as relative copy number of mtDNA per diploid nuclear genome using quantitative PCR with primers specific to β-globin and mtDNA, as described [26,27].
Statistical Analyses
For all characteristic data, independent t-tests were used. For HSkMC comparisons of 24hFA across body size, repeated measures ANOVA was used to test for effect of BMI, effect of treatment, or for interaction. For HSkMC FENO concentration/time course experiments, three separate univariate analyses of variance (ANOVAs) were used to compare across 24hFA pre-treatments and across FENO concentrations. Bonferroni corrections were used for post-hoc analyses. Separate independent t-tests were used to compare 24hFA with all other treatments for FENO mechanism experiments. Paired t-tests were used to test FENO treatment effects in human skeletal muscle tissue experiments. Statistical difference is indicated at p≤0.05. Data are expressed as the mean ± SEM. Statistical analyses were performed using SPSS Statistics (SPSS, IBM Corp., Armonk, NY).
Results
HSkMC subject characteristics
The characteristics of lean and obese subjects for the HSkMC experiments are presented in Table 1. By design, the obese group had a higher BMI and body fat percentage. While fasting glucose levels were similar between groups, the obese individuals had higher fasting insulin levels and HOMA-IR scores (p<0.05), suggesting insulin resistance.
Table 1.
Patient Characteristics for HSkMC and Skeletal Muscle Tissue Experiments
| HSkMC Subjects | Pre-Diabetic Subjects | |||
|---|---|---|---|---|
| Lean (n=10) |
Obese (n=10) |
Pre-FENO (n=11) |
Post-FENO -- |
|
| Age (y) | 22.4 ± 0.6 | 24.2 ± 1.1 | 48.8 ± 3.0 | -- |
| Sex (M/F) | (10/0) | (10/0) | 6/5 | -- |
| BMI (kg/m2) | 21.6 ± 0.7 | 36.4 ± 1.7* | 35.9 ± 1.3 | 37.0 ± 1.5# |
| Body Fat (%) | 17.8 ± 3.1 | 41.1 ± 1.8* | 38.1 ± 3.2 | 38.6 ± 2.7 |
| Glucose (mmol/L) | 4.9 ± 0.1 | 5.0 ± 0.1 | 5.7 ± 0.3 | 5.7 ± 0.3 |
| Insulin (μIU/L) | 4.6 ± 0.7 | 16.0 ±1.5* | 12.7 ± 2.8 | 10.9 ± 2.0 |
| HOMA-IR | 1.0 ±0.2 | 3.5 ± 0.3* | 3.4 ± 1.0 | 2.9 ± 0.6 |
| Triglycerides (mg/dL) | 103.4 ± 14.2 | 134.6 ± 23.2 | 161.8 ± 33.5 | 92.1 ± 11.8# |
| Hemoglobin A1c (%) | -- | -- | 5.8 ±0.2 | 5.9 ± 0.2 |
| SI (10−4min−1/μU/mL) | -- | -- | 1.5 ± 0.2 | 1.9 ± 0.3 |
indicates significant difference from Lean.
indicates significant difference from Pre-FENO.
Responses to 24h lipid incubation
FAO increased with 24hFA (p<0.05), though the lean subjects exhibited a 4-fold greater response to 24hFA than obese (+40% vs. +9% from CTRL to 24hFA conditions in lean vs. obese, respectively; Fig. 1A; p<0.05), indicating greater metabolic flexibility. We did not observe differences in mitochondrial content, as estimated by mitochondrial DNA copy number (Fig. 1B). To further investigate the pathogenesis of metabolic inflexibility, mitochondrial function was evaluated in similar conditions in permeabilized HSkMCs. Notably, in permeabilized HSkMC, state 3 respiration supported by palmitoyl carnitine, or PCM3, was increased with 24hFA conditions in lean subjects but decreased in HSkMC from obese individuals (+34% and −17%, respectively; Fig. 1B, inset; p<0.05). However, mitochondrial respiration in all other conditions tested were not different between groups, nor did respiration change with 24hFA in either group for SR3, Olig, or FCCP (Fig. 1C). Mitochondrial respiratory control ratios (mean = 2.78 ± 0.26) and state 4 respiration (Fig. 1C) were similar between groups and not altered with 24hFA.
Figure 1.
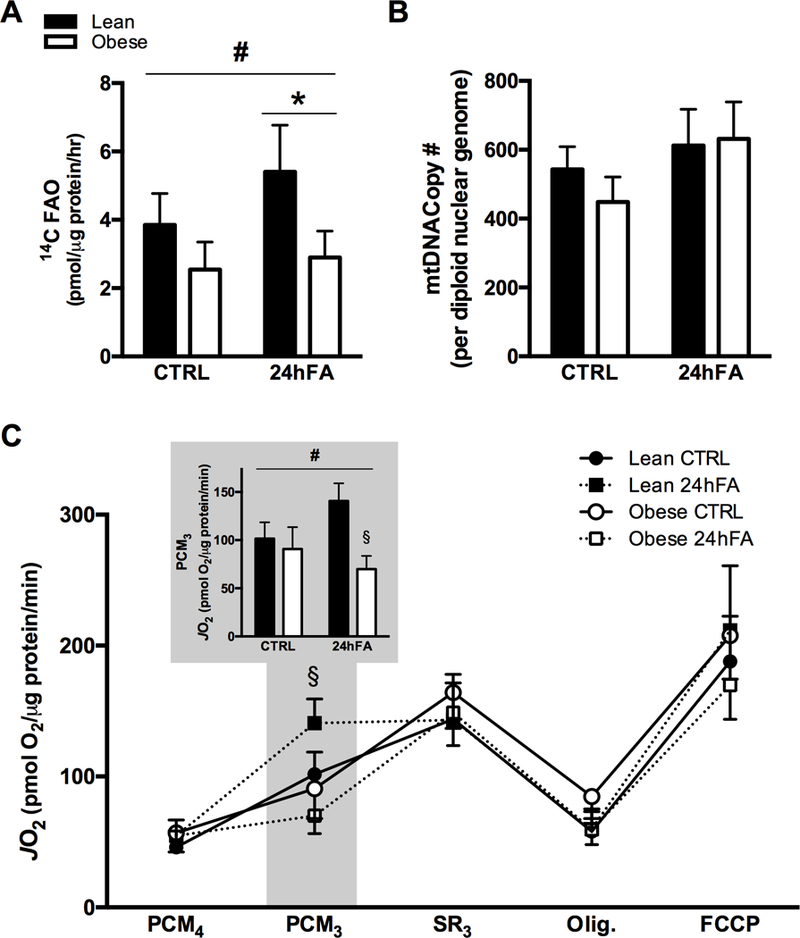
24hFA exposure increases lipid oxidation in HSkMC from lean but not obese subjects. HSkMC from lean (open bars) and obese (filled bars) were incubated with or without 200 μM fatty acid for 24 h (24hFA) prior to measures of FAO from 14C-labeled palmitate:oleate or mitochondrial respiration of permeabilized HSkMC. Complete FAO was increased with 24hFA treatment in both lean and obese HSkMC, though the response was more robust in lean HSkMC (A). There were no differences between groups, nor for 24hFA, in mitochondrial DNA Copy number (B). In permeabilized HSkMC, subsequent compound additions in the Seahorse from basal (PCM4; palmitoyl carnitine + malate) were ADP (PCM3), succinate + rotenone (SR3), oligomycin, and FCCP (C). Only PCM3 showed differences, where a significant ANOVA interaction revealed 24hFA-induced increases in PCM3 in HSkMC from lean, and PCM3 decreases in HSkMC from obese subjects (C, inset). Data are mean ± SEM. # indicates significant repeated measures ANOVA interaction. * indicates significant effect of 24hFA treatment for both lean and obese, combined. § indicates significant difference from lean in 24hFA condition.
Acute FENO treatment increases FAO
Acute (2h) FENO incubation stimulated a 4 to 5-fold increases in FAO for both the CTRL and 24hFA conditions in a dose dependent manner (Fig. 2A; p<0.05). A 12 μM FENO treatment also increased FAO to a similar magnitude as the 6 μM FENO, though this condition resulted in considerable cell death (data not shown). Longer duration FENO incubations were not effective for increasing FAO (Fig. 2B). Longer duration FENO incubations were also performed for 3 μM and 12 μM FENO concentrations, with no significant increase in FAO (data not shown). Thus, 2h, 6 μM FENO was chosen for subsequent experiments.
Figure 2.
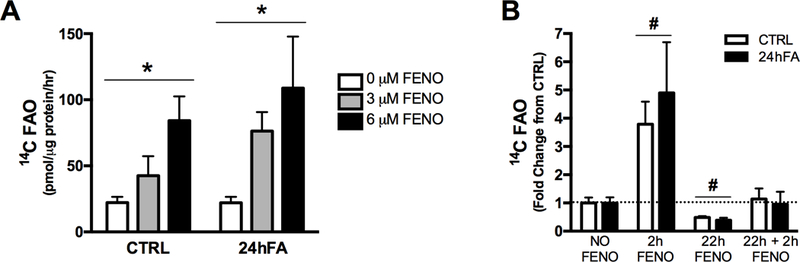
Acute FENO treatment increases FAO in HSkMC with lipid metabolic inflexibility. Concentration courses of FENO treatment show that 6 μM FENO treatment is most robust for increasing 14C labeled palmitate:oleate FAO in obese, metabolically inflexible HSkMC (A). More prolonged treatment with 6 μM FENO for acute (2h), prolonged (22h), or acute + prolonged (2h + 22h) FENO treatments did not show similar robust increase in FAO (B). Data represent metabolically inflexible HSkMC from n=2–4 obese subjects, measured in triplicate. Data are mean ± SEM. * indicates significant dose effect for FENO concentration. # indicates significant difference from NO FENO for CTRL and 24hFA combined.
FENO may act through PPARα and AMPK phosphorylation
To test the specificity of the effect of FENO on FAO, we used GW6471 (a PPARα antagonist) or compound C (an AMPK inhibitor). FENO-mediated increases in FAO, were abolished with GW6471 co-incubation (Fig. 3A; p<0.05), indicating that FENO-mediated increases in FAO depend on PPARα activity. Likewise, co-incubation of FENO with Compound C, a competitive inhibitor of AMPK, abolished the effect of FENO on FAO and reduced FAO to below basal rates (CTRL or 24hFA alone; Fig. 3A, p<0.05). To corroborate these metabolic effects, Fig. 3B shows that AMPK phosphorylation was increased in the cells treated with 24hFA + 2h, 6 μM FENO (p<0.05), to a similar extent as observed with 0.5 μM AICAR (p<0.05). These effects of FENO were eliminated when GW6471 or Compound C were added (Fig. 3B).
Figure 3.
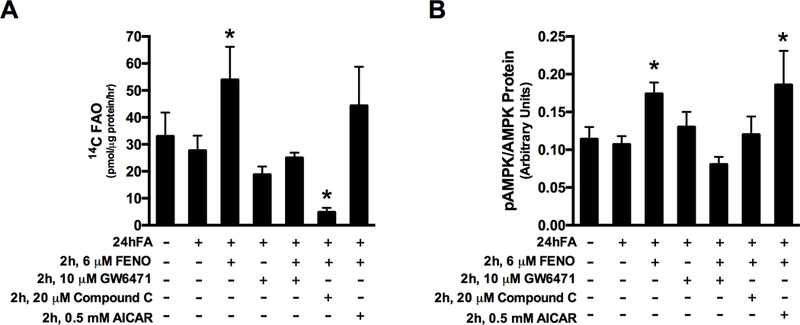
FENO may act through PPARα and AMPK phosphorylation. Treatment of obese, metabolically inflexible HSkMC with FENO in combination with GW6471, Compound C, or AICAR as indicated, show that FENO-induced increases in FAO are inhibited by co-incubation with PPARα antagonist or AMPK inhibitor, though FENO treatment with AMPK activation by AICAR does not increase FAO beyond that of FENO alone (A). Similar observations are made for phosphorylated AMPK/Total AMPK protein levels (Thr172) (B). Data are mean ± SEM. * indicates significant difference from 24hFA condition.
Effects of FENO in individuals with pre-diabetes
Baseline characteristics and clinical outcomes of the 10-week FENO treatment in obese, pre-diabetic patients are summarized in Table 1. As expected, FENO treatment lowered triglyceride levels by 45% (p<0.05). However, insulin sensitivity, when measured by either HOMA-IR or FSIGT, did not change. There were no differences in citrate synthase activity (Fig. 4A), mitochondrial ETS enzyme activity (CI, CII+CIII, or CIV; Fig. 4B), or in mRNA content of PPARα-responsive genes (Fig. 4C). Nevertheless, in a subset of subjects, FAO of fresh skeletal muscle tissue homogenates increased by 10–80% from Pre- to Post-FENO treatment (n=3, Fig. 4D, p<0.05).
Figure 4.
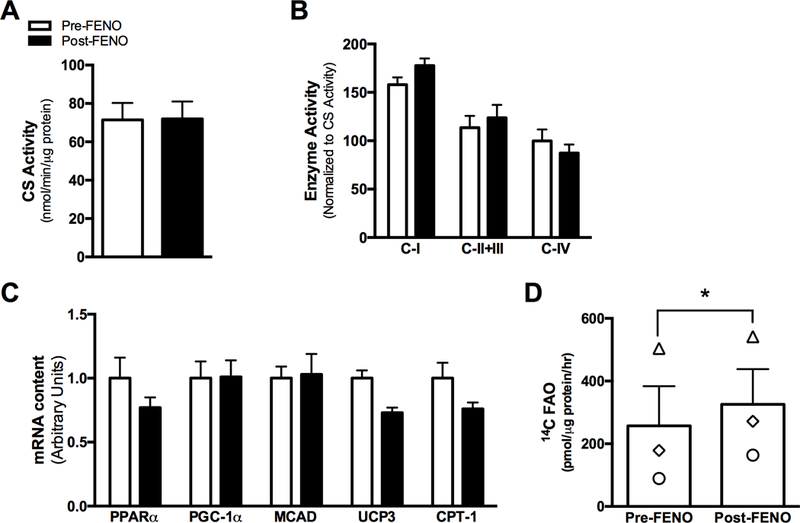
In vivo FENO treatment does not affect skeletal muscle mitochondrial enzyme activity or genes or proteins linked with PPARα. Skeletal muscle citrate synthase activity (A) mitochondrial ETS complex enzyme activities (B), mRNA content of genes linked with PPARα activation (C) were not different after 10 weeks of FENO treatment, though complete FAO from (14C-labeled palmitate) was increased in fresh skeletal muscle tissue homogenates of a subset of pre-diabetic patients treated with FENO for 10-weeks (A). Data are mean ± SEM. * indicates significant difference from Pre-FENO condition.
Discussion
Metabolic flexibility, defined as the ability to appropriately adjust oxidation to changes in nutrient availability, is a hallmark of metabolic health. In skeletal muscle specifically, metabolic flexibility is related to higher insulin sensitivity, reduced adiposity, and greater cardiovascular fitness [3]. The present data indicate that the ability to increase FAO upon exposure to a physiologically relevant lipid mixture (oleate:palmitate) was significantly dampened in skeletal muscle cells from obese compared to lean subjects, which was not explained by differences in mitochondrial content (Fig. 1B). When measuring mitochondrial respiration in permeabilized cells, we did not find differences in basal or maximal respiration rates, nor for state 3 respiration supported by succinate + rotenone, which tests flux through ETS complex CII (Fig. 1C). These results suggest that lipid metabolic inflexibility in HSkMC from obese humans with respect to lipid oxidation does not involve deficits in ETS complex function. Therefore, differences in the response to 24hFA likely occur either at the tricarboxylic acid or TCA cycle, where the CO2 is produced, or at upstream events in fatty acid metabolism. Previous reports in skeletal muscle tissue and cells from humans with obesity or type 2 diabetes have also shown normal electron transport function, particularly when data are normalized for mitochondrial content[28,29]. Likewise, we and others have shown deficits in TCA cycle and fatty acid transport/metabolism specifically in humans and animal models of obesity [6,8,30–32].
FENO was used to determine if FAO can be increased in metabolically inflexible HSkMC via PPARα activation [14,33]. Results in animal studies have been mixed, with some findings suggestive of FENO-induced increases in fatty acid oxidation [34–36] and others not [37]. In the present study, we found that short-term exposure to FENO (2h) increased FAO in metabolically inflexible HSkMC, indicating that the effects of FENO are fast-acting and transient. Based on these findings, we hypothesized that the effects of FENO on FAO were independent of PPARα, perhaps acting through a more short-term metabolic activation, such as AMPK phosphorylation, which has been previously reported [38,39]. AMPK is sensitive to the cellular energy state and is a potent activator of FAO, shifting substrate choice to favor lipd over carbohydtates [40]. However, co-incubation of HSkMC with FENO and either the PPARα inhibitor GW6471 or the AMPK inhibitor Compound C eliminated FENO-mediated increases in FAO (Fig. 3A), suggesting that FENO may be acting through both PPARα and AMPK. Several studies have reported increased activity of AMPK by FENO [38,39] but the interactions between AMPK and PPARα, and the activation of these proteins themselves, are complex and often contradictory [41–43], including some evidence of AMPK inhibition of PPARα [43]. In understanding these interactions, the utility of the HSkMC system lies in the ability to incubate human skeletal muscle under conditions which would be difficult to replicate in vivo; the finding of FENO functioning through AMPK in terms of modulating FAO thus provides important information relative to the PPARα-AMPK axis.
Due to the potential effectiveness of the intervention, we investigated whether in vivo FENO treatment in individuals with pre-diabetes increased FAO in a manner similar to that in the primary muscle cell cultures. While some studies have shown that FENO improves peripheral insulin sensitivity in patients with metabolic syndrome [16,17], our findings confirm many others in terms of there being no effect [14,18–20]. Neither did we observe FENO-induced effects on mRNA content of PPARα-responsive genes or for mitochondrial enzyme activity. However, in a subset of subjects, we observed increases in FAO in skeletal muscle tissue after FENO treatment (Fig. 4D), which appeared to be induced independently of changes in mitochondrial content (as indicated by citrate synthase activity) or the expression of PPARα responsive genes. The discrepancy between PPARα response to FENO in the patient biopsies versus FENO treatment in the primary muscle cell cultures could be the timing of the exposure to FENO as the last dose of FENO was over 24h prior to skeletal muscle sample collection. Based on our results in primary muscle cells, the effects of FENO are relatively short-lived and, therefore, may not be detected a moreprolonged period after administration. However, when isolated mitochondria from these patients were metabolically stimulated to oxidize fatty acids, FAO was increased at the Post-FENO timepoint, suggesting some residual effect or metabolic adaptation to chronic FENO treatment when tested in vivo. Future investigations should aim to clarify the timing of sample collection in relation to FENO dosing, as well as the acute vs. chronic effects of FENO treatment in humans.
In summary, the current data suggest that lipid metabolic inflexibility observed in HSkMC from obese subjects appears not to be a function of differences in electron transport flux, but rather to events upstream of electron transport, such as decrements in TCA cycle flux or β-oxidation. Short-term FENO increased FAO through both PPARα and AMPK activation in vitro. Taken together, these novel results suggest that PPARα activators may improve metabolic flexibility acutely, but their role in skeletal muscle metabolism in vivo requires further investigation.
Acknowledgments
Funding: These studies were supported by NIH/NCATS Colorado CTSA Grant Number UL1 TR001082, the Colorado Nutrition and Obesity Research Center (NORC), NIH P30 DK048520; VA Merit Grant (N.R.), NIH DK056112 (J.A.H.); K.E.B. was supported by NIH F32 DK089743 and K12 HD057022.
Footnotes
Disclosure: The authors have no conflict of interest
University of Colorado SOM: Mail stop 8106, 12631 East 17th Avenue, Aurora, CO 80045. Office: AO1 #7508, Phone: 303 724 4651
VA ECHCS: 1055 Clermont Street 111H, Denver, CO 80220. Phone: 303-399-8020x4314
References
- 1.Muoio DM. Metabolic Inflexibility: When Mitochondrial Indecision Leads to Metabolic Gridlock. Cell 2014; 159: 1253–1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Corpeleijn E, Saris WHM, Blaak EE. Metabolic flexibility in the development of insulin resistance and type 2 diabetes: effects of lifestyle. Obes Rev 2009; 10: 178–193 [DOI] [PubMed] [Google Scholar]
- 3.Kelley DE. Skeletal muscle fat oxidation: timing and flexibility are everything. J Clin Invest 2005; 115: 1699–1702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kelley DE, Goodpaster B, Wing RR, Simoneau JA. Skeletal muscle fatty acid metabolism in association with insulin resistance, obesity, and weight loss. Am J Physiol 1999; 277: E1130–E1141 [DOI] [PubMed] [Google Scholar]
- 5.Anderson EJ, Lustig ME, Boyle KE, Woodlief TL, Kane DA, Lin C-T, Price JW, Kang L, Rabinovitch PS, Szeto HH, Houmard JA, Cortright RN, Wasserman DH, Neufer PD. Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J Clin Invest 2009; 119: 573–581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Battaglia GM, Zheng D, Hickner RC, Houmard JA. Effect of exercise training on metabolic flexibility in response to a high-fat diet in obese individuals. Am J Physiol Endocrinol Metab 2012; 303: E1440–E1445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mogensen M, Sahlin K, Fernström M, Glintborg D, Vind BF, Beck-Nielsen H, Højlund K. Mitochondrial respiration is decreased in skeletal muscle of patients with type 2 diabetes. Diabetes 2007; 56: 1592–1599 [DOI] [PubMed] [Google Scholar]
- 8.Hulver MW, Berggren JR, Cortright RN, Dudek RW, Thompson RP, Pories WJ, MacDonald KG, Cline GW, Shulman GI, Dohm GL, Houmard JA. Skeletal muscle lipid metabolism with obesity. AJP: Endocrinology and Metabolism 2003; 284: E741–E747 [DOI] [PubMed] [Google Scholar]
- 9.Boyle KE, Zheng D, Anderson EJ, Neufer PD, Houmard JA. Mitochondrial lipid oxidation is impaired in cultured myotubes from obese humans. Int J Obes (Lond) 2012; 36: 1025–1031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boyle KE, Canham JP, Consitt LA, Zheng D, Koves TR, Gavin TP, Holbert D, Neufer PD, Ilkayeva O, Muoio DM, Houmard JA. A high-fat diet elicits differential responses in genes coordinating oxidative metabolism in skeletal muscle of lean and obese individuals. J Clin Endocrinol Metab 2011; 96: 775–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Contreras AV, Torres N, Tovar AR. PPAR-α as a key nutritional and environmental sensor for metabolic adaptation. Adv Nutr 2013; 4: 439–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Auwerx J, Schoonjans K, Fruchart JC, Staels B. Transcriptional control of triglyceride metabolism: fibrates and fatty acids change the expression of the LPL and apo C-III genes by activating the nuclear receptor PPAR. Atherosclerosis 1996; 124 Suppl: S29–S37 [DOI] [PubMed] [Google Scholar]
- 13.Sugden MC, Zariwala MG, Holness MJ. PPARs and the orchestration of metabolic fuel selection. Pharmacol Res 2009; 60: 141–150 [DOI] [PubMed] [Google Scholar]
- 14.Perreault L, Bergman BC, Hunerdosse DM, Howard DJ, Eckel RH. Fenofibrate administration does not affect muscle triglyceride concentration or insulin sensitivity in humans. Metabolism 2011; 60: 1107–1114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bergman BC, Hunerdosse DM, Kerege A, Playdon MC, Perreault L. Localisation and composition of skeletal muscle diacylglycerol predicts insulin resistance in humans. Diabetologia 2012; 55: 1140–1150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Koh KK, Han SH, Quon MJ, Yeal Ahn J, Shin EK. Beneficial effects of fenofibrate to improve endothelial dysfunction and raise adiponectin levels in patients with primary hypertriglyceridemia. Diabetes Care 2005; 28: 1419–1424 [DOI] [PubMed] [Google Scholar]
- 17.Idzior-Walus B, Sieradzki J, Rostworowski W, Zdzienicka A, Kawalec E, Wójcik J, Zarnecki A, Blane G. Effects of comicronised fenofibrate on lipid and insulin sensitivity in patients with polymetabolic syndrome X. Eur J Clin Invest 2000; 30: 871–878 [DOI] [PubMed] [Google Scholar]
- 18.Fabbrini E, Mohammed BS, Korenblat KM, Magkos F, McCrea J, Patterson BW, Klein S. Effect of fenofibrate and niacin on intrahepatic triglyceride content, very low-density lipoprotein kinetics, and insulin action in obese subjects with nonalcoholic fatty liver disease. J Clin Endocrinol Metab 2010; 95: 2727–2735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Belfort R, Berria R, Cornell J, Cusi K. Fenofibrate reduces systemic inflammation markers independent of its effects on lipid and glucose metabolism in patients with the metabolic syndrome. J Clin Endocrinol Metab 2010; 95: 829–836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Anderlová K, Dolezalová R, Housová J, Bosanská L, Haluzíková D, Kremen J, Skrha J, Haluzík M. Influence of PPAR-alpha agonist fenofibrate on insulin sensitivity and selected adipose tissue-derived hormones in obese women with type 2 diabetes. Physiol Res 2007; 56: 579–586 [DOI] [PubMed] [Google Scholar]
- 21.Rasouli N, Kern PA, Elbein SC, Sharma NK, Das SK. Improved insulin sensitivity after treatment with PPARγ and PPARα ligands is mediated by genetically modulated transcripts. Pharmacogenetics and Genomics 2012; 22: 484–497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Peroxisome Proliferator–Activated Receptor-γ Coactivator-1α Overexpression Increases Lipid Oxidation in Myocytes From Extremely Obese Individuals. 2010; 59: 1407–1415 Available from: http://eutils.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&id=20200320&retmode=ref&cmd=prlinks [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Boston RC, Stefanovski D, Moate PJ, Sumner AE, Watanabe RM, Bergman RN. MINMOD Millennium: a computer program to calculate glucose effectiveness and insulin sensitivity from the frequently sampled intravenous glucose tolerance test. Diabetes Technol Ther 2003; 5: 1003–1015 [DOI] [PubMed] [Google Scholar]
- 24.Berggren JR, Boyle KE, Chapman WH, Houmard JA. Skeletal muscle lipid oxidation and obesity: influence of weight loss and exercise. AJP: Endocrinology and Metabolism 2008; 294: E726–E732 [DOI] [PubMed] [Google Scholar]
- 25.Boyle KE, Newsom SA, Janssen RC, Lappas M, Friedman JE. Skeletal muscle MnSOD, mitochondrial complex II, and SIRT3 enzyme activities are decreased in maternal obesity during human pregnancy and gestational diabetes mellitus. J Clin Endocrinol Metab 2013; 98: E1601–E1609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Menshikova EV, Ritov VB, Toledo FGS, Ferrell RE, Goodpaster BH, Kelley DE. Effects of weight loss and physical activity on skeletal muscle mitochondrial function in obesity. AJP: Endocrinology and Metabolism 2005; 288: E818–E825 [DOI] [PubMed] [Google Scholar]
- 27.Consitt LA, Boyle KE, Houmard JA. Exercise as an Effective Treatment for Type 2 Diabetes In: Type 2 Diabetes Mellitus. Totowa, NJ: Humana Press, 2008: 135–150 [Google Scholar]
- 28.Boushel R, Gnaiger E, Schjerling P, Skovbro M, Kraunsøe R, Dela F. Patients with type 2 diabetes have normal mitochondrial function in skeletal muscle. Diabetologia 2007; 50: 790–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fisher-Wellman KH, Weber TM, Cathey BL, Brophy PM, Gilliam LAA, Kane CL, Maples JM, Gavin TP, Houmard JA, Neufer PD. Mitochondrial respiratory capacity and content are normal in young insulin-resistant obese humans. Diabetes 2014; 63: 132–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boyle KE, Zheng D, Anderson EJ, Neufer PD, Houmard JA. Mitochondrial lipid oxidation is impaired in cultured myotubes from obese humans. Int J Obes (Lond) 2012; 36: 1025–1031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Noland RC, Koves TR, Seiler SE, Lum H, Lust RM, Ilkayeva O, Stevens RD, Hegardt FG, Muoio DM. Carnitine insufficiency caused by aging and overnutrition compromises mitochondrial performance and metabolic control. J Biol Chem 2009; 284: 22840–22852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Seiler SE, Martin OJ, Noland RC, Slentz DH, DeBalsi KL, Ilkayeva OR, An J, Newgard CB, Koves TR, Muoio DM. Obesity and lipid stress inhibit carnitine acetyltransferase activity. J Lipid Res 2014; 55: 635–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cree MG, Newcomer BR, Read LK, Sheffield-Moore M, Paddon-Jones D, Chinkes D, Aarsland A, Wolfe RR. Plasma triglycerides are not related to tissue lipids and insulin sensitivity in elderly following PPAR-alpha agonist treatment. Mech Ageing Dev 2007; 128: 558–565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yoon M, Jeong S, Lee H, Han M, Kang JH, Kim EY, Kim M, Oh GT. Fenofibrate improves lipid metabolism and obesity in ovariectomized LDL receptor-null mice. Biochem Biophys Res Commun 2003; 302: 29–34 [DOI] [PubMed] [Google Scholar]
- 35.Jeong S, Han M, Lee H, Kim M, Kim J, Nicol CJ, Kim BH, Choi JH, Nam K-H, Oh GT, Yoon M. Effects of fenofibrate on high-fat diet-induced body weight gain and adiposity in female C57BL/6J mice. Metabolism 2004; 53: 1284–1289 [DOI] [PubMed] [Google Scholar]
- 36.Furuhashi M, Ura N, Murakami H, Hyakukoku M, Yamaguchi K, Higashiura K, Shimamoto K. Fenofibrate improves insulin sensitivity in connection with intramuscular lipid content, muscle fatty acid-binding protein, and beta-oxidation in skeletal muscle. J Endocrinol 2002; 174: 321–329 [DOI] [PubMed] [Google Scholar]
- 37.Mancini FP, Lanni A, Sabatino L, Moreno M, Giannino A, Contaldo F, Colantuoni V, Goglia F. Fenofibrate prevents and reduces body weight gain and adiposity in diet-induced obese rats. FEBS Lett 2001; 491: 154–158 [DOI] [PubMed] [Google Scholar]
- 38.Murakami H, Murakami R, Kambe F, Cao X, Takahashi R, Asai T, Hirai T, Numaguchi Y, Okumura K, Seo H, Murohara T. Fenofibrate activates AMPK and increases eNOS phosphorylation in HUVEC. Biochem Biophys Res Commun 2006; 341: 973–978 [DOI] [PubMed] [Google Scholar]
- 39.Hong YA, Lim JH, Kim MY, Kim TW, Kim Y, Yang KS, Park HS, Choi SR, Chung S, Kim HW, Kim HW, Choi BS, Chang YS, Park CW. Fenofibrate Improves Renal Lipotoxicity through Activation of AMPK-PGC-1α in db/db Mice. PLoS ONE 2014; 9: e96147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ju H, Shin H, Son C, Park K, Choi I. 3-Iodothyronamine-mediated metabolic suppression increases the phosphorylation of AMPK and induces fuel choice toward lipid mobilization. Horm Metab Res 2015; 47: 605–610 [DOI] [PubMed] [Google Scholar]
- 41.Lee WJ, Kim M, Park H-S, Kim HS, Jeon MJ, Oh KS, Koh EH, Won JC, Kim M-S, Oh GT, Yoon M, Lee K-U, Park J-Y. AMPK activation increases fatty acid oxidation in skeletal muscle by activating PPARalpha and PGC-1. Biochem Biophys Res Commun 2006; 340: 291–295 [DOI] [PubMed] [Google Scholar]
- 42.Lee SK, Lee JO, Kim JH, Kim N, You GY, Moon JW, Sha J, Kim SJ, Lee YW, Kang HJ, Park SH, Kim HS. Coenzyme Q10 increases the fatty acid oxidation through AMPK-mediated PPARα induction in 3T3-L1 preadipocytes. Cell Signal 2012; 24: 2329–2336 [DOI] [PubMed] [Google Scholar]
- 43.Sozio MS, Lu C, Zeng Y, Liangpunsakul S, Crabb DW. Activated AMPK inhibits PPAR-{alpha} and PPAR-{gamma} transcriptional activity in hepatoma cells. Am J Physiol Gastrointest Liver Physiol 2011; 301: G739–G747 [DOI] [PMC free article] [PubMed] [Google Scholar]