

Abstract
Real‐world evidence provides important information about the effects of medicines in routine clinical practice. To engender trust that evidence generated for regulatory purposes is sufficiently valid, transparency in the reasoning that underlies study design decisions is critical. Building on existing guidance and frameworks, we developed the Structured Preapproval and Postapproval Comparative study design framework to generate valid and transparent real‐world Evidence (SPACE) as a process for identifying design elements and minimal criteria for feasibility and validity concerns, and for documenting decisions. Starting with an articulated research question, we identify key components of the randomized controlled trial needed to maximize validity, and pragmatic choices are considered when required. A causal diagram is used to justify the variables identified for confounding control, and key decisions, assumptions, and evidence are captured in a structured way. In this way, SPACE may improve dialogue and build trust among healthcare providers, patients, regulators, and researchers.
Real‐world evidence (RWE) provides important information about the effects of medicines in routine clinical practice, as actually prescribed by physicians and taken by patients. For many years, observational designs and real‐world data (RWD) sources have been used to characterize patient populations, describe the natural history of diseases, and assess postapproval safety and effectiveness as part of the drug development lifecycle. More recently, the use of RWE for pharmacovigilance, in particular, has evolved as a result of advances in data linkage and computing power, the greater availability of analytic tools and methodologies, and established best practice. These advances have also made possible the potential use of RWE for other purposes, such as product approval and label expansions. In the United States and Europe, RWD comparators are already being used for selected approval of new molecular entities in rare disease and oncology.1 Under the 21st Century Cures Act and the Prescription Drug User Fee Act (PDUFA) VI, the US Food and Drug Administration (FDA) has issued a framework for evaluating the use of RWE to augment the insights of randomized controlled trials (RCTs), which includes forthcoming guidance for designing and conducting these studies.2 The European Medicine Agency (EMA) adaptive pathways, which is an accelerated approval approach to permit earlier patient access to new medicines, has also emphasized the importance of data from real‐world clinical practice.3 However, to realize the opportunity to use RWE for regulatory decisions related to product effectiveness, the approach to RWE designs and methods must be sound and “fit for purpose.”
Advocates for the expanded use of RWE to support regulatory decisions have called for greater transparency in the design and conduct of research using RWD,1, 4, 5, 6 with specific guidance about what this constitutes.7 Over time, with transparency about how data are selected, curated, analyzed, and validated, it is thought that decision‐makers will have greater trust in the evidence generated. For many, one of the first steps is standardized design and methods reporting across journals, providing much more detail than is currently the case. For others, one of the first steps would involve the public posting of protocols, which the EMA already requires for postauthorization safety studies sponsored by pharmaceutical companies. These actions are intended to facilitate replication by other researchers and transparency for multiple stakeholders. Best‐practice guidance is also essential. The recently released RWE framework from the FDA highlights that guidance on the use of RWD sources and best methodological practice already exists.5, 8, 9, 10, 11, 12, 13 Likewise, there are general pharmacoepidemiology checklists designed to promote study quality (e.g., see the European Network of Centres for Pharmacoepidemiology and Pharmacovigilance (ENCePP) Checklist for Study Protocols14), along with other checklists for studies intended to generate RWE for regulatory use,15, 16 which together can be used to show the series of decisions that led to a particular study design and data source being selected. Although these guidance and checklist documents are useful for technical experts, they are not meant to be decision aids or to bring to the surface the underlying rationale and assumptions of design and methods decisions taken by researchers.
Other authors have provided guidance on the decision‐making underlying the generation of RWE from electronic healthcare databases for regulatory purposes. Schneeweiss6 and Schneeweiss et al.17 proposed a framework for generating evidence fit for decision making, which focuses on rapid analysis of healthcare databases (much of which can be applied beyond studies conducted in existing data).6, 17 Their “MVET framework” refers to meaningful, valid, expedited, and transparent evidence and is proposed as a tool to help researchers and stakeholders converge on the optimal solution for a particular research question about a medicine's net benefit. Franklin et al.4 proposed a structured process to design and implement database analyses that consists of a sequence of considerations and actions related to data fitness, completion of a statistical analysis plan, feasibility analyses, protocol registration, analyses, and structured reporting, with emphasis on transparency and allowance for regulatory feedback.4
In what follows, we propose a framework complementary to these previous examples. Our proposal focuses on two components of the MVET framework: validity, and the “trust” component of transparency. Our proposed Structured Preapproval and Postapproval Comparative study design framework to generate valid and transparent real‐world Evidence (SPACE) can be thought of as a tool to elicit and elucidate these two components—validity and trust—by exposing the assumptions and reasoning behind design decisions, thus enabling a priori sponsor–regulator agreement on design decisions to ensure the highest quality of evidence that can be practicably achieved. Importantly, SPACE uses a structured format that is not wedded to particular types of data, unlike other published frameworks. SPACE begins with articulation of a very specific research question. From there, we specify key components of the RCT that we would use to maximize validity, as suggested by Hernán18 and Hernán and Robins.19 More pragmatic choices are then incorporated as needed. A causal diagram (such as a directed acyclic graph20, 21) is drawn to justify the identification of variables for confounding control, and key decisions, assumptions, and evidence are captured in a structured way, ideally in real time. In the next section, we discuss why this framework is needed, describe step‐by‐step how to use the tool, and then draw on past examples to illustrate its use.
Why do we need SPACE?
As regulators and researchers grapple with the specific scientific and operational standards needed to incorporate RWE in regulatory decisions, we look at the many decades of experience using RWD to conduct postapproval comparative safety studies. Schneeweiss et al.17 rightly pointed out that “there is no generalizable advice regarding where optimal information for a given question can be found.” However, there is an opportunity to draw on principles from experiences with RWE in the pharmacoepidemiology field. This experience spans more than 20 years; is broad in its use of study designs, RWD sources, end points, and analytic tools; includes successes and many high‐profiles failures; and, importantly, benefits from being shared knowledge across many academics, regulators, and pharmaceutical researchers. A key principle that emerges from these experiences is the importance of first articulating the optimal design regardless of known constraints (whether these be operational or financial), instead of starting with an available data source. Beginning this way forces researchers to clearly articulate the trade‐offs they make between validity and efficiency when designing and executing a study. Our aim here is to illuminate, in general and using specific examples, how we go about finding the best information on a case‐by‐case basis, based on the minimal criteria for validity that arise from a given research question.
Like others, we refer to “regulatory‐quality” RWE as arising from studies that enable estimation of a realized causal effect (i.e., the effect the treatment actually had on the outcome in the study population).6 Valid effect estimation requires that internal validity violations are avoided through the design and/or analysis.22, 23 This task is most feasible in an RCT, in which bias is reduced via experimental design features, such as randomization and regimented measurement and follow‐up; for this reason, in practice, the RCT is considered the gold standard for measuring causal effects. In RWE studies, many of these RCT design features are absent. As the study becomes more pragmatic, there is more residual uncertainty around the impact of bias on the study results. Thus, we suggest real‐world conditions are allowed when of specific interest or needed to improve study feasibility, and potential biases can arguably be minimized. To assure an interpretable effect, the researcher should be explicit in his/her rationale for any decisions that take the design further away from the RCT standard, so that this uncertainty is minimized to an acceptable level. To frame the decision‐making process, we find it helpful to articulate high‐level priorities based on the stakes involved. For example, if we are concerned about missing the small effect of a treatment on decreased mortality, we might have less tolerance for uncertainty around bias and use more experimental features. In contrast, if we are concerned about confirming a large effect or we are conducting a postapproval study to confirm safety of long‐term use, we may be able to tolerate more uncertainty around bias in favor of a less controlled study that can be completed more quickly. Many of these parameters are shown in Schneeweiss et al.'s17 Figure 4 and Table 2. Other considerations might include the severity of the outcome (e.g., mortality vs. improvement of a moderate disease symptom) and the overall benefit–risk profile of the medicine (i.e., if a nonnull study result improves or tips the balance to the negative). Stakeholder input (e.g., regulators, patient groups, and clinical experts) is critical in determining what is clinically meaningful, allowably uncertain, and how uncertainty will be characterized or quantified post hoc. A priori quantification of potential biases (see examples in refs. 24, 25, 26, 27, 28) may also be useful, in particular for discussions with regulators around the likelihood of effect estimate interpretability.
Once it is determined that the study result(s) are likely to be meaningful and interpretable, we can begin the study design process. When building RWE for regulatory decisions, transparency around decision making is critical. Although final decisions around data source and design are typically recorded in a protocol or analysis plan, the decision‐making process and evidence to support those decisions is recorded less consistently and with less detail. It is important that these decisions are not only transparent but also structured for stakeholders, such as regulators, patients, and physicians. Given the increasing complexity of RWE studies in terms of the number of institutions, data providers, academics, sponsors, etc. involved, transparency facilitates common understanding of why design decisions have been made the way they have. It also guides conversations about what constitutes fit‐for‐purpose evidence and why is potentially a tool for collating different stakeholder views, and addresses varied views by planning sensitivity or secondary analyses up‐front.
How should SPACE be used?
The structured study design framework described here (see Figures 1 , 2 , 3 , 4 , 5 , 6 ) aims to enable experienced scientific and medical decision‐makers to increase transparency around the assumptions and trade‐offs underlying their decisions about what constitutes fit‐for‐purpose RWE (e.g., trade‐offs between validity and generalizability, or between validity and feasibility). The framework is intended to precede and supplement a prespecified protocol and analytic plan. Together these documents ensure that primary, secondary, and sensitivity analyses are specified prior to data collection (or extraction if secondary data) and statistical analyses. SPACE is intended to reflect the natural thinking process of technical experts, by organizing decision points, evidence, and criteria, and aiding iterative and evidence‐based decision making.
Figure 1.
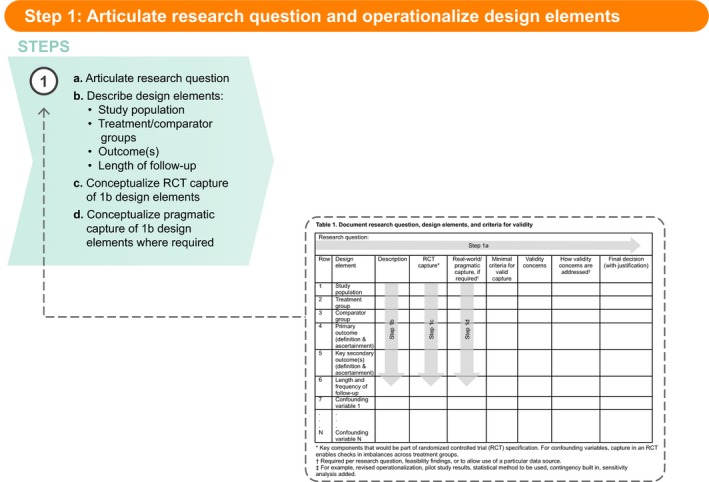
Step 1: Articulate research question and operationalize design elements. RCT, randomized controlled trial
Figure 2.
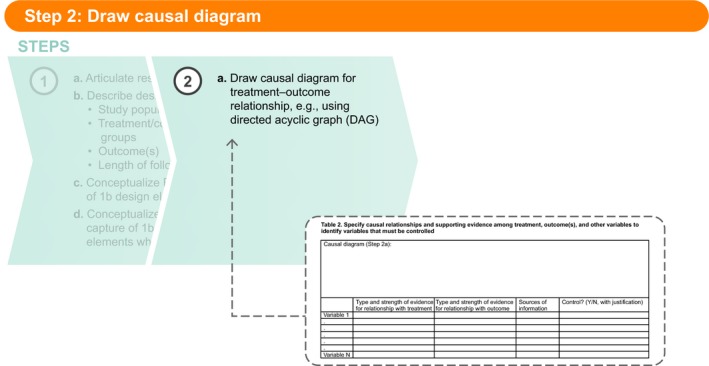
Step 2a: Draw causal diagram.
Figure 3.

Step 2b: Describe and conceptualize potential confounder capture.
Figure 4.
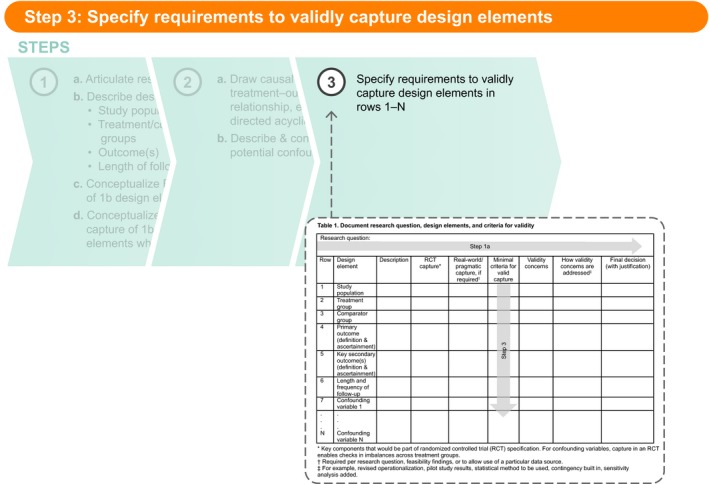
Step 3: Specify requirements to validly capture design elements.
Figure 5.
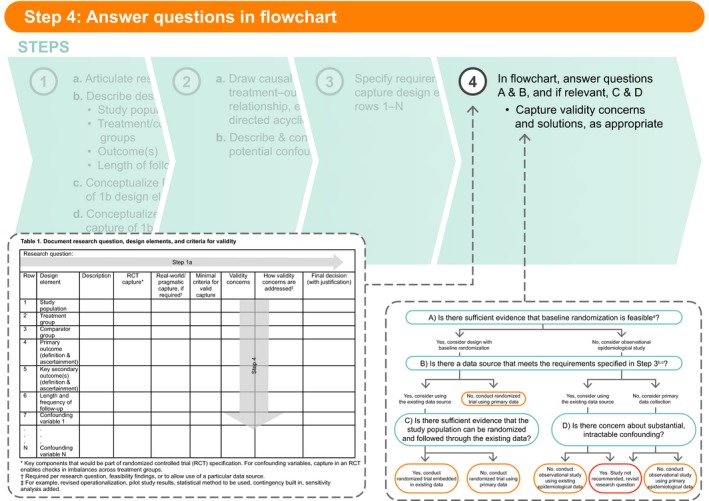
Step 4: Answer questions in flowchart.
Figure 6.
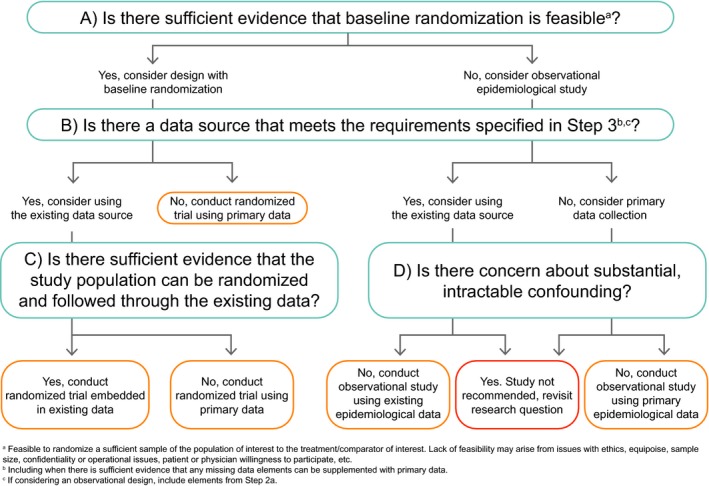
Structured Preapproval and Postapproval Comparative study design framework to generate valid and transparent real‐world Evidence (SPACE) flowchart.
Ideally, the intent to measure a safety effect or effectiveness for regulatory decision making will trigger use of SPACE. Its use is envisioned as connecting that intent to systematic feasibility assessment, and ultimately to a final protocol (with analysis plan). The protocol/analysis plan is where the final, fully operationalized definitions (e.g., an algorithm of diagnoses, medications, and procedures used to identify the occurrence of a clinical end point) are recorded. These definitions should be specific enough to allow another researcher with access to the same underlying data source to exactly replicate that design element, as per the reproducibility component of the “T” in the MVET framework.4
Once the research question is formulated, utilizing SPACE can help organize and document one's thinking. This structured framework follows typical steps required for design decisions, reminding the user to document decisions and evidence at each step. In the following paragraphs, we walk through the SPACE steps one by one. We then introduce a postapproval comparative safety study example, and describe the considerations that led to the final design decisions captured in Table S1 .
Step 1: Articulate research question and design elements
The first step in any good research study is to clearly and specifically articulate the research question (Table 1 , Step 1a). This should include specification of the study population, treatment, and comparator groups, outcome(s) of interest, and the length of follow‐up, such that the design elements clearly arise from the research question (Table 1 , Step 1b).9 If the design elements are not easily identified, we suggest further clarification of the research question (which may require discussion with stakeholders/decision‐makers) before moving on to Step 1c, in which the ideal capture for each element is conceptualized. To help ensure validity requirements are met, it has been suggested that we consider which RCT we would carry out, if feasible and ethical,18, 19 then consider whether the design elements in that conceptualized RCT should be less controlled and more pragmatic. Indeed, RCT evidence is the basis for most drug approvals and label changes, and we believe that using this as the starting place will lead to better transparency and acceptance of RWE. To realize the potential for RWE with randomization, the framework challenges the user to consider RCT designs on a continuum from most controlled to most pragmatic. In practice, given the purpose of generating RWE, and prior to selecting an observational design, the user will focus on the feasibility of an RCT with the most pragmatic elements. The capture of the key components of the conceptualized RCT is Step 1c (Table 1 ).
Table 1.
Document research question, design elements, and criteria for validity
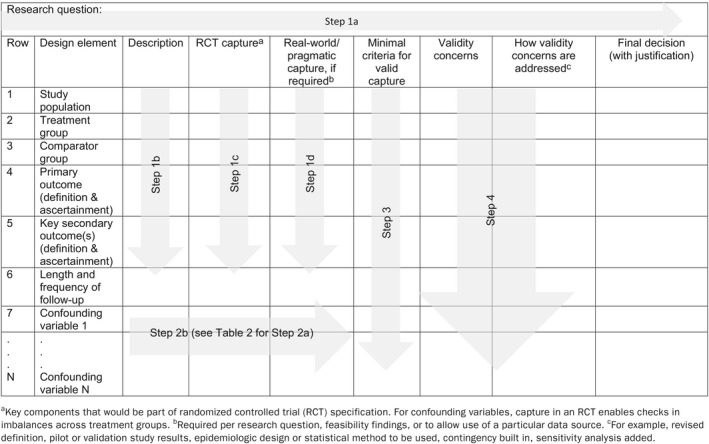
The next part of Step 1 is to determine whether a less controlled and more pragmatic, real‐world conceptualization should be considered for any of these design elements (Table 1 , Step 1d). The need for pragmatic design elements in comparative studies typically arise for one of two reasons. First, pragmatic design elements arise when specific aspects of routine clinical practice are critical to address the question of interest. For example, “is drug X effective when used by patients with more comorbidities than those studied in the pivotal phase III trial?,” which could be addressed preapproval or postapproval. It should be clear from the fully articulated research question whether this is the case. Some elements of routine use that might be of interest are shown in Figure S1 , overlaid on a simple diagram of a typical RCT.
Second, the need for more pragmatic elements can arise due to ethical or feasibility considerations. Figures S2 and S3 show routinely used elements that might arise as a result of these considerations. Although pragmatic elements arising from the research question can be captured in Step 1d (Table 1 ), pragmatic elements required because of feasibility or ethical concerns will often become clear later in the SPACE process. For instance, we might realize during the feasibility assessment for an observational study (where randomization cannot be used) that the primary end point cannot be captured according to the RCT conceptualization (which, for example, requires a monthly procedure for patients who, under routine care, are only seen approximately every 6 months). In this decision path, we would consider the more pragmatic version of the end point (capture procedures conducted during routine visits) in the “Real‐world/pragmatic capture” column.
Step 2: Draw causal diagram
After the completion of Step 1, the next step (in Table 2 , Step 2a) is to draw a causal diagram for each treatment–comparator–outcome relationship specified in the research question and described in Table 1 , Steps 1b and 1c. If, for example, the treatment will be compared with two comparators for one primary outcome, two causal diagrams will be needed (unless a strong argument can be made that the causes of the treatment vs. each comparator are exactly the same). Likewise, a study comparing a treatment with one comparator but with two key outcomes would require two causal diagrams. Directed acyclic graphs20, 21 and single‐world intervention graphs29 are two options that allow the user to specify the theorized causal relationships among the treatment, outcome(s), and other variables considered for control of confounding (e.g., causes shared by treatment and outcome). This is not a trivial task and may lead to a complicated diagram, but it is a critical step in identifying variables that should and should not be controlled to validly estimate the effect. Table 2 provides a structure for capturing the causal diagram, and, importantly, the strength of evidence for each relationship. The user can also include key references for these decisions. The information captured in Table 2 should indicate which variables are potential confounders and should, therefore, be controlled.
Table 2.
Specify causal relationships and supporting evidence among treatment, outcome(s), and other variables to identify variables that must be controlled
| Causal diagram (Step 2a): | ||||
|---|---|---|---|---|
| Type and strength of evidence for relationship with treatment | Type and strength of evidence for relationship with outcome | Sources of information | Control? (Y/N, with justification) | |
| Variable 1 | ||||
| . | ||||
| . | ||||
| . | ||||
| . | ||||
| . | ||||
| Variable N | ||||
Once potential confounders are identified, in Step 2b these variables should be described and conceptualized similarly to the other design elements in Table 1 . Again, the intent is to capture the key components that could be measured most rigorously in an RCT. Similarly to Step 1, if a more pragmatic definition of a confounder is shown to be sufficiently valid, that alternative conceptualization should be captured in the appropriate column in Table 1 .
One might wonder why, at this point, we would spend time drawing a causal diagram and identifying potential confounders. Transparency in the researcher's assumptions about the causal relationships among study variables is critical to decision making about the necessity of randomization and designing the confounding control approach if randomization is not used. Even in a randomized study, accurate identification of potentially confounding variables and their valid measurement allows the researcher to confirm that randomization achieved sufficient balance across treatment groups.
Step 3: Specify requirements to validly capture design elements
In Step 3, the user specifies the minimal criteria that must be met to validly capture each design element in Table 1 , rows 1−N. These criteria will become the basis for data‐source (and, if needed, primary data collection) feasibility evaluation and should be as specific as needed for another researcher or stakeholder to agree or disagree with a particular criterion and to reach the same conclusion about feasibility if applying these same criteria. For example, a study in which the research question led to specification of a study population that includes a sufficient number of older people (to fill a post–phase III knowledge gap), a criterion could be: Data source does/can include at least X people aged 65–75 and 76–85 years. However, a study in which the primary end point is measured by biopsy could include the criterion: Data source captures biopsy procedure codes and results. For some rows of Table 1 , the minimal criteria listed will be absolute requirements; in other cases, the criteria might be desirable but not required. As an example, for a study with an intended long follow‐up period, 5 years of patient follow‐up might be preferred, but 3 years of follow‐up might be set as the minimum criterion for feasibility. Any criterion that is preferred but not required should be noted as such in Step 3.
Moving from Step 3 to Step 4 (Table 1 ), and certainly following Step 4, typically requires systematic feasibility assessment, which is often also an iterative process (e.g., narrowing down options based on a few key criteria and then performing more detailed feasibility using the remaining criteria on the remaining options). Qualities of a good feasibility assessment are described elsewhere (for example, see the ENCePP Guide on Methodological Standards in Pharmacoepidemiology9). Thoughtful feasibility is, in our opinion, key to designing an optimized study. In our own work, we follow a formal process for systematic feasibility assessment conduct based on best practices established over time. Although SPACE leads to the minimal criteria that form the basis of such a feasibility assessment, this process is beyond the scope of this paper. In general, the minimal criteria set in Table 1 , Step 3 can be ranked such that a user quickly minimizes the list of potential options (e.g., restricted to collecting primary data for valid capture of the primary end point) or potential data sources (e.g., by limiting to data sources with >40 million enrollees, laboratory results, or in‐hospital prescription data).
Step 4: Answer questions in flowchart
After completion of Steps 1–3, the questions in the flowchart (Figure 6 ) should be answered, as appropriate. The responses to these questions, along with relevant feasibility results, should be captured in Table 1 , Step 4. As a user considers the minimal criteria needed for valid capture in Step 3, they will likely note validity concerns along the way. For example, when considering the minimal criteria for valid measure of the primary end point, the user could be concerned that if the study hypothesis is known to the investigator (i.e., under primary data collection) and the treatment is not masked, misclassification bias due to differential screening could occur. In this case, the concern might be addressed by imposing mandatory screening or masked end point adjudication. This should be noted in the “Validity concerns” and “How validity concerns are addressed” columns.
In Figure 6 , Question A asks whether there is sufficient evidence that baseline randomization is feasible. This question refers to the feasibility of randomizing a sufficient sample of the study population to the treatment/comparator of interest. Lack of randomization feasibility may arise from issues with ethics, equipoise, sample size, confidentiality or operational issues, and patient or physician willingness to participate, etc. Other than for reasons of ethical violations, which will likely be known to the researcher at the start of the design process, sample‐size calculations (typically taking the largest of a reasonable range of minimal sample sizes estimated by varying assumptions) will be needed to answer this question.
If the answer to Question A is yes, the user should consider a design with baseline randomization and will continue down the left side of Figure 6 . If the answer to Question A is no, the user should consider an observational epidemiological design and continue down the right side of Figure 6 . Question B then asks whether there is a data source that meets the requirements specified in Step 3. (Note that if the answer to Question A was no, the potential confounders specified in Table 1 , rows 7−N should be included in the feasibility assessment conducted to answer Question B.) Again, the responses are yes or no. A yes response indicates that the user should consider using the existing data. By “existing data” we mean a data source in which the types of data needed for the study are already collected into an electronic system, even if the specific data needed for the study are not yet in the dataset (i.e., will accumulate in the future). For instance, if a data source included all the necessary variables but does not yet include people exposed to the treatment of interest, we would still consider the data source feasible if we have evidence that a sufficient number of people exposed to the treatment will become captured in the data in the time frame needed for the study. The response to Question B should also consider whether an existing data source that includes the majority of necessary data can be supplemented. In this circumstance, we would typically evaluate the feasibility of adding the field to the existing data‐source collection structure (which would change the response to yes), or collecting the missing information de novo for study participants and merging it with the electronic data. If it is feasible to supplement the existing data, the answer to Question B can be yes.
On the left side of Figure 6 , if the answer to Question B is no, a randomized trial to collect primary data will be needed. If the answer to Question B is yes, the user should continue to Question C, which asks whether there is sufficient evidence that the study population can be randomized and followed through the existing data. This should also be evaluated during data‐source feasibility. The user might ask the data owner whether randomization of people included in the data source has been done previously (including how many times, under what circumstances, success of the randomized study, etc.), and whether willing participants not currently captured in the data source can be added to the ongoing data collection (again, asking how many times, under what circumstances, success of the randomized study, etc.). If a successful embedded randomized trial has not been previously done, the data owner should be asked to provide evidence of feasibility.
If on the right side of Figure 6 , the user will continue to Question D, while staying in the “lane” indicated based on their response to Question B. Question D asks whether there is concern about substantial, intractable confounding. If the answer to Question D is yes, conducting a study is not recommended. The user might revisit the research question and consequent design elements to determine if there is an alternative acceptable research question that will terminate elsewhere in the flowchart. If the answer to Question D is no and the answer to Question B was yes, then an observational study using the existing epidemiological data source can be conducted. If the answer to Question D is no and the answer to Question B was no, then a primary data‐collection observational study can be conducted.
What constitutes substantial, intractable confounding (relevant to Question D) is, of course, not black and white. Concerns about intractable confounding could arise from prior studies, stakeholder concerns, baseline or feasibility evidence, etc. Considerations will include the potential for unknown confounding (typically in cases where the underlying causal structure is not well understood); uncontrolled confounding due to missing data on, or misclassification of, one or more confounders or concerns about the ability to control confounding due to positivity violations (i.e., when there are no/few people in a treatment group at some strata of the confounders). The answer to this question should be evidence‐based; evidence for likely confounding is documented in Table 2 , and the means by which this confounding will be controlled is documented in Table 1 .
Revisit Steps 1–4 as needed: The steps are inherently iterative
As new evidence, algorithms, stakeholder views, etc. become available, the design elements in Steps 1 and 2 may be revised. These changes, in turn, might make it necessary to revisit the requirements specified in Step 3, and the responses to the flowchart questions and validity considerations in Step 4. In our experience, the first pass through these steps is usually based on limited, well‐known information, and we add evidence, and further specify and refine as we work through the steps and necessary feasibility. For instance, we might specify a basic preliminary outcome algorithm that is then refined after systematic review of the published studies that evaluated the outcome of interest.
In addition, if one is doing a randomized study in which pragmatic elements are not required per the research question or feasibility, they might still be evaluated and incorporated, if required, to take advantage of a data source that is otherwise sufficient. For example, if the research question for a particular study did not specify “routine follow‐up” but an existing electronic health record database is being considered that meets the requirements identified in Table 1 rows 1–6, the user can capture the pragmatic operationalization of outcome measurement and length of follow‐up necessitated by routine follow‐up and describe any concerns they have regarding the impact on validity that they might have (e.g., in terms of bias in outcome). If there are no concerns or the concerns can be sufficiently addressed (e.g., with pilot work, validation, end point adjudication, etc.), the researcher might continue with the RCT embedded in the existing data source, articulating any changes to the original research question.
By the time the user is at the point of preparing a final protocol and analysis plan, the far right column of Table 1 , “Final decision (with justification),” should be complete and include the necessary detail for a reviewer to understand how the final choices were made, under what assumptions, and based on what evidence.
Examples of how the SPACE framework can be used
The following section illustrates the application of the SPACE framework using Ziprasidone Observational Study of Cardiac Outcomes (ZODIAC), a pragmatic clinical trial designed to address a safety question for ziprasidone.
Example 1: ZODIAC
Ziprasidone (Geodon; Pfizer, New York, NY; approved in 1998 by the Swedish Medical Products Agency (MPA) and in 2001 by the FDA) is an atypical antipsychotic for the treatment of patients with schizophrenia. Concerns about its safety, specifically whether the corrected QT (QTc) prolongation caused by the drug would translate into increased mortality from sudden death in patients using it in the real world, remained at the time of ziprasidone's approval by regulatory authorities in the United States and Europe. QTc prolongation is of clinical concern because of its potential to induce Torsades de Pointes and other serious ventricular arrhythmias, which could result in sudden death. Before approval, the sponsor (Pfizer) completed a randomized comparison of six antipsychotics, examining their effects on the QTc interval at and around the time of estimated peak plasma/serum concentrations in the absence and presence of metabolic inhibition.30 The study found that the mean QTc prolongation was ~ 9–14 ms greater for ziprasidone than for several others tested but ~ 14 ms lower than thioridazine, a drug with reports of sudden death at high doses. Mean QTc intervals did not exceed 500 ms for any patient taking any antipsychotic, a threshold generally accepted as important for the development of Torsades de Pointes. Although drugs associated with the risk of a greater degree of QTc prolongation than ziprasidone had been shown to increase the risk of sudden death, the precise relationship between QTc prolongation and the risk of serious adverse cardiac events was unknown at the time of ziprasidone's approval. Despite these concerns, it was approved first by the Swedish MPA (as part of the European Union mutual recognition procedure) and the FDA due to its demonstrated effectiveness and the need for additional treatments for schizophrenia. In addition, premarketing clinical trials indicated that ziprasidone might be associated with improved lipids and lower incidence of weight gain, potential benefits that were also suggested in the Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) real‐world study, which added a ziprasidone arm in 2002.31
It is in this context that Pfizer made a commitment to the FDA and Swedish MPA to design and conduct a real‐world study for postmarketing safety evaluation. Detailed information about the design, operational conduct, and results of the ZODIAC study, including the names of epidemiological, psychiatric, and cardiovascular experts involved, are published elsewhere.32, 33, 34 The study was unprecedented in psychiatric research, both in size and design.35 We selected this example to illustrate how using the SPACE framework results in the selection of a pragmatic randomized study with observational follow‐up, the design ultimately agreed with the FDA and Swedish MPA. Tables S1 and S2 provide the main text Tables 1 and 2 completed for the ZODIAC example. Additional background details for each step are provided here.
Step 1: Articulate research question and operationalize design elements
The research question underlying ZODIAC (Step 1a) was “Does ziprasidone's modest QTc‐prolonging effect cause an increased risk of sudden death when compared with another atypical antipsychotic without this effect and when used among patients with schizophrenia diagnosed and treated in usual care settings in the US, EU, and other regions of the world?” Examination of this question reveals the basic design elements described in Step 1b (see Table S1 ).
Study population: Patients with schizophrenia diagnosed and treated in usual care settings in the United States, European Union, and other regions of the world.
Treatment: Ziprasidone
Comparator: Another atypical antipsychotic without a modest QTc‐prolonging effect
Primary outcome: Sudden death
Length of follow‐up: Time required to examine a clinically relevant (if due to QTc‐prolonging effect of ziprasidone) and measurable outcome in the real world
From these design elements, general descriptions were added per Step 1b and key components of the RCT conceptualization were described per Step 1c (see Table S1 ). Next, a more pragmatic conceptualization of each element was specified (rows 1–6). For the study population, treatment, and comparator groups, more pragmatic capture was required per the research question (i.e., “when used among patients with schizophrenia diagnosed and treated in usual care settings”). As opposed to the structured interview with Diagnostic and Statistical Manual of Mental Disorders criteria specified as the RCT‐like capture of schizophrenia, here, a physician's reported diagnosis of schizophrenia was deemed preferable to reflect usual care practice and may have included schizoaffective disorders or other schizophrenia spectrum diagnoses, depending on the enrolling physician's practice. Given the specific interest in usual care treatment, for both ziprasidone and comparator, routine follow‐up postrandomization (e.g., allowing medication changes and usual care visits) was preferred.
For the primary and secondary outcomes and length/frequency of follow‐up, a more pragmatic conceptualization was added as a result of the earlier identified need for usual care follow‐up. For instance, although preapproval trials of atypical antipsychotics for patients with schizophrenia were typically 6 weeks and rarely up to 12–16 weeks in length, preliminary sample‐size calculations indicated that a minimum of 6 months’ follow‐up for tens of thousands of patients would be needed. Even with frequent contact, follow‐up in this patient population faces numerous challenges for that duration. Under usual care visits, this required capture of potential end points at routine visits to healthcare providers or at the end of the study contact. This included following up patients by contacting next of kin, analyses of national and regional mortality indices, and adjudication by an expert end point committee masked to treatment status.
Step 2: Draw causal diagram
Table S2 contains the directed acyclic graph for considering the causal effect of ziprasidone (vs. olanzapine) on sudden death, with the justification for considering high risk of cardiovascular disease to be a strong potential confounder.
Step 3: Specify requirements to validly capture design elements
In Table S1 , the minimal criteria needed to validly capture the design elements in rows 1−N were specified. Of these, ability to identify and randomize a large number of people with a schizophrenia diagnosis, capture vital status reports, and collect medical records and other information necessary for adjudication were key criteria for feasibility.
Step 4: Answer questions in flowchart
From preliminary discussions with treating physicians and clinical experts in the countries under consideration, there was no reason to believe that randomization (to ziprasidone and one of the comparators under consideration) of a large number of people with a schizophrenia diagnosis was infeasible. Thus, the answer to Question A in Figure 6 was yes and a study with baseline randomization was planned. At the time, there were no existing data sources that met the criteria identified in Step 3. The answer to Question B was no, and it was determined that primary data collection was needed. Because routine follow‐up was required to address the research question, the optimal design was a large simple (or pragmatic) trial with baseline randomization and entirely routine follow‐up. In addition, to better mimic real‐world care, treatment allocation was not masked, and physicians and patients were free to change dosing based on the patient's response to the assigned medication. This was important to ensure efficacious and comparable treatments because the approved label for ziprasidone in the United States recommended a low starting dose with dose escalation as required, reflecting potential cardiovascular concerns about high doses.
Despite cardiovascular outcomes being elevated in patients with schizophrenia compared with the general population, it became clear that even a very large sample size would not permit a statistically powered evaluation of the differences in the incidence of sudden death. Nonsuicide mortality was chosen as the primary end point (for which the study was powered), because even a larger increase in an uncommon cause of death, such as sudden death, could be counterbalanced by a small decrease in a more common cause of death, such as atherosclerotic events. The all‐cause, nonsuicide mortality‐aggregate measure was, therefore, deemed to be the most important and appropriate primary outcome measure. In discussion with the FDA and Swedish MPA, cardiovascular mortality and sudden death were identified as key secondary outcomes. An end‐point committee, composed of academic experts, adjudicated primary and secondary end points based on a review of medical and hospital records and death certificates, masked to treatment status.
To achieve the necessary sample size, other countries were added as more approvals of ziprasidone were obtained worldwide. Ultimately, >18,000 patients from 18 countries were randomized.
Additional examples
As another illustration of our framework, see Tables S3 and S4 . These tables are completed for an RWD postapproval safety study designed to evaluate the relationship between phosphodiesterase type 5 inhibitors (PDE5is), which are used to treat erectile dysfunction (ED), and nonarteritic anterior ischemic optic neuropathy (NAION). Tables S3 and S4 illustrate how a primary data collection case‐crossover study design was determined to be the optimal design to address the research question.
Real‐world aspects were not part of the research question but were instead driven by feasibility considerations. Even at the earliest stages of protocol development, randomization was out of the question based on infeasibility due to the large number of patients needed to study a very rare event (and clearly supported by the regulatory agency, which asked for an observational study). Through discussion with experts in epidemiology, ophthalmology, and urology, the case‐crossover design was identified as the optimal design because it was: (i) uniquely suited to a research question involving intermittent drug exposure (PDE5i are taken as needed) and an end point with abrupt onset (timing of NAION symptom onset is easily identified by the patient), (ii) a solution to the feasibility problem of studying a rare end point (patients are selected by end point status), and (iii) an ideal control for confounding (self‐matching controls for the confounding effects of time‐invariant personal characteristics, including the known risk factors shared between NAION and ED as well as unknown factors). The information in Tables S3 and S4 , therefore, represents a retroactive conceptualization of the RCT, but nonetheless illustrates what a real‐time articulation of the study design decision making would have looked like. Ultimately, an observational case‐crossover study with primary data collection was selected, as existing data did not provide sufficiently valid capture of the end point or timing of drug exposure. Given the media attention to the study hypothesis, steps were taken to mitigate potential bias in the measurement of the drug exposure and outcome. Additional background on the genesis of the study, which was completed in 2013, is provided below.
The PDE5i products first approved for as‐needed use to treat ED were sildenafil citrate (Viagra; Pfizer; approved in 1998 by the FDA/EMA), followed by tadalafil (Cialis; Eli Lilly, Indianapolis, IN; approved in 2002/2003 by the FDA/EMA), and vardenafil (Levitra; Bayer Healthcare Pharmaceuticals, Wayne, NJ; approved in 2003 by the FDA/EMA). Since approval, cases of NAION in men recently using PDE5i have been reported. NAION is a rare visual disorder estimated to occur in 2–12 per 100,000 men aged 50+ years, believed to be caused by hypoperfusion in the optic nerve. It typically presents as sudden, painless, partial vision loss in one eye, usually noticed upon wakening. The visual loss varies in magnitude and persistence with no effective treatment. Many of the case reports noted the presence of risk factors that are shared by NAION and ED, such as diabetes and hypertension, making their interpretation difficult. Internal review of data from Pfizer‐sponsored clinical trials and postmarketing surveillance studies of PDE5i had not indicated an association with NAION, although none of these trials or studies was designed to specifically evaluate this risk. Class labeling addressed the risk of NAION, noting the difficulty in separating the effect of patient risk factors from a drug‐induced effect. To address the limitations of the data available at the time, the FDA requested—and Pfizer committed to perform—a class‐level study to evaluate whether as‐needed use of PDE5i increases the risk of acute NAION within five half‐lives of drug ingestion.36
Finally, Tables S5 and S6 are completed for an ongoing RWD postapproval safety study designed to evaluate whether use of conjugated estrogens with bazedoxifene (CE/BZA; Duavive; Pfizer; approved in 2013 by the FDA and 2014 by the EMA), used to treat postmenopausal estrogen deficiency symptoms, increases the risk of endometrial cancer.37 Conduct of this postapproval study was a condition for EMA approval, with the overall aim to monitor the safety profile of CE/BZA in comparison to estrogen and progestin combination hormone replacement therapies. Some pragmatic aspects were part of the research question (e.g., wider variety of users, using the study medications, and being followed for a longer period of time). Others were driven by feasibility considerations. For instance, because the intention was to evaluate endometrial safety in a real‐world population and detection of the primary end points requires several years of follow‐up, an observational cohort study was conducted in the United States, where CE/BZA was commercially available more than a year earlier than in the European Union. Tables S5 and S6 illustrate how a health insurance claims–based cohort study, combined with end point validation and machine learning, was determined to be sufficient to address the research question.
Conclusions
SPACE provides a structured process for identifying study design elements, minimal criteria for feasibility and validity concerns, and for documenting decisions, to enable sponsor–regulator (and other stakeholder) agreement that a particular RWE study is fit for the intended regulatory purpose. From the articulation of a very specific research question, through identifying key components of the randomized trial that one would do to maximize validity, to considering pragmatic choices when required, SPACE is complementary to existing guidance and expands upon the validity and transparency components of the MVET framework. To achieve RWE that meets thresholds for causal interpretations (i.e., “regulatory quality” RWE), we must strive to estimate causal effects. When trade‐offs are required, internal validity has to trump generalizability and efficiency to meet this goal. Thus, our focus on internal validity is intended to aid sound decision making, to expose the assumptions and reasoning behind design decisions, and ultimately to improve dialogue and build trust among healthcare providers, patients, regulators, and researchers.
Documenting the evidence and justification for design decisions can highlight other available options, making clear where and what type of sensitivity analyses will be most useful. Additionally, this documentation allows decision‐makers to better understand how a particular design, method, or data source was chosen, and determine whether the rationale underlying those decisions applies to their circumstance (e.g., the clinical practice of their health system, or their regional or national population).
Although SPACE was developed based on our experience designing postapproval comparative studies for regulatory purposes, we find it works equally well when considering RWE to support new indications and label expansions (i.e., preapproval). As others consider the SPACE framework and use the tools (a downloadable and fully editable version of Tables 1 and 2 are included (as Tables S7 and S8 ), we hope that both researchers using the tools and stakeholders reviewing the content (which, for instance, could be included in a protocol appendix) will share their feedback. We expect SPACE to evolve as we all become more experienced using RWE to successfully support new and expanded indications.
Funding
Editorial support for this work was sponsored by Pfizer.
Conflict of Interest
N.M.G., R.F.R, and U.B.C. are full‐time employees and stock/shareholders of Pfizer.
Supporting information
Figure S1. Conceptualize the RCT you would do (if feasible/ethical) to address the research question.
Figure S2. Identify real‐world/pragmatic elements critical to the research question.
Figure S3. Identify real‐world/pragmatic elements required for feasibility.
Table S1. ZODIAC example: Document research question, design elements, and criteria for validity.
Table S2. ZODIAC example: Specify causal relationships and supporting evidence among treatment, outcome(s), and other variables to identify variables that must be controlled.
Table S3. NAION example: Document research question, design elements, and criteria for validity.
Table S4. NAION example: Specify causal relationships and supporting evidence among treatment, outcome(s), and other variables to identify variables that must be controlled.
Table S5. CE/BZA example: Document research question, design elements, and criteria for validity.
Table S6. CE/BZA example: Specify causal relationships and supporting evidence among treatment, outcome(s), and other variables to identify variables that must be controlled.
Table S7. Document research question, design elements, and criteria for validity.
Table S8. Specify causal relationships and supporting evidence among treatment, outcome(s), and other variables to identify variables that must be controlled.
Acknowledgment
Editorial assistance was provided by Paul Hassan, PhD, CMPP, of Engage Scientific Solutions. The authors thank Peter Honig, MD, MPH, and an anonymous reviewer for very helpful comments that led to important improvements to the manuscript.
References
- 1. Dreyer, N.A. Advancing a framework for regulatory use of real‐world evidence: when real is reliable. Ther. Innov. Regul. Sci. 52, 362–368 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. US Food and Drug Administration (FDA) . Framework for FDA's Real‐World Evidence Program. 1−37 <https://www.fda.gov/downloads/ScienceResearch/SpecialTopics/RealWorldEvidence/UCM627769.pdf> (2018). Accessed April 15, 2019
- 3. European Medicines Agency . Adaptive Pathways Workshop: Report on a Meeting with STAKEHOLDERS at EMA on 8 December 2016 (European Medicines Agency, London, England, 2017). [Google Scholar]
- 4. Franklin, J.M. , Glynn, R.J. , Martin, D. & Schneeweiss, S. Evaluating the use of nonrandomized real‐world data analyses for regulatory decision making. Clin. Pharmacol. Ther. 105, 2019 (2019). [DOI] [PubMed] [Google Scholar]
- 5. Berger, M.L. et al Good practices for real‐world data studies of treatment and/or comparative effectiveness: recommendations from the joint ISPOR‐ISPE Special Task Force on real‐world evidence in health care decision making. Pharmacoepidemiol. Drug Saf. 26, 1033–1039 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schneeweiss, S. Improving therapeutic effectiveness and safety through big healthcare data. Clin. Pharmacol. Ther. 99, 262–265 (2016). [DOI] [PubMed] [Google Scholar]
- 7. European Medicines Agency (EMA) European Network of Centres for Pharmacoepidemiology and Pharmacovigilance (ENCePP) . The ENCePP Code of Conduct for scientific independence and transparency in the conduct of pharmacoepidemiological and pharmacovigilance studies <http://www.encepp.eu/code_of_conduct/documents/ENCePPCodeofConduct_Rev3.pdf>. Accessed March 7, 2019.
- 8. Council for International Organizations of Medical Sciences (CIOMS) . International Ethical Guidelines for Epidemiological Studies issued by the Council for International Organizations of Medical Sciences (CIOMS) <http://ieaweb.org/wp-content/uploads/2012/06/cioms.pdf>. Accessed March 7, 2019.
- 9. European Medicines Agency (EMA) European Network of Centres for Pharmacoepidemiology and Pharmacovigilance (ENCePP) . ENCePP Guide on Methodological Standards in Pharmacoepidemiology <http://www.encepp.eu/standards_and_guidances/methodologicalGuide.shtml;>. Accessed March 7, 2019.
- 10. US Food and Drug Administration (FDA) . FDA Guidance for Industry: Good Pharmacovigilance Practices and Pharmacoepidemiologic Assessment <https://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm071696.pdf>. Accessed March 7, 2019.
- 11. US Food and Drug Administration (FDA) . FDA Guidance for Industry and FDA Staff: Best Practices for Conducting and Reporting Pharmacoepidemiologic Safety Studies Using Electronic Healthcare Data <http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM243537.pdf>. Accessed March 7, 2019.
- 12. International Epidemiological Association (IEA) . Good Epidemiological Practice (GEP) guidelines issued by the International Epidemiological Association (IEA) <http://ieaweb.org/2010/04/good-epidemiological-practice-gep/>. Accessed March 7, 2019.
- 13. International Society for Pharmacoepidemiology (ISPE) . Guidelines for Good Pharmacoepidemiology Practices (GPP) issued by the International Society for Pharmacoepidemiology (ISPE) <https://www.pharmacoepi.org/resources/guidelines_08027.cfm>. Accessed March 7, 2019.
- 14. European Medicines Agency (EMA) European Network of Centres for Pharmacoepidemiology and Pharmacovigilance (ENCePP) . ENCePP Checklist for Study Protocols <http://www.encepp.eu/standards_and_guidances/documents/ENCePPChecklistforStudyProtocolsRevision4_000.doc>. Accessed March 7, 2019.
- 15. Girman, C.J. , Ritchey, M.E. , Zhou, W. & Dreyer, N.A. Considerations in characterizing real‐world data relevance and quality for regulatory purposes: a commentary. Pharmacoepidemiol. Drug Saf. 28, 439–442 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Miksad, R.A. & Abernethy, A.P. Harnessing the power of real‐world evidence (RWE): a checklist to ensure regulatory‐grade data quality. Clin. Pharmacol. Ther. 103, 202–205 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Schneeweiss, S. et al Real world data in adaptive biomedical innovation: a framework for generating evidence fit for decision‐making. Clin. Pharmacol. Ther. 100, 633–646 (2016). [DOI] [PubMed] [Google Scholar]
- 18. Hernán, M.A. With great data comes great responsibility: publishing comparative effectiveness research in epidemiology. Epidemiology 22, 290–291 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hernán, M.A. & Robins, J.M. Using big data to emulate a target trial when a randomized trial is not available. Am. J. Epidemiol. 183, 758–764 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Greenland, S. , Pearl, J. & Robins, J.M. Causal diagrams for epidemiologic research. Epidemiology 10, 37–48 (1999). [PubMed] [Google Scholar]
- 21. Pearl, J. Causal diagrams for empirical research. Biometrika 82, 669–688 (1995). [Google Scholar]
- 22. Schwartz, S. , Campbell, U.B. , Gatto, N.M. & Gordon, K. Toward a clarification of the taxonomy of “bias” in epidemiology textbooks. Epidemiology 26, 216–222 (2015). [DOI] [PubMed] [Google Scholar]
- 23. Rothman, K.J. , Greenland, S. & Lash, T.L. Modern Epidemiology, 3rd edn (Lippincott Williams & Wilkins, Philadelphia, PA, 2008). [Google Scholar]
- 24. Lash, T.L. , Fox, M.P. , MacLehose, R.F. , Maldonado, G. , McCandless, L.C. & Greenland, S. Good practices for quantitative bias analysis. Int. J. Epidemiol. 43, 1969–1985 (2014). [DOI] [PubMed] [Google Scholar]
- 25. Munafò, M.R. , Tilling, K. , Taylor, A.E. , Evans, D.M. & Davey, Smith G. Collider scope: when selection bias can substantially influence observed associations. Int. J. Epidemiol. 47, 226–235 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lash, T.L. , Fox, M.P. & Fink, A.K. Spreadsheets from Applying Quantitative Bias Analysis to Epidemiologic Data <https://sites.google.com/site/biasanalysis/> (2014). Accessed February 23, 2018.
- 27. VanderWeele, T.J. & Ding, P. Sensitivity analysis in observational research: introducing the E‐value. Ann. Intern. Med. 167, 268–274 (2017). [DOI] [PubMed] [Google Scholar]
- 28. Greenland, S. Multiple‐bias modelling for analysis of observational data. J. R. Stat. Soc. Ser. A Stat. Soc. 168, 267–306 (2005). [Google Scholar]
- 29. Richardson, T.S. & Robins, J.M. Single world intervention graphs (SWIGs): a unification of the counterfactual and graphical approaches to causality. Working Paper Number 128, Center for Statistics and the Social Sciences, University of Washington <https://www.csss.washington.edu/Papers/wp128.pdf> (2013). Accessed March 7, 2019.
- 30. Harrigan, E.P. et al A randomized evaluation of the effects of six antipsychotic agents on QTc, in the absence and presence of metabolic inhibition. J. Clin. Psychopharmacol. 24, 62–69 (2004). [DOI] [PubMed] [Google Scholar]
- 31. Lieberman, J.A. et al Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N. Engl. J. Med. 353, 1209–1223 (2005). [DOI] [PubMed] [Google Scholar]
- 32. Kolitsopoulos, F.M. , Strom, B.L. , Faich, G.A. , Eng, S.M. , Kane, J.M. & Reynolds, R.F. Lessons learned in the conduct of a global, large simple trial of treatments indicated for schizophrenia. Contemp. Clin. Trials 34, 239–247 (2013). [DOI] [PubMed] [Google Scholar]
- 33. Strom, B.L. et al Comparative mortality associated with ziprasidone and olanzapine in real‐world use among 18,154 patients with schizophrenia: the Ziprasidone Observational study of Cardiac Outcomes (ZODIAC). Am. J. Psychiatry 168, 192–201 (2011). [DOI] [PubMed] [Google Scholar]
- 34. Strom, B.L. et al The Ziprasidone Observational Study of Cardiac Outcomes (ZODIAC): design and baseline subject characteristics. J. Clin. Psychiatry 69, 114–121 (2008). [DOI] [PubMed] [Google Scholar]
- 35. Stroup, T.S. What can large simple trials do for psychiatry? Am. J. Psychiatry 168, 117–119 (2011). [DOI] [PubMed] [Google Scholar]
- 36. Campbell, U.B. et al Acute nonarteritic anterior ischemic optic neuropathy and exposure to phosphodiesterase type 5 inhibitors. J. Sex. Med. 12, 139–151 (2015). [DOI] [PubMed] [Google Scholar]
- 37. European Medicines Agency (EMA) European Network of Centres for Pharmacoepidemiology and Pharmacovigilance (ENCePP) . Post‐Authorization Safety Study (PASS) of conjugated estrogens/bazedoxifene (CE/BZA) in the United States (posted protocol) <http://www.encepp.eu/encepp/viewResource.htm?id=26516;>. Accessed March 7, 2019.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Conceptualize the RCT you would do (if feasible/ethical) to address the research question.
Figure S2. Identify real‐world/pragmatic elements critical to the research question.
Figure S3. Identify real‐world/pragmatic elements required for feasibility.
Table S1. ZODIAC example: Document research question, design elements, and criteria for validity.
Table S2. ZODIAC example: Specify causal relationships and supporting evidence among treatment, outcome(s), and other variables to identify variables that must be controlled.
Table S3. NAION example: Document research question, design elements, and criteria for validity.
Table S4. NAION example: Specify causal relationships and supporting evidence among treatment, outcome(s), and other variables to identify variables that must be controlled.
Table S5. CE/BZA example: Document research question, design elements, and criteria for validity.
Table S6. CE/BZA example: Specify causal relationships and supporting evidence among treatment, outcome(s), and other variables to identify variables that must be controlled.
Table S7. Document research question, design elements, and criteria for validity.
Table S8. Specify causal relationships and supporting evidence among treatment, outcome(s), and other variables to identify variables that must be controlled.