

Summary
Background
Nonalcoholic fatty liver disease (NAFLD) is a prevalent disorder associated with obesity and diabetes. Few treatment options are effective for patients with NAFLD, but connections between the gut microbiome and NAFLD and NAFLD‐associated conditions suggest that modulation of the gut microbiota could be a novel therapeutic option.
Aim
To examine the effect of the gut microbiota on pathophysiologic causes of NAFLD and assess the potential of microbiota‐targeting therapies for NAFLD.
Methods
A PubMed search of the literature was performed; relevant articles were included.
Results
The composition of bacteria in the gastrointestinal tract can enhance fat deposition, modulate energy metabolism and alter inflammatory processes. Emerging evidence suggests a role for the gut microbiome in obesity and metabolic syndrome. NAFLD is often considered the hepatic manifestation of metabolic syndrome, and there has been tremendous progress in understanding the association of gut microbiome composition with NAFLD disease severity. We discuss the role of the gut microbiome in NAFLD pathophysiology and whether the microbiome composition can differentiate the two categories of NAFLD: nonalcoholic fatty liver (NAFL, the non‐progressive form) vs nonalcoholic steatohepatitis (NASH, the progressive form). The association between gut microbiome and fibrosis progression in NAFLD is also discussed. Finally, we review whether modulation of the gut microbiome plays a role in improving treatment outcomes for patients with NAFLD.
Conclusions
Multiple pathophysiologic pathways connect the gut microbiome with the pathophysiology of NAFLD. Therefore, therapeutics that effectively target the gut microbiome may be beneficial for the treatment of patients with NAFLD.
1. INTRODUCTION
Nonalcoholic fatty liver disease (NAFLD) is characterised by fat accumulation (or steatosis) in >5% of hepatocytes in individuals who either consume little alcohol or have no other secondary causes of steatosis such as viral hepatitis, lipodystrophy or medications associated with the development of steatosis.1, 2 The increasing prevalence of NAFLD parallels rises in the incidence of obesity and insulin resistance.3, 4 NAFLD is among the most common causes of liver disease and liver transplantation in the Western hemisphere.4, 5 NAFLD can be sub‐classified into two categories: the non‐progressive form, nonalcoholic fatty liver (NAFL) and the progressive form, nonalcoholic steatohepatitis (NASH).1, 6 NASH is a clinic‐pathologic entity that is typically characterised by the presence of zone 3 steatosis, ballooning and lobular inflammation; perisinusoidal fibrosis may or may not be also present.1, 7 Fibrosis progression rate is estimated to be higher in NASH than in NAFL and progression to cirrhosis may take up to 30 years; however, rapid progression to cirrhosis may occur in a small subset of patients.8 In addition, NASH is associated with increased risk of hepatocellular carcinoma (HCC) and all‐cause (cardiovascular and liver‐related) mortality.9, 10, 11
NAFLD pathogenesis is related to multiple insults that occur simultaneously and may act synergistically,12, 13 including accumulation of triglycerides (TGs),12 mitochondrial dysfunction and increased oxidative stress,12, 13, 14 altered mechanisms of apoptosis and autophagy,15, 16 increased levels of toxic lipid‐related factors (eg, free fatty acids)12 and liver inflammation.12 Genetics (eg, mutations in the patatin‐like phospholipase domain‐containing 3 gene)17 and adverse consequences of dietary habits and sedentary lifestyle (eg, insulin resistance, central obesity, dyslipidaemia and hypertriglyceridemia) likely contribute to overall NAFLD pathophysiology and augment the underlying mechanisms of liver insult.12
Accumulating evidence also implicates the gut microbiota in the development and progression of NAFLD18, 19 and suggests that therapeutic agents that target the gut microbiota may be beneficial. This review examines the pathophysiologic implications of altered gut microbiota in NAFLD and highlights recent progress in the development of microbiota‐targeting therapies for patients with liver disease.
2. MATERIALS AND METHODS
A PubMed search was performed for English language articles published between January 1, 2002 and January 31, 2019. Search terms included “cirrhosis” or “insulin resistance” or “liver disease” or “metabolic syndrome” or “NAFLD” or “obesity” or “steatohepatitis” and “microbiome.” The search was initially limited to primary publications and those of human subjects; articles reporting animal studies were later retrieved to allow more thorough discussion of pathophysiology. An additional nonsystematic search was performed to gather data on the use of prebiotics, probiotics, symbiotics, synbiotics and antibiotics for NAFLD. A total of 230 articles were retrieved; of these, 60 articles were reviewed. In addition, the bibliographies of these 60 articles were reviewed for additional relevant articles to consider for inclusion.
3. MICROBIOTA, MICROBIOME AND THE GUT
The term “microbiota” refers to a community of microbes in an organism, whereas “microbiome” is used to refer to both the microbiota and the collective genomes and gene products of the microbiota living in or on an organism. The gastrointestinal (GI) tract of humans contains 10 trillion to 100 trillion bacteria, with approximately 15 000‐36 000 species.20, 21 Composition of the gut microbiota varies among individuals as the result of multiple intrinsic (eg, age22, 23) and extrinsic (eg, method of feeding after birth24, 25 and geographic region inhabited26) aspects.
4. ROLE OF GUT MICROBIOME IN MAINTAINING HOMEOSTASIS
Progressing along the human GI tract from the jejunum to the colon, the number and the diversity of bacteria increases,27 and the predominant bacterial species change. In the upper GI tract (oesophagus and proximal small bowel), Streptococcus species predominate,28, 29 whereas in the colon, Firmicutes and Bacteroidetes are most prevalent.20, 21, 30, 31, 32 These locational alterations likely reflect the overall functionality of the dominant species (eg, Firmicutes and Bacteroidetes convert dietary complex carbohydrates and insoluble oligosaccharides to short‐chain fatty acids [SCFAs], which can be absorbed by the host within the intestines).33
The interconnection between the gut microbiome and the host is complex. The host provides both a suitable environment and nutrients for bacterial growth, and the host's diet, disease states and medications affect gut bacteria.33 The gut microbiota can, in turn, affect host nutrient and drug metabolism, contribute to maintaining the mucosal barrier of the GI tract, affect mucosal immunity and contribute to disease states.34 Bacteria in the GI tract synthesise host nutrients, such as vitamins and amino acids and conjugate primary bile acids (BAs) to form secondary BAs, such as deoxycholic acid and lithocholic acids.35, 36 The gut microbiota themselves derive sustenance mainly through the fermentation of dietary complex carbohydrates and indigestible oligosaccharides ingested by the host. Bacterial metabolism of these complex carbohydrates produces SCFAs (eg, butyrate, propionate and acetate), which the host can subsequently use as an energy source.34, 37, 38 A study of 15 healthy women given diets with varying levels of choline for 2 months found that the composition of the gut microbiome assessed by pyrosequencing of 16S ribosomal RNA bacterial genes in stool samples was altered from baseline with varying levels of dietary choline.39 Choline depletion was associated both with variations in the levels of Gammaproteobacteria and Erysipelotrichia and variations in amount of liver fat.39 The investigators hypothesised that the tendency to develop hepatic steatosis with a choline‐deficient diet could be predicted by a model based on bacterial levels, presence of a single nucleotide polymorphism affecting choline metabolism and change in hepatic steatosis.39
Alterations in either the number or function of the tight junctions found on the GI epithelium can result in increased intestinal permeability, allowing for passage of antigens and microbes into systemic circulation. A growing body of research indicates that BA metabolism via epidermal growth factor signalling may affect these tight junctions.40, 41 Deoxycholic acid and chenodeoxycholic acid have been shown to interact with and phosphorylate the endothelial growth factor receptor, ultimately resulting in tight junction rearrangement (through alterations in occludin, a structural protein found in tight junctions) and increased paracellular permeability.41 Interestingly, lithocholic acid has been shown to increase the integrity of tight junctions and to attenuate the production of reactive oxygen species, tumour necrosis factor alpha (TNF‐α), interleukin‐1β and interferon‐γ.42
Gut microbiota play a key immunomodulatory role, interacting closely with macrophages, dendritic cells, gut‐associated lymphoid tissues, B cells and T cells.43 For example, a healthy gut microbiome is integral to the proper development and function of T regulatory cells through a variety of cellular signalling mechanisms,44, 45, 46, 47 such as Clostridium butyricum inducing transforming growth factor‐β1 expression via toll‐like receptor (TLR)‐2 activation.48 Bacteria in the GI tract also participate in maintaining intestinal villous function34 and preventing intestinal epithelial cell apoptosis.49 In addition, alteration of the gut microbiota appears to have a role in intestinal disease (eg, inflammatory bowel disease [IBD]50, 51) and extraintestinal disease (eg, obesity,52, 53 diabetes43, 52, 53 and chronic liver disease54, 55). Furthermore, accumulating evidence supporting the role of the gut microbiota in drug metabolism (and corresponding effects on efficacy or adverse events) suggests that assessing microbiome activity could impact pharmaceutical drug development.56, 57
5. GUT MICROBIOME AND METABOLIC SYNDROME
Metabolic syndrome is defined as the presence of any three of the following conditions: central obesity, hypertension, impaired glucose tolerance (or overt diabetes mellitus), hypertriglyceridemia and low levels of high‐density lipoproteinemia (HDL).58 Given the close association between NAFLD and obesity, insulin resistance and TG levels,59 NAFLD is often considered the hepatic manifestation of metabolic syndrome.60 Because gut microbiota play a key role in metabolism and energy production from dietary intake,61 it stands to reason that they are closely related to components of metabolic syndrome (eg, obesity and diabetes).
One of the main factors in the development of obesity, diabetes and NAFLD is diet. Diet affects the composition of GI bacteria, which then influences host metabolism and inflammation.62 Turnbaugh and colleagues demonstrated the effect of diet on the gut microbiota in a mouse model.63 They humanised the gut microbiota of these mice by transplanting them with human faeces, and then they fed the mice either a high‐fat, high‐sugar diet (“Western diet”) or a plant‐based, low‐fat diet.63 The group that was fed a high‐fat diet had a lower percentage of Bacteroidetes spp in its gut microbiota and a higher percentage of Firmicutes compared with mice fed a plant‐based diet. Studies in humans and mice have shown an increased Firmicutes/Bacteroidetes ratio in individuals who are overweight or obese,64, 65 and a reduction in the Firmicutes/Bacteroidetes ratio with weight loss.32
In addition, higher colonic levels of SCFAs have been observed in obese individuals compared with their nonobese counterparts.64, 66, 67 The higher production of SCFAs may result in energy accumulation and subsequent weight gain.68 In a comparison between individuals who were morbidly obese and individuals of normal weight, increased numbers of both the H2‐producing bacteria Prevotellaceae and H2‐oxidising Archaea microorganisms were observed in the morbidly obese participants.69 The authors hypothesised that interspecies H2 transfer accelerated carbohydrate fermentation and the production of acetate, with an ensuing increased energy uptake by the host. An increase in SCFA levels may also alter other metabolic pathways (eg, lipid and glucose metabolism via activation of peroxisome proliferator‐activated receptor gamma70 and glucagon‐like peptide‐1 [GLP‐1]).71
The gut microbiota can also directly affect factors that control adiposity. For example, alterations in the gut microbiota affect the epithelial cell product, fasting‐induced adipocyte factor (FIAF, also known as angiopoietin‐like protein 4), which inhibits lipoprotein lipase. Preclinical models have demonstrated that microbial suppression of the FIAF gene increases lipogenesis,72 whereas administration of the bacterial strain Lactobacillus paracasei ssp paracasei F19 both induced FIAF gene expression and reduced body fat.73 The gut microbiota may also inhibit activity of the enzyme, adenosine monophosphate‐activated protein kinase, in muscle and liver, resulting in reduction of fatty acid oxidation and increased fat storage.74, 75
The gut microbiome also has a role in type 2 diabetes mellitus (T2DM). For example, a correlation between the Firmicutes/Bacteroidetes ratio and plasma glucose concentrations was reported in patients with T2DM.76 In addition, as stated above, the gut microbiota affect the production and release of GLP‐1, which affects pancreatic β‐cell function. SCFAs also appear to be closely interrelated with insulin resistance. Compared with patients without insulin resistance, patients with T2DM have a decrease in butyrate‐producing bacteria.77 SCFAs bind to G‐protein (GPR)‐coupled receptors (eg, GPR41 and GPR43),78 which may lead to the secretion of factors such as protein YY that affect satiety, gastric motility and pancreatic function.79, 80, 81
SCFAs also regulate various aspects of GI inflammation, such as neutrophil migration, T‐cell differentiation and macrophage expression of proinflammatory cytokines.82 Insulin resistance is often accompanied by low‐grade inflammation.83, 84 The movement of the GI bacterial product lipopolysaccharides [LPS] into intestinal capillaries may contribute to inflammation and insulin resistance.84 TLRs are a type of innate immune receptor that are thought to recognise LPS and other products of invading pathogenic bacteria.83 TLR knockout studies in mice are helping to elucidate the role of the gut microbiota in metabolic syndrome and insulin resistance. For example, mice deficient in TLR‐5 developed features of metabolic syndrome, including insulin resistance.85 The authors of this report suggested that changes in the composition of gut microbiota, resulting from loss of TLR‐5, induced the low‐grade inflammation that contributed to the symptoms of metabolic syndrome. Another report described mice with elimination of TLR‐4 specifically in hepatocytes.86 When these mice received a high‐fat diet, they became obese, but compared with control mice, these mice displayed enhanced insulin sensitivity and reduced hepatic steatosis. Thus, hepatocytes are centrally involved in the effects of inflammation on metabolic control, with the gut microbiota making up a significant part of that inflammatory signal.86
6. GUT MICROBIOME AND NAFLD
The major NAFLD risk factors (ie, diet, obesity and insulin resistance) are closely connected with the gut microbiome (Figure 1).14, 62, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96 It is reasonable to speculate that the gut microbiota and the pathophysiology of NASH are closely intertwined. One study found that the gut microbiota play a large role in the development of NAFLD, by transplanting the gut microbiota from mice with diet‐induced NAFLD into germ‐free mice; NAFLD developed in the initially germ‐free mice.18 In humans, characterisation of the faecal microbiomes of 86 patients with biopsy‐proven NAFLD (n = 72, stages 0‐2 fibrosis; n = 14, stages 3‐4 [advanced] fibrosis) revealed that patients with NAFLD and advanced fibrosis had increased levels of Proteobacteria, whereas patients with mild fibrosis had increased levels of Firmicutes.19 In addition, 37 of 40 features that were predictors of advanced fibrosis in patients with NAFLD were related to the gut microbiota.
Figure 1.

Role of the gut microbiome in NAFLD progression.14, 62, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96 DAMPS, damage‐associated molecular patterns; LPL, lipoprotein lipase; LPS, lipopolysaccharides; NAFLD, nonalcoholic fatty liver disease; NASH, nonalcoholic steatohepatitis; PAMPs, pathogen‐associated molecular patterns; TG, triglyceride; TLR, toll‐like receptors; TMAO, trimethylamine‐N‐oxide
Patients with NAFLD have increased intestinal permeability related to disrupted tight junctions.97 As mentioned previously, increased GI “leakiness” may allow bacterial translocation and entry of bacteria‐derived products into the portal circulation.98, 99 Once in the liver, these factors may initiate proinflammatory cascades (eg, production of interleukin‐6 [IL‐6] and TNF‐α) via interaction with the TLR present on a variety of cell types (ie, Kupffer cells, stellate cells and hepatocytes).100 In fact, more than half of patients with NAFLD may have small‐intestinal bacterial overgrowth,97 and the presence of this comorbid condition parallels cirrhosis severity (ie, Child‐Turcotte‐Pugh class).101, 102
Inflammation is a key factor in the development of NASH, and LPS produced by GI bacteria trigger proinflammatory cytokine cascades that involve TLR‐4 and nuclear factor kappa B (NFκB).92 LPS are also, as mentioned earlier, a key factor in the activation of Kupffer cells. Kupffer cells may release inflammatory cytokines in response to leptin, a hormone associated with adipocytes, thereby indirectly activating hepatic stellate cells and potentially perpetuating liver fibrosis.103 In a mouse model, obesity‐induced leptin increased liver responsiveness to LPS and enhanced progression of NASH.104 Results from a meta‐analysis of patients with NAFLD or NASH showed that circulating levels of leptin were higher in patients with NAFLD compared with healthy controls (standardised mean difference 0.64; 95% CI 0.42‐0.86) and in patients with NASH compared with patients with simple steatosis (standardised mean difference 0.21; 95% CI, 0.02‐0.40).105
Non‐inflammatory bacterial products also have been implicated in the development of hepatic steatosis. Monosaccharides produced by microbial fermentation of carbohydrates in the GI tract may activate carbohydrate‐responsive element‐binding protein (ChREBP) and sterol‐response element‐binding protein 1 (SREBP1) pathways, which regulate lipid accumulation. In an obese mouse model, deficiency of ChREBP reduced hepatic fat levels, suggesting that inhibiting ChREBP could be beneficial in patients with hepatic steatosis.106 Using stool samples and 16S ribosomal RNA gene pyrosequencing, Zhu et al88 examined the gut bacteria of three groups of paediatric patients—healthy, obese without NASH and those with biopsy‐proven NASH. The study found an association between health status and gut microbiome composition (at the phylum, family and genus levels). Both the obese and NASH groups demonstrated increased abundance of Bacteroidetes (specifically species of the genus Prevotella) and decreased abundance of Firmicutes compared with the healthy group. In addition, levels of species in the Proteobacteria phylum increased with progression from the healthy to obese to NASH groups, while the abundance of species in the Actinobacteria phylum (specifically those of the genus Bifidobacterium) decreased with worsening health status. The gut microbiome composition of obese patients and patients with NASH was similar, except for increased levels of Proteobacteria (specifically those of the Enterobacteriaceae family and the genus Escherichia; P < 0.05 for all three levels of classification) in the NASH group compared with the obese group. Increased abundance of ethanol‐producing bacteria in the NASH microbiome prompted the investigators to measure serum alcohol levels in the three groups. While little to no difference was noted in ethanol levels between the healthy and obese groups, ethanol levels were significantly increased in the NASH vs the obese groups (P < 0.01). Combined with the demonstration of increased abundance of Escherichia in patients with NASH, the authors theorised a pathophysiologic mechanism linking the altered microbiome in NASH and the development of hepatic inflammation.
Although other studies have demonstrated increased blood ethanol levels associated with NASH and obesity,94, 107 the study conducted by Zhu et al was the first to demonstrate that patients with NASH have higher blood ethanol levels than obese patients, and related this finding to alcohol‐producing bacteria in the gut microbiome of NASH patients.88 The results from this study suggest that patients with NASH may be differentiated from healthy and obese patients by assessing the gut microbiome using stool samples. The authors postulated that the constant presence of bacteria‐derived ethanol in the patients with NASH supplied a source of reactive oxygen species that could, in turn, increase inflammation and fibrosis.88 Figure 2 summarises the effects of various factors involved in the development of fibrosis and cirrhosis in patients with NAFL.19, 40, 41, 88, 108, 109
Figure 2.
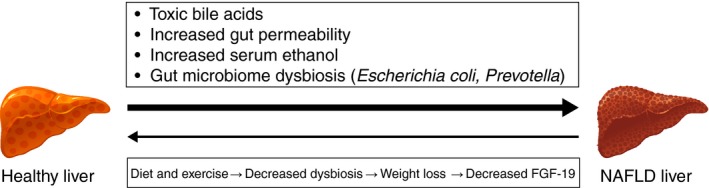
Association between gut microbiome and NAFLD. Patients with NAFL can progress to fibrosis and cirrhosis through different mechanisms, including toxic bile acids,40, 41, 124 increased gut permeability,41 increased endogenous ethanol88 and gut microbiome dysbiosis19, 88 (with higher levels of Escherichia coli and Prevotella). However, patients can have an improvement in hepatic inflammation and fibrosis with lifestyle modifications that include exercise and diet (which improves gut microbiome dysbiosis) and weight loss, as both of these conditions decrease FGF‐19.108, 109 FGF‐19, fibroblast growth factor‐19; NAFL, nonalcoholic fatty liver; NAFLD, nonalcoholic fatty liver disease
7. ROLE OF THE GUT MICROBIOME IN DIFFERENTIATING NAFL FROM NASH AND ADVANCED FIBROSIS
Although animal studies have linked gut dysbiosis to the severity of hepatic inflammation and/or fibrosis, the findings of preclinical studies do not necessarily translate to humans; however, few human studies have examined this connection in the NAFLD setting.110, 111 In a study comparing the gut microbiomes of 50 patients (healthy controls, patients with NAFL/simple steatosis and patients with NASH), patients with NASH had lower levels of Bacteroidetes and higher levels of Clostridium coccoides compared with both healthy controls and patients with NAFL (P < 0.05).110 In another study, examining the taxonomic composition of gut microbiota using 16S ribosomal RNA gene sequencing with stool samples from 57 patients with biopsy‐proven NAFLD, 30 patients had F0/1 fibrosis stage disease and 27 patients had advanced fibrosis (defined as ≥ F2 fibrosis stage).111 Ten patients with F0/1 stage and 25 patients with ≥ F2 stage had NASH. Patients with NASH had higher levels of Bacteroides and a lower abundance of Prevotella compared with patients without NASH. In comparison to patients with F0/1 fibrosis stage, patients with advanced fibrosis (≥F2 fibrosis stage) had increased levels of Bacteroides and Ruminococcus and lower levels of Prevotella. A multivariate analysis revealed that NASH was associated with increased Bacteroides, whereas findings of increased Ruminococcus were associated with advanced fibrosis.
Loomba et al19 characterised the gut microbiome of patients with biopsy‐proven NAFLD using whole‐genome shotgun sequencing for stool samples. Of 86 patients in the study, 72 had early fibrosis (F0/2) and the remaining 14 patients had F3/4 fibrosis. The median abundance for species in patients with early fibrosis was 2.5% Eubacterium rectale and 1.7% Bacteroides vulgatus, while E coli (1.0%) and B vulgatus (2.2% were predominant in patients with advanced fibrosis. Interestingly, a statistically significant increase in Proteobacteria levels was apparent as patients progressed to advanced fibrosis. Given that the increase in E coli levels preceded any clinical measures of fibrosis, they postulated that dysbiosis precedes the development of portal hypertension. However, larger studies are needed to determine if this finding is simply a correlation, or if a causal association is possible.
Exposure of liver Kupffer cells to bacterial LPS results in a release of proinflammatory cytokines through activating pathways that involve TLRs, myeloid differentiation factor 88 and NFκB,100, 112, 113 which may activate stellate cells and fibrogenesis. These factors are integral in the promotion of inflammation and the progression of fibrosis to cirrhosis in many diseases, such as viral hepatitis, biliary liver disease and NAFLD.112, 114 In addition, a case‐control/cross‐sectional study has shown that trimethylamine‐N‐oxide, the liver product of a bacterial metabolite of choline, is associated with hepatic steatosis and inflammation.87
Neither of these studies had any subgroup analysis of patients with and without insulin resistance or other confounding variables that may also affect gut microbiota.19, 110, 111 However, there may be a future role for the gut microbiome to be used as a non‐invasive marker to determine the presence of NASH and advanced fibrosis. However, further studies, with larger cohorts, are needed before this marker can be recommended for diagnosis and prognosis of NASH. Thus, while additional studies are warranted to validate the promise of gut microbial profiling, the gut microbiome in these patient populations may one day serve as an emerging tool for non‐invasive diagnosis of disease severity and monitoring progression.
8. GUT MICROBIOTA AND BILE ACIDS
BA composition is influenced by gut microbiota, and BAs are also thought to play a role in the development of NAFLD.115 Compared with healthy controls, patients with NASH have been shown to have higher concentrations of total faecal BA, cholic acid, chenodeoxycholic acid and BA synthesis, and a higher ratio of primary BA to secondary BA.95 BAs such as chenodeoxycholic acid bind to the farnesoid X receptor (FXR) in the intestines.116, 117 FXR is a member of the nuclear receptor superfamily and plays a key role in the absorption and transport of BA into the liver, as well as de novo hepatic lipogenesis, very low‐density lipoprotein transport and TG metabolism.96, 115, 118 Mice deficient in FXR demonstrated increased hepatic TG and cholesterol content,119 whereas FXR stimulation has been seen to suppress NFκB signalling, leading to decreased hepatic inflammation.120 Of note, findings using the high‐fat diet murine model of NAFLD demonstrated that intestinal antagonism of FXR through the manipulation of gut microbiota resulted in decreased hepatic lipogenesis.115 Stimulation of FXR has also been shown to alter carbohydrate metabolism, phospho‐enolpyruvate carboxykinase gene expression and gluconeogenesis regulation.121
Glucose homeostasis is also governed by GLP‐1, which is stimulated by G‐protein coupled receptor 5 (TGR5). Because the ligands for TGR5 are gut bacteria‐derived secondary BAs, the gut microbiome may play a large part in both lipid metabolism (through FXR) and glucose homeostasis (through TGR5).96, 122 Animal models of NAFLD have shown reductions in hepatic steatosis after exposure to BA derivatives that are FXR agonists123; in the FXR Ligand Obeticholic Acid in NASH Treatment trial, improvement of hepatic steatosis and inflammation was observed in patients with NAFLD who received obeticholic acid.124
9. TREATMENTS FOR NAFLD THAT TARGET GUT MICROBIOTA
Historically, the mainstay of treatment for patients with NAFLD has been lifestyle modification (eg, diet, exercise and weight loss) and the correction of underlying risk factors (eg, tight control of T2DM).108 Most pharmacologic treatments for NAFLD are designed to improve insulin sensitivity (eg, metformin, thiazolidinediones liraglutide and sitagliptin), reduce oxidative stress (vitamin E, ursodeoxycholic acid and pentoxifylline) or downregulate fibrosis mechanisms (angiotensin receptor blockers).125 Unfortunately, these medications have not demonstrated consistent improvement in liver fibrosis.125, 126, 127 Alternatively, data are accumulating on the potential role of therapies that alter gut microbiota in the treatment of patients with NAFLD and NASH.
9.1. Prebiotics
Prebiotics are indigestible foods that promote the growth of beneficial GI bacteria through the fermentation of the prebiotic into SCFA.128 Preclinical studies of prebiotics have shown improvement in biochemical and histologic markers of NAFLD.129 One randomised trial (placebo crossover design) has been published to date.130 In patients with biopsy‐proven NASH (n = 7), prebiotic administration (ie, oligofructose 16 g/d) significantly reduced hepatic levels of aspartate aminotransferase (AST; P < 0.05 vs placebo) and nonsignificantly decreased TG concentrations compared with placebo after 8 weeks of treatment. However, a systematic review that included four clinical studies of patients with obesity‐related NAFLD did not support the use of prebiotics, due to a lack of study quality.131
9.2. Probiotics
Probiotics are living microorganisms that are ingested and improve the mucosal integrity of the GI tract through alteration of the gut microbiota (via competitive colonisation and by acidification of the GI lumen).128 To date, six double‐blind, randomised controlled trials,132, 133, 134, 135, 136, 137 one open‐label, randomised controlled trial138 and one open‐label, single‐treatment trial139 have examined the effect of probiotics in patients with NAFLD (Table 1). The studies have reported improvement in several biochemical markers (eg, alanine aminotransferase [ALT], AST and TNF‐α). A systematic review that included three clinical studies examining the efficacy of probiotics in patients with NAFLD did not support their use in this patient population, due to a lack of high‐quality studies.131
Table 1.
Human studies of probiotics for NAFLD
| Publication | Study population | Study design/treatments | Primary outcomes |
|---|---|---|---|
| Miccheli et al 2015132 | Obese children with elevated ALT and ultrasonographic and histologic evidence of NAFLD | DB, RCT; patients received placebo or the probiotic medical food VSL#3a qd (1 package for patients aged < 10 y and 2 packets for patients aged > 10 y) for 4 mo |
|
| Alisi et al 2014133 | Obese children (median age, 10‐11 y) with histologically diagnosed NAFLD | DB, RCT; patients received placebo or VSL#3a (1 sachet/d for patients aged < 10 y or 2 sachets/d for patients aged > 10 y) for 4 mo |
|
| Nabavi et al 2014134 | Patients with NAFLD (unspecified diagnosis) | DB, RCT; patients received 300 g/d of conventional yogurt containing L bulgaricus and S thermophilus, or yogurt enriched with B lactis Bb12 and L acidophilus La5 for 8 wk |
|
| Shavakhi et al 2013135 | Adults with histologically confirmed NASH, persistent elevation of ALT, and alcohol consumption < 20 mg in men or < 10 g in women | DB, RCT; patients received 2 tablets of metformin 500 mg and either probiotic supplement daily or placebo for 6 mo |
|
| Wong et al 2013138 | Adults with histology‐proven NASH | OL, RCT; patients received “usual care” or 1 sachet of Lepicol® b b.i.d. for 6 mo |
|
| Aller et al 2011136 | Patients with biopsy‐proven NAFLD | DB, RCT; patients received placebo or 1 tablet containing L bulgaricus and S thermophilus qd for 3 mo |
|
| Vajro et al 2011137 | Paediatric patients with a BMI > 95th percentile for their age and sex who had liver abnormalities (eg, increased ALT levels) associated with ultrasound evidence of fatty liver (n = 20) | DB, RCT; patients received either Lactobacillus GG 12 billion CFU/d (n = 10) or placebo (n = 10) for 8 wk |
|
| Loguercio et al 2005139 | Patients with biopsy‐proven NAFLD (n = 22) alcoholic cirrhosis (n = 20), HCV (n = 20) or HCV‐related cirrhosis (n = 16) | OL; patients received VSL#3a for 3 mo |
|
Abbreviations: 4‐HNE, 4‐hydroynonenal; AAA, aromatic amino acids; ALT, alanine aminotransferase; AST, aspartate aminotransferase; b.i.d., twice daily; BCAA, branched chain amino acids; BMI, body mass index; CFU, colony‐forming unit; CRP, C‐reactive protein; DB, double‐blind; GGT, gamma‐glutamyl transferase; GLP‐1, glucagon‐like peptide 1; HCV, hepatitis C virus; HDL, high‐density lipoprotein; HDL‐C, high‐density lipoprotein cholesterol; HOMA‐IR, homeostasis model assessment of insulin resistance; IHTG, intrahepatic triglycerides; LDL, low‐density lipoprotein; LDL‐C, low‐density lipoprotein cholesterol; MDA, malondialdehyde; NAFLD, nonalcoholic fatty liver disease; NASH, nonalcoholic steatohepatitis; OL, open label; qd, once daily; RCT, randomised controlled trial; S‐NO, S‐nitrosothiol; TC, total cholesterol; TG, triglyceride; TNF‐α, tumour necrosis factor alpha.
VSL#3 (Alfasigma USA, Inc; Covington, LA, USA) is a probiotic mixture containing S thermophilus, B breve, B infantis, B longum, L acidophilus, L plantarum, L paracasei, and L delbrueckii ssp bulgaricus.
Lepicol® (Healthy Bowels Company Ltd; Birmingham, UK at the time of the study) contains L plantarum, L delbrueckii ssp bulgaricus, L acidophilus, L rhamnosus, B bifidum, and fructooligosaccharides.
Unfortunately, few studies have examined the effect of probiotics on histologic markers of NAFLD and NASH. In a 2013 meta‐analysis of randomised controlled trials, only four studies were available when patient inclusion was limited to those with histologically or radiologically diagnosed NAFLD.140 However, Alisi et al133 reported that obese children with histologically diagnosed NAFLD who received sachets of eight probiotic strains daily for 4 months (n = 22) had a significantly lower risk of “more severe” steatosis (vs “less severe” steatosis) compared with children who received placebo (n = 22).
9.3. Synbiotics
Synbiotics (or symbiotics) are a combination of both a prebiotic and a probiotic, and represent an emerging area of therapeutic research in NAFLD. Malaguarnera et al141 evaluated 66 patients with histologically diagnosed NASH who were randomly assigned to receive 24 weeks of a synbiotic (Bifidobacterium longum plus a prebiotic [fructooligosaccharides]) or placebo. Both groups underwent lifestyle modification and a B vitamin regimen. Compared with the placebo arm, the active treatment arm had significantly lower TNF‐α and C‐reactive protein (CRP) levels, as well as histologic improvement (decreased hepatocellular injury, inflammation and steatosis) after treatment (P < 0.05).
In the largest double‐blind, placebo‐controlled trial to date, 80 patients with ultrasound‐diagnosed NAFLD were randomly assigned to receive either a synbiotic (probiotics [Lactobacillus casei, Lactobacillus acidophilus, Lactobacillus rhamnosus, Lactobacillus bulgaricus, Bifidobacterium breve, B longum and Streptococcus thermophilus] and fructooligosaccharides) or placebo for 8 weeks. At the end of the intervention period, patients who received synbiotics had significantly reduced steatosis (as measured by ultrasound) vs baseline, whereas no significant improvement was observed in patients who received placebo.142 No significant differences in CRP, ALT or AST levels were observed between groups (adjusted for energy intake). In contrast, a study of 50 lean patients (ie, low or normal body mass index [BMI]) with NAFLD (patients had steatosis and elevated ALT) demonstrated significant reductions in fibrosis and hepatic steatosis, fasting blood sugar, TG levels and markers of inflammation after 28 weeks of synbiotic supplementation compared with placebo (P < 0.05).143
9.4. Antibiotics
Several small trials, mostly in animal models, have analysed the effect of antibiotics on NAFLD. The mechanism of action for antibiotics is multifactorial and may include alterations in the gut microbiota composition, bacterial virulence and/or bacterial metabolic function,128 although the specific pathway differs with each antibiotic. In a murine model, improvement in NAFLD was observed after the administration of an antibiotic cocktail (bacitracin, neomycin and streptomycin).115 This improvement was hypothesised to be the result of alterations in gut microbiota and BA metabolism and reductions in intestinal FXR signalling, serum ceramides and fatty acid synthesis. Reduced levels of bile salt hydrolase, a bacterial enzyme that metabolises the BA, result in the retention of tauro‐beta‐muricholic acid in the ileum, which then inhibits FXR signalling within the intestinal wall, leading to decreased ceramide production.115 Decreased ceramide production causes hepatic SREBP1C inhibition and decreased hepatic fatty acid accumulation. These findings were replicated in a rat study that showed alterations in the tissue BA profile, steroid biosynthesis and FXR signalling pathways after streptomycin and penicillin administration.144 As in the previous study, elevated levels of tauro‐beta‐muricholic acid were observed, although this time in the liver.
Several human studies of antibiotics in NAFLD have assessed the effect of rifaximin, a nonsystemic antibiotic (Table 2).145, 146, 147 Rifaximin is currently indicated in the United States to prevent overt hepatic encephalopathy recurrence and to treat travellers’ diarrhoea and irritable bowel syndrome (IBS) with diarrhoea.148, 149 Although one may hypothesise that rifaximin exerts its effect by altering the composition of the gut microbiota, data have shown only modest changes in the components of the gut microbiome in patients with cirrhosis and hepatic encephalopathy after rifaximin treatment.150 Additionally, preclinical studies have indicated that the efficacy of rifaximin may be attributable to its beneficial effects on host cell physiology and bacterial gene expression.151, 152, 153
Table 2.
Human studies of nonsystemic antibiotics for NAFLD
| Publication | Study population | Study design/treatments | Primary outcomes |
|---|---|---|---|
| Cobbold et al 2017145 | Adults with histologically confirmed NAFLD | OL rifaximin 400 mg b.i.d. for 6 wk (n = 15) |
|
| Gangarapu et al 2015146 | Adults with histologically confirmed NAFLD (steatosis, n = 15; NASH, n = 27) | OL rifaximin 1200 mg/d for 4 wk (n = 42) |
|
| Kakiyama et al 2013147 | Adult patients with “early” cirrhosis (Child‐Pugh Class A without history of decompensation) | Longitudinal sub study; rifaximin 550 mg b.i.d. for 8 wk (n = 6) |
|
Abbreviations: ALT, alanine aminotransferase; AST, aspartate aminotransferase; b.i.d., twice daily; BA, bile acid; BMI, body mass index; GGT, gamma‐glutamyl transferase; HDL, high‐density lipoprotein; HOMA‐IR, homeostasis model assessment‐insulin resistance; IL, interleukin; LDL, low‐density lipoprotein; NAFLD, nonalcoholic fatty liver disease; NASH, nonalcoholic steatohepatitis; OL, open label; TLR, toll‐like receptor; TNF‐α, tumour necrosis factor alpha.
9.5. Faecal transplantation
Faecal transplantation has been used successfully in the treatment of patients with refractory and recurrent Clostridium difficile.154, 155 Although no human studies have examined the role of faecal transplantation for NAFLD, this strategy may be a potential avenue for exploration. In mouse models of NAFLD, animals that underwent a faecal transplantation from wild‐type mice donors showed decreased hepatic gluconeogenesis156 and reduced intestinal permeability157 (Table 3). However, faecal transplantation is not without risks. For example, a 2015 case report documented that a previously lean patient with recurrent C difficile infection developed obesity after a faecal transplant from an obese donor.158
Table 3.
Animal studies of faecal microbiota transplantation for NAFLD
| Publication | Study population | Study design/treatments | Primary outcomes |
|---|---|---|---|
| Nicolas et al 2017156 |
Donors:
Recipients: WT mice |
Recipients were gavaged with gut microbiota obtained from cecum of (a) WT mice fed a normal diet; (b) WT mice fed a high‐fat diet; (c) genetically obese mice |
|
| Li et al 2015157 |
Donors: WT mice Recipients: WT mice that received ceftriaxone b.i.d. for 7 d to induce gut microbiota dysbiosis |
Recipients gavaged for 3 d with faecal microbiota from WT mice or cultured bacteria initially isolated from donor mice faeces |
|
Abbreviations: b.i.d., twice daily; WT, wild type.
Both murine knockout studies85, 86 and population‐based studies19, 88 have examined alterations in gut microbiota in both NAFLD/NASH and in non‐NAFLD subjects with risk factors for NAFLD. In the murine model, faecal transplantation from human obese adult twins into germ‐free lean mice resulted in the development of obesity in these mice,159 indicating that dysbiosis may lead to the development of obesity even in the absence of poor diet or genetic predisposition.
Studies in humans indicate that even in healthy individuals who donate stool for a faecal microbiota transplant (FMT), faeces from a select few patients (called “super donors”) may yield more FMT success than stool obtained from other healthy donors.160, 161 FMTs have been studied in the setting of C. difficile infection162 and in chronic illnesses, such as IBD,160, 161 IBS,163 constipation164 and neurologic conditions.165 Two separate studies assessing the efficacy of FMT in the treatment of IBD found that patients who had received stool transplants from a particular donor had a higher success rate in inducing clinical and endoscopic remission compared with patients who did not receive stool from the “super donors.”160, 161 FMTs using stool obtained from lean donors transplanted into patients with metabolic syndrome led to a greater degree of improvement in peripheral insulin sensitivity compared with autologous FMT.166, 167 Thus far, the only factor that seems to predict a successful FMT is the diversity of gut microbiota in the donor; conversely, recipients who are able to increase their faecal microbiome diversity to a higher degree in response to FMT were more likely to have successful outcomes in treatment of the underlying disease.168, 169
10. CONCLUSIONS
The GI tract and the liver develop from the same embryologic origins in the foregut, and this close interrelationship is maintained. Obesity, diet and insulin resistance are common risk factors for the development of NAFLD,15, 83, 84 and these risk factors seem to have a strong connection with the gut microbiome.12 While diet and obesity play a role in the modification of bacteria in the gut microbiota, the bacteria, in turn, affect the ability of host cells to produce and absorb nutrient‐derived energy.34 SCFAs and the ratios of the different fatty acids produced by bacteria are affected by the predominant type of bacteria in the GI lumen. Excess production of certain SCFAs can lead to the accumulation of excess energy in the form of adipose tissue, and obese patients are found to have altered ratios of SCFAs compared with their lean counterparts.64, 66, 67, 68, 69 These GI bacteria also affect host lipid metabolism and insulin sensitivity.77, 80, 170
Obesity and insulin resistance are also associated with increased intestinal permeability171, 172, 173 and, therefore, increased rates of bacterial translocation, which activates proinflammatory cascades.84, 170, 174, 175 Kupffer cell activation by bacterial products such as LPS results in oxidative stress and the development of hepatocyte inflammation and fibrosis. LPS also promote the development of hepatic TG accumulation and hepatic steatosis.12, 100, 112, 113 There is a strong association between bacterial translocation and the degree of hepatic decompensation in the setting of cirrhosis of any aetiology,174 and among gut microbiota, BA metabolism, FXR and hepatic steatosis and inflammation.115, 120, 121, 124 In addition, activation of TLRs by bacterial products results in increased systemic and hepatic inflammation, a major stimulus in the development of NASH and the progression to fibrosis.100, 112, 113 Thus, treatment options aimed at targeting the gut microbiome, or the downstream cell‐signalling effects of the microbiome, continue to be therapeutic targets for the treatment of patients with NAFLD.
AUTHORSHIP
Guarantor of the article: Dr Rohit Loomba.
Author contributions: RL was involved in conception of the idea for the review. SJ wrote the first draft of the manuscript. RL critically reviewed and revised the manuscript for important intellectual content. All authors approved the final version of the manuscript.
ACKNOWLEDGEMENTS
Declaration of personal interests: S. Jayakumar reports no conflicts of interest. R. Loomba is supported in part by the American Gastroenterological Association Foundation, Sucampo Pharmaceuticals, Inc., ASP Designated Research Award in Geriatric Gastroenterology, and by a T. Franklin Williams Scholarship Award.
Jayakumar S, Loomba R. Review article: emerging role of the gut microbiome in the progression of nonalcoholic fatty liver disease and potential therapeutic implications. Aliment Pharmacol Ther. 2019;50:144–158. 10.1111/apt.15314
The Handling Editor for this article was Professor Stephen Harrison, and this uncommissioned review was accepted for publication after full peer‐review.
Funding information
Technical editorial assistance was provided under the direction of the authors by Synchrony Medical Communications, LLC, West Chester, PA. Funding for this support was provided by Salix Pharmaceuticals, Bridgewater, NJ, USA.
REFERENCES
- 1. European Association for the Study of the Liver (EASL), European Association for the Study of Diabetes (EASD), European Association for the Study of Obesity (EASO) . EASL‐EASD‐EASO Clinical Practice Guidelines for the management of non‐alcoholic fatty liver disease. J Hepatol. 2016;64:1388–1402. [DOI] [PubMed] [Google Scholar]
- 2. Masuoka HC, Chalasani N. Nonalcoholic fatty liver disease: an emerging threat to obese and diabetic individuals. Ann N Y Acad Sci. 2013;1281:106–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lazo M, Hernaez R, Eberhardt MS, et al. Prevalence of nonalcoholic fatty liver disease in the United States: the Third National Health and Nutrition Examination Survey, 1988–1994. Am J Epidemiol. 2013;178:38–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pais R, Barritt AS, Calmus Y, et al. NAFLD and liver transplantation: current burden and expected challenges. J Hepatol. 2016;65:1245–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Loomba R, Sanyal AJ. The global NAFLD epidemic. Nat Rev Gastroenterol Hepatol. 2013;10:686–690. [DOI] [PubMed] [Google Scholar]
- 6. Goh GB, McCullough AJ. Natural history of nonalcoholic fatty liver disease. Dig Dis Sci. 2016;61:1226–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Puri P, Sanyal AJ. Nonalcoholic fatty liver disease: definitions, risk factors, and workup. Clin Liver Dis. 2012;1:99–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Singh S, Allen AM, Wang Z, Prokop LJ, Murad MH, Loomba R. Fibrosis progression in nonalcoholic fatty liver vs nonalcoholic steatohepatitis: a systematic review and meta‐analysis of paired‐biopsy studies. Clin Gastroenterol Hepatol. 2015;13:643–654.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Adams LA, Lymp JF, St. Sauver J, et al. The natural history of nonalcoholic fatty liver disease: a population‐based cohort study. Gastroenterology. 2005;129:113–121. [DOI] [PubMed] [Google Scholar]
- 10. Ekstedt M, Franzén LE, Mathiesen UL, et al. Long‐term follow‐up of patients with NAFLD and elevated liver enzymes. Hepatology. 2006;44:865–873. [DOI] [PubMed] [Google Scholar]
- 11. Matteoni CA, Younossi ZM, Gramlich T, Boparai N, Liu YC, McCullough AJ. Nonalcoholic fatty liver disease: a spectrum of clinical and pathological severity. Gastroenterology. 1999;116:1413–1419. [DOI] [PubMed] [Google Scholar]
- 12. Buzzetti E, Pinzani M, Tsochatzis EA. The multiple‐hit pathogenesis of non‐alcoholic fatty liver disease (NAFLD). Metabolism. 2016;65:1038–1048. [DOI] [PubMed] [Google Scholar]
- 13. Takaki A, Kawai D, Yamamoto K. Multiple hits, including oxidative stress, as pathogenesis and treatment target in non‐alcoholic steatohepatitis (NASH). Int J Mol Sci. 2013;14:20704–20728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cusi K. Role of insulin resistance and lipotoxicity in non‐alcoholic steatohepatitis. Clin Liver Dis. 2009;13:545–563. [DOI] [PubMed] [Google Scholar]
- 15. Czaja MJ. Function of autophagy in nonalcoholic fatty liver disease. Dig Dis Sci. 2016;61:1304–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Alkhouri N, Carter‐Kent C, Feldstein AE. Apoptosis in nonalcoholic fatty liver disease: diagnostic and therapeutic implications. Expert Rev Gastroenterol Hepatol. 2011;5:201–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Xu R, Tao A, Zhang S, Deng Y, Chen G. Association between patatin‐like phospholipase domain containing 3 gene (PNPLA3) polymorphisms and nonalcoholic fatty liver disease: a HuGE review and meta‐analysis. Sci Rep. 2015;5:9284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Le Roy T, Llopis M, Lepage P, et al. Intestinal microbiota determines development of non‐alcoholic fatty liver disease in mice. Gut. 2013;62:1787–1794. [DOI] [PubMed] [Google Scholar]
- 19. Loomba R, Seguritan V, Li W, et al. Gut microbiome‐based metagenomic signature for non‐invasive detection of advanced fibrosis in human nonalcoholic fatty liver disease. Cell Metab. 2017;25:1054–62.e1‐e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Frank DN, St. Amand AL, Feldman RA, Boedeker EC, Harpaz N, Pace NR. Molecular‐phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci USA. 2007;104:13780–13785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Turnbaugh PJ, Ley RE, Hamady M, Fraser‐Liggett C, Knight R, Gordon JI. The human microbiome project: exploring the microbial part of ourselves in a changing world. Nature. 2007;449:804–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Claesson MJ, Cusack S, O'Sullivan O, et al. Composition, variability, and temporal stability of the intestinal microbiota of the elderly. Proc Natl Acad Sci USA. 2011;108:4586–4591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. van Tongeren SP, Slaets J, Harmsen H, Welling GW. Fecal microbiota composition and frailty. Appl Environ Microbiol. 2005;71:6438–6442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Stark PL, Lee A. The microbial ecology of the large bowel of breast‐fed and formula‐fed infants during the first year of life. J Med Microbiol. 1982;15:189–203. [DOI] [PubMed] [Google Scholar]
- 25. Schwartz S, Friedberg I, Ivanov IV, et al. A metagenomic study of diet‐dependent interaction between gut microbiota and host in infants reveals differences in immune response. Genome Biol. 2012;13:r32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Prideaux L, Kang S, Wagner J, et al. Impact of ethnicity, geography, and disease on the microbiota in health and inflammatory bowel disease. Inflamm Bowel Dis. 2013;19:2906–2918. [DOI] [PubMed] [Google Scholar]
- 27. Kalser MH, Cohen R, Arteaga I, et al. Normal viral and bacterial flora of the human small and large intestine. N Engl J Med. 1966;274:500–505. [DOI] [PubMed] [Google Scholar]
- 28. Pei Z, Bini EJ, Yang L, Zhou M, Francois F, Blaser MJ. Bacterial biota in the human distal esophagus. Proc Natl Acad Sci USA. 2004;101:4250–4255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shanahan ER, Zhong L, Talley NJ, Morrison M, Holtmann G. Characterisation of the gastrointestinal mucosa‐associated microbiota: a novel technique to prevent cross‐contamination during endoscopic procedures. Aliment Pharmacol Ther. 2016;43:1186–1196. [DOI] [PubMed] [Google Scholar]
- 30. Eckburg PB, Bik EM, Bernstein CN, et al. Diversity of the human intestinal microbial flora. Science. 2005;308:1635–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang X, Heazlewood SP, Krause DO, Florin T. Molecular characterization of the microbial species that colonize human ileal and colonic mucosa by using 16S rDNA sequence analysis. J Appl Microbiol. 2003;95:508–520. [DOI] [PubMed] [Google Scholar]
- 32. Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature. 2006;444:1022–1023. [DOI] [PubMed] [Google Scholar]
- 33. Selber‐Hnatiw S, Rukundo B, Ahmadi M, et al. Human gut microbiota: toward an ecology of disease. Front Microbiol. 2017;8:1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jandhyala SM, Talukdar R, Subramanyam C, Vuyyuru H, Sasikala M, Nageshwar RD. Role of the normal gut microbiota. World J Gastroenterol. 2015;21:8787–8803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Laparra JM, Sanz Y. Interactions of gut microbiota with functional food components and nutraceuticals. Pharmacol Res. 2010;61:219–225. [DOI] [PubMed] [Google Scholar]
- 36. Fukiya S, Arata M, Kawashima H, et al. Conversion of cholic acid and chenodeoxycholic acid into their 7‐oxo derivatives by Bacteroides intestinalis AM‐1 isolated from human feces. FEMS Microbiol Lett. 2009;293:263–270. [DOI] [PubMed] [Google Scholar]
- 37. Macfarlane S, Macfarlane GT. Regulation of short‐chain fatty acid production. Proc Nutr Soc. 2003;62:67–72. [DOI] [PubMed] [Google Scholar]
- 38. Morrison DJ, Mackay WG, Edwards CA, Preston T, Dodson B, Weaver LT. Butyrate production from oligofructose fermentation by the human faecal flora: what is the contribution of extracellular acetate and lactate? Br J Nutr. 2006;96:570–577. [PubMed] [Google Scholar]
- 39. Spencer MD, Hamp TJ, Reid RW, Fischer LM, Zeisel SH, Fodor AA. Association between composition of the human gastrointestinal microbiome and development of fatty liver with choline deficiency. Gastroenterology. 2011;140:976–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Svegliati‐Baroni G, Ridolfi F, Hannivoort R, et al. Bile acids induce hepatic stellate cell proliferation via activation of the epidermal growth factor receptor. Gastroenterology. 2005;128:1042–1055. [DOI] [PubMed] [Google Scholar]
- 41. Raimondi F, Santoro P, Barone MV, et al. Bile acids modulate tight junction structure and barrier function of Caco‐2 monolayers via EGFR activation. Am J Physiol Gastrointest Liver Physiol. 2008;294:G906–G913. [DOI] [PubMed] [Google Scholar]
- 42. Sarathy J, Detloff SJ, Ao M, et al. Yin and Yang of bile acid action on tight junctions in a model colonic epithelium. Physiol Rep. 2017;5:e13294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wu HJ, Wu E. The role of gut microbiota in immune homeostasis and autoimmunity. Gut Microbes. 2012;3:4–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Geuking M, Cahenzli J, Lawson M, et al. Intestinal bacterial colonization induces mutualistic regulatory T cell responses. Immunity. 2011;34:794–806. [DOI] [PubMed] [Google Scholar]
- 45. Smith PM, Howitt MR, Panikov N, et al. The microbial metabolites, short‐chain fatty acids, regulate colonic Treg cell homeostasis. Science. 2013;341:569–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Arpaia N, Campbell C, Fan X, et al. Metabolites produced by commensal bacteria promote peripheral regulatory T cell generation. Nature. 2013;504:451–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Valentini M, Piermattei A, Di Sante G, Migliara G, Delogu G, Ria F. Immunomodulation by gut microbiota: role of Toll‐like receptor expressed by T cells. J Immunol Res. 2014;2014:586939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kashiwagi I, Morita R, Schichita T, et al. Smad2 and Smad3 inversely regulate TGF‐beta autoinduction in Clostridium butyricum‐activated dendritic cells. Immunity. 2015;43:65–79. [DOI] [PubMed] [Google Scholar]
- 49. Yan F, Cao H, Cover TL, et al. Colon‐specific delivery of a probiotic‐derived soluble protein ameliorates intestinal inflammation in mice through an EGFR‐dependent mechanism. J Clin Invest. 2011;121:2242–2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ferreira CM, Vieira AT, Ramirez Vinolo MA, Oliveira FA, Curi R, dos Santos MF. The central role of the gut microbiota in chronic inflammatory diseases. J Immunol Res. 2014;2014:689492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hold GL, Smith M, Grange C, Watt ER, El‐Omar EM, Mukhopadhya I. Role of the gut microbiota in inflammatory bowel disease pathogenesis: what have we learnt in the past 10 years? World J Gastroenterol. 2014;20:1192–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kelsen JR, Wu GD. The gut microbiota, environment and diseases of modern society. Gut Microbes. 2012;3:374–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Baothman OA, Zamzami MA, Taher I, Abubaker J, Abu‐Farha M. The role of gut microbiota in the development of obesity and diabetes. Lipids Health Dis. 2016;15:108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Tilg H, Cani PD, Mayer EA. Gut microbiome and liver diseases. Gut. 2016;65:2035–2044. [DOI] [PubMed] [Google Scholar]
- 55. Llorente C, Schnabl B. The gut microbiota and liver disease. Cell Mol Gastroenterol Hepatol. 2015;1:275–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Clayton TA, Baker D, Lindon JC, Everett JR, Nicholson JK. Pharmacometabonomic identification of a significant host‐microbiome metabolic interaction affecting human drug metabolism. Proc Natl Acad Sci USA. 2009;106:14728–14733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wallace BD, Wang H, Lane KT, et al. Alleviating cancer drug toxicity by inhibiting a bacterial enzyme. Science. 2010;330:831–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Alberti K, Eckel RH, Grundy SM, et al. Harmonizing the metabolic syndrome: a joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation. 2009;120:1640–1645. [DOI] [PubMed] [Google Scholar]
- 59. Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease—meta‐analytic assessment of prevalence, incidence, and outcomes. Hepatology. 2016;64:73–84. [DOI] [PubMed] [Google Scholar]
- 60. Marchesini G, Bugianesi E, Forlani G, et al. Nonalcoholic fatty liver, steatohepatitis, and the metabolic syndrome. Hepatology. 2003;37:917–923. [DOI] [PubMed] [Google Scholar]
- 61. Velagapudi VR, Hezaveh R, Reigstad CS, et al. The gut microbiota modulates host energy and lipid metabolism in mice. J Lipid Res. 2010;51:1101–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Graham C, Mullen A, Whelan K. Obesity and the gastrointestinal microbiota: a review of associations and mechanisms. Nutr Rev. 2015;73:376–385. [DOI] [PubMed] [Google Scholar]
- 63. Turnbaugh PJ, Ridaura VK, Faith JJ, Rey FE, Knight R, Gordon JI. The effect of diet on the human gut microbiome: a metagenomic analysis in humanized gnotobiotic mice. Sci Transl Med. 2009;1:6ra14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Rahat‐Rozenbloom S, Fernandes J, Gloor GB, Wolever TM. Evidence for greater production of colonic short‐chain fatty acids in overweight than lean humans. Int J Obes (Lond). 2014;38:1525–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Murphy EF, Cotter PD, Healy S, et al. Composition and energy harvesting capacity of the gut microbiota: relationship to diet, obesity and time in mouse models. Gut. 2010;59:1635–1642. [DOI] [PubMed] [Google Scholar]
- 66. Schwiertz A, Taras D, Schäfer K, et al. Microbiota and SCFA in lean and overweight healthy subjects. Obesity (Baltimore). 2010;18:190–195. [DOI] [PubMed] [Google Scholar]
- 67. Fernandes J, Su W, Rahat‐Rozenbloom S, Wolever T, Comelli EM. Adiposity, gut microbiota and faecal short chain fatty acids are linked in adult humans. Nutr Diabetes. 2014;4:e121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Payne AN, Chassard C, Banz Y, Lacroix C. The composition and metabolic activity of child gut microbiota demonstrate differential adaptation to varied nutrient loads in an in vitro model of colonic fermentation. FEMS Microbiol Ecol. 2012;80:608–623. [DOI] [PubMed] [Google Scholar]
- 69. Zhang H, DiBaise JK, Zuccolo A, et al. Human gut microbiota in obesity and after gastric bypass. Proc Natl Acad Sci USA. 2009;106:2365–2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. den Besten G, Bleeker A, Gerding A, et al. Short‐chain fatty acids protect against high‐fat diet‐induced obesity via a PPARγ‐dependent switch from lipogenesis to fat oxidation. Diabetes. 2015;64:2398–2408. [DOI] [PubMed] [Google Scholar]
- 71. Tolhurst G, Heffron H, Lam YS, et al. Short‐chain fatty acids stimulate glucagon‐like peptide‐1 secretion via the G‐protein‐coupled receptor FFAR2. Diabetes. 2012;61:364–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Bäckhed F, Ding H, Wang T, et al. The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci USA. 2004;101:15718–15723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Aronsson L, Huang Y, Parini P, et al. Decreased fat storage by Lactobacillus paracasei is associated with increased levels of angiopoietin‐like 4 protein (ANGPTL4). PLoS ONE. 2010;5:e13087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Bäckhed F, Manchester JK, Semenkovich CF, Gordon JI. Mechanisms underlying the resistance to diet‐induced obesity in germ‐free mice. Proc Natl Acad Sci USA. 2007;104:979–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Khan MJ, Gerasimidis K, Edwards CA, Shaikh MG. Role of gut microbiota in the aetiology of obesity: proposed mechanisms and review of the literature. J Obes. 2016;2016:7353642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Larsen N, Vogensen FK, van den Berg F, et al. Gut microbiota in human adults with type 2 diabetes differs from non‐diabetic adults. PLoS ONE. 2010;5:e9085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Qin J, Li Y, Cai Z, et al. A metagenome‐wide association study of gut microbiota in type 2 diabetes. Nature. 2012;490:55–60. [DOI] [PubMed] [Google Scholar]
- 78. Brown AJ, Goldsworthy SM, Barnes AA, et al. The Orphan G protein‐coupled receptors GPR41 and GPR43 are activated by propionate and other short chain carboxylic acids. J Biol Chem. 2003;278:11312–11319. [DOI] [PubMed] [Google Scholar]
- 79. Musso G, Gambino R, Cassader M. Obesity, diabetes, and gut microbiota: the hygiene hypothesis expanded? Diabetes Care. 2010;33:2277–2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Scheithauer T, Dallinga‐Thie GM, de Vos WM, Nieuwdorp M, van Raalte DH. Causality of small and large intestinal microbiota in weight regulation and insulin resistance. Mol Metab. 2016;5:759–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Sam AH, Gunner DJ, King A, et al. Selective ablation of peptide YY cells in adult mice reveals their role in beta cell survival. Gastroenterology. 2012;143:459–468. [DOI] [PubMed] [Google Scholar]
- 82. Sun M, Wu W, Liu Z, Cong Y. Microbiota metabolite short chain fatty acids, GPCR, and inflammatory bowel diseases. J Gastroenterol. 2017;52:144–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Wellen KE, Hotamisligil GS. Inflammation, stress, and diabetes. J Clin Invest. 2005;115:1111–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Cani PD, Amar J, Iglesias MA, et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes. 2007;56:1761–1772. [DOI] [PubMed] [Google Scholar]
- 85. Vijay‐Kumar M, Aitken JD, Carvalho FA, et al. Metabolic syndrome and altered gut microbiota in mice lacking Toll‐like receptor 5. Science. 2010;328:228–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Jia L, Vianna CR, Fukuda M, et al. Hepatocyte Toll‐like receptor 4 regulates obesity‐induced inflammation and insulin resistance. Nat Commun. 2014;5:3878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Chen Y‐M, Liu Y, Zhou R‐F, et al. Associations of gut‐flora‐dependent metabolite trimethylamine‐N‐oxide, betaine and choline with non‐alcoholic fatty liver disease in adults. Sci Rep. 2016;6:19076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Zhu L, Baker SS, Gill C, et al. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: a connection between endogenous alcohol and NASH. Hepatology. 2013;57:601–609. [DOI] [PubMed] [Google Scholar]
- 89. Ridlon JM, Kang DJ, Hylemon PB, Bajaj JS. Bile acids and the gut microbiome. Curr Opin Gastroenterol. 2014;30:332–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Softic S, Cohen DE, Kahn CR. Role of dietary fructose and hepatic de novo lipogenesis in fatty liver disease. Dig Dis Sci. 2016;61:1282–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Parker R. The role of adipose tissue in fatty liver diseases [published online ahead of print February 21, 2018]. Liver Res. 2018;2:35‐42. [Google Scholar]
- 92. Ye D, Li F, Lam K, et al. Toll‐like receptor‐4 mediates obesity‐induced non‐alcoholic steatohepatitis through activation of X‐box binding protein‐1 in mice. Gut. 2012;61:1058–1067. [DOI] [PubMed] [Google Scholar]
- 93. Gao B. Innate immunity and steatohepatitis: a critical role of another toll (TLR‐9). Gastroenterology. 2010;139:27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Volynets V, Küper MA, Strahl S, et al. Nutrition, intestinal permeability, and blood ethanol levels are altered in patients with nonalcoholic fatty liver disease (NAFLD). Dig Dis Sci. 2012;57:1932–1941. [DOI] [PubMed] [Google Scholar]
- 95. Mouzaki M, Wang AY, Bandsma R, et al. Bile acids and dysbiosis in non‐alcoholic fatty liver disease. PLoS ONE. 2016;11:e0151829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Aron‐Wisnewsky J, Gaborit B, Dutour A, Clement K. Gut microbiota and non‐alcoholic fatty liver disease: new insights. Clin Microbiol Infect. 2013;19:338–348. [DOI] [PubMed] [Google Scholar]
- 97. Miele L, Valenza V, La Torre G, et al. Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology. 2009;49:1877–1887. [DOI] [PubMed] [Google Scholar]
- 98. Rai R, Saraswat VA, Dhiman RK. Gut microbiota: its role in hepatic encephalopathy. J Clin Exp Hepatol. 2015;5:S29–S36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Mao JW, Tang HY, Zhao T, et al. Intestinal mucosal barrier dysfunction participates in the progress of nonalcoholic fatty liver disease. Int J Clin Exp Pathol. 2015;8:3648–3658. [PMC free article] [PubMed] [Google Scholar]
- 100. Kesar V, Odin JA. Toll‐like receptors and liver disease. Liver Int. 2014;34:184–196. [DOI] [PubMed] [Google Scholar]
- 101. Pande C, Kumar A, Sarin SK. Small‐intestinal bacterial overgrowth in cirrhosis is related to the severity of liver disease. Aliment Pharmacol Ther. 2009;29:1273–1281. [DOI] [PubMed] [Google Scholar]
- 102. Jun DW, Kim KT, Lee OY, et al. Association between small intestinal bacterial overgrowth and peripheral bacterial DNA in cirrhotic patients. Dig Dis Sci. 2010;55:1465–1471. [DOI] [PubMed] [Google Scholar]
- 103. Metlakunta A, Huang W, Stefanovic‐Racic M, Dedousis N, Sipula I, O'Doherty RM. Kupffer cells facilitate the acute effects of leptin on hepatic lipid metabolism. Am J Physiol Endocrinol Metab. 2017;312:E11–E18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Imajo K, Fujita K, Yoneda M, et al. Hyperresponsivity to low‐dose endotoxin during progression to nonalcoholic steatohepatitis is regulated by leptin‐mediated signaling. Cell Metab. 2012;16:44–54. [DOI] [PubMed] [Google Scholar]
- 105. Polyzos SA, Aronis KN, Kountouras J, Raptis DD, Vasiloglou MF, Mantzoros CS. Circulating leptin in non‐alcoholic fatty liver disease: a systematic review and meta‐analysis. Diabetologia. 2016;59:30–43. [DOI] [PubMed] [Google Scholar]
- 106. Denechaud PD, Dentin R, Girard J, Postic C. Role of ChREBP in hepatic steatosis and insulin resistance. FEBS Lett. 2008;582:68–73. [DOI] [PubMed] [Google Scholar]
- 107. Cope K, Risby T, Diehl AM. Increased gastrointestinal ethanol production in obese mice: implications for fatty liver disease pathogenesis. Gastroenterology. 2000;119:1340–1347. [DOI] [PubMed] [Google Scholar]
- 108. Neuschwander‐Tetri BA, Caldwell SH. Nonalcoholic steatohepatitis: summary of an AASLD single topic conference. Hepatology. 2003;37:1202–1219. [DOI] [PubMed] [Google Scholar]
- 109. Bozadjieva N, Heppner KM, Seeley RJ. Targeting FXR and FGF19 to treat metabolic diseases‐lessons learned from bariatric surgery. Diabetes. 2018;67:1720–1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Mouzaki M, Comelli EM, Arendt BM, et al. Intestinal microbiota in patients with nonalcoholic fatty liver disease. Hepatology. 2013;58:120–127. [DOI] [PubMed] [Google Scholar]
- 111. Boursier J, Mueller O, Barret M, et al. The severity of nonalcoholic fatty liver disease is associated with gut dysbiosis and shift in the metabolic function of the gut microbiota. Hepatology. 2016;63:764–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Henao‐Mejia J, Elinav E, Thaiss CA, Flavell RA. The intestinal microbiota in chronic liver disease. Adv Immunol. 2013;117:73–97. [DOI] [PubMed] [Google Scholar]
- 113. Henao‐Mejia J, Elinav E, Thaiss CA, Licona‐Limon P, Flavell RA. Role of the intestinal microbiome in liver disease. J Autoimmun. 2013;46:66–73. [DOI] [PubMed] [Google Scholar]
- 114. Luedde T, Schwabe RF. NF‐κB in the liver—linking injury, fibrosis and hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol. 2011;8:108–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Jiang C, Xie C, Li F, et al. Intestinal farnesoid X receptor signaling promotes nonalcoholic fatty liver disease. J Clin Invest. 2015;125:386–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Makishima M, Okamoto AY, Repa JJ, et al. Identification of a nuclear receptor for bile acids. Science. 1999;284:1362–1365. [DOI] [PubMed] [Google Scholar]
- 117. Modica S, Gofflot F, Murzilli S, et al. The intestinal nuclear receptor signature with epithelial localization patterns and expression modulation in tumors. Gastroenterology. 2010;138:636–48.e1‐e12. [DOI] [PubMed] [Google Scholar]
- 118. Tu H, Okamoto AY, Shan B. FXR, a bile acid receptor and biological sensor. Trends Cardiovasc Med. 2000;10:30–35. [DOI] [PubMed] [Google Scholar]
- 119. Sinal CJ, Tohkin M, Miyata M, Ward JM, Lambert G, Gonzalez FJ. Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell. 2000;102:731–744. [DOI] [PubMed] [Google Scholar]
- 120. Wang YD, Chen WD, Wang M, Yu D, Forman BM, Huang W. Farnesoid X receptor antagonizes NF‐κB in hepatic inflammatory response. Hepatology. 2008;48:1632–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Stayrook KR, Bramlett KS, Savkur RS, et al. Regulation of carbohydrate metabolism by the farnesoid X receptor. Endocrinology. 2005;146:984–991. [DOI] [PubMed] [Google Scholar]
- 122. Thomas C, Gioiello A, Noriega L, et al. TGR5‐mediated bile acid sensing controls glucose homeostasis. Cell Metab. 2009;10:167–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Cipriani S, Mencarelli A, Palladino G, Fiorucci S. FXR activation reverses insulin resistance and lipid abnormalities and protects against liver steatosis in Zucker (fa/fa) obese rats. J Lipid Res. 2010;51:771–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Neuschwander‐Tetri BA, Loomba R, Sanyal AJ, et al. Farnesoid X nuclear receptor ligand obeticholic acid for non‐cirrhotic, non‐alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo‐controlled trial. Lancet. 2015;385:956–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Said A, Akhter A. Meta‐analysis of randomized controlled trials of pharmacologic agents in non‐alcoholic steatohepatitis. Ann Hepatol. 2017;16:538–547. [DOI] [PubMed] [Google Scholar]
- 126. Singh S, Khera R, Allen AM, Murad MH, Loomba R. Comparative effectiveness of pharmacological interventions for nonalcoholic steatohepatitis: a systematic review and network meta‐analysis. Hepatology. 2015;62:1417–1432. [DOI] [PubMed] [Google Scholar]
- 127. Sawangjit R, Chongmelaxme B, Phisalprapa P, et al. Comparative efficacy of interventions on nonalcoholic fatty liver disease (NAFLD): a PRISMA‐compliant systematic review and network meta‐analysis. Medicine (Baltimore). 2016;95:e4529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Anand G, Zarrinpar A, Loomba R. Targeting dysbiosis for the treatment of liver disease. Semin Liver Dis. 2016;36:37–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Parnell JA, Raman M, Rioux KP, Reimer RA. The potential role of prebiotic fibre for treatment and management of non‐alcoholic fatty liver disease and associated obesity and insulin resistance. Liver Int. 2012;32:701–711. [DOI] [PubMed] [Google Scholar]
- 130. Daubioul CA, Horsmans Y, Lambert P, Danse E, Delzenne NM. Effects of oligofructose on glucose and lipid metabolism in patients with nonalcoholic steatohepatitis: results of a pilot study. Eur J Clin Nutr. 2005;59:723–726. [DOI] [PubMed] [Google Scholar]
- 131. Tarantino G, Finelli C. Systematic review on intervention with prebiotics/probiotics in patients with obesity‐related nonalcoholic fatty liver disease. Future Microbiol. 2015;10:889–902. [DOI] [PubMed] [Google Scholar]
- 132. Miccheli A, Capuani G, Marini F, et al. Urinary (1)H‐NMR‐based metabolic profiling of children with NAFLD undergoing VSL#3 treatment. Int J Obes (Lond). 2015;39:1118–1125. [DOI] [PubMed] [Google Scholar]
- 133. Alisi A, Bedogni G, Baviera G, et al. Randomised clinical trial: the beneficial effects of VSL#3 in obese children with non‐alcoholic steatohepatitis. Aliment Pharmacol Ther. 2014;39:1276–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Nabavi S, Rafraf M, Somi MH, Homayouni‐Rad A, Asghari‐Jafarabadi M. Effects of probiotic yogurt consumption on metabolic factors in individuals with nonalcoholic fatty liver disease. J Dairy Sci. 2014;97:7386–7393. [DOI] [PubMed] [Google Scholar]
- 135. Shavakhi A, Minakari M, Firouzian H, Assali R, Hekmatdoost A, Ferns G. Effect of a probiotic and metformin on liver aminotransferases in non‐alcoholic steatohepatitis: a double blind randomized clinical trial. Int J Prev Med. 2013;4:531–537. [PMC free article] [PubMed] [Google Scholar]
- 136. Aller R, De Luis DA, Izaola O, et al. Effect of a probiotic on liver aminotransferases in nonalcoholic fatty liver disease patients: a double blind randomized clinical trial. Eur Rev Med Pharmacol Sci. 2011;15:1090–1095. [PubMed] [Google Scholar]
- 137. Vajro P, Mandato C, Licenziati MR, et al. Effects of Lactobacillus rhamnosus strain GG in pediatric obesity‐related liver disease. J Pediatr Gastroenterol Nutr. 2011;52:740–743. [DOI] [PubMed] [Google Scholar]
- 138. Wong V, Won G, Chim A, et al. Treatment of nonalcoholic steatohepatitis with probiotics. A proof‐of‐concept study. Ann Hepatol. 2013;12:256–262. [PubMed] [Google Scholar]
- 139. Loguercio C, Federico A, Tuccillo C, et al. Beneficial effects of a probiotic VSL#3 on parameters of liver dysfunction in chronic liver diseases. J Clin Gastroenterol. 2005;39:540–543. [DOI] [PubMed] [Google Scholar]
- 140. Ma YY, Lin L, Yu CH, Shen Z, Chen LH, Li YM. Effects of probiotics on nonalcoholic fatty liver disease: a meta‐analysis. World J Gastroenterol. 2013;19:6911–6918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Malaguarnera M, Vacante M, Antic T, et al. Bifidobacterium longum with fructo‐oligosaccharides in patients with non alcoholic steatohepatitis. Dig Dis Sci. 2012;57:545–553. [DOI] [PubMed] [Google Scholar]
- 142. Asgharian A, Askari G, Esmailzade A, Feizi A, Mohammadi V. The effect of symbiotic supplementation on liver enzymes, C‐reactive protein and ultrasound findings in patients with non‐alcoholic fatty liver disease: a clinical trial. Int J Prev Med. 2016;7:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Mofidi F, Poustchi H, Yari Z, et al. Synbiotic supplementation in lean patients with non‐alcoholic fatty liver disease: a pilot, randomised, double‐blind, placebo‐controlled, clinical trial. Br J Nutr. 2017;117:662–668. [DOI] [PubMed] [Google Scholar]
- 144. Swann JR, Want EJ, Geier FM, et al. Systemic gut microbial modulation of bile acid metabolism in host tissue compartments. Proc Natl Acad Sci USA. 2011;108:4523–4530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Cobbold J, Atkinson S, Marchesi JR, et al. Rifaximin in non‐alcoholic steatohepatitis: an open‐label pilot study. [published online ahead of print April 20, 2017]. Hepatol Res. 2017;48:69–77. [DOI] [PubMed] [Google Scholar]
- 146. Gangarapu V, Ince AT, Baysal B, et al. Efficacy of rifaximin on circulating endotoxins and cytokines in patients with nonalcoholic fatty liver disease. Eur J Gastroenterol Hepatol. 2015;27:840–845. [DOI] [PubMed] [Google Scholar]
- 147. Kakiyama G, Pandak WM, Gillevet PM, et al. Modulation of the fecal bile acid profile by gut microbiota in cirrhosis. J Hepatol. 2013;58:949–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Xifaxan® (rifaximin) tablets, for oral use [package insert]. Bridgewater, NJ: Salix Pharmaceuticals; 2018. Accessed August 16, 2018.
- 149. Shayto RH, Abou Mrad R, Sharara AI. Use of rifaximin in gastrointestinal and liver diseases. World J Gastroenterol. 2016;22:6638–6651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Bajaj JS, Heuman DM, Sanyal AJ, et al. Modulation of the metabiome by rifaximin in patients with cirrhosis and minimal hepatic encephalopathy. PLoS ONE. 2013;8:e60042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Brown EL, Xue Q, Jiang ZD, Xu Y, DuPont HL. Pretreatment of epithelial cells with rifaximin alters bacterial attachment and internalization profiles. Antimicrob Agents Chemother. 2010;54:388–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Jiang ZD, Ke S, DuPont HL. Rifaximin‐induced alteration of virulence of diarrhoea‐producing Escherichia coli and Shigella sonnei . Int J Antimicrob Agents. 2010;35:278–281. [DOI] [PubMed] [Google Scholar]
- 153. Schrodt C, McHugh EE, Gawinowicz MA, DuPont HL, Brown EL. Rifaximin‐mediated changes to the epithelial cell proteome: 2‐D gel analysis. PLoS ONE. 2013;8:e68550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Burke KE, Lamont JT. Fecal transplantation for recurrent Clostridium difficile infection in older adults: a review. J Am Geriatr Soc. 2013;61:1394–1398. [DOI] [PubMed] [Google Scholar]
- 155. Rohlke F, Stollman N. Fecal microbiota transplantation in relapsing Clostridium difficile infection. Therap Adv Gastroenterol. 2012;5:403–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Nicolas S, Blasco‐Baque V, Fournel A, et al. Transfer of dysbiotic gut microbiota has beneficial effects on host liver metabolism. Mol Syst Biol. 2017;13:921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Li M, Liang P, Li Z, et al. Fecal microbiota transplantation and bacterial consortium transplantation have comparable effects on the re‐establishment of mucosal barrier function in mice with intestinal dysbiosis. Front Microbiol. 2015;6:692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Alang N, Kelly CR. Weight gain after fecal microbiota transplantation. Open Forum Infect Dis. 2015;2:ofv004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Ridaura VK, Faith JJ, Rey FE, et al. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science. 2013;341:1241214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Moayyedi P, Surette MG, Kim PT, et al. Fecal microbiota transplantation induces remission in patients with active ulcerative colitis in a randomized controlled trial. Gastroenterology. 2015;149:102–109 e6. [DOI] [PubMed] [Google Scholar]
- 161. Paramsothy S, Kamm MA, Kaakoush NO, et al. Multidonor intensive faecal microbiota transplantation for active ulcerative colitis: a randomised placebo‐controlled trial. Lancet. 2017;389:1218–1228. [DOI] [PubMed] [Google Scholar]
- 162. Youngster I, Russell GH, Pindar C, Ziv‐Baran T, Sauk J, Hohmann EL. Oral, capsulized, frozen fecal microbiota transplantation for relapsing Clostridium difficile infection. JAMA. 2014;312:1772–1778. [DOI] [PubMed] [Google Scholar]
- 163. Halkjær SI, Christensen AH, Lo B, et al. Faecal microbiota transplantation alters gut microbiota in patients with irritable bowel syndrome: results from a randomised, double‐blind placebo‐controlled study. Gut. 2018;67:2107–2115. [DOI] [PubMed] [Google Scholar]
- 164. Ding C, Fan W, Gu L, et al. Outcomes and prognostic factors of fecal microbiota transplantation in patients with slow transit constipation: results from a prospective study with long‐term follow‐up. Gastroenterol Rep (Oxf). 2018;6:101–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Xu MQ, Cao HL, Wang WQ, et al. Fecal microbiota transplantation broadening its application beyond intestinal disorders. World J Gastroenterol. 2015;21:102–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Kootte RS, Levin E, Salojarvi J, et al. Improvement of insulin sensitivity after lean donor feces in metabolic syndrome is driven by baseline intestinal microbiota composition. Cell Metab. 2017;26:611–619 e6. [DOI] [PubMed] [Google Scholar]
- 167. Vrieze A, Van Nood E, Holleman F, et al. Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology. 2012;143:913–916.e7. [DOI] [PubMed] [Google Scholar]
- 168. Vaughn BP, Vatanen T, Allegretti JR, et al. Increased intestinal microbial diversity following fecal microbiota transplant for active Crohn's disease. Inflamm Bowel Dis. 2016;22:2182–2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Vermeire S, Joossens M, Verbeke K, et al. Donor species richness determines faecal microbiota transplantation success in inflammatory bowel disease. J Crohns Colitis. 2016;10:387–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Cani PD, Bibiloni R, Knauf C, et al. Changes in gut microbiota control metabolic endotoxemia‐induced inflammation in high‐fat diet‐induced obesity and diabetes in mice. Diabetes. 2008;57:1470–1481. [DOI] [PubMed] [Google Scholar]
- 171. Everard A, Cani PD. Diabetes, obesity and gut microbiota. Best Pract Res Clin Gastroenterol. 2013;27:73–83. [DOI] [PubMed] [Google Scholar]
- 172. Moreno‐Navarrete JM, Sabater M, Ortega F, Ricart W, Fernández‐Real JM. Circulating zonulin, a marker of intestinal permeability, is increased in association with obesity‐associated insulin resistance. PLoS ONE. 2012;7:e37160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Frazier TH, DiBaise JK, McClain CJ. Gut microbiota, intestinal permeability, obesity‐induced inflammation, and liver injury. JPEN J Parenter Enteral Nutr. 2011;35:14S–20S. [DOI] [PubMed] [Google Scholar]
- 174. Cirera I, Martin Bauer T, Navasa M, et al. Bacterial translocation of enteric organisms in patients with cirrhosis. J Hepatol. 2001;34:32–37. [DOI] [PubMed] [Google Scholar]
- 175. Llovet JM, Bartolí R, March F, et al. Translocated intestinal bacteria cause spontaneous bacterial peritonitis in cirrhotic rats: molecular epidemiologic evidence. J Hepatol. 1998;28:307–313. [DOI] [PubMed] [Google Scholar]